

Abstract
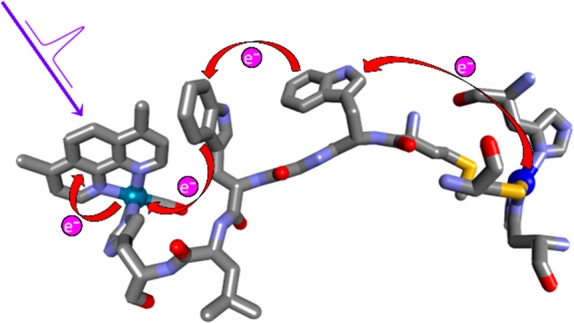
We have constructed and structurally characterized a Pseudomonas aeruginosa azurin mutant Re126WWCuI, where two adjacent tryptophan residues (W124 and W122, indole separation 3.6–4.1 Å) are inserted between the CuI center and a Re photosensitizer coordinated to the imidazole of H126 (ReI(H126)(CO)3(4,7-dimethyl-1,10-phenanthroline)+). CuI oxidation by the photoexcited Re label (*Re) 22.9 Å away proceeds with a ∼70 ns time constant, similar to that of a single-tryptophan mutant (∼40 ns) with a 19.4 Å Re–Cu distance. Time-resolved spectroscopy (luminescence, visible and IR absorption) revealed two rapid reversible electron transfer steps, W124 → *Re (400–475 ps, K1 ≅ 3.5–4) and W122 → W124•+ (7–9 ns, K2 ≅ 0.55–0.75), followed by a rate-determining (70–90 ns) CuI oxidation by W122•+ ca. 11 Å away. The photocycle is completed by 120 μs recombination. No photochemical CuI oxidation was observed in Re126FWCuI, whereas in Re126WFCuI, the photocycle is restricted to the ReH126W124 unit and CuI remains isolated. QM/MM/MD simulations of Re126WWCuI indicate that indole solvation changes through the hopping process and W124 → *Re electron transfer is accompanied by water fluctuations that tighten W124 solvation. Our finding that multistep tunneling (hopping) confers a ∼9000-fold advantage over single-step tunneling in the double-tryptophan protein supports the proposal that hole-hopping through tryptophan/tyrosine chains protects enzymes from oxidative damage.
Short abstract
Insertion of two tryptophan residues between a rhenium photooxidant and the copper(I) center in Pseudomonas aeruginosa azurin accelerates electron flow by almost 4 orders of magnitude.
Introduction
Single-step tunneling (ET) in proteins can move electrons between donor and acceptor sites separated by about 25 Å on a millisecond time scale.1−4 Inserting redox-active groups between the terminal donor and acceptor accelerates electron transport (EThop) by splitting the reaction pathway into shorter tunneling steps,1,2,4−9 achieving much higher charge migration rates and extending the charge separation range. Many natural redox systems employ multistep tunneling (hopping), transferring an electron sequentially along a series of redox proteins or cofactors. A case in point is the Ralstonia eutropha O2-tolerant [NiFe]-hydrogenase, where electrons travel from the active site to the protein surface through a series of Fe–S clusters involving tunneling steps of 10.7, 9.7, and 8.7 Å;10 even more striking is the respiratory complex I, where an electron is transported over 90 Å through a redox chain consisting of a flavin mononucleotide and a series of Fe–S clusters.11,12 Electron hopping also takes place in photosynthesis—both within reaction centers and when moving the photoseparated holes and electrons along the chloroplast membrane.6
Tryptophan and/or tyrosine residues are of special importance as hole hopping intermediates. In the prototypal radical enzyme ribonucleotide reductase,13−15 substrate reaction is triggered by 35-Å hole transfer across a chain of Tyr residues to the nucleotide binding site;5,16−20 and in photolyases and cryptochromes, a photogenerated hole moves over ∼15 Å in ∼30 ps from a photoexcited flavin through a chain of three precisely positioned tryptophans.21,22 We recently proposed that hole transfer through Trp/Tyr chains protects oxidases and oxygenases by moving potentially destructive oxidizing equivalents (holes) to protein surfaces where they can be disarmed by cellular reductants.1,23−27 While our hypothesis is well supported by bioinformatics analysis of the structures of redox enzymes, it calls for deeper mechanistic investigations of the hopping mechanism.
The blue copper protein Pseudomonas aeruginosa azurin is an excellent platform to investigate ET mechanisms, owing to the presence of a reversible CuII/I redox couple in a robust structure that allows for multiple mutations and covalent attachment of a RuII or ReI photooxidant to a surface histidine (H) at a defined position.7,8,28−33 Although azurin does not contain chains of aromatic amino acids,27 tryptophan7,34,35 and nitrotyrosine8 residues can be introduced into the redox pathways by site mutations with retention of the protein structure. In particular, replacing a lysine (K122) by tryptophan (W) results in dramatic (>100×) acceleration of CuI oxidation by a photoexcited Re metallolabel in Re124W122CuI azurin (Re = ReI(CO)3(dmp)(H124)+; CT excited state (*Re) = ReII(CO)3(dmp•–)(H124)+; dmp = 4,7-dimethyl-1,10-phenanthroline).7,9 The reaction involves W122 oxidation as the first ET step in the mechanism (Scheme 1A). Electron (hole) hopping also can occur across a hydrophobic protein–protein interface, as was observed in {Re126T124W122CuI}2, where Re excitation in one subunit leads to oxidation of the tryptophan and CuI in the neighboring subunit.34 Expanding the azurin electron hopping system to include mutants with two closely spaced aromatic amino acid residues provides a well-characterized minimal model to investigate multiple hopping (Scheme 1B). We report here on a series of structurally characterized azurin mutants labeled with a Re photooxidant at H126 and containing either tryptophan or phenylalanine (F) at positions 124 and 122. (Mutants are abbreviated Re126WWCu, Re126WFCu, and Re126FWCu, where the first and second letters specify the 124 and 122 residues, respectively. All other naturally occurring Trp and Tyr residues were replaced by Phe.) EThop reactions were studied in CuI azurins, whereas the corresponding CuII and ZnII forms were used to evaluate redox reactivity of the Re···W···W moiety in isolation. Systematic spectroscopic and kinetics investigations of photoinduced EThop in these mutants have shed light on factors that control multiple hole hopping along tryptophan chains.
