

SUMMARY
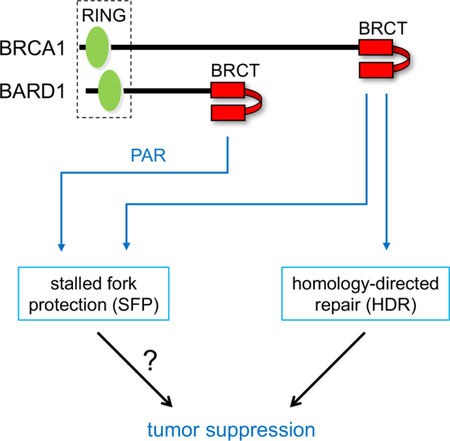
The BRCA1 tumor suppressor preserves genome integrity through both homology-directed repair (HDR) and stalled fork protection (SFP). In vivo, BRCA1 exists as a heterodimer with the BARD1 tumor suppressor, and both proteins harbor a phosphate-binding BRCT domain. Here we compare mice with mutations that ablate BRCT phospho-recognition by Bard1 (Bard1S563F and Bard1K607A) or Brca1 (Brca1S1598F). Brca1S1598F abrogates both HDR and SFP, suggesting that both pathways are likely impaired in most BRCA1-mutant tumors. Although not affecting HDR, the Bard1 mutations ablate poly(ADP-ribose)-dependent recruitment of BRCA1/BARD1 to stalled replication forks, resulting in fork degradation and chromosome instability. Nonetheless, Bard1S563F/S563F and Bard1K607A/K607A mice, unlike Brca1S1598F/S1598F mice, are not tumor-prone, indicating that HDR alone is sufficient to suppress tumor formation in the absence of SFP. Nevertheless, since SFP, unlike HDR, is also impaired in heterozygous Brca1/Bard1-mutant cells, SFP and HDR may contribute to distinct stages of tumorigenesis in BRCA1/BARD1 mutation carriers.
Keywords: BARD1, BRCA1, familial breast cancer, tumor suppression, BRCT domain, genome instability, stalled replication forks, poly(ADP-ribose), PARP inhibition
Graphical Abstract
eTOC Blurb:
Billing et al. describe the mechanism by which the BRCA1/BARD1 tumor suppressor complex is recruited to stalled DNA replication forks and identify molecular features of the complex required for maintenance of genome integrity though homology-directed repair (HDR) and stalled fork protection (SFP).
INTRODUCTION
Germline mutations of the BRCA1 tumor suppressor gene are a common cause of hereditary breast and ovarian cancer (Foulkes, 2008). The BRCA1 protein preserves genome integrity by promoting homology-directed repair of double-strand DNA breaks (Moynahan et al., 1999) and protecting stalled DNA replication forks from nucleolytic degradation (Schlacher et al., 2012). Although not formally proven, homology-directed repair (HDR) and stalled fork protection (SFP) are both thought to be key elements of BRCA1 tumor suppression (Jiang and Greenberg, 2015; Kolinjivadi et al., 2017a; Moynahan and Jasin, 2010; Nagaraju and Scully, 2007). In vivo, BRCA1 exists in association with BARD1 (Jin et al., 1997; Wu et al., 1996), and multiple lines of evidence indicate that these proteins function as an obligate stoichiometric heterodimer (Laufer et al., 2007; McCarthy et al., 2003; Westermark et al., 2003). Importantly, the tumor suppression activity of BRCA1 appears to be mediated by the BRCA1/BARD1 heterodimer, since mammary-specific inactivation of either Brca1 or Bard1 elicits mammary carcinomas in mice that resemble the basal-like triple-negative breast tumors of human BRCA1 mutation carriers (Shakya et al., 2008). In addition, germline mutations of human BARD1 have been identified as the pathogenic lesion in some families with hereditary breast and ovarian cancer (De Brakeleer et al., 2016). Thus, BARD1 makes unknown, but essential, contributions to the tumor suppression activity of the BRCA1/BARD1 heterodimer.
