

Abstract
Introduction
Pheochromocytoma (PHEO) is a rare catecholamine-producing tumour arising from chromaffin cells in the sympatho-adrenal system, and can present as asymptomatic adrenal incidentaloma (AI).
Patient
We describe the case of a 61-year-old woman with a right adrenal mass incidentally discovered, who was biochemically characterized with subclinical hypercortisolism (SH). The patient was scheduled for adrenalectomy because of increasing seizure of the right adrenal gland with a haemorrhagic and focal pseudocystic appearance macroscopically, incidental histological and immunohistochemical PHEO, and micronodular cortico-adrenal hyperplasia.
Discussion
This report describes a rare case of incidental non-functioning PHEO coexisting with corticomedullary hyperplasia and SH.
LEARNING POINTS
Rare presentation of coexisting common (adrenal incidentaloma and subclinical Cushing’s syndrome) and uncommon (pheochromocytoma) endocrinological conditions.
Keywords: Adrenal incidentaloma, subclinical hypercortisolism, pheochromocytoma, adrenal hyperplasia
INTRODUCTION
Pheochromocytoma (PHEO) is a rare catecholamine-producing tumour arising from chromaffin cells in the sympatho-adrenal system, and has a prevalence of 0.1–0.6%[1]. Hypertension, either paroxysmal or sustained, is the most consistent finding in PHEO, and the classic triad of symptoms (headache, palpitations and diaphoresis) is present in more than 50% of patients[2]. However, PHEO can present as asymptomatic adrenal incidentaloma (AI) revealed during abdominal imaging.
CASE REPORT
In May 2016, a 61-year-old woman was referred to our centre for evaluation of a right adrenal mass (diameter 44 mm) and arterial hypertension. She did not complain of any relevant symptoms. In July 2010, the patient had been evaluated in another hospital with an incidental finding of a right adrenal mass (diameter 28 mm) (Fig. 1), with biochemical and hormonal corticomedullary findings in the normal range (Table 1). The patient was treated with ramipril (10 mg/day) and amlodipine (10 mg/day).
Figure 1.
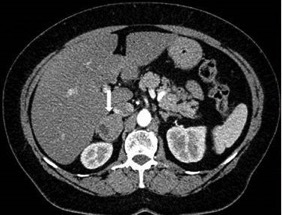
Computed tomography of the abdomen showing an incidental finding of a right adrenal mass (diameter 28 mm)
Table 1.
Endocrinological data of the patient 6 years before diagnosis, at diagnosis and 6 months after laparoscopic removal of the tumour
| July 2010 | May 2016 | December 2016 | Normal Values | |
|---|---|---|---|---|
| Serum | ||||
| Plasma aldosterone (pg/ml) | 128 | 106 | 86 | 30–160 |
| Plasma cortisol (mg/dl) | 11 | 15.6 | 14.7 | 6–23 |
| Plasma renin activity (ng/ml/h) | 3 | 0.4 | 1.2 | 0.5–3 |
| ACTH (pg/ml) | 19 | 7 | 22 | 8–50 |
| Plasma cortisol after 1 mg of dexamethasone (overnight test) | - | 3.5 | - | <1.8 |
| Urine | ||||
| Urinary free cortisol (mg/24 h) | 160 | 256.8 | 198.2 | 38–208 |
| Urinary mandelic acid (mg/24 h) | 5 | 6.6 | 5.3 | 0–10.6 |
| Urinary total catecholamine (mg/24 h) | 70 | – | – | – |
| Urinary aldosterone secretion (mg/24 h) | – | 13.9 | 8.6 | 2.84–34 |
| Urinary total metanephrines (mg/24 h) | – | 264 | 55 | 20–350 |
On physical examination, the patient’s heart rate was 61 bpm and her blood pressure (BP) was 150/90 mmHg without postural drop, her BMI was 30.7 kg/m2, and she had a waist circumference of 107 cm. No signs of hypercortisolism, such as buffalo hump, moon face, striae or skin abnormalities, were seen. Ambulatory blood pressure monitoring (ABPM) revealed a global mean BP of 135/85 mmHg without a drop in nocturnal BP (non-dipping profile). The EKG was unremarkable. Routine blood tests were normal. Hormonal evaluation was performed after adequate preparation (Table 1). In particular, urinary free cortisol (UFC) and plasma cortisol (PC) following an overnight dexamethasone test were high and associated with a low plasma ACTH value. A diagnosis of subclinical hypercortisolism (SH) was made, and considering the increasing diameter of the adrenal mass, the patient underwent a laparoscopic adrenalectomy. Intraoperatively, the patient did not experience any haemodynamic alterations. The post-operative course was uneventful, and the patient was discharged from hospital on a cortisol substitute and attended regular follow-up appointments.