Scheme 1. (A) Photoinduced ET Cycle of Re124W122CuI (ref (7)) and (B) Analogous Photoinduced ET Cycle of Re126WWCuI.
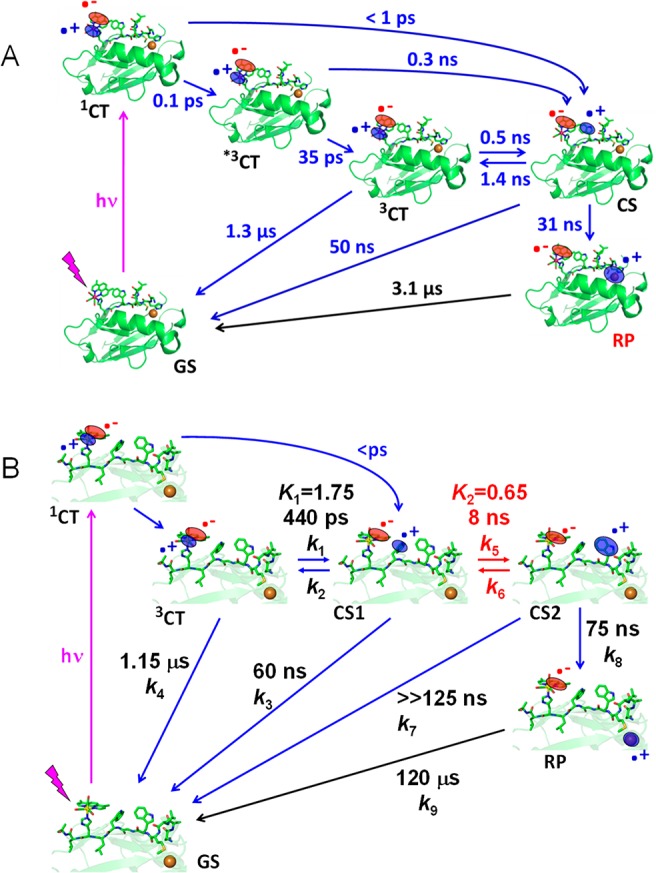
(A) *3CT and 3CT denote hot and relaxed excited states of the Re label, respectively. The photocycle starts with optical excitation to the 1CT state *ReII(CO)3(dmp•–)H124W122AzCuI followed by several relaxation steps, establishing an equilibrium between 3CT and the charge-separated (CS) state ReI(CO)3(dmp•–)H124(W122•+)CuI. The oxidized tryptophan intermediate W122•+ then undergoes ∼30 ns reduction by CuI over a ∼11 Å distance, forming the redox product (RP) ReI(CO)3(dmp•–)H124W122CuII. The cycle is completed by ∼3 μs dmp•– → CuII back electron transfer across 19.4 Å (refs (7 and 9)). (B) The time constants were determined in this work. The Re–Cu charge separation takes place over 23 Å via hopping through two Trp residues. The hot *3CT state and its relaxation were omitted for clarity. 3CT is a mixed Re → dmp MLCT/dmp-intraligand state.
Results and Discussion
Structures
X-ray crystal structures of Re126WWCuII (PDB ID: 6MJS), Re126WFCuII (6MJT), and Re126FWCuII (6MJR) were determined (Table S1) to resolutions of 1.85, 1.9, and 2.0 Å, respectively, and the regions of the redox cofactors are shown in Figure 1. The shortest ET-relevant distances are reported in Table 1. The Re–W124–W122 hopping sequence is characterized by multiple short (3.5–4.0 Å) contacts between mutually T-oriented aromatic groups, and the dmp methyl groups are in close proximity to the W124 indole. Structures of all three mutants are superimposable, and replacement of either of the two tryptophans in Re126WWCuII with phenylalanine switches off one of the hopping steps without altering the overall geometry or length of the Re–Cu EThop pathway. In particular, the Re chromophore becomes redox-isolated in Re126FWCuI, where the W122 residue is too far from the Re site for ET to compete with 3CT decay. The W122–Cu pathway (∼11 Å) is the same in Re126WWCuII as in single-tryptophan mutants Re126FWCuII, Re126T124W122CuII,34 and Re124W122CuII(7) (Figure S1). In Re126WFCuI, the photocycle is largely limited to ET in the Re126W124 unit, owing to the long W124–Cu distance.
Figure 1.

Structures of ReH126-azurin mutants showing intramolecular distances between the redox cofactors. WW: Re126WWCuII (PDB ID: 6MJS). FW: Re126FWCuII (6MJR). WF: Re126WFCuII – chain B (6MJT; in chain A, the W122-indole is oriented backward and the Re(CO)3(dmp) unit is tilted leftwards). Lower right: Superposition of ET-relevant regions and protein folds of Re126WWCuII (green), Re126FWCuII (pink), and Re126WFCuII – chain B (light blue) demonstrates their structural similarity. Packing of Re126WWCuII and Re126FWCuII in the respective asymmetric unit is shown in Figure S2.
Table 1. Shortest Atom–Atom Intramolecular Distances between Redox-Active Sitesa.
| distance | Re126WWCuII | Re126WFCuIIb | Re126FWCuII | Re124W122CuIIh |
|---|---|---|---|---|
| Re–W124 | 6.9 | 7.6 | ||
| dmp–W124 | 3.5c | 3.7d | ||
| W124–W122 | 3.9e | |||
| Re–W122 | 11.4 | 11.1 | 6.3 | |
| dmp–W122 | 7.8 | 7.1 | 3.4 | |
| Cu–W122 | 10.7 | 15.7f | 10.7 | 10.8 |
| Cu–dmp | 20.6 | 19.9 | 20.2 | 16.0g |
| Cu–Re | 22.9 | 22.7 | 23.3 | 19.4 |
| angle (deg) | ||||
| dmp–W124 | 67.7 | 20.8i | ||
| W124–W122 | 78.7 |
Only aromatic C and N atoms, as well as Re and Cu, are considered. Values averaged over the molecules comprising the unit cell.
