

Abstract
We have recently demonstrated that catalase content in mouse cardiac mitochondria is selectively elevated in response to high dietary fat, a nutritional state associated with oxidative stress and loss in insulin signaling. Catalase and various isoforms of glutathione peroxidase and peroxiredoxin each catalyze the consumption of H2O2. Catalase, located primarily within peroxisomes and to a lesser extent mitochondria, has a low binding affinity for H2O2 relative to glutathione peroxidase and peroxiredoxin. As such, the contribution of catalase to mitochondrial H2O2 consumption is not well understood. In the current study, using highly purified cardiac mitochondria challenged with micromolar concentrations of H2O2, we found that catalase contributes significantly to mitochondrial H2O2 consumption. In addition, catalase is solely responsible for removal of H2O2 in nonrespiring or structurally disrupted mitochondria. Finally, in mice fed a high-fat diet, mitochondrial-derived H2O2 is responsible for diminished insulin signaling in the heart as evidenced by reduced insulin-stimulated Akt phosphorylation. While elevated mitochondrial catalase content (∼50%) enhanced the capacity of mitochondria to consume H2O2 in response to high dietary fat, the selective increase in catalase did not prevent H2O2-induced loss in cardiac insulin signaling. Taken together, our results indicate that mitochondrial catalase likely functions to preclude the formation of high levels of H2O2 without perturbing redox-dependent signaling.
Keywords: H2O2, catalase, mitochondria, heart, insulin signaling
NEW & NOTEWORTHY
Catalase contributes to H2O2 consumption in cardiac mitochondria, particularly when mitochondria are compromised. Overexpression of catalase (50-fold) within mitochondria prevents diet-induced loss in insulin-stimulated Akt phosphorylation implicating mitochondrial-derived H2O2. Endogenous increases in mitochondrial catalase (∼50%) do not, however, prevent this loss. Catalase likely limits H2O2 toxicity without perturbing redox-dependent signaling.
the pro-oxidant molecule H2O2 has been implicated in redox regulation of multiple signal transduction pathways (12, 19, 20, 27) and in oxidative damage associated with disease. The level of H2O2 must therefore be tightly regulated, particularly in the mitochondria, a major intracellular source. H2O2 can be enzymatically consumed by catalase, glutathione peroxidases (Gpx), and the peroxiredoxins (Prdx). The mechanism by which catalase metabolizes H2O2 differs from both Gpx and Prdx in several ways (1, 17, 21). 1) Catalase utilizes a prosthetic heme group to reduce H2O2 whereas Gpx and Prdx rely on catalytic thiol groups on cysteine residues. 2) Catalase is reduced by oxidizing a second molecule of H2O2 while reduction of Gpx and Prdx requires the cofactors glutathione and thioredoxin, respectively. Replenishment of reduced glutathione and thioredoxin requires NADPH. 3) The binding affinity of catalase for H2O2 is in the low millimolar range vs. the low micromolar range for Gpx and Prdx (2, 8, 11, 14). 4) The maximum rate of catalysis for catalase is orders of magnitude greater than Gpx and Prdx (2, 8, 11, 14). 5) Catalase is largely present within peroxisomes and, to a lesser extent, in the mitochondria, while Gpx and Prdx isoforms are distributed throughout the cell. Given the kinetic properties and intracellular localization of catalase, the role of the enzyme in mitochondrial H2O2 homeostasis has been questioned (2, 5).
We have recently demonstrated that, in mice, catalase is present in the matrix of cardiac mitochondria (23), consistent with previous reports indicating localization of catalase in rat heart mitochondria (2, 18). In addition, we found that mitochondrial catalase content in the heart is selectively elevated relative to other antioxidant proteins within days of providing mice a high-fat diet (23), a nutritional state associated with oxidative stress (7, 22, 24, 28) and insulin resistance (6, 22, 26, 29). The current study sought to determine the contribution of catalase to H2O2 consumption by cardiac mitochondria and evaluate the physiological relevance of high-fat diet-induced increases in the expression of mitochondrial catalase in the heart. The study employed highly purified cardiac mitochondria challenged with micromolar concentrations of H2O2, inhibitors of antioxidant pathways, and catalase overexpressing and knockout mice. Our results indicate that while Gpx and/or Prdx play a predominant role in mitochondrial H2O2 consumption, catalase contributes significantly, particularly under conditions when mitochondrial function is compromised. In addition, increased expression of mitochondrial catalase in response to high dietary fat enhances the rate of mitochondrial H2O2 consumption but does not prevent H2O2-induced loss in cardiac insulin signaling as judged by insulin-stimulated Akt phosphorylation. Mitochondrial catalase may therefore function to maintain H2O2 concentrations within a range that prevents free radical damage while preserving redox signaling.
MATERIALS AND METHODS
Animals, dietary model, and sample preparation.
