

Summary:
While the preponderance of morbidity and mortality in medulloblastoma patients are due to metastatic disease, most research focuses on the primary tumor due to a dearth of metastatic tissue samples and model systems. Medulloblastoma metastases are found almost exclusively on the leptomeningeal surface of the brain and spinal cord; dissemination is therefore thought to occur through shedding of primary tumor cells into the cerebrospinal fluid followed by distal re-implantation on the leptomeninges. We present evidence for medulloblastoma circulating tumor cells (CTCs) in therapy naïve patients, and demonstrate in vivo through flank xenografting and parabiosis that medulloblastoma CTCs can spread through the blood to the leptomeningeal space to form leptomeningeal metastases. Medulloblastoma leptomeningeal metastases express high levels of the chemokine CCL2, and expression of CCL2 in medulloblastoma in vivo is sufficient to drive leptomeningeal dissemination. Hematogenous dissemination of medulloblastoma offers a new opportunity to diagnose and treat lethal disseminated medulloblastoma.
Keywords: pediatric cancer, brain tumors, metastases, medulloblastoma, circulating tumor cells
Graphical Abstract
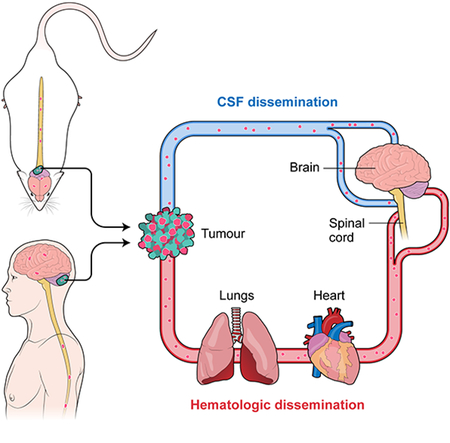
Introduction
Medulloblastoma, a malignant embryonal tumor of the developing cerebellum, is an important cause of childhood morbidity and mortality. The overwhelming majority of medulloblastoma research has focused on studies of therapy naïve primary tumor, or models of the therapy naïve primary tumor. Nearly all clinically relevant medulloblastoma metastases are found in the leptomeningeal space, attached to the pia mater, underneath the arachnoid mater, and bathed in the cerebrospinal fluid. Distant systemic metastases have been reported, but are usually confined to rare patients with very advanced, highly treated disease. This pattern of metastases that is restricted to the leptomeningeal space has led to an assumption in the literature, poorly supported by empirical evidence, that medulloblastoma metastasizes through direct shedding of tumor cells from the primary tumor into the cerebrospinal fluid, survival and migration within the CSF, followed by distal implantation and growth on the pial surface of the central nervous system (Chang et al., 1969).
Mortality among medulloblastoma patients is rarely due to the primary tumor or to recurrence at the primary site, but rather due to metastases at the time of recurrence (Morrissy et al., 2016; Ramaswamy et al., 2013). While imaging evidence of medulloblastoma metastases are detected in about 35% of patients at presentation, all patients are presumed to have diffuse leptomeningeal metastases. As such, irradiation of the entire brain and spinal cord in all patients over three years of age as prophylaxis against metastases is both the standard of care, and the major source of morbidity to survivors, with resultant significant detrimental effects on intelligence, neurological and endocrine function, and development of secondary radiation induced neoplasms (Moxon-Emre et al., 2014). The clear role of metastatic disease in driving the mortality and morbidity for children with medulloblastoma is underlined by the fact that medulloblastoma metastases are highly biologically divergent from their matched primary tumor, with only very few rational targets for therapy shared between the metastatic and the primary compartment (Wang et al., 2015; Wu et al., 2012). Although there are currently no therapies approved or in clinical trials for the specific treatment or prevention of medulloblastoma leptomeningeal metastases, clearly the metastatic compartment should be the focus of research in order to decrease patient mortality and morbidity.
Results:
To determine the genes and pathways driving leptomeningeal dissemination of medulloblastoma, we undertook whole genome sequencing (WGS) of simultaneously collected primary tumor, leptomeningeal metastatic medulloblastoma, and peripheral blood. We observed multiple seminal somatic single nucleotide variants (sSNV) that were clonal in the metastasis, but only subclonal in the matched primary tumor, consistent with the metastasis arising through clonal selection from a pre-existing clone in the primary tumor (Figure 1a). After removing sequencing artifacts, we also observed support for some somatic variants at a very low variant allele frequency (VAF) in the peripheral blood sample, consistent with either circulating tumor cells (CTCs) or circulating tumor DNA (Figure 1a, Table S1, Figure S1a). We performed an in-depth analysis by deep-sequencing selected SNVs, from human tumor/metastasis/peripheral blood trios from three patients. Selected sSNVs displayed heterogeneous VAF in the primary tumors and in the metastasis, with some sSNVs present at the same VAF in primary tumors and metastasis, and some showing specificity for the metastasis or for the primary tumors (Figure 1b and Figure S1b left panels). Deep sequencing also revealed that a fraction of the sSNVs sequenced was detectable in the blood compartment with a low VAF (Figure 1b and Figure S1b). This subset of sSNVs present in the peripheral blood were either shared with both the primary and the metastatic disease (high AF in both compartment), or were specific to the metastatic compartment (high VAF in metastasis and not detectable in primary) (Figure 1b right panels and Figure S1b). These data are consistent with the presence of either CTCs or circulating tumor DNA in human patients with therapy naïve metastatic medulloblastoma, and are suggestive of a contribution of CTCs to the clonal composition of the metastatic compartment.
Figure 1: Circulating Medulloblastoma Tumor Cells in Therapy-Naïve Humans.

a) Read counts from whole genome sequencing of simultaneously collected, patient matched primary tumor, metastatic tumor, and blood in one representative metastatic patient identifies a somatic mutation (C-T) found to be heterozygous in the metastasis (VAF 47.5%), subclonal in the primary tumor (12.5%), and extremely subclonal (1.3%) in the patient matched blood, suggesting the presence of circulating tumor cells. b) Clonality comparison of somatic mutations identified by whole genome sequencing of peripheral blood as compared to primary tumor versus patient matched metastases reveals that putative circulating tumor cells may carry mutations restricted to the metastatic compartment and mutations shared between the primary tumor and the metastasis. See also Figure S1 and Table S1. c) ImageStream analysis shows NCAM+ve/CD45-ve cells in the blood of MB patients sampled at diagnosis suggestive of CTC cells. d) Representative high power images of MB CTC in one patient display NCAM positivity and abnormal morphology. e) the same patient also shows diffuse NCAM positivity in MB (spinal metastasis pre-treatment), scale bars 50μm left, 10μm right panel. See also Figure S1.
To determine the presence of CTCs in metastatic medulloblastoma cases we collected peripheral blood from 6 patients at diagnosis, purified the PBMCs and analyzed by imaging flow cytometry (ImageStream). To improve the specificity and sensitivity of CTC detection we stained the patient derived PBMCs with NCAM (CD56) and CD45. CD56 was has been previously used for differential diagnose of extra-neural disseminated CTCs from other malignancies (Lou et al., 2011; Amhad et al., 2002; Etzell et al., 2006), and therefore constitutes a sensitive marker to identify potential MB CTCs at diagnosis. We found evidence of nucleated, CD45-ve, NCAM+ve, morphologically abnormal cells in 3 out of 6 patients with MB. These cells are MB CTCs that are circulating in the blood of MB patients at the time of diagnosis (Figure 1c–d, Figure S1c). NCAM+ve CD45-ve CTC were found in the order of 16 out of 51805, 30 out of 54785 and 5 out of 272613 total cells examined, however these numbers are likely to be an underestimate of the actual CTC frequency as CD56 is sometimes focally expressed (Fig 1E, Figure S1d). To study the interplay of medulloblastoma CTC and leptomeningeal metastasis we used well-characterized mouse models of Shh metastatic medulloblastoma which harbors random transposon induced GOF and LOF mutations in the context of PTCH1 loss (Wu et al., 2012), and three human patient derived medulloblastoma xenografts labeled with EGFP, which were grafted into the cerebellum of immune deficient NSG mice (Figure 2a). At the time of endpoint, blood sampling and flow-cytometry revealed rare EGFP+ve cells in the peripheral blood of mice implanted with fluorescent xenografts in contrast to non-implanted naïve NSG mice (Figure 2a and Figure S2). We conclude that human and murine therapy naïve medulloblastoma can exhibit circulating tumor cells.
Figure 2: Circulating Medulloblastoma Cells Generate Leptomeningeal Medulloblastoma and Vice Versa.

a) Peripheral blood from a murine GEMM model of metastatic MB, as well as immune deficient mice grafted with three patient derived xenograft models of human metastatic medulloblastoma analyzed by flow cytometry demonstrates GFP+ve cells in the blood consistent with the presence of rare circulating medulloblastoma cells (1 in 1000 to 1 in 100000). Prevalence of CTC in mice injected with different lines is summarized in inlet table. (Scale bars, 1mm) b) Primary human PDX medulloblastoma cells or matched spinal metastasis were serially isolated by FACS and grafted into the cerebellum of NSG mice, (100 cells each) followed by observation and autopsy at clinical end-point. c) There is no significant difference in survival between metastatic derived grafts (n=22) and primary tumor derived grafts (n=17) (Log Rank test p=0.719). d) Mice grafted in the cerebellum with metastatic derived cells (n=15) have a greatly increased burden of spinal leptomeningeal metastases at endpoint as compared to mice grafted with primary tumors (n=7), as assessed by flow-cytometry. (Mann-Whitney U test p=0.040) In the box plots center lines show the medians; box limits indicate the 25th and 75th percentiles; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles e) Xenografting of patient derived medulloblastoma xenografts, or allografting of GEMM murine medulloblastomas into the flank of NSG mice was followed by survival surgery to remove the flank mass at a predetermined size, followed by post-operative care and monitoring reveals leptomeningeal metastases (scale bars, 200μm). f) Some, but not all mice develop leptomeningeal metastases in a delayed fashion after removal of their flank medulloblastoma graft (*ONS76 flank implants were sub-totally resected).