Two molecules with different Re/W122-indole orientations are present. The listed distances are pertinent to the molecule with closer contacts.
An additional close contact (3.9 Å) exists between the W124 indole ring and C(CH3-dmp).
Closest distance between the indole ring and C(CH3-dmp) = 3.5 Å.
The distances in the four molecules comprising the asymmetric unit are in the range 3.6–4.1 Å.
Cu–W124 distance.
Closest distance between Cu and C(CH3-dmp) = 15.3 Å.
PDB ID: 2I7O; see Figure S1.
dmp-W122.
Re126WWCuII and Re126FWCuII pack in the asymmetric unit so that redox cofactors on different protein monomers interact with each other (Figure S2). Assuming that similar dimerization occurs in solution,34,36 it is likely that intermolecular EThop will be observed at higher protein concentrations.
Photoinduced Electron Transport
The EThop kinetics of Re126WWCuI and its variants were studied following the protocol established for Re124W122CuI (Scheme 1).7 Pulsed laser excitation of the Re label at 400 or 355 nm triggers a sequence of ET steps whose kinetics were followed by measuring the decay of *Re luminescence at 560 nm and absorption-time profiles at 500 (*Re and ReH126(CO)3(dmp•–)) and 632.8 nm (CuII formation and decay). Time-resolved IR (TRIR) spectroscopy in the range of CO stretching vibrations was used to distinguish ground, excited, and reduced forms of the Re label (negative (bleach) bands and positive features shifted to higher and lower frequencies upon excitation, respectively).37 In all UV–visible transient spectroscopic experiments, the protein concentration was kept below 40 μM to minimize contributions from intermolecular EThop.34 Additional insights were provided by measurements with other mutants: all reactivity is confined to the Re126WW moiety in ReH126WWZnII and ReH126WWCuII; ET between *Re and the proximal Trp in isolation was probed in ReH126WFCuI, and the Re label is effectively removed from the redox pathway by phenylalanine in ReH126FWCuI. Results are summarized in Figure 2 and Table 2, and Scheme 1B outlines the mechanism together with elementary rate constants extracted from kinetics simulations (vide infra).
Figure 2.

Transient absorption and luminescence time profiles measured on dilute (<40 μM) Re-azurin solutions. (A) Transient absorption of Re126 azurins at 632.8 nm: Re126WWCuI, 32 μM; Re126WWZn, 17 μM; Re126FWCuI, 30 μM; Re126FWCuI, 24 μM. (B) Comparison of the CuII transient absorption signals (632.8 nm) for Re126WWCuI (22 μM, red) and Re124W122CuI (27 μM, black, scaled by a factor of 22/27) measured under virtually identical excitation conditions (1–1.5 mJ/pulse). (C) Luminescence decay of Re126 azurins at 560 nm. (D) Multiexponential luminescence decay of Re124W122CuI in the pico-nanosecond range.
Table 2. Kinetics Fitting Parameters from Time-Resolved Luminescence and Transient Absorbance Measurements on Re126 Azurins with 355 nm Excitation.
| τ1 /ps | τ2 /ns | τ3 /ns | τ4 /μs | τ5 /μs | τ6 /ms | |
|---|---|---|---|---|---|---|
| Re126WWCuI | ||||||
| luminescence τ | 270 ± 20 | 4 ± 1 | 81 ± 6 | |||
| (% amplitude) | (61 ± 3) | (15 ± 2) | (23 ± 1) | |||
| TA τ | 68 ± 5 | 1.2 ± 0.1 | 123 ± 10 | 4.6 ± 0.5 | ||
| amplitude (632.8 nm) | –0.011 | 0.005 | 0.020 | 0.002 | ||
| amplitude (500 nm) | 0.004 | 0.003 | 0.008 | 0.0008 | ||
| Re126WWCuII | ||||||
| luminescence | 290 ± 10 | 4 ± 2 | 79 ± 7 | |||
| (% amplitude) | (71 ± 1) | (9 ± 1) | (20 ± 2) | |||
| Re126WWZnII | ||||||
| luminescence | 430 ± 40 | 10 ± 3 | 100 ± 7 | |||
| (% amplitude) | (45 ± 2) | (18 ± 2) | (36 ± 2) | |||
| TA (500 nm) τ | 125 ± 30 | 4 ± 3a | ||||
| Re126WFCuI | ||||||
| luminescence | 200 ± 10 | 3 ± 1 | 71 ± 8 | |||
| (% amplitude) | (73 ± 2) | (14 ± 1) | (13 ± 1) | |||
| TA (500 nm) τ | 0.9 ± 0.1 | |||||
| Re126FWCuI | ||||||
| luminescence | 340 ± 150 | 3 ± 2 | 120 ± 100 | 1.15 ± 0.2 | ||
| (% amplitude) | (36 ± 6) | (14 ± 6) | (5 ± 4) | (45 ± 5) | ||
| TA (500 nm) τ | 1.10 ± 0.15 | 20 ± 10a |
Minor component.
*Re luminescence is strongly quenched by W124 in Re126WWCuI, Re126WWZnII, and Re126WFCuI, decaying with fast multiexponential kinetics (Table 2). On the other hand, Re126FWCuI (Figure 2C) exhibits a long luminescence decay time (1.15 μs) consistent with an unquenched 3CT excited state. (Similar values were found for redox-inactive Re126T124X122CuI (X = K or F, 730 ns) and Re124F122CuI (1.3 μs).7,34) Pico- and nanosecond TRIR spectra (Figure 3) demonstrate that the *Re 3CT excited state decays to produce a charge-separated (CS) state with a reduced Re complex, ReI(H126)(CO)3(dmp•–).37,38 Luminescence decay kinetics obtained for all mutants containing W124 are similar (Table 2), indicating that *Re reduction by W124 is the common reaction step, regardless of the metal (CuI, CuII, ZnII) or the 122 amino acid (W, F).