C57BL6/J mice were purchased from Jackson Laboratories and catalase global knockout mice (9) and mitochondrial targeted catalase overexpressing (mCAT) mice (25) were acquired from Eugene Chen at the University of Michigan and Peter Rabinovitch at the University of Washington, respectively. Mice were maintained on normal chow (LabDiet 5053 stock diet) or, for dietary studies, 6-wk-old mice were fed either a high-fat diet or a low-fat diet (60 vs. 10% total kcal from lard) purchased from Research Diets (D12492 and D12450B) for 1 wk. At the designated time points, mice were euthanized by cervical dislocation and the hearts were homogenized in buffer A (buffer A = 10 mM MOPS, 1 mM EDTA, 210 mM mannitol, and 70 mM sucrose, pH 7.4). Whole heart lysates were centrifuged at 550 g for 5 min and decanted through cheesecloth. Protein concentrations were determined using the BCA Protein Assay Reagent (Thermo Scientific) according to manufacturer's protocol. C57BL6/J mice (Jackson Laboratories) were used for all nondietary studies and hearts were prepared as described above. All experiments were approved by the Oklahoma Medical Research Foundation Institutional Animal Care and Use Committee.
Mitochondrial isolation.
Cardiac mitochondria were pelleted from whole heart lysates via centrifugation at 10,000 g for 10 min at 4°C. The supernatant was discarded and the mitochondrial pellets resuspended in ice cold buffer A. Mitochondria were further purified by differential centrifugation using a self-generating Percoll gradient (GE Healthcare Life Sciences). Isolated mitochondria were layered onto a 40% isotonic Percoll solution and centrifuged at 66,000 g for 40 min at 4°C. The purified mitochondria formed a single distinct layer within the gradient that was collected and diluted in 12 ml of ice-cold buffer A. The purified mitochondria were then separated from any residual Percoll by pelleting at 10,000 g for 15 min at 4°C. This final purified mitochondrial pellet was resuspended in ice-cold buffer A. As previously demonstrated, the purified mitochondria were free of peroxisomal contamination as judged by Western blot analysis (23).
Mitochondrial H2O2 consumption.
Isolated mitochondria (125 μg protein) were incubated at 37°C in a 250-μl reaction containing buffer B (buffer B = 10 mM MOPS, 210 mM mannitol, 70 mM sucrose, 5 mM K2HPO4, and 0.05% BSA, pH 7.4) in the presence or absence of 25 μM palmitoyl-carnitine and 100 μM malate, 100 μM sodium azide, or 1.0 mM iodoacetamide. All reagents were mixed with mitochondria 30 s before the addition of H2O2 to a final concentration of 25 μM. At designated time points, 20 μl of the reaction were mixed with 2.0 ml of buffer C (buffer C = 25 mM MOPS, 20 μM Amplex red, and 4 U horseradish peroxidase, pH 7.4). The concentration of H2O2 present at each time point was determined by measuring resorufin fluorescence with excitation and emission wavelengths of 570 and 585 nm, respectively (Horiba Fluoromax-4 spectrofluorometer). The rate of H2O2 consumption was calculated over the linear range (n = 3 separate mice for each condition).
Western blot analysis.
Whole heart lysates were immunoblotted with antibodies for pan Akt and pAkt Thr308 (Cell Signaling). Protein samples were incubated for 10 min at 70°C in the presence of 70 mM SDS, 100 mM DTT, and 50 μM MG132, resolved by gel electrophoresis (NuPAGE 10% Bis-Tris gel; Invitrogen), and transferred to PVDF membrane (Bio-Rad). Antibody incubations were carried out in 10 mM K2HPO4, 150 mM NaCl, 0.05% Tween-20, and 5% nonfat dry milk at pH 7.4. Secondary antibodies conjugated to horseradish peroxidase (Pierce) were visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Quantification of GSH and GSSG.
Mitochondria were allowed to respire for indicated amounts of time followed by extraction of GSH and GSSG into 5% metaphosphoric acid. Proteins were pelleted by centrifugation (10 min at 16,000 g, 4°C). The supernatant was filtered (0.45-μm syringe filters). GSH and GSSG were quantified by ion pairing reverse-phase HPLC and electrochemical detection (Shimadzu HPLC system, ESA Coularray electrochemical detector 5600A set at 750 mV). GSH and GSSG were eluted through a C18 column [Phenomenex Luna C18(2), 100 Å, 3 μM, 150 × 4.6 mm] at 0.5 ml/min using an isocratic mobile phase consisting of 25 mM NaH2PO4, 0.5 mM 1-octane sulfonic acid, and 4% acetonitrile, pH 2.7. GSH and GSSG concentrations were calculated employing GSH and GSSG standard curves constructed from peak areas.
Quantification of NADPH.