The C-terminal sequences of both BRCA1 and BARD1 contain two tandem copies of the “BRCA1 C-terminal” (BRCT) repeat (Figure S1A), an amino acid motif found in over twenty human proteins (Glover et al., 2004; Wu et al., 2015a). Tandem BRCT repeats can form a phospho-recognition surface that preferentially binds peptides containing phosphoserine (Manke et al., 2003; Yu et al., 2003). For example, the BRCT domain of BRCA1 interacts specifically with certain phosphorylated isoforms of several proteins implicated in HDR, including Abraxas/CCDC98, BACH1/BRIP1/FANCJ, CtIP, and UHRF1 (Jiang and Greenberg, 2015) (Figure S1). Most pathogenic BRCA1 lesions are frameshift or nonsense mutations that would eliminate or grossly disrupt the BRCT domain. However, in some families, tumor susceptibility can be attributed to a single amino acid substitution in BRCA1, often involving residues within its BRCT domain. Moreover, structural studies have shown that one such residue (S1655) forms a hydrogen bond with the phosphate group of BRCA1 phospho-ligands, and that the pathogenic S1655F mutation disrupts the interaction of BRCA1 with its known BRCT phospho-ligands (Botuyan et al., 2004; Clapperton et al., 2004; Shiozaki et al., 2004; Varma et al., 2005; Williams et al., 2004). Previously, we showed that the corresponding mutation in murine Brca1 (S1598F) abrogates HDR and elicits basal-like triple-negative mammary tumors in mice (Shakya et al., 2011). These observations indicate that BRCT phospho-recognition is required for BRCA1-mediated tumor suppression and suggest that HDR is a critical component of this process.
Although the BARD1 BRCT domain contains a hydrophilic cleft analogous to the BRCT phosphate-binding pocket of BRCA1 (Birrane et al., 2007; Edwards et al., 2008), proteins that bind the BARD1 BRCT domain in a phospho-dependent manner have not yet been reported. Instead, Li and Yu (Li and Yu, 2013) showed that the BARD1 BRCT domain specifically recognizes poly(ADP-ribose) (PAR) and that this interaction is specifically required for early recruitment of the BRCA1/BARD1 heterodimer to sites of DNA damage.
To determine how the BARD1 BRCT domain contributes to BRCA1/BARD1 function, we have characterized mice with mutations (S563F and K607A) that disrupt its phosphate-binding pocket. Unlike a comparable mutation in the Brca1 BRCT domain (S1598F) (Shakya et al., 2011), these Bard1 mutations do not impair HDR or increase the tumor susceptibility of mice. Instead, they disrupt the recruitment of Brca1/Bard1 heterodimers to stalled replication forks, render stalled forks vulnerable to nucleolytic degradation, and promote chromosomal instability in the face of replication stress. Moreover, stalled fork protection (SFP) is also impaired in cells expressing the Brca1-S1598F mutant, implying that most pathogenic BRCA1 mutations associated with human cancer abrogate both HDR and SFP. These observations indicate that Brca1 BRCT phospho-recognition is essential for both HDR and SFP, while Bard1 BRCT phospho-recognition is only required for SFP. Moreover, since SFP and chromosomal stability are impaired in cells that are heterozygous for the various BRCT-mutant alleles (i.e., Bard1S563F, Bard1K607A, and Brca1S1598F), our data support a model of pathogenesis in which defects in SFP and HDR promote successive stages of tumor development in human BRCA1/BARD1 mutation carriers.