Figure 3.

Difference TRIR spectra of Re126WWCuI measured at selected time delays after 400 nm, 50 fs excitation. Measured in ∼1.8 mM/D2O solution, 20 mM KPi (pD ≅ 7.1). Blue and red labels denote features due to the 3CT state (*Re) and reduced ReI(H126)(CO)3(dmp•–) in the two CS states (and RP at later time delays). Negative bands correspond to depleted ground-state population. The spectral features evolve in the directions of the arrows. Time evolution of the highest CT band is largely determined by excited-state relaxation (ref (38); the simultaneous decay and rise of 3CT and CS features on late-picosecond and early nanosecond time scales confirm (ultra)fast reduction of the excited Re label. Because of the high concentration used (∼1.8 mM), the kinetics correspond to a combination of intra- and intermolecular processes.
Nanosecond transient absorption (TA) measurements revealed large absorbance increases indicative of CuII formation (632.8 nm) only in Re126WWCuI and Re124W122CuI (Figure 2); CuII formation was not observed in low-concentration solutions of Re126WFCuI, Re126FWCuI, or Re126WWZnII, whose much weaker transient absorption at 632.8 nm (Figure 2A) originates from the 3CT and CS states. Hence, fast CuI photooxidation requires the presence of both W124 and W122 in the EThop pathway. The ∼68 ns rise of Re126WWCuI 632.8 nm TA parallels that observed7 for Re124W122CuI (∼40 ns), despite different Re–Cu distances (22.9 and 19.4 Å, respectively), indicating an analogous rate-determining step (the ∼11 Å W122•+→CuI “hole hop”). TA kinetics (632.8 nm) measured under virtually identical excitation conditions show that the CuII yield for Re126WWCuI is 1.5–2.4 times lower than for Re124W122CuI, where EThop involves a single W122 intermediate (Figure 2B and SI - Section S3).
The ReI(H126)(CO)3(dmp•–) → CuII recombination reaction closes the photocycle; simultaneous fitting of the 632.8 and 500 nm kinetics gives a ∼120 μs time constant for this process. The back-reaction time constant accords with the estimate (∼150 μs) for single-step dmp•– → CuII tunneling (SI-Section S2). The almost 2000-fold difference in the charge-separation and recombination time scales reflects the different mechanisms: multistep and single-step tunneling, respectively. The charge-separation/recombination advantage increases with EThop range: Re124W122CuI shows an ∼80-fold difference over 19.4 Å.
The overall performance of the Re126WWCuI photocycle can be assessed by comparing CuII formation kinetics (monitored at 632.8 nm) with the time constant of single-step CuI → *Re ET (∼630 μs) estimated from the value reported30 for Re83WT-azurinCuI (770 ns, r = 16.8 Å) by correcting for the longer Cu–Re distance (22.9 Å) in Re126WWCuI (see SI-Section S2). Given an unquenched 3CT lifetime of about 1 μs,7 photoinduced CuI oxidation should not be observable if single-step tunneling were the only operative mechanism. Instead, 355 nm, ∼8 ns laser-pulse excitation of ≤40 μM Re126WWCuI solutions led to CuII (RP, Scheme 1) formation (∼68 ns time constant), followed by ∼120 μs ground-state recovery. Remarkably, hole hopping through the two intervening tryptophan residues accelerates CuI oxidation by a factor of 9000 compared to single-step tunneling.
The solution to the rate law for the kinetics model outlined in Scheme 1B (beginning from3CT) is a 4-exponential function. One of the empirical rate constants in the solution is equal to the elementary rate constant k9. The remaining empirical rate constants are functions of k1–8, given by the roots of a third-order polynomial. To obtain estimates for these elementary rate constants, we solved the rate law numerically (see SI, Section S3). Values used for k3 and k4 were fixed, based on measurements in Re124W122CuI and Re126FWCuI, respectively.7 We simulated the kinetics for 4.8 × 107 combinations of the remaining parameters (see SI, Section S3) and retained those combinations in which the empirical rate constants were in satisfactory agreement with the observed luminescence rate constants and relative amplitudes, the TA kinetics, and the relative CuII yield (Table 3).
Table 3. Results from Numerical Solutions to the Rate Law Implied by Scheme 1B.
| Re126WWCuI | Inputs: k3–1 = 60 ns; k4–1 = 1.15 μs; k9–1 = 120 μs | |||||
|---|---|---|---|---|---|---|
| elementary rate constants | k1–1 /ps | K1 | k5–1 /ns | K2 | k7–1 /ns | k8–1 /ns |
| 400–475 | 1.5–2.0 | 7–9 | 0.55–0.75 | 125–750 | 60–90 | |
| yield and empirical time constants | τ1 /ps | τ2 /ns | τ3 /ns | τ4 /μs | Φ124/Φ126 | |
| 260–280 | 3.5–4 | 70–90 | 120 | 2.0–2.2 | ||
| Re126WWCuII | Inputs: k3–1 = 60 ns;k4–1 = 1.15 μs; k8 = k9 = 0 | ||||
|---|---|---|---|---|---|
| elementary rate constants | k1–1 /ps | K1 | k5–1 /ns | K2 | k7–1 /ns |
| 375–425 | 2.25–3.25 | 9–21 | 0.25–0.75 | 100–350 | |
| empirical time constants | τ1 /ps | τ2 /ns | τ3 /ns | ||
| 280–300 | 3.5–4.5 | 70–90 | |||
| Re126WFCuI | Inputs: k4–1 = 1.15 μs; k5 = k6 = k8 = k9 = 0 | ||
|---|---|---|---|
| elementary rate constants | k1–1 /ps | K1 | k3–1 /ns |
| 234 | 5.7 | 61 ns | |
| empirical time constants | τ1 /ps | τ2 /ns | |
| 200 | 71 | ||
The kinetics simulations of Re126WWCuI revealed that W124 and W122 are distinct hopping intermediates (if there were a single highly delocalized {W124,W122}•+ intermediate, the relative CuII yield would be much higher). To account for the low CuII yield, the CS1 ⇌ CS2 equilibrium must be shifted to the left (K2 = 0.55–0.75), indicating that hole-localization on W124 proximal to Re is thermodynamically preferred. The simulations suggest that the W122 → W124•+ ET time constant (k5–1) can be constrained to 7–9 ns, but the ReI(H126)(CO)3(dmp•–) → W122•+ (CS2 → GS) ET time constant is less well-defined (k5–1 = 125–750 ns).