Isolated mitochondria were incubated for 5 min at 0.5 mg/ml in 10 mM MOPS, 210 mM mannitol, 70 mM sucrose, 5 mM K2HPO4, and 0.05% BSA at pH 7.4 with 25 μM palmitoyl-carnitine and 100 μM malate as respiratory substrates in the presence or absence of 100 μM sodium azide or 1.0 mM iodoacetamide. NADPH was extracted from mitochondria using an equal volume of 250 mM KOH. Protein was precipitated on ice (20 min) and pelleted by centrifugation (10 min at 16,000 g), and the supernatant was filtered (0.45-μM syringe filters) before analysis. High-performance liquid chromatography (Shimadzu LC-20A High Precision Binary Gradient HPLC system) and a UV/VIS diode array spectrometer were used to resolve and detect NADPH. The mobile phase consisted of 100 mM KH2PO4 and 1.0 mM tetrabutylammonium sulfate (TBAS) at pH 6.0 (buffer A) and CH3CN (buffer B) with a flow rate of 1.0 ml/min over an Eclipse Plus C18 column with 5-μM diameter beads, 4.6 × 150 mM in length (Agilent). NADPH was resolved (100-μl injection) using the following step-wise gradients of buffer A/B: 100%/0% for 2.5 min, 95%/5% for 5 min, and 85%/15% for 7.5 min. NADPH was detected by absorption at 340 nm and quantified based on the integrated area of standards.
Insulin stimulation.
Mice were fed ad libitum for 1 wk. Insulin (0.05 U/g body wt) was administered intraperitoneally 5 min before death and hearts were prepared as described above.
RESULTS AND DISCUSSION
Cardiac mitochondria consume H2O2 through multiple mechanisms.
We sought evidence for the contribution of catalase within the mitochondria to H2O2 consumption relative to other antioxidants. Highly purified cardiac mitochondria (23) were treated with 25 μM H2O2, an experimental condition previously shown to induce transient and reversible H2O2-mediated functional alterations rather than global damage to the mitochondria (3, 4, 13, 15). Where indicated, sodium azide and/or iodoacetamide were added to inhibit catalase and/or thiol-dependent antioxidant systems, respectively. In the presence of the respiratory substrates palmitoyl-carnitine and malate, mitochondria consumed H2O2 at a rate of 13.6 ± 2.3 nmol H2O2·min−1·mg protein−1 (Fig. 1A). Nonrespiring mitochondria consumed H2O2 at a rate less than half (5.0 ± 0.6 nmol H2O2·min−1·mg protein−1) that observed in respiring mitochondria (Fig. 1, A and B). Disruption of mitochondria by sonication increased the rate to 10.7 ± 1.2 nmol H2O2·min−1·mg protein−1 (Fig. 1C). Glutathione peroxidase and thioredoxin reductase require NADPH, a cofactor produced during mitochondrial respiration, to regenerate glutathione and thioredoxin, respectively. Thus the reduced rate of H2O2 consumption in the absence of respiratory substrate likely reflects diminished contribution from the glutathione peroxidase/glutathione reductase and/or peroxiredoxin/thioredoxin/thioredoxin reductase systems. Catalase therefore appears to contribute to mitochondrial H2O2 consumption, particularly in the absence of respiratory substrate and in disrupted mitochondria.
Fig. 1.
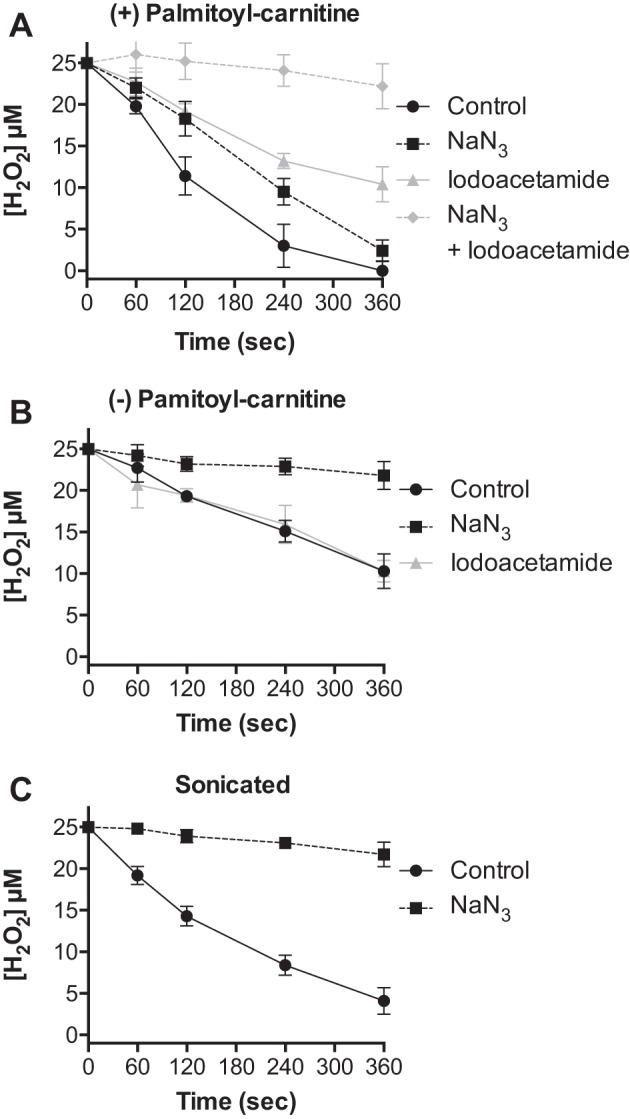
Catalase contributes to cardiac mitochondrial H2O2 consumption. The concentration of H2O2 was measured at indicated times following the addition of 25 μM H2O2 to isolated cardiac mitochondria (0.5 mg/ml): A: respiring on palmitoyl-carnitine (25 μM) and malate (100 μM) in the absence (black circles) or presence of sodium azide (black squares, 100 μM), iodoacetamide (gray triangles, 1.0 mM), or sodium azide and iodoacetamide (gray diamonds). B: with no respiratory substrate in the absence (black circles) or presence of sodium azide (black squares) or iodoacetamide (gray triangles). C: disrupted by sonication in the absence (black circles) or presence of sodium azide (black squares). Values are presented as the mean ± SD (n = 3).