RESULTS
Mice homozygous for either the Bard1S563F or Bard1K607A allele are viable, but the males are sterile
The four amino acids of the human BRCA1 BRCT domain that form direct contacts with phosphoserine (S1655, G1656, T1700, K1702) are all conserved in both human (S575, G576, T617, K619) and mouse (S563, G564, T605, K607) BARD1 (Ayi et al., 1998; Wu et al., 1996) (see Figure S1B). Therefore, we introduced the S563F and K607A missense mutations into the Bard1 gene of mice to generate the Bard1S563F and Bard1K607A alleles, respectively (Figures S2 and S3). The Bard1-S563F mutation is analogous to the pathogenic Brca1-S1598F mutation that ablates the HDR and tumor suppression activities of Brca1 (Shakya et al., 2011), while the Bard1-K607A mutation corresponds to the human BARD1-K619A mutation that abrogates BARD1 recognition of PAR (Li and Yu, 2013). To ascertain the effect of the S563F and K607A mutations on phospho-recognition by the mouse Bard1 BRCT domain, purified GST-Bard1 fusion proteins containing the BRCT domain (mouse Bard1 residues 377–765) were evaluated for their ability to interact with PAR in vitro. Although wildtype GST-Bard1, but not GST alone, readily bound PAR chains, this interaction was ablated by both the S563F and K607A mutations (Figure S1C). For convenience, the Bard1S563F and Bard1K607A alleles are abbreviated hereafter as Bard1SF and Bard1KA, respectively, and the Brca1S1598F allele (Shakya et al., 2011) as Brca1SF.
Upon intercrossing heterozygous mutant animals (Bard1SF/+ or Bard1KA/+), we obtained homozygous mutant pups (Bard1SF/SF or Bard1KA/KA) at the expected Mendelian ratio (~25%).Thus, in contrast to Bard1-null animals (Bard1–/–), which undergo embryonic lethality in a manner indistinguishable from Brca1-null mice (McCarthy et al., 2003), Bard1SF/SF and Bard1KA/KA mice appear to develop normally except that the males, but not the females, are sterile. Although Brca1SF/SF males are also sterile (Shakya et al., 2011), the Bard1SF/SF and Bard1KA/KA mice do not display other developmental abnormalities characteristic of Brca1SF/SF mice, such as growth retardation, kinked tails, and white spots on the belly and hind feet. As shown in Figure S4A, the testes of Bard1SF/SF, Bard1KA/KA and Brca1SF/SF mice are markedly smaller than those of their wildtype and heterozygous-mutant littermates. The seminiferous tubules of Bard1SF/SF and Bard1KA/KA testes display a mosaic pattern (Figure S4B) in which roughly half of the tubules are largely devoid of germ cells, apart from a ring of spermatogonia adjacent to the basal membrane (Figures S4Cii and S4Cv), while the remaining tubules show maturation arrest at the pachytene stage of spermatogenesis (Figures S4Ciii and S4Cvi). This phenotype is distinct from that of Brca1SF/SF testes, in which nearly all tubules display a uniform pattern of maturation arrest at a later stage of spermatogenesis (Figure S4B and S4Civ). These observations imply that BRCT phospho-recognition by Bard1 and Brca1 mediates functions in spermatogenesis that are at least partly distinct.
Bard1SF/SF and Bard1KA/KA cells are hypersensitive to a subset of genotoxic agents
To evaluate Bard1 function at the cellular level, isogenic panels of mouse embryonic fibroblasts (MEFs) were derived. As shown in Figure S5A, comparable expression of Bard1 protein was observed in the nuclear fractions of wildtype (Bard1+/+) and mutant (Bard1SF/SF and Bard1KA/KA) cells. Brca1 and its associated protein Ctip were efficiently co-immunoprecipitated with Bard1 from nuclear extracts of both wildtype (Bard1+/+) and mutant (Bard1SF/SF and Bard1KA/KA) MEFs (Figure S5B), indicating that the mutant Bard1 proteins retain the ability to form heterodimers with Brca1. Also, these mutations do not alter the association of Bard1 with either Mre11 (Figure S5C) or the heterochromatin protein HP1 (Wu et al., 2015b) (Figure S5D).