The experimental kinetics (Table 2) show several minor components (e.g., the 1.2 μs and 4.6 ms TA decays in Re126WWCuI) that are not recovered by simulations. These features can be attributed to intermolecular EThop in azurin dimers, as observed for Re126T124W122CuI,34 and supported by observations of concentration-dependent luminescence decay kinetics in Re126WWCuII and photoredox activity in Re126FWCuI at higher concentrations.
Further insight into the nature of the electronic states and intermediates of Re126WWCuI was obtained by QM/MM molecular dynamics (MD) simulations of the solvated protein,39,40 where the quantum part (QM) consisted of -Re(H126)(CO)3(dmp)L125W124G123W122-, and the rest of the system was treated by MM (Figure S12). The QM calculations employed density functional theory (DFT) techniques with the PBE0 functional41,42 and D3 dispersion correction;43 see the Supporting Information for computational details. For each case, several QM/MM/MD trajectories, which differed in initial conditions, were run for up to 10 ps after equilibration.
In agreement with the proposed mechanism, TDDFT MD simulations found 3CT to be the lowest excited state of solvated Re126WWCuI. As usual for Re carbonyl-diimines, it arises from Re → dmp metal-to-ligand charge transfer (MLCT) and dmp-intraligand excitations44−48 whose relative contributions vary in time (Figure S13). The 3CT state is closely followed in energy by several CS states. Whereas distances and angles among redox cofactors (Re, dmp, indoles) do not exhibit any major or systematic differences in the 3CT, CS1, and CS2 states (Figure S14), indole solvation was found to be very sensitive to the actual charge distribution and appears to be a dominant component of the ET reorganization. Each indole NH is strongly solvated by a single water molecule at about 2 Å with other water molecules lying farther away (Figures 4, S15). Oxidation of either one of the two tryptophans is accompanied by H-bonding and tighter solvation. In particular, W124 solvation tightens upon oxidation to W124•+ in the CS1 state where the NH---OH2 shortens by about 0.1 Å relative to 3CT, owing to a ca. + 0.15 e– increase in charge on the indole N atom. Restoring uncharged W124 in CS2 relaxes its solvation and shifts water molecules toward W122. Contrasting behavior was found for W122, whose solvation is similar in the ground, 3CT, and CS1 states but tightens in CS2, where the W122•+ NH group is strongly bound to a single water molecule (Figure 4). Comparing the two tryptophans reveals that the W122 indole is generally solvated less tightly than W124. Surprisingly, in CS2, the distances between W124 and W122•+ indoles and their respective closest water molecules are comparable despite different charges (Figure 4). The generally weaker W122 solvation could be related to its steric shielding by the -S118A119L120- backbone 3.4–4.4 Å away (Figure 1). Such an asymmetric environment makes W124 a slightly stronger reductant than W122, favors single-indole hole localization in CS1 and CS2 over a delocalized {W124; W122}•+ intermediate, and shifts the CS1 ⇌ CS2 equilibrium to the left (K2 < 1), limiting the CuII formation yield.
Figure 4.
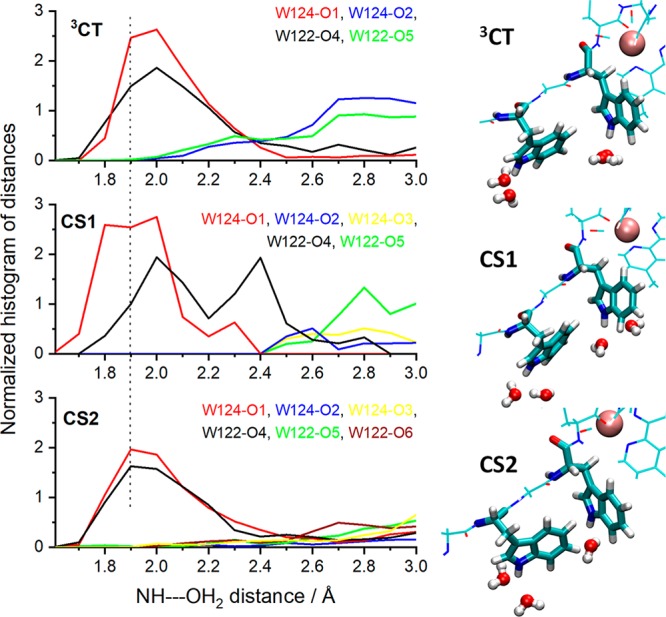
Left: Distribution function of individual water molecules around W124 and W122 indole NH groups. The dotted line at 1.9 Å helps to visualize the differences. Right: Snapshots showing water molecules within 2.5 Å of the indole-NH hydrogen atoms (the brown sphere represents the Re atom).
Concluding Remarks
Multistep electron tunneling dramatically increases the range over which electrons can be transported through proteins. Our prior study revealed that hole hopping through a single intervening tryptophan residue could accelerate electron transport by a factor of ∼102.7 Our present study demonstrates that electron hopping through two adjacent tryptophan residues in Re126WWCuI accelerates CuI oxidation by a factor of ∼104 relative to single-step CuI → *Re tunneling. The timetable for electron tunneling/transport (Figure 5) illustrates the advantage of multistep tunneling in the Re-azurin construct: hopping through Trp•+ intermediates enables sub-microsecond electron transport across more than 20 Å.