Catalase contributes to cardiac mitochondrial H2O2 consumption.
We performed experiments to further investigate the contribution of catalase relative to antioxidant enzymes that rely on thiol oxidation and reduction (Gpx and Prdx) to mitochondrial H2O2 removal. Respiring cardiac mitochondria were treated with sodium azide to inhibit catalase or the thiol-reactive compound iodoacetamide to inhibit antioxidant pathways reliant on thiol chemistry. As shown in Fig. 1A, treatment with either sodium azide or iodoacetamide significantly reduced the rate of H2O2 consumption from 13.6 ± 2.3 to 7.8 ± 0.7 and 6.0 ± 0.6 nmol H2O2·min−1·mg protein−1, respectively. Treatment with both inhibitors nearly abolished the rate of H2O2 clearance. The rate of consumption in nonrespiring mitochondria was solely sensitive to sodium azide (Fig. 1B). In mitochondria disrupted by sonication, H2O2 consumption was largely blocked by sodium azide (Fig. 1C). These data indicate that catalase activity plays a substantial role in mitochondrial H2O2 consumption, particularly when mitochondrial respiration and/or structural integrity are impaired.
We determined whether the inhibitory effects of sodium azide were specific to catalase. The levels of oxidized and reduced glutathione were measured upon incubation of respiring mitochondria with 25 μM H2O2 in the presence and absence of sodium azide. In untreated mitochondria, H2O2 induced a significant drop in reduced glutathione (GSH) and an increase in oxidized glutathione (GSSG) that returned to baseline within 8 min (Fig. 2). As shown in Fig. 2, sodium azide treatment had no effects on glutathione oxidation in response to H2O2 or subsequent regeneration of GSH upon H2O2 consumption. In contrast, iodoacetamide significantly diminished GSH levels throughout the experimental protocol. While sodium azide is known to inhibit complex IV of the electron transport chain and the rate of respiration, sodium azide had no effect on the level of mitochondrial NADPH (2.85 ± 0.05 vs. 2.89 ± 0.09 nmol/mg in the absence and presence of NaN3, n = 3). Thus sodium azide does not appear to affect antioxidant enzymes dependent on thiol (Fig. 2) or NADPH oxidation and reduction. Iodoacetamide (1.0 mM) had no effect on respiration (not shown) or the level of NADPH (2.85 ± 0.05 vs. 2.70 ± 0.12 nmol/mg in the absence and presence of iodoacetamide, n = 3). We next evaluated the ability of sodium azide to influence the rate of H2O2 consumption in cardiac mitochondria isolated from mice lacking catalase. Respiring cardiac mitochondria from catalase knockout mice displayed a rate of H2O2 consumption that was not affected by sodium azide and nearly abolished by iodoacetamide treatment (Fig. 3A). Furthermore, the rate of H2O2 consumption by cardiac mitochondria from catalase knockout mice was nearly the same as that for mitochondria from wild-type mice treated with sodium azide (7.7 ± 0.4 vs. 7.8 ± 0.7 nmol H2O2·min−1·mg protein−1). Nonrespiring mitochondria from catalase knockout mice as well as mitochondria disrupted by sonication displayed no significant rate of H2O2 consumption (Fig. 3, B and C). These data indicate that the sodium azide-sensitive rate of mitochondrial H2O2 consumption is specific to catalase activity. Taken together, our findings provide evidence for a significant role of catalase in the consumption of H2O2 in cardiac mitochondria.
Fig. 2.