To determine the biological importance of medulloblastoma CTCs, we orthotopically transplanted NSG mice with D425S medulloblastoma cells that had been isolated by flow cytometry from the primary tumor or from the spinal leptomeningeal space (Figure 2b). There was no significant difference in survival between mice grafted with cells from the metastatic compartment or the primary compartment (Figure 2b). The cause of death in all cases could be ascribed to the primary tumor in the cerebellum. Removal of the spinal cord at the time of autopsy, followed by single cell dissociation and flow cytometry analysis to identify EGFP+ve medulloblastoma cells revealed a significantly increased incidence of leptomeningeal metastases in the animals grafted in the cerebellum with metastatic cells as compared to animals grafted in the cerebellum with primary tumor derived cells (Figure 2d). These data support a model in which metastatic medulloblastoma cells are more capable to give rise to leptomeningeal metastases as compared to cells derived from the primary tumor.
Prior publications have always assumed that medulloblastoma disseminates to the leptomeninges through cell shedding from the primary tumor into the cerebrospinal fluid (CSF), followed by distal implantation and growth (Chang et al., 1969). This assumption of a CSF mediated route is likely based on the pattern of metastasis which is largely limited to the leptomeninges, but the support for which is largely free of experimental empirical evidence. The correlative relationship that we observed in humans with metastatic medulloblastoma between circulating tumor cells and leptomeningeal disease suggests an alternative model in which medulloblastoma cells enter the blood circulation, and then home to the leptomeningeal space. Other types of human malignancy such as leukemia that originate in the blood are known to show a preference to home to the leptomeningeal space (Gladdy et al., 2003). We hypothesize therefore that medulloblastoma might be able to disseminate through the blood circulation, to seed the leptomeninges, and give rise to leptomeningeal metastatic disease.
To test this disruptive hypothesis we used a GEMM model of medulloblastoma (Wu et al., 2012), an allograft model of medulloblastoma (Pei et al., 2012), and five different patient derived xenografts of human medulloblastoma and grafted them into the flank of NSG mice (Figure 2e,f). These flank grafts were not contiguous with the central nervous system and therefore have no direct route to the cerebrospinal fluid or the leptomeningeal space. Once the flank grafts reached the maximal acceptable size as outlined by the institutional animal care committee, the animals were placed under general anesthetic and the flank tumor was carefully surgically removed in an aseptic manner. Post-operatively the animals were monitored for regrowth of the flank tumor or the development of novel symptoms, including neurological symptoms and signs. No animal reached endpoint due to recurrence in the primary tumor location in the flank. All medulloblastomas tested except for MB002 resulted in leptomeningeal metastasis (Figure 2e,f). At the time of necropsy, many animals also had liver metastases, although no additional extra-CNS sites of metastases were observed. We conclude that medulloblastoma cells can enter the blood circulation as circulating tumor cells, home to the leptomeningeal space, and give rise to leptomeningeal metastases.
To rigorously test the role of the blood system in the leptomeningeal dissemination of medulloblastoma, we established a medulloblastoma parabiosis model in NSG mice. Young female sibling NSG mice pairs were chosen, and one sister (hereafter the donor) was grafted in the cerebellum with 50,000 Med-411FH, MMB or MMS medulloblastoma cells using stereotaxis. One to three days after cerebellar grafting and recovery, the sister NSG mice underwent parabiosis surgery (Figure 3a). Ten days after the parabiosis surgery, the mice were again placed under general anesthesia and one sister had Evans blue injected into the tail vein to demonstrate the passage of the blue dye to the other sister mouse, thus proving the patency of the parabiotic connection (Figure 3b). When the donor mouse became symptomatic (approximately one month), the joining surgery was reversed, the donor mouse was sacrificed, and the recipient sister was survived and observed. The donor mice are shown to develop medulloblastoma in the cerebellum, as well as leptomeningeal metastases. Surviving recipient sisters were observed until endpoint, where 3/6 recipients were found to have leptomeningeal metastases of disseminated Med-411FH cells by NCAM staining (Figure 3c–e, Figure S3a). MMB and MMS cells colonized the leptomeninges of 2/4 and 1/4 recipient mice, respectively (Figure 3d–e). Metastatic foci of Med-411FH cells were also observed in the liver and lungs of two recipient mice, as summarized in Figure S3b. Through this stringent test, we conclude that medulloblastoma is able to spread through the circulation to penetrate and populate the leptomeninges resulting in leptomeningeal metastases.
Figure 3: Hematogenous Dissemination of Medulloblastoma Between Parabiont Mice.

a) Schematic of the workflow to assess hematogenous dissemination of medulloblastoma using parabiont NSG twins. b) Evans blue administered by tail vein injection under general anesthesia demonstrates patency of the vascular anastomosis in the parabionts. c) Primary medulloblastoma is observed in the donor twin sacrificed after twin separation at the time of symptoms. The recipient is observed until endpoint, at which time leptomeningeal metastases were observed in three out of six parabiont pairs for PDX line Med-411FH. See also Figure S3. d) PDX lines MMB and MMS also display hematogenous tropism to leptomeninges when used implanted in parabiont mice, scale bars 50μm. e) Frequency of hematogenous dissemination in MB parabiont, see also figure S3.
Medulloblastoma metastases are only seldom surgically removed from human children, as they are not usually focal, there is uncertain or no clinical benefit to their removal, and the underlying assumption was that they are similar to the primary tumor. To determine the mechanism(s) driving hematogenous dissemination of leptomeningeal medulloblastoma, we compared the gene expression profiles of human surgical samples from a very limited set of matched primary and metastatic medulloblastomas (Wang et al., 2015). The chemokine CCL2 is expressed at a much higher level in the metastases than their matched primary tumor (Figure 4a). CCL2 is the second most differentially expressed gene when comparing primary grp3 tumors and patient matched metastases (Table S2). RNAseq analysis of 70 cases of human MB Group 3 primary tumors demonstrates that patients with higher M stage are significantly (p=0.0214) more likely to express CCL2 (Figure 4b). CCL2 binds to its receptor CCR2, and is known to play an important role in the diapedesis of leukocytes between endothelial cells at the blood-brain-barrier so that they can enter the central nervous system, such as in multiple sclerosis or bacterial meningitis. The human CCL2 gene resides on chromosome 17q, which is frequently somatically genetically gained in Group 3 and Group 4 medulloblastoma. Activation of PI3-kinase signaling due to deletion of PTEN on chromosome 10q is a known driver of medulloblastoma leptomeningeal dissemination (Wu et al., 2012). Gain of chromosome 17q, is associated with a metastatic phenotype among patients with Group 3 and Group 4 medulloblastoma, but not in SHH medulloblastoma (Figure 4c, Figure S4a–b). Indeed, high M stages are very rare in Group 3 patients who do not have gain of chromosome 17q and loss of chromosome 10q (Figure S4b). Gain of chromosome 17q is also positively correlated with leptomeningeal dissemination in Group 4 medulloblastoma, where there is no contribution of 10q loss to higher M stages (Figure 4c, Figure 4Sc).
Figure 4: CCL2 Overexpression in Human Leptomeningeal Metastases.

a) CCL2 is differentially expressed between human primary and metastatic Group 3 medulloblastoma surgical samples as compared by expression microarray (p=0.041). See also Table S2. b) Expression of CCL2 is associated with metastatic status 2 and 3 in a cohort of 70 Grp3 MB with clinical annotation, 46.6% of M2/3 express CCL2 vs 20% in the M0/1 group. c) Human CCL2 resides on chromosome 17q, a region frequently gained in Group 3. Gain of chromosome 17q is significantly more common in patients with leptomeningeal dissemination for both Group 3 and Group 4 cohorts. In contrast, CCL2 expression is only marginally correlated with M2/3 in Shh patients, and the trend (23.5% CCL2 positive M1/0 vs 43.5% CLL2 positive) does not reach statistical significance (p=0.2462 Fisher’s exact test). d) 10q and 17q status as determined by 450K methylation array and 250K array (ONS76*)
To test the mechanistic importance of CCL2 and its receptor CCR2 in the process of leptomeningeal dissemination of medulloblastoma, we used lentiviral transfer to express CCL2, CCR2, or both CCL2 and CCR2 in combination in the infrequently metastatic ONS76 and MB002 medulloblastoma lines (Figure S5a–b). Where-as control ONS76 and MB002 are only poorly metastatic when xenografted into the cerebellum of NSG mice (5/10 and 2/11 mice respectively), expression of CCL2 increases the incidence (9/10 and 8/13) or the size of spinal leptomeningeal metastases (Figure 5a–b, e; Figure 5Sc–d). Over-expression of the receptor CCR2 alone, or the combination of CCL2 and CCR2 was tested in ONS76, where 5/5 mice showed leptomeningeal metastases in both instances. The CCL2/CCR2 axis has been implicated in the metastatic process of several adult cancers, and it likely plays a role in tumor cells transiting into, and out of the blood circulation rather than specifically targeting cells to the leptomeningeal space. We conclude that CCL2 and/or CCR2 can be sufficient to drive leptomeningeal dissemination of medulloblastoma.