Figure 5.
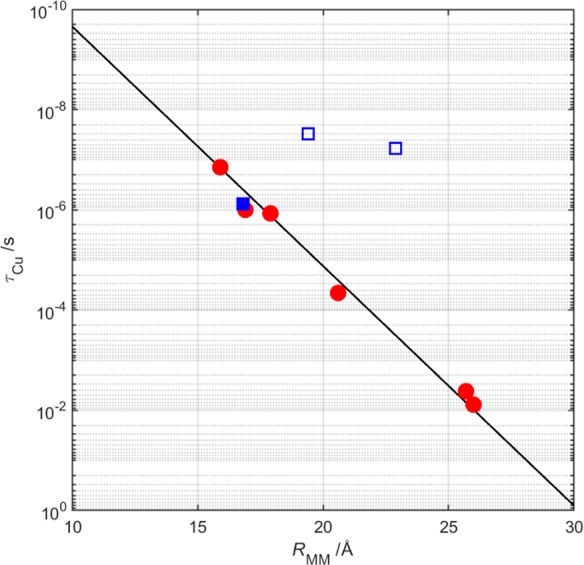
Plot of CuI oxidation time constant (τCu) as a function of the metal–metal separation (RMM) in constructs of Ru- (circles) and Re-derivatized (squares) azurins. Reactions of all Ru-azurins and Re83-azurin (solid square) involve single-step tunneling. The two open squares illustrate the reactions of Re124WCuI and Re126WWCuI.
The advantage of hopping over single-step tunneling is sensitive to the structure of the hopping system and the driving forces associated with the individual ET steps. Taking the Re124WCuI and Re126WWCuI constructs as models, we simulated the EThop kinetics for all possible values of redox-site distances (Figure 6). A key component in the hopping advantage is the single-step tunneling distance. The longer distance in the Re126WWCuI model gives an optimum double-hop advantage ∼50 times greater than the single-hop advantage in the Re124WCuI model. The optimum advantage of a double-hop over a single-hop (via Trp) increases as the single-step distance increases. In a comparison of single- and double hopping through Trp over a 20-Å separation between Re and Cu, optimized double-hopping provides just a 2.5-fold advantage over a single hop; at a 23-Å Re–Cu separation, optimized double-hopping is 10 times better than a single hop.
Figure 6.

Simulated single-Trp (A) and double-Trp (B) hopping advantages for constructs modeled on Re124WCuI (d(Re–Cu) = 20 Å) and Re126WWCuI (d(Re–Cu) = 23 Å). Optimum positioning of the intervening Trp in the Re124WCuI construct (r1 = 7 Å, r2 = 13 Å, colinear) leads to a predicted 103.8 hopping advantage. In the Re126WWCuI model, optimum positioning of the two Trp residues (r1 = 6 Å, r2 = 7 Å, r3 = 10 Å, colinear) produces a 105.5 hopping advantage over single-step tunneling.
Our observations suggest that protein constructs containing multiple closely spaced Trp (or Tyr) residues could support transport of high potential holes across distances of 30 Å or more. Such facile movement of holes through polypeptides necessitates careful placement of oxidizable residues in enzymes that operate at high potentials. We have found that chains of three or more Trp and Tyr residues separated by ≤5 Å are relatively common in the structures of redox enzymes, particularly those that participate in reactions with oxygen.23−25,27 These Trp/Tyr chains may play protective antioxidant roles by disarming highly oxidizing intermediates when reactions with intended substrates are disrupted.25,26 Kinetics measurements with Re124WCuI and Re126WWCuI demonstrate that once a hole is injected into the indole ring of a Trp residue, it can rapidly migrate to a nearby indole, even when environmental disparities produce an unfavorable free-energy gradient. Current efforts are aimed at elucidating whether Trp/Tyr chains play functional as well as protective roles in high-potential enzymatic redox catalysis.
Acknowledgments
We thank Martin Pižl (JH Institute) for his help analyzing the TRIR spectra. Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number R01DK019038. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional support was provided by the Arnold and Mabel Beckman Foundation, the Czech Science Foundation (GAČR) Grant 17-011375, and the STFC Rutherford Appleton Laboratory (UK). X-ray crystallography data were collected on SSRL Beamline 12-2 through the support of the Caltech Molecular Observatory, funded by the Gordon and Betty Moore Foundation, Beckman Institute, and the Sanofi-Aventis Bioengineering Research Program. Operations at SSRL are supported by U.S. DOE and NIH.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00882.
X-ray data refinement statistics and validation, figures of the Re124W122CuII structure and packing of the neighboring chains of Re126WWCuII and Re126FWCuII, estimates of single-step tunneling rates, details and summary of kinetics modeling, QM/MM dynamics simulations: structure of the modeled system, trajectories of structural parameters and of indole NH-water distances (PDF)
The authors declare no competing financial interest.
Notes
Safety Statement: No unexpected or unusually high safety hazards were encountered.