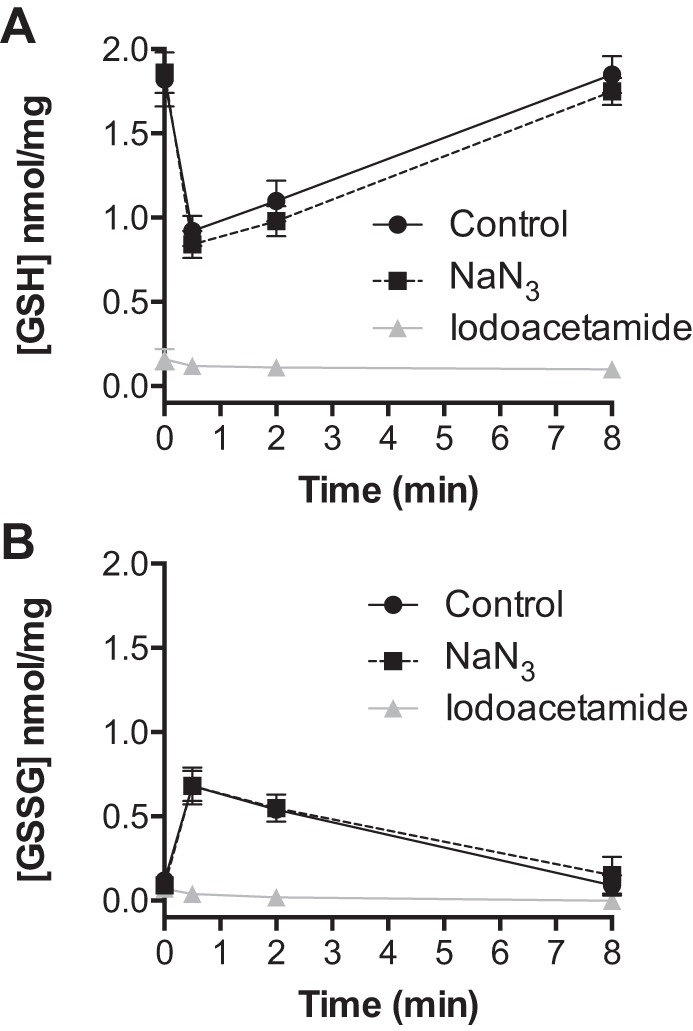
Treatment of respiring mitochondria with sodium azide has no effect on glutathione metabolism following exposure to H2O2. The concentrations of reduced (GSH) and oxidized (GSSG) glutathione were measured at indicated times following the addition of 25 μM H2O2 to isolated cardiac mitochondria (0.5 mg/ml) respiring on palmitoyl-carnitine (25 μM) and malate (100 μM). GSH (A) and GSSG (B) concentrations in the absence (black circles) or presence of sodium azide (black squares) or iodoacetamide (gray triangles). Values are presented as the mean ± SD (n = 3).
Fig. 3.
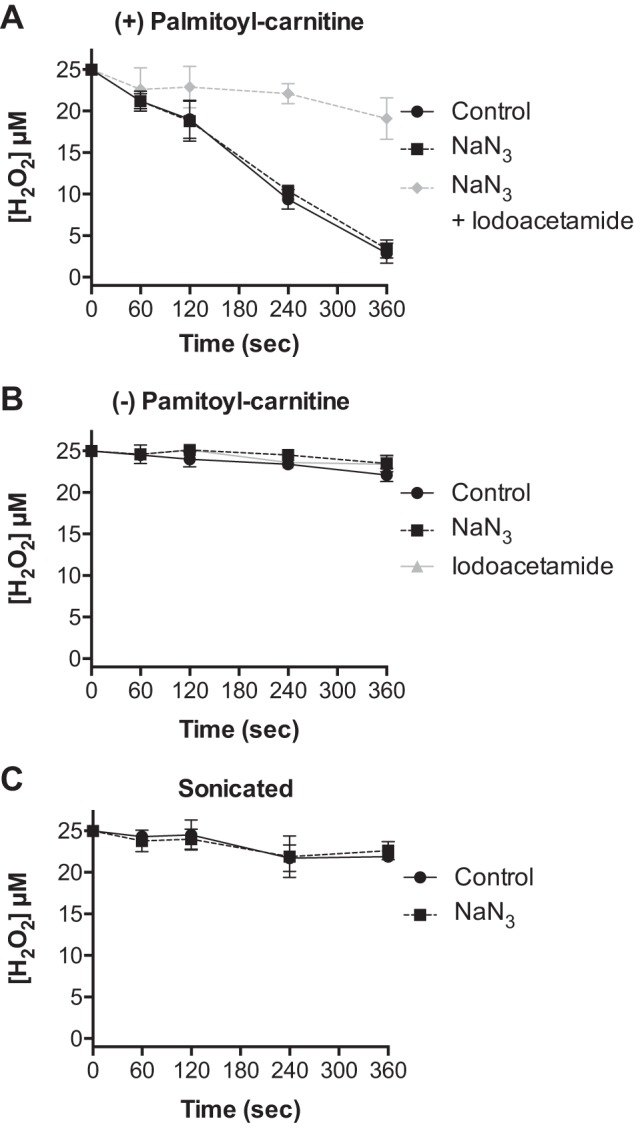
H2O2 consumption by cardiac mitochondria isolated from catalase knockout mice requires respiratory substrate and is insensitive to sodium azide. The concentration of H2O2 was measured at indicated times following the addition of 25 μM H2O2 to cardiac mitochondria (0.5 mg/ml) isolated from catalase knockout mice: A: respiring on palmitoyl-carnitine (25 μM) and malate (100 μM) in the absence (black circles) and presence of sodium azide (black squares) or sodium azide and iodoacetamide (gray diamonds). B: with no respiratory substrate in the absence (closed circles) or presence of sodium azide (closed squares) or iodoacetamide (gray triangles). C: disrupted by sonication in the absence (black circles) or presence of sodium azide (black squares). Values are presented as the mean ± SD (n = 3).