Figure 5: The CCL2/CCR2 Axis drives Medulloblastoma Leptomeningeal Dissemination.

a) The ONS76 line was infected with CCL2 alone or in combination with CCR2 expressing or control lentiviruses. Control ONS76 xenografts are poorly metastatic (5/10 animals) when xenografted into NSG mice, where-as expression of either CCL2 (9/10 animals) or CCR2 (5/5 animals), or both CCL2 and CCR2 (5/5 animals) are able to increase leptomeningeal dissemination (left side photomicrographs, scale bars H&E 50μm, fluorescence 2mm). Metastasis driven by CCL2 or CCR2 overexpression are significantly larger than the metastasis found on the spinal cords of ONS76-CTL xenografted mice (right side box-plot, p=0.023 and p=0.037 respectively, Mann-Withney U-test). In the box plots center lines show the medians; box limits indicate the 25th and 75th percentiles; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles b) Similarly, overexpression of CCL2 the poorly metastatic medulloblastoma patient derived line MB002 significantly increases prevalence of metastasis (18% to 61%, p=0.029, Fisher’s exact test with mid P adjustment α=0.05, left side photmicrographs, scale bars H&E 50μm, fluorescence 2mm) without affecting the size of the metastatic lesions (right side box-plot). In the box plots center lines show the medians; box limits indicate the 25th and 75th percentiles; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles c) Knockdown of CCL2 by lentiviral mediated shRNAs (single hairpin and combination of two hairpins) in highly metastatic human D425S medulloblastoma cells show decreased protein expression of CCL2 by Western blotting and diminished metastatic deposits after in vivo intracranial implantation in NSG mice (scale bars, 1mm). d) Summary Table of the statistical significance of all comparisons (Fisher’s exact test with mid P adjustment, α=0.05). e-f) CCR2 KO in the tumor microenvironment in vivo attenuates the metastatic behavior of MB cells in both CB and flank implants.
Med-411FH were used for orthotopic flank xenografts of CCR2−/−;NSG or CCR2wt;NSG littermates. The absence of CCR2 leads to a trend in size decrease of metastatic deposits in the leptomeninges in intracranial implants, which is more pronounced than in flanks (p=0.051 and p=0.314 respectively, Mann-Withney U-test). Boxes highlight mice with higher LMD. In the box plots center lines show the medians; box limits indicate the 25th and 75th percentiles; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles (Scale bars, 1mm (E and F).
To test the necessity for CCL2 in the dissemination of medulloblastoma, we employed single or pooled (2x) shRNAs delivered by lentivirus to the highly metastatic D425S medulloblastoma line. Where-as control D425S is highly metastatic (Figure 5d), diminished protein expression of CCL2 significantly decreases the incidence of metastases as compared to controls (4/5 controls) such that few mice have any discernible metastases (Figure 5d–e, Figure S5e). We conclude that in some circumstances, CCL2 is necessary for leptomeningeal dissemination.
To determine if CCL2 exerts its role on MB metastasis by activating CCR2 present in the tumor microenvironment (macrophages, glial and endothelial cells), we crossed NSG immunocompromised mice to CCR2 homozygously deleted mice, then used the resulting cross as recipients for MB xenografting, in the brain as well as in the flank (Figure 5e–f). We found no change in the prevalence of metastasis when mice genetically KO for CCR2 were used for intracranial implants, however we observed a change in the distribution of the size of the metastasis in the spinal cords of CCR2−/−;NSG (Figure 5e, p=0.05118). Notably, the very large macrometastasis observable in the spines of CCR2+/+;NSG mice were not detectable in CCR2−/− mice, indicating that CCR2 expression in the tumor microenvironment could play a role in supporting the growth and expansion of leptomeningeal metastasis. In the flank implants shown in Figure 5f, we observe a similar trend with 45% of the CCR2−/−;NSG mice harboring CNS metastasis, versus 66% of prevalence in CCR2+/+;NSG mice (Figure 5f, p=0.314). In absence of CCR2 in the tumor microenvironment metastasis from flank implants also show a trend for reduced size (P=0.34), and lower incidence of large macrometastasis (Figure 5f). We conclude that CCL2 induces MB metastasis by acting in a cell-autonomous as well as a non-cell-autonomous manner.
Group 3 MB tumors are characterized by overexpression of c-Myc. To confirm that the CCL2/CCR2 axis plays a role in the context of c-Myc overexpression we used an allograft model of Group 3, prominin+ve/lin-ve NSCs are infected with c-Myc along with DNp53-Luciferase to establish MPLuc MB (Pei et al., 2012). To this model we added CCL2, CCR2 or the combination of both CCL2 and CCR2. We found that 1 out of 13 (7.7%) control mice developed spinal metastasis luciferase signal, as contrasted with 7 out of 13 (54%, p=0.006) mice in the double transduced CCL2/CCR2. In this system single over expression of CCR2 was sufficient to promote spinal metastasis in 4/6 (67%, p=0.009) mice. We only found 1 mouse implanted with CCL2 overexpressing cells that showed a detectable spinal signal (Figure 6a and Figure S6a). We conclude that the activation of the CCL2/CCR2 axis in a c-Myc driven Group 3 MB allograft model promotes metastatic dissemination.
Figure 6: The CCL2/CCR2 Axis Drives Leptomeningeal Dissemination In Vivo.

NSG mice implanted with MPLuc allografts develop spinal metastasis with higher frequency when both CCL2 and CCR2 are overexpressed (1/13 vs 7/13, p=0.006 Fisher’s Exact Test with mid-P adjustment, α=0.05), the black arrows point to BLI signals from the spinal metastasis observable 15 days after implantations of the tumor cells (See also Figure S6). b) Nestin-TVA transgenic mice were injected with viruses to transfer either Shh alone, Shh+CCL2, Shh+CCR2, Shh+CCL2+CCR2, or CCL2+CCR2 alone. Mice injected with CCL2+CCR2 did not develop medulloblastoma. All other genotypes were observed to have a similar rate of medulloblastoma free survival (Log-Rank test p=0.217). c) While Shh virus alone leads to localized medulloblastoma in vivo, the addition of CCL2 and/or CCR drives the development of leptomeningeal metastases. Expression of CCL2, or CCR2 has no significant impact on the incidence of primary tumor formation (Chi-square test p=0.107), but does have a significant and dramatic effect on the incidence of leptomeningeal metastases (Chi-square test p=0.018). d) Primary and metastatic medulloblastoma sections from Nestin-TVA mice infected with Shh and mCherry-CCL2 viruses demonstrates that CCL2 expression is subclonal in the primary tumor, but is highly clonally selected and ubiquitous in the metastases. e) Primary and metastatic medulloblastoma sections from Nestin-TVA mice infected with Shh, Shh+CCL2, and Shh+ CCL2+CCR2 viruses demonstrates that CCL2 expression - revealed by IHC for CLL2 mcherry tag - is subclonal in the primary tumor, but is clonally selected in the meningeal metastasis. Scale bars 50μm.
To stringently test the role of CCL2 and CCR2 in the dissemination of medulloblastoma in an immune intact, genetic mouse model we used the Nestin-TVA model of medulloblastoma, driven by viruses encoding for the Sonic Hedgehog protein (Rao et al., 2003). In this model, RCAS retroviruses will infect cells in the Nestin expressing compartment as they express the TVA receptor, but not other cells as they lack the viral receptor. This allows for somatic in vivo gene transfer in a cell specific manner. To test the role of CCL2 and CCR2 in dissemination we bred a large cohort of mice (total 363 mice) that were driven to express Shh alone, Shh +CCL2, Shh+CCR2, Shh+CCL2+CCR2, or CCL and CCR2 without Shh (Figure 6b,c). Medulloblastoma was not observed in animals infected with RCAS viruses encoding CCL2 and CCR2 alone (Figure 6b). Transfer of Shh alone results in localized medulloblastoma in 39% of cases, with only rare leptomeningeal metastases. Indeed, there is no significant difference in the incidence of primary medulloblastoma in mice infected with Shh viruses with the addition of CCL2, or CCR2, or both CCL2 and CCR2 suggesting that CCL2 and CCR2 are not drivers in the primary tumor. Conversely, there was a dramatic and significant increase in the incidence of metastases with the addition of CCL2, CCR2, or both genes in combination (Figure 6c). Immunohistochemical analysis of primary and metastatic tumors driven by RCAS viruses encoding Shh and mCherry-CCL2 demonstrate that while CCL2 expression is only subclonal in the primary tumors, CCL2 is expressed in nearly 100% of cells in the leptomeningeal metastases consistent with a model in which CCL2 drives clonal selection of primary medulloblastoma cells that are able to complete the metastatic cascade, and in keeping with the concept that CCL2 is required for medulloblastoma metastases (Figure 6d). Data from this genetic model highly support that both CCL2 and CCR2 are sufficient to promote the leptomeningeal dissemination of medulloblastoma.