As shown in Figures 1A and S6A, Bard1SF/SF and Bard1KA/KA MEFs are hypersensitive to the DNA inter-strand crosslinking agent mitomycin C (MMC), in a manner reminiscent of Brca1SF/SF MEFs (Shakya et al., 2011). These cells are also hypersensitive to the PARP inhibitor olaparib, but to a reproducibly lesser degree than Brca1SF/SF cells (Figures 1B and S6B). However, unlike Brca1SF/SF cells, Bard1SF/SF and Bard1KA/KA MEFs displayed little, if any, hypersensitivity to ionizing radiation (IR) (data not shown). Thus, the two Bard1 mutants share a similar pattern of genotoxin sensitivity, which is partly distinct from that of Brca1SF/SF cells.
Figure 1. Bard1SF/SF cells are hypersensitive to DNA damaging agents and display genotoxin-induced chromosomal instability.

A) Colony survival analysis of MMC-treated isogenic Bard1+/+ and Bard1SF/SF MEFs, along with Brca1+/+ and Brca1SF/SF MEFs. Survival is quantified as percentage of colonies on MMC-treated relative to untreated plates. Each condition was tested in triplicate, and error bars represent standard error of the mean.
B) Colony survival analysis of olaarib-treated Bard1+/+ and Bard1SF/SF MEFs, along with Brca1+/+ and Brca1SF/SF MEFs.
C) Bard1+/+ and Bard1SF/SF primary MEFs were cultured with or without 40 ng/mL MMC for 16 hours and structural chromosome abnormalities were quantified by T-FISH. The mean number of aberrations per cell is denoted by a horizontal red line, and the error bars represent standard error of the mean. P values were calculated by unpaired Student’s T-Test (n.s. = no significance, ** = p<0.01, *** = p<0.001).
D) T-FISH analysis of Bard1+/+ and Bard1KA/KA primary MEFs.
Bard1SF/SF and Bard1KA/KA cells accumulate chromosomal rearrangements in response to genotoxic stress
To assess chromosomal stability, primary Bard1+/+ and Bard1SF/SF MEFs were cultured in the presence or absence of MMC and evaluated by telomere-specific fluorescence in situ hybridization (T-FISH). Few metaphases with chromosomal abnormalities (~5%), each harboring a low burden of cytogenetic defects (< 0.1 per metaphase on average), were observed in untreated Bard1+/+ or Bard1SF/SF cells (Figure 1C). Thus, unlike Brca1SF/SF cells (Shakya et al., 2011), Bard1SF/SF cells do not spontaneously accumulate chromosomal rearrangements, at least to a level detectable by cytogenetic analysis. However, upon MMC treatment, nearly 60% of Bard1SF/SF metaphases displayed cytogenetic abnormalities and the average number of chromosome aberrations observed was significantly higher in Bard1SF/SF cells (1.62 per metaphase) than in Bard1+/+ cells (0.68 per metaphase). Similar results were obtained upon cytogenetic analysis of Bard1KA/KA primary MEFs (Figure 1C). Thus, Bard1 BRCT phospho-recognition is required to suppress the formation of MMC-induced, but not spontaneous, chromosomal rearrangements.
Bard1SF/SF and Bard1KA/KA cells are proficient for HDR
The phospho-recognition property of the Brca1 BRCT domain is essential for HDR (Shakya et al., 2011). To ascertain whether HDR is also dependent on Bard1 BRCT phospho-recognition, we first examined recruitment of the Brca1/Bard1 heterodimer to sites of DNA damage in cells exposed to ionizing radiation (IR). As expected (Shakya et al., 2011), the formation of IR-induced Brca1 (Figure 2A) and Bard1 (Figure 2B) nuclear foci was abrogated in Brca1SF/SF cells relative to isogenic Brca1+/+ control cells. In contrast, the levels of Brca1 and Bard1 focus formation in Bard1SF/SF and Bard1KA/KA cells were indistinguishable from those of wildtype controls (Figure 2A-B). Likewise, IR-induced Rad51 focus formation was markedly reduced in rca1SF/SF cells, but not in Bard1SF/SF or Bard1KA/KA cells (Figure 2C).
Figure 2. Bard1SF/SF and Bard1KA/KA cells are competent for Rad51 focus formation and homology-directed repair (HDR).