High-fat diet enhances mitochondrial H2O2 consumption.
We have previously shown that a high-fat diet increases cardiac mitochondrial catalase content and activity ∼50% (23). We therefore determined the effect of this increase on the rate of H2O2 consumption by the mitochondria. As shown in Fig. 4, cardiac mitochondria isolated from mice fed a high-fat relative to low-fat diet for 1 wk displayed a significantly faster rate of H2O2 consumption (Fig. 4A, 11.9 ± 0.7 vs. 8.8 ± 0.4 nmol H2O2·min−1·mg protein−1, respectively, P ≤ 0.002). Importantly, in the presence of sodium azide, respiring mitochondria from high-fat- and low-fat-fed mice displayed the same rate of H2O2 consumption (Fig. 4B, 7.1 ± 0.8 vs. 7.2 ± 0.4 nmol H2O2·min−1·mg protein−1, respectively). These results demonstrate that the diet-induced increase in catalase expression enhances the rate of H2O2 removal by cardiac mitochondria.
Fig. 4.
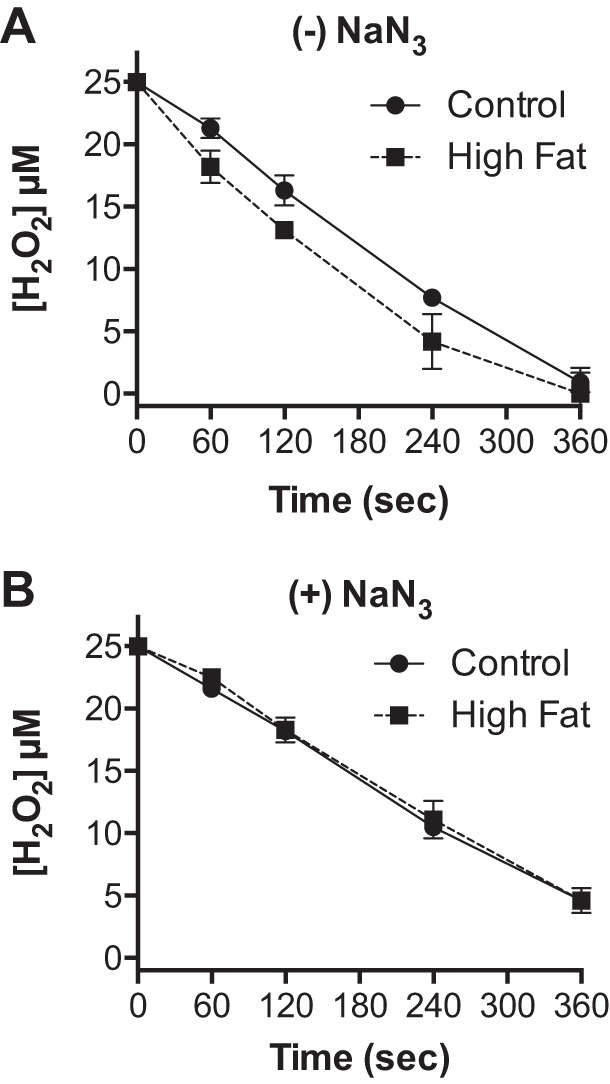
High dietary fat enhances catalase-dependent cardiac mitochondrial H2O2 consumption. Cardiac mitochondria were isolated from mice fed a low (black circles)- or high (black squares)-fat diet for 1 wk. The concentration of H2O2 was measured at indicated times following the addition of 25 μM H2O2 to mitochondria (0.5 mg/ml) respiring on palmitoyl-carnitine (25 μM) and malate (100 μM) in the absence (A) or presence (B) of sodium azide. Values are presented as the mean ± SD (n = 3).
Mitochondrial H2O2 production modulates cardiac insulin signaling.