Supplementary Material
References
- Winkler J. R.; Gray H. B. Long-Range Electron Tunneling. J. Am. Chem. Soc. 2014, 136, 2930–2939. 10.1021/ja500215j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J. R.; Gray H. B. Electron Flow through Metalloproteins. Chem. Rev. 2014, 114, 3369–3380. 10.1021/cr4004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Long-Range Electron Transfer. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 3534–3539. 10.1073/pnas.0408029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Electron Tunneling through Proteins. Q. Rev. Biophys. 1999, 36, 341–372. 10.1017/S0033583503003913. [DOI] [PubMed] [Google Scholar]
- Warren J. J.; Ener M. E.; Jr Vlček A.; Winkler J. R.; Gray H. B. Electron Hopping through Proteins. Coord. Chem. Rev. 2012, 256, 2478–2487. 10.1016/j.ccr.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren J. J.; Winkler J. R.; Gray H. B. Hopping Maps for Photosynthetic Reaction Centers. Coord. Chem. Rev. 2013, 257, 165–170. 10.1016/j.ccr.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih C.; Museth A. K.; Abrahamsson M.; Blanco-Rodriguez A. M.; Di Bilio A. J.; Sudhamsu J.; Crane B. R.; Ronayne K. L.; Towrie M.; Vlček A. Jr.; Richards J. H.; Winkler J. R.; Gray H. B. Tryptophan-Accelerated Electron Flow through Proteins. Science 2008, 320, 1760–1762. 10.1126/science.1158241. [DOI] [PubMed] [Google Scholar]
- Warren J. J.; Herrera N.; Hill M. G.; Winkler J. R.; Gray H. B. Electron Flow through Nitrotyrosinate in Pseudomonas aeruginosa Azurin. J. Am. Chem. Soc. 2013, 135, 11151–11158. 10.1021/ja403734n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Rodríguez A. M.; Di Bilio A. J.; Shih C.; Museth A. K.; Clark I. P.; Towrie M.; Cannizzo A.; Sudhamsu J.; Crane B. R.; Sýkora J.; Winkler J. R.; Gray H. B.; Záliš S.; Vlček A. Jr. Phototriggering Electron Flow through ReI-Modified Pseudomonas aeruginosa Azurins. Chem. - Eur. J. 2011, 17, 5350–5361. 10.1002/chem.201002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch J.; Scheerer P.; Frielingsdorf S.; Kroschinsky S.; Friedrich B.; Lenz O.; Spahn C. M. T. The Crystal Structure of an Oxygen-Tolerant Hydrogenase Uncovers a Novel Iron-Sulphur Centre. Nature 2011, 479, 249–253. 10.1038/nature10505. [DOI] [PubMed] [Google Scholar]
- Hayashi T.; Stuchebrukhov A. A. Electron Tunneling in Respiratory Complex I. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 19157–19162. 10.1073/pnas.1009181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J.; Roessler M. M. Energy Conversion, Redox Catalysis and Generation of Reactive Oxygen Species by Respiratory Complex I. Biochim. Biophys. Acta, Bioenerg. 2016, 1857, 872–883. 10.1016/j.bbabio.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenberg A.; Reichard P. Electron Spin Resonance of the Iron-Containing Protein B2 from Ribonucleotide Reductase. J. Biol. Chem. 1972, 247 (11), 3485–3488. [PubMed] [Google Scholar]
- Sjöberg B. M.; Reichard P. Nature of the Free Radical in Ribonucleotide Reductase from Escherichia Coli. J. Biol. Chem. 1977, 252 (2), 536–541. [PubMed] [Google Scholar]
- Larsson A.; Sjoberg B. M. Identification of the Stable Free-Radical Tyrosine Residue in Ribonucleotide Reductase. EMBO J. 1986, 5 (8), 2037–2040. 10.1002/j.1460-2075.1986.tb04461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnihan E. C.; Nocera D. G.; Stubbe J. Reversible, Long-Range Radical Transfer in E. coli Class Ia Ribonucleotide Reductase. Acc. Chem. Res. 2013, 46 (11), 2524–2535. 10.1021/ar4000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshansky L.; Greene B. L.; Finkbeiner C.; Stubbe J.; Nocera D. G. Photochemical Generation of a Tryptophan Radical within the Subunit Interface of Ribonucleotide Reductase. Biochemistry 2016, 55, 3234–3240. 10.1021/acs.biochem.6b00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshansky L.; Stubbe J.; Nocera D. G. Charge-Transfer Dynamics at the Α/Β Subunit Interface of a Photochemical Ribonucleotide Reductase. J. Am. Chem. Soc. 2016, 138, 1196–1205. 10.1021/jacs.5b09259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger M. J. Electron Relay in Proteins. Science 2008, 320 (5884), 1730–1731. 10.1126/science.1160001. [DOI] [PubMed] [Google Scholar]
- Sjoberg B. M.Ribonucleotide Reductases - a Group of Enzymes with Different Metallosites and a Similar Reaction Mechanism. In Metal Sites in Proteins and Models: Iron Centres; Hill H. A. O.; Sadler P. J.; Thomson A. J., Eds.; Springer-Verlag Berlin: Berlin, 1997; Vol. 88, pp 139–173. [Google Scholar]
- Lukacs A.; Eker A. P. M.; Byrdin M.; Brettel K.; Vos M. H. Electron Hopping through the 15 Å Triple Tryptophan Molecular Wire in DNA Photolyase Occurs within 30 ps. J. Am. Chem. Soc. 2008, 130, 14394–14395. 10.1021/ja805261m. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Tan C.; Guo X.; Li J.; Wang L.; Sancar A.; Zhong D. Determining Complete Electron Flow in the Cofactor Photoreduction of Oxidized Photolyase. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 12966–12971. 10.1073/pnas.1311073110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Hole Hopping through Tyrosine/Tryptophan Chains Protects Proteins from Oxidative Damage. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10920–10925. 10.1073/pnas.1512704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J. R.; Gray H. B. Electron Flow through Biological Molecules: Does Hole Hopping Protect Proteins from Oxidative Damage?. Q. Rev. Biophys. 