Discussion:
Most patients with metastatic medulloblastoma are observed to have metastases confined to the leptomeningeal space, undoubtedly leading to the assumption that medulloblastoma spreads through the cerebrospinal space to colonize the distal leptomeninges. However, the vast majority of human cancers metastasize primarily through the blood; indeed some cancers that were thought to undergo an extreme form of local spread such as peritoneal spread of ovarian cancer are now thought in at least some cases to occur hematogenously (Pradeep et al., 2014). There are other extra-CNS malignancies such as leukemia and breast cancer that in some circumstances can show a marked preference to penetrate and populate the leptomeninges resulting in leptomeningeal cancer (Gladdy et al., 2003). Patients with end stage, highly treated medulloblastoma do present on occasion with systemic non-CNS metastases (Rudin et al., 2009), a scenario that might become more common with more effective treatment of CNS disease. While our data provide high-level support for the hematogenous dissemination of medulloblastoma, we cannot prove that spread does not also occur through a CSF route due to the difficulty in proving a negative. It is indeed possible that both hematogenous and CSF spread are occurring in some patients, and that both are clinically relevant. Similarly, both lymphatic and hematogenous dissemination of breast cancer are known to occur, and both are clinically important.
The CCL2/CCR2 signaling axis is well known to play an important role in the transit of leukocyte populations into the central nervous system in a number of infectious and inflammatory diseases (Chu et al., 2014; Semple et al., 2010). CCL2 has also been imputed in other types of non-leptomeningeal and non-CNS metastases including systemic metastases from breast and colon cancer (Qian et al., 2011; Wolf et al., 2012). Our data support a model in which expression of CCL2 allows medulloblastoma to metastasize, but in which CCL2 probably does not play an exclusive role in MB cells homing to the leptomeninges. The mechanism(s) for leptomeningeal specificity for medulloblastoma cells are unknown. Drugs and therapeutic antibodies to block the CCL2/CCR2 signaling axis are being tested in several human clinical trials for both neoplastic and non-neoplastic conditions. There is some early evidence of efficacy of CCL2 blockade to prevent breast metastases, although cessation of therapy leads to a metastatic rebound if therapy is ceased (Bonapace et al., 2014).
The discovery of a hematogenous route for medulloblastoma metastases offers opportunities for disease diagnosis and monitoring of response to therapy. There are currently no specific therapies to prevent or treat medulloblastoma metastases. Many medulloblastoma patients who do not have metastases at presentation, subsequently present with de novo metastatic disease at the time of recurrence. Future experiments should examine the role of CCL2/CCR2 blockade in the prevention of metastases, as well as in the prevention of progression in patients with established metastases, as the CCL2/CCR2 signaling axis represents the first biological target for medulloblastoma metastases. The discovery of a hematogenous route for medulloblastoma metastases offers opportunities for disease diagnosis and monitoring of response to therapy.
STAR METHODS:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael D. Taylor (mdtaylor@sickkids.ca).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice:
Ptch+/−/Math1-SB11/T2Onc were previously generated from crossing Ptch1 heterozygous (Ptch1tm1Mps/J, #003081 Jackson Laboratories) with T2Onc (TgTn(sb-T2/Onc)76Dla) and Math1-SB11 mice (Wu et al., 2012), these mice were crossed with Math1-GFP (Lumpkin et al., 2003) reporter mice to generate quadruple transgenic mice. Mice were bred and housed accordingly to the guidelines from the Canadian Animal Care Committe at the Toronto Centre for Phenogenomics and The University Health Network, MaxBell Facility (Toronto, ON). Ccr2 deficient mice were purchased from Jackson Laboratories (B6.129S4-Ccr2tm1Ifc/J, Stock No: 004999) and genotyped accordingly to vendor suggested protocols. CCR2 homozygously deleted mice were bred to NSG mice to generate F1 triple heterozygotes mice. Breeding pairs were established from CCR2wt;NSG and CCR2KO;NSG, and males and females littermates were used as CTLs and experimental mice, respectively. NSG mice were purchased from Jackson Laboratories (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, stock no: 005557), females and males were used in the study for flank and CB tumor cells injections. Parabiosis surgeries were only performed between female littermates of matching weight. Clinical endpoints were assessed by veterinary technicians blinded to the experimental groups.
Human medulloblastoma tumour specimens:
All tumour specimens were obtained in accordance with the Research Ethics Board at the Hospital for Sick Children (Toronto, Canada).
Cell lines:
Medulloblastoma cell line ONS76 was grown in DMEM plus 10%FBS. The D425S and MB002 were a kind gift from S. Mitra (1). D425S and MB002 cells were cultured in serum free expansion media composed of DMEM/F12, EGF 20 ng/mL, bFGF 10 ng/mL, B27, N2, Glutamine, LIF. Med-411FH were purchased from the Brain Tumor Resource Laboratory (Olson Lab, Fred Hutchinson Cancer Research) and propagated exclusively orthotopically in NSG mice. The line derives from a 3yrs old male with anaplastic MB and genomic and immunohistological features of grp3 medulloblastoma (www.btrl.org). Myc driven grp3 MB allograft line MPGFPluc was a kind gift from Dr. R. Wechesler-Reya and was exclusively propagated in vivo in NSG mice. The allograft line SB;Ptc−/+ was derived from murine primary tumors from the Ptch+/−/Math1-SB11/T2Onc/Tg(Math1-GFP) or Ptch+/−/Math1-SB11/T2Onc/Tg(Math1-GFP), these cells are derived from a highly metastatic MB mouse model (Wu et al., 2012) crossed with Math1-GFP (MMB line was derived from an autoptic specimen from a brain metastasis of a young male with grp3 high Myc recurrent medulloblastoma. MMS line was derived from a spinal metastasis from the same patient as the MMB line, this line also retains grp3 high Myc features. MMB and MMS line were also exclusively passaged orthotopically in NSG mice.
PDX lines were exclusively kept in culture for time required to infect them with fluorescent proteins coding lentiviruses (12hrs), in serum free NSC media composed of DMEM/F12, EGF 20 ng/mL, bFGF 10 ng/mL, B27, N2, Glutamine, LIF. The identity of all cell lines was periodically confirmed by STR genotyping (Geneprint, Promega) or 450K methylation array (Illumina), and it matched the original patient profile, the received clone (MB002), or deposited data.
METHOD DETAILS
Whole Genome and RNA Sequencing:
All patients gave informed consent to the samples collection; the samples were sequenced and analyzed at Canada’s Michael Smith Genome Sciences Centre at the BC Cancer Agency (GSC). Libraries for whole genome sequencing were constructed using either the plate-based or SPRI-TE library construction protocol, as previously described (Morrissy at al., 2016). Bam files were sorted with SAMTools (version 0.1.13) and merged using Picard MarkDuplicates.jar (version 1.71). The merged bam files were subsequently indexed with SAMTools index (version 0.1.17). SNVs from WGS data were analyzed using Samtools mpileup and Strelka (Saunders et al., 2012) as described previously (Morrissy et al., 2016). To verify SNVs, samples were subjected to targeted deep amplicon sequencing of the tumour and normal DNA as described previously (Morrissy et al 2016). Briefly, primers were designed with the Primer3 software with a GC clamp, an optimal Tm of 64°C and tested with a combination of UCSC’s in-silico PCR tools to obtain unique hits. All primer pairs were designed such that the variant is located within a maximum of 250bp of the 5’or 3’ amplicon end. Illumina adapters were added to the primers to allow for pooled direct sequencing. Genomic DNA templates or library construction intermediates were used as starting material to generate PCR products using Phusion DNA polymerase (Fisher Scientific, catalogue # F-540L). After two PCR rounds (amplification and indexing) PCR products of the desired size range were purified using 8% PAGE gels, QC’ed and pooled together for sequencing on the Illumina MiSeq platform with paired-end 250bp reads using v2 reagents. Reads were aligned using BWA-SW, and SNVs called with Samtools mpileup with the following parameters: -d 1000000 -B -C50 -DES. Indels were called using VarScan and the following parameters: mpileup2indel --min-var-freq 0 --p-value 1 --strand-filter 0.
Messenger RNA library construction and sequencing and alignment of strand-specific RNA-seq data was performed as described in (Morrissy et al., 2016). Briefly, PolyA mRNA was purified from two micrograms of total RNA samples using the 96-well MultiMACS mRNA isolation kit on the MultiMACS 96 separator (Miltenyi Biotec, Germany). First-strand cDNA was synthesized using the Superscript cDNA Synthesis kit (Life Technologies, USA) and random hexamer primers at a concentration of 5μM along with a final concentration of 1ug/uL Actinomycin D, followed by Ampure XP SPRI beads on a Biomek FX robot (Beckman-Coulter, USA). The second strand cDNA was synthesized following the Superscript cDNA Synthesis protocol by replacing the dTTP with dUTP in dNTP mix, followed by UNG digestion (Uracil-N-Glycosylase, Life Technologies, USA) in the post-adapter ligation reaction. After QC and fragmentation, paired-end (PE) plate-based libraries were prepared on a Biomek FX robot (Beckman-Coulter, USA). Library protocol included end-repair and phosphorylation in a single reaction, 3’ A-tailing by Klenow fragment (3’ to 5’ exo minus) and cleanup using Ampure XP SPRI beads. The adapter-ligated products were purified using Ampure XP SPRI beads, then PCR-amplified with Phusion DNA Polymerase (Thermo Fisher Scientific Inc. USA) using Illumina’s PE primer set, with cycle conditions of 98°C 30sec followed by 10–15 cycles of 98°C 10sec, 65°C 30sec and 72°C 30sec, and then 72°C 5min. The PCR products were purified using Ampure XP SPRI beads, and checked with a Caliper LabChip GX for DNA samples using the High Sensitivity Assay (PerkinElmer, Inc. USA). PCR products with a desired size range were purified, QC’ed on an Agilent DNA 1000 series II assay and Quant-iT dsDNA HS Assay Kit using Qubit fluorometer (Invitrogen), then diluted to 8nM. The final concentration was verified by Quant-iT dsDNA HS Assay .The libraries, 2×100 PE lanes, were sequenced on the Illumina HiSeq 2000/2500 platform using v3 chemistry and HiSeq Control Software version 2.0.10. Illumina paired-end RNA sequencing data was aligned to GRCh37-lite genome-plus-junctions using BWA (version 0.5.7) as previously described (Morrissy et al., 2016). BWA “aln” and “sample” were run with default parameters but for disabling the Smith-Waterman alignment. Finally, reads failing the Illumina chastity filter are flagged with a custom script, and duplicated reads were flagged with Picard Tools (version 1.31). Primary MB were considered positive for CCL2 if expression values (RPKM) was greater than 10.