High dietary fat is associated with oxidative stress and the development of insulin resistance in various tissues (6, 7, 16, 22–24, 28). Insulin signaling has been reported to be inhibited by H2O2 (7, 10, 16). As shown in Fig. 5A, despite a 50% increase in mitochondrial catalase content, wild-type mice fed a high-fat diet for 1 wk exhibited diminished insulin-stimulated Akt phosphorylation within the heart. In contrast, mice that overexpress human catalase targeted to the mitochondria (mCat) did not exhibit diet-induced decreases in insulin signaling (Fig. 5B). Cardiac mitochondria from mCat mice exhibit rapid and near complete consumption of a 25-μM bolus of H2O2 within 15 s (not shown), a far greater rate than that seen with cardiac mitochondria from high-fat fed wild-type mice (Fig. 4). This rapid rate of H2O2 removal reflects the 50-fold enrichment of catalase in cardiac mitochondria from mCat mice (25) relative to the 50% increase in wild-type mice fed a high-fat diet (23). Thus diminished insulin signaling induced by high dietary fat appears mediated by increases in mitochondrial H2O2 production. However, the magnitude of catalase upregulation is not sufficient to block H2O2-dependent loss of insulin signaling. We previously reported that insulin-stimulated Akt phosphorylation is depressed in the heart of wild-type mice fed a high-fat diet for 1 wk, with no change evident at 3 days (6). As shown in Fig. 5, C and D, catalase knockout mice exhibited a similar reduction in insulin-stimulated Akt phosphorylation at 1 wk of high dietary fat, with no change at 3 days. Thus endogenous catalase does not affect H2O2-induced loss in insulin signaling in response to high dietary fat.
Fig. 5.

Increased mitochondrial H2O2 production in response to high dietary fat impairs cardiac insulin signaling independent of endogenous changes in catalase. The ratio of phosphorylated (Thr308) to total Akt was determined in cardiac tissue from wild-type (A), mitochondrial targeted catalase overexpressing (mCAT; B), and catalase knockout (C and D) mice fed a low or high-fat diet for 1 wk or 3 days, as indicated. Values are presented as the mean ± SE. *P < 0.05, low vs. high-fat diet (n = 5).
Conclusions.
Our results offer evidence that catalase functions in concert with Gpx and Prdx to regulate mitochondrial H2O2 concentrations. Despite a relatively low binding affinity for H2O2, catalase has a high catalytic activity that is insensitive to mitochondrial respiration or changes in structural integrity (Fig. 1). As observed for mCat mice, 50-fold overexpression of human catalase (25) targeted to the mitochondria prevents diet-induced loss in insulin signaling (Fig. 5). These data provide in vivo evidence that mitochondrial derived H2O2 acts as a redox signal to inhibit cardiac insulin signaling. The endogenous increase in mitochondrial catalase (∼0.5-fold), previously observed in wild-type mice fed a high-fat diet for 1 wk (23), results in an increase in the rate of catalase-dependent H2O2 consumption (Fig. 4) but does not prevent diet-induced loss of cardiac insulin signaling (Fig. 5). We propose that selective induction of catalase expression in response to high-fat diet coupled with the enzyme's low binding affinity provides protection from H2O2-induced damage while permitting H2O2 concentrations that impact insulin signaling. As such, catalase may function to prevent toxic spikes in H2O2 concentration under normal and pathological conditions. In contrast, Prdx and Gpx, with binding affinities in the range of physiological concentrations of H2O2 (2, 8, 11), are likely responsible for regulating mitochondrial H2O2 concentrations conducive for redox-derived signaling.
GRANTS
This work was supported in part by National Institute of General Medical Sciences Grant GM-104934. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
P.M.R., M.K., and L.I.S. conception and design of research; P.M.R., A.C., M.K., and L.I.S. performed experiments; P.M.R., A.C., M.K., and L.I.S. analyzed data; P.M.R., A.C., M.K., and L.I.S. interpreted results of experiments; P.M.R. and L.I.S. prepared figures; P.M.R. drafted manuscript; P.M.R., M.K., and L.I.S. edited and revised manuscript; P.M.R., A.C., M.K., and L.I.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Melinda West for work with the care of the animals used in this study. We also thank Drs. Peter Rabinovitch (University of Washington) and Eugene Chen (University of Michigan) for providing the mCAT and catalase knockout mice, respectively.