A) Brca1 focus formation in isogenic Bard1+/+ and Bard1SF/SF MEFs and Brca1+/+ and Brca1SF/SF MEFs was measured 1 hour after exposure to 10 Gy. Each bar graph is an average of three independent experiments, and the error bars represent standard error of the mean. Statistical analyses were conducted using unpaired Student’s T-Test (** = p<0.01, **** = p<0.0001).
B) Bard1 focus formation evaluated as in panel A
C) Rad51 focus formation evaluated as in panel A.
D) HDR efficiency was measured in Brca1+/+ and Brca1SF/SF embryonic stem (ES) cell subclones containing an integrated DR-GFP reporter. Cells were transfected with an empty (EV) or I-SceI-expressing (I-SceI) vector and the percentage of GFP-positive cells quantified by flow cytometry. Each ES cell subclone was analyzed in triplicate transfections. Error bars represent standard error of the mean.
E) HDR efficiency was measured in independent subclones of isogenic Bard1+/+ and Bard1SF/SF ES cells (left) and Bard1+/+ and Bard1KA/KA ES cells (right) containing an integrated DR-GFP reporter.
To determine the impact of Bard1 BRCT phospho-recognition on HDR directly, we derived isogenic lines of embryonic stem (ES) cells from Bard1+/+, Bard1SF/SF, and Bard1KA/KA blastocysts and then generated ES subclones that possess a DR-GFP recombination reporter integrated into the Pim1 locus (Pierce et al., 2001). To measure repair of an I-SceI-induced chromosomal DSB, these subclones were transfected with an I-SceI expression vector, and GFP-positive ES cells were quantified by flow cytometry. As expected (Shakya et al., 2011), the proportion of GFP-positive cells was markedly reduced (9-fold) in Brca1SF/SF cells relative to Brca1+/+ cells (Figure 2D). In contrast, no significant difference was observed upon analysis of multiple independent subclones of wildtype (Bard1+/+) and Bard1-mutant (Bard1SF/SF and Bard1KA/KA) ES cells (Figures 2E). Thus, HDR is dependent on phospho-recognition by the Brca1 BRCT domain, but not by the Bard1 BRCT domain.
Bard1 BRCT phospho-recognition is required to protect stalled forks from nucleolytic degradation
Since BRCA1/2-mediated stalled fork protection (SFP) is also critical for genome integrity (Schlacher et al., 2011; Schlacher et al., 2012), we next examined SFP in Bard1SF/SF and Bard1KA/KA cells. MEFs were subjected to successive 20-minute pulses with the nucleoside analogs 5’-iodo-2-deoxyuridine (IdU) and 5’-chloro-2-deoxyuridine (CldU) (Figure 3A), and the track lengths of single DNA fibers were measured following treatment with hydroxyurea (HU), a reversible inhibitor of ribonucleotide reductase that stalls DNA replication forks by reducing cellular deoxynucleotide pools. Although the ratio of the lengths of adjacent IdU and CldU replication tracts approximate unity in HU-treated wildtype (Bard1+/+) cells, the CldU/IdU ratios were significantly reduced in HU-treated Bard1SF/SF and Bard1KA/KA cells (Figures 3B and3C), indicating a defect in SFP. Importantly, SFP was restored by culturing these cells with mirin, an inhibitor of Mre11 nuclease activity (Dupre et al., 2008). Identical results were obtained upon analysis of either immortalized (Figure 3B and3C) or primary (Figure 3D) MEFs. Thus, phospho-recognition by the Bard1 BRCT domain is required to protect stalled forks from Mre11-dependent nucleolytic degradation.
Figure 3. Bard1SF/SF, Bard1KA/KA, and Brca1SF/SF cells have a defect in stalled fork protection.

A) Schematic of the DNA fiber assay. MEFs were exposed to sequential 20-minute pulses of IdU and CldU and then harvested immediately (–HU control) or after a 90-minute incubation with 2 mM hydroxyurea (HU). As indicated, cells were exposed to 50 µM mirin to inhibit Mre11 nuclease activity.