PATHOLOGICAL EXAMINATIONS
Macroscopically, the right adrenal gland measured 6×4×3.2 cm and was a rusty-coloured nodule with a haemorrhagic and focal pseudocystic appearance. The remnant adrenal gland had a cortex with a micronodular pattern around a widened greyish medulla. Histology revealed a lesion with tumour cells arranged in well-defined nests. The nuclei were round or oval with prominent nucleoli, occasionally containing inclusion-like striatures (cytoplasmic invaginations). The mitotic rate was 3/10 HPF (Fig. 2). No necrosis or vascular invasion was recorded. A multinodular pattern was observed in the residual cortex, with an obvious zona fasciculata. The medulla of the normal gland showed widening (cortex/medulla ratio <10:1), with cells arranged in a nest. Immunohistochemistry using antibody against chromogranin, synaptophysin and neuron-specific enolase showed positive results, but anti-inhibin was negative. S-100 protein immunoreactivity showed a conspicuous number of substentacular cells. Immunohistochemical studies using antibody against ACTH were positive in the adrenal cortex and medulla (Fig. 3). A final diagnosis was made of adrenal PHEO accompanied by micronodular cortico-medullary hyperplasia. The patient was examined 6 and 12 months post-operatively and showed normal adrenal hormone values (Table 1). No further cortisol treatment was given.
Figure 2.
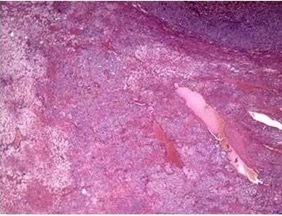
Histological results for the adrenal cortex showing a multinodular pattern in the zona fasciculata
Figure 3.
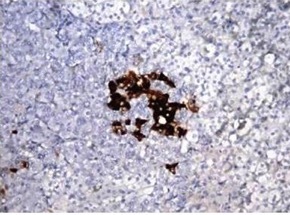
Immunohistochemical results showing positive antibody against ACTH in the adrenal cortex and medulla
DISCUSSION
This report describes a rare case of incidental non-functioning PHEO coexisting with corticomedullary hyperplasia and SH. Approximately 5–6.5% of AI are PHEO, and a few rare patients with PHEO are completely asymptomatic[1, 2]. In our patient, none of the specific signs and symptoms of hyperproduction of catecholamines were present at diagnosis. The clinical and biohumoral presentation of PHEO depends on the ability of the neoplasm to synthetize, metabolize and release different catecholamines and their metabolites. A tumour predominantly secreting epinephrine is usually associated with paroxysmal hypertension, while the norepinephrine variant is associated with sustained hypertension[3]. The lack of classic features of PHEO in our patient can be explained by the repeated normal urinary metanephrines. Hormonal evaluation revealed autonomous glucocorticoid production without specific signs and symptoms of hypercortisolism[4]. Histopathological examination of the adrenal mass revealed PHEO associated with corticomedullary hyperplasia.
The results of this study suggest that the adrenal medulla is not only influenced by the cortex but also influences the cortex directly or through the systemic circulation[5]. In particular, we hypothesize that the presence of immunoreactive ACTH in PHEO may be a possible cause of adrenocortical hyperplasia.
In conclusion, we present a rare case of silent adrenal PHEO incidentally discovered and associated with SH with corticomedullary hyperplasia.
Footnotes
Conflicts of Interests: The Authors declare that there are no competing interests.
REFERENCES
- 1.Tischler AS, Pacak K, Eisenhofer G. The adrenal medulla and extra-adrenal paraganglia: then and now. Endocr Pathol. 2014;25:49–58. doi: 10.1007/s12022-013-9286-3. [DOI] [PubMed] [Google Scholar]
- 2.Cotesta D, Petramala L, Serra V, Pergolini M, Crescenzi E, Zinnamosca L, et al. Clinical experience with pheochromocytoma in a single centre over 16 years. High Blood Press Cardiovasc Prev. 2009;16:183–193. doi: 10.2165/11530430-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.Mannelli M, Lenders JW, Pacak K, Parenti G, Eisenhofer G. Subclinical phaeochromocytoma. Best Pract Res Clin Endocrinol Metab. 2012;26:507–515. doi: 10.1016/j.beem.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petramala L, Cavallaro G, Galassi M, Marinelli C, Tonnarini G, Concistrè A, et al. Clinical benefits of unilateral adrenalectomy in patients with subclinical hypercortisolism due to adrenal incidentaloma: results from a single center. High Blood Press Cardiovasc Prev. 2017;24:69–75. doi: 10.1007/s40292-017-0182-7. [DOI] [PubMed] [Google Scholar]
- 5.Lefebvre H, Thomas M, Duparc C, Bertherat J, Louiset E. Role of ACTH in the interactive/paracrine regulation of adrenal steroid secretion in physiological and pathophysiological conditions. Front Endocrinol (Lausanne) 2016;7:98. doi: 10.3389/fendo.2016.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]