Detection of rare somatic mutations in the blood using DeepSeq data:
To find evidence for somatic mutations in the blood that have a low VAF, we generated base quality (baseQ) distributions supporting the reference and all alternate alleles in the primary (and the recurrent) compartments for each sSNV profiled using deep sequencing, as well as for each of the 10bp upstream and downstream of the mutation position. A minimum of 100 reads in each compartment was required to consider an event (i.e. primary, metastasis, and blood). Due to our amplification and sequencing strategy, all reads start at the same position, and the target SNV is always at a specific position in the read (i.e. a given mutation covered by 2000 reads will be at base position 40 in all reads). Thus, unlike shotgun protocols where read starts are random, the SNVs are never affected by sequencing errors at the end of the read (where errors tend to happen more often), and cumulative sequencing error rates for whole reads are not applicable in estimating local error rates at a specific base. Instead, detection of a real mutation is only confounded by the subset of sequencing errors at the same position in the read that causes a base change to match the mutation; sequencing errors matching the other two possible bases (i.e. non-reference and non-mutation) are a non-ambiguous measure of the error rate at a particular position. Thus, to distinguish sequencing errors from real mutations with low VAF, for each allele (i.e. the reference allele and all three alternate alleles), we generated base quality (baseQ) distributions from all reads covering the position of the mutation (as well as flanking positions); the reference base was further used as the benchmark distribution of a base without appreciable sequencing errors. The non-reference alleles that had the highest (1) mean baseQ value, (2) max baseQ value, and (3) highest number of reads with baseQ values > 30, were considered real events. When all three criteria were not matched, the presence of the mutation at a low frequency could not be confirmed. At positions where these criteria were matched, the baseQ distributions of the alternate allele closely matched the baseQ distribution of the positive control reference base, could be easily distinguished from sequencing errors, and nearly always matched the expected mutation at that position, confirming the presence of the mutation in the diagnostic sample. To further increase stringency and account for sequence context, we compared the baseQ distributions of every mutation to the distribution of nucleotide-matched neighboring bases without somatic mutations. i.e. for an A>G mutation, we compared the baseQ distribution of the G allele at other A bases within 10bp flanking sequence, which were not somatically mutated in either the diagnostic or metastatic sample. Cases where flanking A>G baseQ distributions were high (i.e. qualities near 40, or near the peak of the bases supporting the reference base) may be examples of sequence context-specific PCR errors, and were excluded from analysis.
ImageStream sample preparation and analysis for Medulloblastoma patients:
Patient samples were prepared from 3–5ml PB, PBMC separated by lympholyte and resuspended in PBS/2%BSA. Samples were stained with biotinylated anti hCD56 (Biotin Mouse Anti-Human CD56 Clone B159, BD bioscience) (1:200) for 45’ on ice, washed and stained with APC-streptavidin conjugates for 30’ on ice. Negative CTLs samples were prepared omitting the anti hCD56 antibody (APC-strep only CTLs). Samples were resuspended in a volume of 100ul PBS −/− 1% BSA in a 1.5ml low retention microfuge tube (Sigma-cat#T4816). 10ul of Mouse-anti- human CD45 PE-CF594 (clone HI30– BD Biosciences cat# 562279) was then added and the sample incubated for 30 minutes at 4°C. Following incubation 900ml of PBS -/- 1% FBS was added along with 2ul of the nuclear dye Hoechst333342 (BD Biosciences cat# 561908) and incubated for an additional 30min at 4°C. Following incubation, the sample was spun down at 400G, supernatant removed and pellet washed in an additional 1ml of PBS −/− 1% FBS. The sample was then spun down again at 400G and the supernatant removed leaving a residual 50ul of supernatant to resuspend the pellet. Samples were acquired on a 5 laser 12 channel ImageStream MK2 imaging flow cytometer at 60 X magnification following ASSIST calibration (Amnis Corporation). Channels 1,4,7,9 and 11 we used for acquisition along with lasers 405nm (20mw), 488nm(200mw), 592nm (300mw) and 642nm (150mw) used for excitation. A bright-field (BF) area lower limit of 50um2 was used to eliminate debris and speed beads during acquisition. The entire sample was collected in a series of 100×103 event Raw Image Files which ranged from 4–10 separate files depending on patient. Image analysis was carried out using the IDEAS software (Amnis Corporation). The analysis strategy was as follows: 1.Focused cells were identified using the Gradient RMS feature (channel 1BF – default mask M01) 2. Single cells were identified using the features Area vs. Aspect Ratio of channel 1 (BF/default mask M01). 3. Nucleated cells were positively gated using Hoechest nuclear dye staining (channel 07 intensity feature –default mask M07). 4. The CD45 negative cell population was gated using the channel 04 (PE-CF594) intensity feature – default mask M04. 5. Finally NCAM (CD56) positive cells were gated using channel 11(APC) intensity feature – default mask M011. Potential circulating tumor cell candidates were visually verified. Unstained cells were used to help establish gates based on intensity features and compensation was verified using single stained controls.
Orthotopic injection of MB tumor cells:
MB PDXs lines (100 cells Figure 2D; 50,000 cells Figure 3; 1,000 cells in Figure 5A,B,C; 50,000 cells in Figure 5E) were xenografted by stereotactic injection into posterior fossas of immunodeficient 6 to 8 weeks old NODscid gamma mice females and males (NSG, Jackson lab) using the following stereotactic coordinates: 2mm posterior to lambda, 2mm lateral and 2mm deep. Tumors were allowed to develop until neurological endpoint. Survival of mice was estimated using a Kaplan-Meier curves and differences evaluated using a Log-Rank test.
Flank implantation of MB tumor cells:
500,000 MB tumor cells were resuspended 1:1 in PBS/Matrigel and injected subcutaneously in the flanks of 6 to 8 weeks old females and males NODscid gamma mice (NSG, Jackson lab). When the tumors reach 1700mm3 the mice are aseptically prepared for removal surgery. Meloxicam was used for pre and post surgical analgesia, the flank tumors are removed with sterile scissors and the incision is sealed with surgical staples. Staples are removed 14 days after surgery. The mice are then monitored for any residual disease recurring at the flank site and for the development of metastases at other sites throughout their body up to 18 months after surgery.
Parabiosis surgery:
The surgeon prepares the animals aseptically and provides warmth with a heating pad. Preemptive analgesia is administered (buprenorphine, 0.1mg.kg, s.c or tramadol, 20 mg/kg IP or SQ every 6–12 hours). Mice are anaesthetized using isoflurane (2–3% mixed with oxygen). After shaving the corresponding lateral aspects of each mouse, matching skin incisions are made from the olecranon to the knee joint of each mouse, and the subcutaneous fascia is bluntly dissected to create about 0,5cm of free skin. The latissimus dorsi and abdominal external oblique muscles on each mouse are split. The peritoneal cavities are also joined to increase stability of the pairs. The olecranon joints are attached by a single internal monofilament suture and tie, and the dorsal and ventral skins are sutured by continuous suture. This method gives a firm support to both animals, preventing the strain on the sutures of the skin and abdominal walls, and the formation of a pocket between the skin of the animals and the cervical and thoracic regions, thus decreasing the chance of infection, and gives all the advantages of fast (10 days) coelio-anastomosis.