B) DNA fiber analysis of isogenic immortalized Bard1+/+ and Bard1SF/SF MEFs. For each condition, the CldU/IdU ratios of at least 150 individual DNA fibers are presented as a dot plot, and the median CldU/IdU ratio is denoted by a horizontal red line. Statistical analyses were conducted using the Mann-Whitney rank sum test (**** p<0.0001).
C) DNA fiber analysis of immortalized Bard1+/+ and Bard1KA/KA MEFs.
D) DNA fiber analysis of primary Bard1+/+, Bard1SF/SF, and Bard1KA/KA MEFs.
E) DNA fiber analysis of immortalized Brca1+/+ and Brca1SF/SF MEFs.
PAR-dependent recruitment of the BRCA1/BARD1 heterodimer to stalled replication forks is impaired in Bard1SF/SF and Bard1KA/KA cells
A potential function for BRCA1 at stalled replication forks first emerged from studies of the nuclear distribution of BRCA1 and BARD1 in S phase cells (Nagaraju and Scully, 2007; Scully et al., 1997). Using immunoflourescent microscopy, Scully et al. (Scully et al., 1997) showed that upon HU treatment BRCA1/BARD1 heterodimers localize to the PCNA-staining DNA replication structures of late S phase cells. Recently, Dungrawala et al. (2015) used iPOND (isolation of proteins on nascent DNA) technology to establish biochemically that BRCA1 and BARD1 are highly enriched at HU-stalled DNA replication forks (Dungrawala et al., 2015). To ascertain whether the SFP defect of Bard1SF/SF and Bard1KA/KA cells reflects a failure of mutant BRCA1/BARD1 heterodimers to mobilize to stalled forks, we first examined HU-induced recruitment of Brca1 and Bard1 to PCNA-staining replication foci. Thus, cells were cultured in the presence or absence of HU, co-stained with PCNA- and Bard1-specific antibodies, and visualized by immunoflourescent microscopy. Cells that exhibit the late S phase pattern of nodular PCNA staining were then identified and examined for the presence (PCNA+Bard1+) or absence (PCNA+Bard1–) of Bard1-staining foci, and the co-staining PCNA+Bard1+ cells were further classified into those in which the PCNA and Bard1 foci were largely co-localized (PCNA+Bard1+ co-localized) or spatially independent (PCNA+Bard1+ non-co-localized) (Figure 4A). As expected (Scully et al., 1997), the proportion of PCNA-staining late S-phase nuclei that harbor co-localizing PCNA and Bard1 foci dramatically increased (from <5% to 80–90%) upon HU treatment of wildtype Bard1+/+ cells (Figures 4B and4C). Significantly, however, we observed a marked reduction (from 80–90% to 20–30%) in the proportion of nuclei with co-localizing PCNA and Bard1 foci in HU-treated Bard1SF/SF and Bard1KA/KA MEFs (Figures 4B and4C). Likewise, HU-induced co-localization of Brca1 with PCNA is also impaired in Bard1SF/SF and Bard1KA/KA cells (Figures 4D and4E). These results indicate that Brca1/Bard1 recruitment to sites of replication fork stalling is impaired by the Bard1-S563F and Bard1-K607A mutations. Moreover, the proportion of co-localizing PCNA/Bard1 foci (Figures 4B and4C) and PCNA/Brca1 foci (Figures 4D and4E) was reduced to similar levels (20–30%) in HU-treated Bard1+/+ cells by PARP1 inhibition with low-dose olaparib, indicating that Brca1/Bard1 recruitment to replication factories by the Bard1 BRCT domain is dependent on the formation of poly(ADP-ribose).
Figure 4. The recruitment of Brca1/Bard1 heterodimers to PCNA replication factories is defective in HU-treated Bard1SF/SF and Bard1KA/KA cells.