REFERENCES
- 1.Alfonso-Prieto M, Biarnes X, Vidossich P, Rovira C. The molecular mechanism of the catalase reaction. J Am Chem Soc : 11751–11761, 2009. [DOI] [PubMed] [Google Scholar]
- 2.Antunes F, Han D, Cadenas E. Relative contributions of heart mitochondria glutathione peroxidase and catalase to H(2)O(2) detoxification in in vivo conditions. Free Radic Biol Med : 1260–1267, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Bulteau AL, Ikeda-Saito M, Szweda LI. Redox-dependent modulation of aconitase activity in intact mitochondria. Biochemistry : 14846–14855, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Bulteau AL, O'Neill HA, Kennedy MC, Ikeda-Saito M, Isaya G, Szweda LI. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science : 242–245, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev : 527–605, 1979. [DOI] [PubMed] [Google Scholar]
- 6.Crewe C, Kinter M, Szweda LI. Rapid inhibition of pyruvate dehydrogenase: an initiating event in high dietary fat-induced loss of metabolic flexibility in the heart. PLoS One : e77280, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fisher-Wellman KH, Neufer PD. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol Metab : 142–153, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaetani GF, Ferraris AM, Rolfo M, Mangerini R, Arena S, Kirkman HN. Predominant role of catalase in the disposal of hydrogen peroxide within human erythrocytes. Blood : 1595–1599, 1996. [PubMed] [Google Scholar]
- 9.Ho YS, Xiong Y, Ma W, Spector A, Ho DS. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J Biol Chem : 32804–32812, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Iwakami S, Misu H, Takeda T, Sugimori M, Matsugo S, Kaneko S, Takamura T. Concentration-dependent dual effects of hydrogen peroxide on insulin signal transduction in H4IIEC hepatocytes. PLoS One : e27401, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manta B, Hugo M, Ortiz C, Ferrer-Sueta G, Trujillo M, Denicola A. The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch Biochem Biophys : 146–154, 2009. [DOI] [PubMed] [Google Scholar]
- 12.Marinho HS, Real C, Cyrne L, Soares H, Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol : 535–562, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McLain AL, Cormier PJ, Kinter M, Szweda LI. Glutathionylation of alpha-ketoglutarate dehydrogenase: the chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radic Biol Med : 161–169, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicholls P, Fita I, Loewen PC. Enzymology and structure of catalases. Adv Inorganic Chem : 51–106, 2000. [Google Scholar]
- 15.Nulton-Persson AC, Szweda LI. Modulation of mitochondrial function by hydrogen peroxide. J Biol Chem : 23357–23361, 2001. [DOI] [PubMed] [Google Scholar]
- 16.Paglialunga S, Ludzki A, Root-McCaig J, Holloway GP. In adipose tissue, increased mitochondrial emission of reactive oxygen species is important for short-term high-fat diet-induced insulin resistance in mice. Diabetologia : 1071–1080, 2015. [DOI] [PubMed] [Google Scholar]
- 17.Prabhakar R, Vreven T, Morokuma K, Musaev DG. Elucidation of the mechanism of selenoprotein glutathione peroxidase (GPx)-catalyzed hydrogen peroxide reduction by two glutathione molecules: a density functional study. Biochemistry : 11864–11871, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Radi R, Turrens JF, Chang LY, Bush KM, Crapo JD, Freeman BA. Detection of catalase in rat heart mitochondria. J Biol Chem : 22028–22034, 1991. [PubMed] [Google Scholar]
- 19.Reczek CR, Chandel NS. ROS-dependent signal transduction. Curr Opin Cell Biol : 8–13, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science : 1882–1883, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS. Controlled elimination of intracellular H(2)O(2): regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal : 619–626, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Rindler PM, Crewe CL, Fernandes J, Kinter M, Szweda LI. Redox regulation of insulin sensitivity due to enhanced fatty acid utilization in the mitochondria. Am J Physiol Heart Circ Physiol : H634–H643, 2013. [DOI] [PubMed] [Google Scholar]
- 23.Rindler PM, Plafker SM, Szweda LI, Kinter M. High dietary fat selectively increases catalase expression within cardiac mitochondria. J Biol Chem : 1979–1990, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sainz N, Rodriguez A, Catalan V, Becerril S, Ramirez B, Gomez-Ambrosi J, Fruhbeck G. Leptin administration downregulates the increased expression levels of genes related to oxidative stress and inflammation in the skeletal muscle of ob/ob mice. Mediators Inflamm : 784343, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science : 1909–1911, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Ussher JR, Koves TR, Jaswal JS, Zhang L, Ilkayeva O, Dyck JR, Muoio DM, Lopaschuk GD. Insulin-stimulated cardiac glucose oxidation is increased in high-fat diet-induced obese mice lacking malonyl CoA decarboxylase. Diabetes : 1766–1775, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell : 1–14, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Vincent HK, Taylor AG. Biomarkers and potential mechanisms of obesity-induced oxidant stress in humans. Int J Obes (Lond) : 400–418, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Zhang L, Ussher JR, Oka T, Cadete VJ, Wagg C, Lopaschuk GD. Cardiac diacylglycerol accumulation in high fat-fed mice is associated with impaired insulin-stimulated glucose oxidation. Cardiovasc Res : 148–156, 2011. [DOI] [PubMed] [Google Scholar]