CCL2 and CCR2b expression studies in ONS76 and MB002SU:
CCL2 and CCR2b cDNA was amplified by PCR from human ependymoma cDNA pool. To generate pLVX-CCL2-IRES-mCherry and pLVX-CCR2-IRES-mCherry, CCL2 and CCR2 fragments were ligated with pLVX-IRES-mCherry (Clontech) at SpeI and NotI. To generate pLVX-CCL2-IRES-zsGreen, IRES in pLVX-IRES-zsGreen (Clontech) was replaced with CCL2-IRES (excised from pLVX-CCL2-IRES-mCherry) at NotI and MluI. To generate RCAS-CCL2-IRES-AcGFP and RCAS-CCR2-IRES-AcGFP, CCL2 and CCR2 were amplified from pLVX-CCL2-IRES-mCherry and pLVX-CCR2-IRES-mCherry, respectively, and inserted to RCAS-IRES-AcGFP (modified by Fults DW by inserting AcGFP to RCAS-Y) at NotI and PacI. To generate RCAS-CCL2-IRES-mCherry, IRES-AcGFP in RCAS-IRES-AcGFP was replaced with CCL2-IRES-mCherry (amplified from pLVX-CCL2-IRES-mCherry) at PacI and ClaI. Sequences of PCR products were confirmed by Sanger sequencing. Sequences of all primers are available upon requests. To generate CCL2 overexpressing lines, ONS76 and MB002 cells were transduced with lentiviral particles (multiplicity of infection of 0.3 = 25%). 72hrs after infection fluorescent cells were purified by FACS and expanded. Verification of CCL2 over expression was performed by western blot using: CCL2 anti MCP1 Antibody (2D8) Novus Biologicals (NBP2–22115) (1:500 in PBS-BSA) and CCR2 Antibody (E68) Novus Biologicals (NB110–55674) (1:500), long term overexpression of the proteins of interest during passaging in animals was monitored by routine check of fluorescence tagging on whole tissues at dissection by fluorescence stereoscope.
CCL2 silencing:
To generate CCL2 knockout lines, D425SGFP cells were transduced with lentiviral particles (multiplicity of infection of 0.3 = 25%) corresponding to different short hairpin RNA (shRNA) constructs targeted to the CCL2 gene (Mission® shRNA TRCN0000006279 (A6), TRCN0000006282 (A9), TRCN0000006283 (A10). Cell were bulk selected in 0.5μg/mL puromycin and expanded in vitro. Six individual shRNA lentiviral particle constructs were tested and A6, A9 and A10 that achieved CCL2 expression inhibition were used in the in vivo experiments. CCL2 downregulation was tested by western blot using CCL2 anti MCP1 Antibody (2D8) Novus Biologicals (NBP2–22115) (1:500 in PBS-BSA).
Elisa assay:
CCL2 concentrations were determined using Elisa analysis with 100μL of undiluted media supernatant. Briefly, cells were resuspended in FBS free media for 6, 12 or 24h. Cell-free media were collected for analysis and stored at −80°C prior to subsequent analysis. CCL2 concentration in the cell-free media was measured by ELISA (eBioscience, Affimetrix inc. San Diego CA-USA ref. 88–739922) according to the manufacturer’s instructions.
Evaluation of extent of metastasis by direct fluorescence:
Presence or absence of metastatic deposits was performed blindly under a direct fluorescence stereoscope, 4 to 6 images were acquired of dorsal and ventral spinal cord with consistent exposure settings during the experiments. Mice spines were defined as positive if a single metastatic deposit was clearly observable in one of the images. Deposits which showed continuity with CB disease and could represent a local spread were excluded from the analysis. Extent of metastasis was evaluated by the way of measurement of fluorescent areas in ImageJ, all the 4–6 images per mouse were used and total area covered by metastatic deposits in each mouse is reported.
Implantation of transduced cerebellar stem cells:
MP tumors were generated by infecting cerebellar stem cells CD133+ with MYC-IRES-luciferase and DNP53-IRES-CD2 retrovirus (Pei et al., 2012). MP tumors were harvested, and dissociated and infected with lentivirus harboring zs-Green CCL2 or/and mcherry-CCR2B. After overnight incubation, infected tumor cells were purified by fluorescence activated cell sorting (FACS) using a BD Influx cell sorter. 104 cells/per animal are resuspended in a mix of Neurocult Basal medium (STEMCELL Technologies, cat#05700) and Corning Matrigel Matrix Growth Factor Reduced (Thomas Scientific, cat#354230) at a ratio 1:1. Then they were transplanted intracisternally into 6–8 weeks old NSG mouse. A mix 1:1 of cells suspension and matrigel is transplanted per animal. Transplanted mice were subjected to weekly bioluminescence imaging. Mice were given intraperitoneal injections of 150 ng/g D-Luciferin (Caliper Life Sciences, cat#12279) and anesthetized with 2.5% isoflurane in an induction chamber. At 7–8 min after injection, animals were imaged using the Xenogen Spectrum (IVIS-200) imaging system. Animals were euthanized when they showed signs of MB.
In vivo somatic cell gene transfer in transgenic mice:
To induce medulloblastomas in immunocompetent mice, we used a version of the RCAS/tv-a somatic cell gene transfer system to transfer and express the Shh gene in Nestin-expressing cells in the cerebellum (Rao et al., 2003). To initiate gene transfer, we injected retrovirus packaging cells (DF-1 cells transfected with and producing recombinant RCAS retrovirus) into the lateral cerebellum of the mouse from an entry point just posterior to the lambdoid suture of the skull (bilateral injections of 105 cells in 1–2 μl of phosphate-buffered saline). For experiments involving simultaneous transfer of multiple genes, we prepared cell pellets by mixing equal numbers of retrovirus-producing cells for each gene. We injected mice within 72 hours after birth because the number of Nestin + cells decreases progressively during the course of neural differentiation. The mice were sacrificed when signs of increased intracranial pressure became apparent, indicated by enlarging head circumference (a sign of hydrocephalus), head tilt, gait ataxia, or failure to eat or drink. Asymptomatic mice were sacrificed 4 months after injection. The brains were fixed in formalin, and divided into quarters by parallel incisions in the coronal plane. To identify spinal dissemination, we fixed whole spinal column preparations in formalin for 48–72 hours and then removed the spinal cord by microdissection.
Tissue preparation for flow-cytometry:
Mouse showing neurological signs of late stage brain tumours or deemed endpoint by the clinical veterinarian were sacrificed under general anesthesia by exsanguination by the way of cardiac puncture. The collected blood (500 to 700ul) was quickly prepared for flow-cytometry using Lympholite-M (Cedarlane) accordingly to the manufacturer protocol. Brain tumors and spinal cord are quickly dissected and kept on ice until dissociation. Brain and spinal cords were imaged under a fluorescence stereoscope to measure extent of metastasis, the whole dorsal and ventral aspects of the CNS were examined. Upon stereoscopic examination the primary tumor samples as well as the spinal cords were quickly dissociated in Enzyme-Free cell dissociation solution (Thermo-Fisher) accordingly to the manufacturer recommendations. Dissociated cells were resuspended in PBS-1%BSA for flow cytometry stainings.
Flow-cytometry to detect MB CTC in mouse blood:
Mouse PBMC were resuspended in 500ul of PBS-1%BSA, mouse dissociated spinal cords were resuspended in PBS-1%BSA at 5–10×10^6/ml and analyzed by flow cytometry using a LSR II (BectonDickinson). As positive CTL single cell suspensions from the primary tumor were always run at the same time as the test blood or spinal sample. The primary tumor sample was used to set voltages for FSC and SSC and fluorescence, dead cells were excluded by PI, SytoxBlue or Dapi. Flow-cytometry data were analyzed using FlowJo.
Histology for NCAM and H&E:
Mouse showing signs of late stage brain tumours were sacrificed and tissue harvested for histological examination. Brain was cut accordingly to the sagittal plane, while the spinal cord was cut transversally in 4–6 pieces, and sectioned accordingly. After 48–72hrs in formalin the specimens were embedded in paraffin and sectioned for histochemical analysis. Extent and location of primary tumors and metastasis was evaluated by standard hematoxylin and eosin staining. NCAM (CD56) (Thermofisher cat#180152) staining of mouse xenograft samples was performed at the Pathology Research Program (UHN) with a Zymed 18–0152 Clone 123C3, using Tris-EDTA pH9.0 1:100 dilution for 1hr.
Histology for mCherry.
Tissue sections were cut 4 μm thick, mounted on glass slides, deparaffinized with toluene, hydrated through a descending series of ethanol, autoclaved in a citrate-based antigen retrieval solution (Vector Laboratories, Burlingame, CA) for 5 min, and cooled to room temperature. Sections were then treated with H2O2 (1% v/v) for 10 min to quench endogenous peroxidase activity and washed with phosphate-buffered saline. After immersion in normal horse serum (2%), sections were incubated with a goat polyclonal antibody against the mCherry epitope tag (Biorbyt, LLC, San Francisco, CA), diluted 1:250–1:500 in phosphate-buffered saline, in a humid chamber at 4°C overnight. Immunoreactive staining was visualized using a biotin-free reporter enzyme staining system (ImmPRESS, Vector Laboratories), which utilizes a micropolymer of peroxidase and affinity-purified secondary antibodies. Diaminobenzidine was used as the chromogenic substrate and toluidine blue as a nuclear counterstain.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the exact value of n, the definition of center, dispersion, precision measures, statistical test and statistical significance are reported in the Figures and Figure Legends. Data is judged to be statistically significant when p <0.05. The statistical significance of Kaplan-Meier survival estimates was assessed using the log-rank (Mantel-Cox) test. No power calculation was performed to pre-determine the samples sizes.
DATA AND SOFTWARE AVAILABILITY
Study DNA and RNA sequencing data and metadata were deposited at: EGAD00001003907. This dataset contains 9 WGS BAM files 9 targeted sequencing BAM files (Fig. 1a–b), and 70 RNA-Seq BAM files (Fig 4b). WGS paired-end reads were aligned to the GRCh37-lite genome using BWA v0.5.7 and samples sequenced over multiple lanes were merged using Picard. Targeted sequencing paired ends reads were demultiplexed from the pooled flow-cells and aligned to the GRCh37-lite genome using BWA-SW. RNAseq paired-end reads were demultiplexed from the pooled flowcell using bcl2fastq and aligned to GRCh37-lite genome-plus-junctions references using BWA v0.5.7. RNAseq reads that mapped to junction regions were then repositioned back to the genome using JAGuaR.