A) Representative images of late S phase PCNA+ cells. Upon immunoflourescent co-staining with antibodies specific for PCNA (red) and Bard1 (green), three distinct populations of PCNA+ cells were identified: 1) cells displaying ≥5 PCNA foci but no Bard1 foci (PCNA +Bard1–, top); 2) cells displaying both ≥5 PCNA foci and ≥5 Bard1 foci that were spatially non-overlapping (PCNA+Brca1+ non-co-localizing, middle); and 3) cells displaying both ≥5 PCNA foci and ≥5 Bard1 foci in which more than half of the Bard1 foci co-localize with PCNA foci (PCNA+Brca1+ co-localizing, bottom). In panels B and C, the percent co-localization is the number of PCNA+Bard1+ co-localizing cells divided by the total number of PCNA+ late S phase cells (PCNA+Bard1– + PCNA+Bard1+ co-localizing + PCNA+Bard1+ non -co-localizing).
B) The percentage of late S phase cells with co-localizing PCNA and Bard1 foci in isogenic Bard1+/+ and Bard1SF/SF MEFs cultured for 90 minutes in the presence or absence of 2mM hydroxyurea (HU) and/or 100 nM olaparib (PARPi). At least 200 late S phase PCNA+ cells were examined for each condition. The histogram presents the average of three independent experiments and the error bars represent standard error of the mean. Statistical analyses were performed using one-way ANOVA (**** p<0.0001).
C) The percentage of late S phase cells with co-localizing PCNA and Bard1 foci in Bard1+/+ and Bard1KA/KA MEFs cultured in the presence or absence of HU and/or PARPi.
D) The percentage of late S phase cells with co-localizing PCNA and Brca1 foci in Bard1+/+ and Bard1SF/SF MEFs cultured in the presence or absence of HU and/or PARPi.
E) The percentage of late S phase cells with co-localizing PCNA and Brca1 foci in Bard1+/+ and Bard1KA/KA MEFs cultured in the presence or absence of HU and/or PARPi.
To confirm that Bard1 BRCT phospho-recognition is required for recruitment of Brca1/Bard1 heterodimers to stalled replication forks, we employed the iPOND method, which allows biochemical recovery of proteins that reside in physical proximity to DNA containing incorporated EdU (5-ethynyl-2′-deoxyuridine) nucleosides (Sirbu et al., 2012). Therefore, wildtype (Bard1+/+) and mutant (Bard1SF/SF or Bard1KA/KA) MEFs were pulse-labeled with EdU for 10 minutes and harvested immediately or cultured for an additional 90 minutes in the presence of HU (Figure 5A). Cell extracts were then subjected to iPOND purification and the recovered materials evaluated by Western analysis to identify proteins that associate with either unstressed or stalled (i.e., HU treated) DNA replication forks. As expected (Dungrawala et al., 2015), HU induced a dramatic increase in the levels of fork-associated Bard1 and Brca1 in Bard1+/+ cells, reflecting specific recruitment of the Brca1/Bard1 heterodimer to stalled replication forks (Figures 5B and5C). In addition, the Brca1-associated Ctip protein is also recruited to stalled replication forks. Remarkably, however, the association of Bard1, Brca1, and Ctip with stalled replication forks is almost completely ablated in Bard1KA/KA and Bard1SF/SF cells (Figures 5B and5C). Moreover, Brca1/Bard1 recruitment to stalled replication forks is also abrogated in HU-treated Bard1+/+ cells by PARP1 inhibition. These biochemical data, together with the cytological results of Figure 4, indicate that phospho-recognition by the Bard1 BRCT domain is required for PAR-mediated recruitment of Brca1/Bard1 to stalled replication forks.
Figure 5. The assembly of Brca1/Bard1 heterodimers onto stalled replication forks is defective in Bard1KA/KA and Bard1SF/SF cells.

A) For iPOND analysis (Sirbu et al., 2012), cell lysates and iPOND-purified fractions were prepared from untreated cell cultures and from parallel cultures pulsed-labeled for 10 minutes with IdU. The IdU-labeled cultures were harvested immediately or after a subsequent 90-chase with 2mM hydroxyurea (HU) and/or 100 nM olaparib (PARPi).