2015, 48, 411–420. 10.1017/S0033583515000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Living with Oxygen. Acc. Chem. Res. 2018, 51, 1850–1857. 10.1021/acs.accounts.8b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polizzi N. F.; Migliore A.; Therien M. J.; Beratan D. N. Defusing Redox Bombs?. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10821–10822. 10.1073/pnas.1513520112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. The Rise of Radicals in Bioinorganic Chemistry. Isr. J. Chem. 2016, 56, 640–648. 10.1002/ijch.201600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connick W. B.; Di Bilio A. J.; Hill M. G.; Winkler J. R.; Gray H. B. Tricarbonyl(1,10-phenanthroline)(imidazole)rhenium(I): A Powerful Photooxidant for Investigations of Electron Tunneling in Proteins. Inorg. Chim. Acta 1995, 240, 169–173. 10.1016/0020-1693(95)04532-5. [DOI] [Google Scholar]
- Winkler J. R.; Di Bilio A. J.; Farrow N. A.; Richards J. H.; Gray H. B. Electron Tunneling in Biological Molecules. Pure Appl. Chem. 1999, 71, 1753–1764. 10.1351/pac199971091753. [DOI] [Google Scholar]
- Crane B. R.; Di Bilio A. J.; Winkler J. R.; Gray H. B. Electron Transfer in Single Crystals of Pseudomonas aeruginosa Azurins. J. Am. Chem. Soc. 2001, 123, 11623–11631. 10.1021/ja0115870. [DOI] [PubMed] [Google Scholar]
- Miller J. E.; Di Bilio A. J.; Wehbi W. A.; Green M. T.; Museth A. K.; Richards J. R.; Winkler J. R.; Gray H. B. Electron Tunneling in Rhenium-Modified Pseudomonas aeruginosa Azurins. Biochim. Biophys. Acta, Bioenerg. 2004, 1655, 59–63. 10.1016/j.bbabio.2003.06.010. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Petrik I. D.; Chacon K. N.; Hosseinzadeh P.; Chen H. H.; Blackburn N. J.; Lu Y. Effect of Circular Permutation on the Structure and Function of Type 1 Blue Copper Center in Azurin. Protein Sci. 2017, 26 (2), 218–226. 10.1002/pro.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray H. B.; Malmstrom B. G.; Williams R. J. P. Copper Coordination in Blue Proteins. JBIC, J. Biol. Inorg. Chem. 2000, 5 (5), 551–559. 10.1007/s007750000146. [DOI] [PubMed] [Google Scholar]
- Takematsu K.; Williamson H.; Blanco-Rodríguez A. M.; Sokolová L.; Nikolovski P.; Kaiser J. T.; Towrie M.; Clark I. P.; Vlček A. Jr; Winkler J. R.; Gray H. B. Tryptophan-Accelerated Electron Flow across a Protein-Protein Interface. J. Am. Chem. Soc. 2013, 135, 15515–15525. 10.1021/ja406830d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farver O.; Skov L. K.; Young S.; Bonander N.; Karlsson B. G.; Vänngård T.; Pecht I. Aromatic Residues May Enhance Intramolecular Electron Transfer in Azurin. J. Am. Chem. Soc. 1997, 119, 5453–5454. 10.1021/ja964386i. [DOI] [Google Scholar]
- Sokolová L.; Williamson H.; Sýkora J.; Hof M.; Gray H. B.; Brutschy B.; Vlček A. Jr. Mass Spectrometric Characterization of Oligomers in Pseudomonas aeruginosa Azurin Solutions. J. Phys. Chem. B 2011, 115, 4790–4800. 10.1021/jp110460k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlček A.; Kvapilová H.; Towrie M.; Záliš S. Electron-Transfer Acceleration Investigated by Time Resolved Infrared Spectroscopy. Acc. Chem. Res. 2015, 48, 868–876. 10.1021/ar5004048. [DOI] [PubMed] [Google Scholar]
- Blanco-Rodríguez A. M.; Busby M.; Ronayne K. L.; Towrie M.; Grǎdinaru C.; Sudhamsu J.; Sýkora J.; Hof M.; Záliš S.; Di Bilio A. J.; Crane B. R.; Gray H. B.; Vlček A. Jr. Relaxation Dynamics of [ReI(CO)3(phen)(Hisx)]+ (X = 83, 107, 109, 124, 126) Pseudomonas aeruginosa Azurins. J. Am. Chem. Soc. 2009, 131, 11788–11800. 10.1021/ja902744s. [DOI] [PubMed] [Google Scholar]
- Blumberger J. Recent Advances in the Theory and Molecular Simulation of Biological Electron Transfer Reactions. Chem. Rev. 2015, 115, 11191–11238. 10.1021/acs.chemrev.5b00298. [DOI] [PubMed] [Google Scholar]
- Ryde U.QM/MM Calculations on Proteins. In Computational Approaches for Studying Enzyme Mechanism, Pt A; Voth G. A., Ed.; Elsevier Academic Press Inc: San Diego, 2016; Vol. 577, pp 119–158. [Google Scholar]
- Adamo C.; Scuseria G. E.; Barone V. Accurate Excitation Energies from Time-Dependent Density Functionl Theory: Assessing the PBE0Model. J. Chem. Phys. 1999, 111, 2889–2899. 10.1063/1.479571. [DOI] [Google Scholar]
- Adamo C.; Barone V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0Model. J. Chem. Phys. 1999, 110, 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Striplin D. R.; Crosby G. A. Nature of the Emitting 3MLCT Manifold of Re(Cl)(CO)3(diimine). Chem. Phys. Lett. 1994, 221, 426–430. 10.1016/0009-2614(94)00282-7. [DOI] [Google Scholar]
- Striplin D. R.; Crosby G. A. Photophysical Investigations of Rhenium(I)Cl(CO)3(phenanthroline) Complexes. Coord. Chem. Rev. 2001, 211, 163–175. 10.1016/S0010-8545(00)00277-0. [DOI] [Google Scholar]
- Vlček A. Jr. Ultrafast Excited-State Processes in Re(I) Carbonyl-Diimine Complexes: From Excitation to Photochemistry. Top. Organomet. Chem. 2010, 29, 73–114. [Google Scholar]
- Kumar A.; Sun S.-S.; Lees A. J. Photophysics and Photochemistry of Organometallic Rhenium Diimine Complexes. Top. Organomet. Chem. 2010, 29, 1–35. [Google Scholar]
- Cannizzo A.; Blanco-Rodríguez A. M.; El Nahhas A.; Šebera J.; Záliš S.; Vlček A. Jr.; Chergui M. Femtosecond Fluorescence and Intersystem Crossing in Rhenium(I) Carbonyl-Bipyridine Complexes. J. Am. Chem. Soc. 2008, 130, 8967–8974. 10.1021/ja710763w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.