Supplementary Material
Supplementary figure S1 relative to figure 1. A) Clonality comparison of somatic mutations identified by whole genome sequencing of peripheral blood as compared to primary tumor versus patient matched metastases reveals that putative circulating tumor cells may carry mutations restricted to the metastatic compartment and mutations shared between the primary tumor and the metastasis. B) Density plots show the base quality support for the reference base (black) and the alternate allele (red) in each compartment (primary tumor, metastatic sample, and matched germline). Support for the alternate allele is also shown for flanking bases (within 10bp) that match the reference allele (sequence maych), where no mutation is expected (blue line) C) Gating strategy used to identify CTCs in human blood pre-therapy: single, hoechst positive and CD45-ve cells are gated and analyzed for NCAM expression. NCAM positive cells were visually verified to have a morphology compatible with tumor cells. D) Representative images of diffuse NCAM positivity in two primary MBs (scale bar 10 m left, 50 m right).
Supplementary figure S2 relative to figure 2. A) naïve NSG mice blood (0.5 to 0.7ml each mouse) was collected and analyzed for the presence of rare auto-fluorescent cells. A total of 3 single events over 9 mice tested were found, indicating extremely low levels of auto-fluorescent cells, MFI for the recorded events was also extremely low when compared to real CTC from tumor implanted mice (top scatter plots). B) end-point PCR for the GFP transgene on the blood of mice implanted with Med-411FH and MB002, the presence of GFP specific band is visible in the Med-411FH implanted mice.
Supplementary figure S3 relative to figure 3. A) Med-411FH cells implanted in the donor parabiont metastasize to the abdomen in only two cases. B) Necropsy and IHC summary for the parabiosis experiments.
Supplementary figure S4 relative to Figure 4. A) Loss of human chromosome 10q, that contains PTEN, accompanied or not by gain of human chromosome 17q (where CCL2 maps) are not significantly more common in Shh MB patients with leptomeningeal dissemination. B) In contrast, CCL2 gain and PTEN loss are associated with higher metastatic stage for Grp3 tumors (p<0.05 Fisher’s exact test). C) In Grp4 tumors, PTEN loss is associated with lower stages MBs, while the combination of PTEN loss and CCL2 gain (highly prevalent in Grp4 as iso17q) is not correlated with higher M stage.
Supplementary figure S5 relative to figure 5. A) Protein levels of CCL2 in several MB cells in culture, compared with others: ATRT (BT16), GBM (GBM-H118) and breast cancer (MDA-MB-231) (n=3, samples means and SD are shown). B) Western blot showing overexpression of CCL2 and CCR2 in ONS76 cells, and of CCL2 in MB002. C-E) Representative fluorescence images of mouse spinal cord after CB implantations of ONS76 overexpressing CCL2, CCR2 or the combination (C), CCL2 in MB002 (D) or D425S shCCL2 (E) scale bars white, 2mm; yellow 1mm. The images labeled by a star have been included in main figure 5. In few instances of very dim fluorescence signal mcherry only or GFP only images are presented, instead of merged images. When spinal cords have no detectable metastases a representative negative image is shown (as mcherry, GFP or merged image).
Supplementary Figure S6 relative to Figure 6. Mice implanted with MP-Luc cells transduced with CCL2 show comparable levels of metastasis than CTL mice (see figure 6A), co-infection with CCL2 and CCR2 leads to an increase in metastasis prevalence (66%) compared to CTLs (7.3%, p=0.008 Fisher’s Exact Test with mid-P adjustment). B) Mice implanted with MPLuc CTL, overexpressing CCL2, CCR2, or both show similar survival.
Table S1 relative to Figure 1: Summary of Deep-Sequencing analysis A) Somatic mutations from the primary or matched metastatic tumor with low variant allele frequency (VAF) support in the germline. The number and frequency of reads in each compartment supporting the mutation is shown for each shortlisted position. (Pri: primary tumorl Met: metastatic sample; Germ: matched germline). B-D) VAF, read coverage, and baseQ statistics (median value and standard deviation (SD) is shown for a particular patient, at all deeply sequenced bases with somatic mutations in at least one tumor compartment.
Table S2 Relative to Figure 2: Differentially expressed genes in primary grp 3 medulloblastoma vs the matched metastasis. Differentially expressed genes in 2 primary grp3 medulloblastoma and 5 matched metastasis are listed, significantly upregulated genes are ranked accordingly to Met vs Pri Log2FC.
Key Resources Table
| SOURCE | IDENTIFIER | |
|---|---|---|
| Antibodies | ||
| Biotin Mouse Anti-Human CD56 Clone B159 | BD bioscience | Cat# 555515 |
| human CD45 PE-CF594 clone HI30 | BD bioscience | cat# 562279 |
| anti MCP1 Antibody (2D8) | Novus Biologicals | NBP2–22115 |
| Anti CCR2 Antibody (E68) | Novus Biologicals | NB110–55674 |
| CD56, N-CAM Mouse Monoclonal Antibody (clone 123C3 | Thermofisher | Cat# 180152 |
| goat polyclonal antibody mCherry | Biorbyt, LLC, San Francisco, CA | orb11618 |
| Bacterial and Virus Strains | ||
| pLVX-IRES-mCherry | Clontech | 631987 |
| pLVX-IRES-zsGreen1 | Clontech | 632187 |
| Biological Samples | ||
| Patient-Derived Xenografts | www.btrl.org | Med-411FH |
| Patient-Derived Xenografts | This paper | MMB |
| Patient-Derived Xenografts | This paper | MMS |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Critical Commercial Assays | ||
| Deposited Data | ||
| WGS, targeted sequencing and RNAseq | Morrissy et al., 2016 and this paper | EGAD00001003907 |
| Affymetrix expression array | Wang et al., 2015 | GSE63670 |
| Experimental Models: Cell Lines | ||
| Medulloblastoma cell line | Yamada et al., 1989 | ONS76 |
| Medulloblastoma cell line | S. Mitra (Gholamin et al., 2017) | D425 |
| Medulloblastoma cell line | S. Mitra (Gholamin et al., 2017) | MB002 |
| Experimental Models: Organisms/Strains | ||
| SB driven Medulloblastoma mouse model Ptch+/−/Math1-SB11/T2Onc | Wu et al, 2012 | N/A |
| B6.129S4-Ccr2tm1Ifc/J | Jackson lab | Stock No: 004999 |
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | Jackson lab | Stock no: 005557 |
| RCAS/tv-a | Rao et al., 2003 | N/A |
| Oligonucleotides | ||
| Recombinant DNA | ||
| pLKO-SH clone | The RNAi Consortium | TRCN0000006283 |
| pLKO-SH clone | The RNAi Consortium | TRCN0000338418 |
| pLKO-SH clone | The RNAi Consortium | TRCN0000338479 |
| pLKO-SH clone | The RNAi Consortium | TRCN0000381382 |
| pLKO-SH clone | The RNAi Consortium | TRCN0000006279 |
| pLKO-SH clone | The RNAi Consortium | TRCN0000338480 |
| Software and Algorithms | ||
| SAMTools (version 0.1.13) | Li et al., 2009 | samtools.sourceforge.net/ |
| Picard MarkDuplicates.jar (version 1.71) | NA | broadinstitute.github.io/picard/ |
| Strelka | Saunders et al., 2012 | https://github.com/Illumina/strelka |
| FlowJoV10 | https://www.flowjo.com/solutions/flowjo | |
| Ideas Software | Amnis Corporation | http://www.emdmillipore.com/ |
| Other | ||
Highlights:
Medulloblastoma patients show circulating tumor cells in the blood at diagnosis
Medulloblastoma circulating tumor cells can home to the meninges to form metastases
The CCL2-CCR2 axis drives meningeal dissemination of medulloblastoma.
Acknowledgements
MDT is supported by the National Institutes of Health (R01CA148699 and R01CA159859), The Pediatric Brain Tumour Foundation, The Terry Fox Research Institute, The Canadian Institutes of Health Research, The Cure Search Foundation, b.r.a.i.n.child, Meagan’s Walk, Genome Canada, Genome BC, and the Ontario Institute for Cancer Research. MDT is also supported by a Canadian Cancer Society Research Institute Impact grant and by a Stand Up To Cancer (SU2C) St. Baldrick’s Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT1113) and SU2C Canada Cancer Stem Cell Dream Team Research Funding (SU2C-AACR-DT-19–15) provided by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, with supplementary support from the Ontario Institute for Cancer Research through funding provided by the Government of Ontario. Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. Research was also supported by The Terry Fox Research Institute. MDT is supported by The Garron Family Chair in Childhood Cancer Research at the Hospital for Sick Children and The University of Toronto. LG was supported by the Davis M. Ferguson Memorial Fund at ABTA. Alex’s Lemonade Stand Young Investigator Award supported VR. This study was conducted with the support of the Ontario Institute for Cancer Research through funding provided by the Government of Ontario. FMGC is supported by the Stephen Buttrum Brain Tumour Research Fellowship, granted by Brain Tumour Foundation of Canada. VR is supported by a CIHR fellowship and an Alberta Innovates-Health Solutions Clinical Fellowship. The huntsman Cancer Foundation supports D.W.F. We would like to thank S. Archer for technical writing and C. Smith for artwork.
Footnotes
The authors declare no conflict of interest.
References:
- Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, and Bentires-Alj M (2014). Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 515, 130–133. [DOI] [PubMed] [Google Scholar]
- Chang CH, Housepian EM, and Herbert C Jr. (1969). An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology 93, 1351–1359. [DOI] [PubMed] [Google Scholar]
- Chu HX, Arumugam TV, Gelderblom M, Magnus T, Drummond GR, and Sobey CG (2014). Role of CCR2 in inflammatory conditions of the central nervous system. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 34, 1425–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladdy RA, Taylor MD, Williams CJ, Grandal I, Karaskova J, Squire JA, Rutka JT, Guidos CJ, and Danska JS (2003). The RAG-1/2 endonuclease causes genomic instability and controls CNS complications of lymphoblastic leukemia in p53/Prkdc-deficient mice. Cancer Cell 3, 37–50. [DOI] [PubMed] [Google Scholar]
- Gholamin S, Mitra SS, Feroze AH, Liu J, Kahn SA, Zhang M, et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci Transl Med [Internet]. 2017;9:eaaf2968. [DOI] [PubMed] [Google Scholar]
- Lumpkin EA, Collisson T, Parab P, Omer-Abdalla A, Haeberle H, Chen P, Doetzlhofer A, White P, Groves A, Segil N, and Johnson JE (2003). Math1-driven GFP expression in the developing nervous system of transgenic mice. Gene Expr.Patterns 3, 389–395. [DOI] [PubMed] [Google Scholar]
- Morrissy AS, Garzia L, Shih DJ, Zuyderduyn S, Huang X, Skowron P, Remke M, Cavalli FM, Ramaswamy V, Lindsay PE, et al. (2016). Divergent clonal selection dominates medulloblastoma at recurrence. Nature 529, 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moxon-Emre I, Bouffet E, Taylor MD, Laperriere N, Scantlebury N, Law N, Spiegler BJ, Malkin D, Janzen L, and Mabbott D (2014). Impact of Craniospinal Dose, Boost Volume, and Neurologic Complications on Intellectual Outcome in Patients With Medulloblastoma. J Clin Oncol. [DOI] [PubMed] [Google Scholar]
- Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ, Witt H, Korshunov A, Read TA, Sun JL, et al. (2012). An animal model of MYC-driven medulloblastoma. Cancer Cell 21, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradeep S, Kim SW, Wu SY, Nishimura M, Chaluvally-Raghavan P, Miyake T, Pecot CV, Kim SJ, Choi HJ, Bischoff FZ, et al. (2014). Hematogenous metastasis of ovarian cancer: rethinking mode of spread. Cancer Cell 26, 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, and Pollard JW (2011). CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ, Shih DJ, Luu B, Dubuc AM, Northcott PA, et al. (2013). Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 14, 1200–1207.24140199 [Google Scholar]
- Rao G, Pedone CA, Coffin CM, Holland EC, and Fults DW (2003). c-Myc enhances sonic hedgehog-induced medulloblastoma formation from nestin-expressing neural progenitors in mice. Neoplasia 5, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, et al. (2009). Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med 361, 1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders Christopher T.; Wong Wendy; Swamy Sajani; Becq Jennifer; Murray Lisa J.; R. Keira Cheetham Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012; doi: 10.1093/bioinformatics/bts271 [DOI] [PubMed] [Google Scholar]
- Semple BD, Kossmann T, and Morganti-Kossmann MC (2010). Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 30, 459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Shimizu K, Tamura K, Okamoto Y, Matsui Y, Moriuchi S, Park K, Mabuchi E, Yamamoto K, Hayakawa T, et al. (1989) Establishment and biological characterization of human medulloblastoma cell lines. No To Shinkei. Jul;41(7):695–702 [PubMed] [Google Scholar]
- Wang X, Dubuc AM, Ramaswamy V, Mack S, Gendoo DM, Remke M, Wu X, Garzia L, Luu B, Cavalli F, et al. (2015). Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol 129, 449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf MJ, Hoos A, Bauer J, Boettcher S, Knust M, Weber A, Simonavicius N, Schneider C, Lang M, Sturzl M, et al. (2012). Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell 22, 91–105. [DOI] [PubMed] [Google Scholar]
- Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJ, Witt H, Croul S, Bouffet E, Fults DW, Eberhart CG, et al. (2012). Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 482, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure S1 relative to figure 1. A) Clonality comparison of somatic mutations identified by whole genome sequencing of peripheral blood as compared to primary tumor versus patient matched metastases reveals that putative circulating tumor cells may carry mutations restricted to the metastatic compartment and mutations shared between the primary tumor and the metastasis. B) Density plots show the base quality support for the reference base (black) and the alternate allele (red) in each compartment (primary tumor, metastatic sample, and matched germline). Support for the alternate allele is also shown for flanking bases (within 10bp) that match the reference allele (sequence maych), where no mutation is expected (blue line) C) Gating strategy used to identify CTCs in human blood pre-therapy: single, hoechst positive and CD45-ve cells are gated and analyzed for NCAM expression. NCAM positive cells were visually verified to have a morphology compatible with tumor cells. D) Representative images of diffuse NCAM positivity in two primary MBs (scale bar 10 m left, 50 m right).
Supplementary figure S2 relative to figure 2. A) naïve NSG mice blood (0.5 to 0.7ml each mouse) was collected and analyzed for the presence of rare auto-fluorescent cells. A total of 3 single events over 9 mice tested were found, indicating extremely low levels of auto-fluorescent cells, MFI for the recorded events was also extremely low when compared to real CTC from tumor implanted mice (top scatter plots). B) end-point PCR for the GFP transgene on the blood of mice implanted with Med-411FH and MB002, the presence of GFP specific band is visible in the Med-411FH implanted mice.
Supplementary figure S3 relative to figure 3. A) Med-411FH cells implanted in the donor parabiont metastasize to the abdomen in only two cases. B) Necropsy and IHC summary for the parabiosis experiments.
Supplementary figure S4 relative to Figure 4. A) Loss of human chromosome 10q, that contains PTEN, accompanied or not by gain of human chromosome 17q (where CCL2 maps) are not significantly more common in Shh MB patients with leptomeningeal dissemination. B) In contrast, CCL2 gain and PTEN loss are associated with higher metastatic stage for Grp3 tumors (p<0.05 Fisher’s exact test). C) In Grp4 tumors, PTEN loss is associated with lower stages MBs, while the combination of PTEN loss and CCL2 gain (highly prevalent in Grp4 as iso17q) is not correlated with higher M stage.
Supplementary figure S5 relative to figure 5. A) Protein levels of CCL2 in several MB cells in culture, compared with others: ATRT (BT16), GBM (GBM-H118) and breast cancer (MDA-MB-231) (n=3, samples means and SD are shown). B) Western blot showing overexpression of CCL2 and CCR2 in ONS76 cells, and of CCL2 in MB002. C-E) Representative fluorescence images of mouse spinal cord after CB implantations of ONS76 overexpressing CCL2, CCR2 or the combination (C), CCL2 in MB002 (D) or D425S shCCL2 (E) scale bars white, 2mm; yellow 1mm. The images labeled by a star have been included in main figure 5. In few instances of very dim fluorescence signal mcherry only or GFP only images are presented, instead of merged images. When spinal cords have no detectable metastases a representative negative image is shown (as mcherry, GFP or merged image).
Supplementary Figure S6 relative to Figure 6. Mice implanted with MP-Luc cells transduced with CCL2 show comparable levels of metastasis than CTL mice (see figure 6A), co-infection with CCL2 and CCR2 leads to an increase in metastasis prevalence (66%) compared to CTLs (7.3%, p=0.008 Fisher’s Exact Test with mid-P adjustment). B) Mice implanted with MPLuc CTL, overexpressing CCL2, CCR2, or both show similar survival.
Table S1 relative to Figure 1: Summary of Deep-Sequencing analysis A) Somatic mutations from the primary or matched metastatic tumor with low variant allele frequency (VAF) support in the germline. The number and frequency of reads in each compartment supporting the mutation is shown for each shortlisted position. (Pri: primary tumorl Met: metastatic sample; Germ: matched germline). B-D) VAF, read coverage, and baseQ statistics (median value and standard deviation (SD) is shown for a particular patient, at all deeply sequenced bases with somatic mutations in at least one tumor compartment.
Table S2 Relative to Figure 2: Differentially expressed genes in primary grp 3 medulloblastoma vs the matched metastasis. Differentially expressed genes in 2 primary grp3 medulloblastoma and 5 matched metastasis are listed, significantly upregulated genes are ranked accordingly to Met vs Pri Log2FC.
Data Availability Statement
Study DNA and RNA sequencing data and metadata were deposited at: EGAD00001003907. This dataset contains 9 WGS BAM files 9 targeted sequencing BAM files (Fig. 1a–b), and 70 RNA-Seq BAM files (Fig 4b). WGS paired-end reads were aligned to the GRCh37-lite genome using BWA v0.5.7 and samples sequenced over multiple lanes were merged using Picard. Targeted sequencing paired ends reads were demultiplexed from the pooled flow-cells and aligned to the GRCh37-lite genome using BWA-SW. RNAseq paired-end reads were demultiplexed from the pooled flowcell using bcl2fastq and aligned to GRCh37-lite genome-plus-junctions references using BWA v0.5.7. RNAseq reads that mapped to junction regions were then repositioned back to the genome using JAGuaR.