B) Immunoblot analysis of protein abundance in the input cell lysates (left) and the corresponding iPOND-purified fractions (right) from Bard1+/+ and Bard1SF/SF cells.
C) Immunoblot analysis of protein abundance in the input cell lysates (left) and the corresponding iPOND-purified fractions (right) from Bard1+/+ and Bard1KA/KA cells.
Brca1SF/SF cells also fail to protect stalled replication forks
Although stalled fork protection (SFP) is dependent on BRCA1 (Schlacher et al., 2012), the functional domains of BRCA1 that mediate SFP have not yet been defined. To ascertain whether the BRCT phospho-recognition activity of BRCA1 contributes to SFP, isogenic Brca1SF/SF and Brca1+/+ cells were subjected to DNA fiber analysis. As shown in Figure 3E, stalled replication forks were degraded in an Mre11-dependent manner upon HU treatment of Brca1SF/SF, but not Brca1+/+ cells. Thus, phospho-recognition by the Brca1 BRCT domain is required for both HDR (Figure 2D) and SFP (Figure 3E). Since the vast majority of pathogenic BRCA1 lesions associated with human cancer disrupt the BRCT domain, either by deletion, truncation, or mutation, these observations suggest that combined inactivation of both the HDR and SFP activities of BRCA1 may be a common, if not essential, feature of tumor formation in BRCA1 mutation carriers.
Replication stress induces DNA damage in Bard1SF/SF, Bard1KA/KA, and Brca1SF/SF cells
To ascertain whether phospho-recognition by the Bard1 and Brca1 BRCT domains is required to suppress DNA damage in the face of replication stress, isogenic MEFs clones were exposed to hydroxyurea and individual cells were evaluated using the alkaline comet assay, in which damaged DNA is visualized as a “comet tail” and quantified by measuring its tail moment (TM). As shown in Figure 6A, a significant increase in HU-induced DNA damage was observed in each of two Bard1SF/SF clones (average TMs of 10.1, 10.6) relative to their Bard1+/+ control (average TMs of 2.43), while a Brca1SF/SF MEF clone also sustained greater DNA damage than its wildtype control (average TMs of 7.10 and 3.10, respectively). Bard1KA/KA cells also displayed markedly increased levels of HU-induced DNA damage relative to controls (Figure 6B). Thus, replication stress readily induces DNA damage in Bard1-mutant cells (Bard1SF/SF and Bard1KA/KA) that are competent for HDR but defective for SFP.
Figure 6. Although Bard1SF/SF and Bard1KA/KA cells accumulate DNA damage during replication stress, Bard1SF/SF and Bard1KA/KA mice are not tumor prone.

A) The alkaline comet assay was used to assess HU-induced DNA damage in isogenic Bard1+/+ and Bard1SF/SF MEFs, as well as isogenic Brca1+/+ and Brca1SF/SF MEFs. For each condition, the individual tail moments of at least 75 cells are presented as a dot plot; the mean tail moment is denoted by a horizontal red line and the standard error of the mean is indicated by error bars. Statistical analyses were conducted using one-way ANOVA (Bard1 clones) or unpaired Student’s t test (Brca1 clones) (**** p<0.0001).
B) Alkaline comet assay to assess HU-induced DNA damage in Bard1+/+ and Bard1KA/KA MEFs, as well as Brca1+/+ and Brca1SF/SF MEFs.
C) Kaplan-Meier tumor-free survival curves of the Bard1+/+ (n = 28), Bard1SF/SF (n = 39), and Bard1KA/KA (n=34) cohorts. Using the log-rank (Mantel-Cox) test, no statistical significance (defined as p<0.05) was observed between the Bard1SF/SF and Bard1+/+ curves (p = 0.9854) or the Bard1KA/KA and Bard1+/+ curves (p = 0.8387). The Kaplan-Meier survival curve of Brca1SF/SF mice (n=72) from (Shakya et al., 2011) is superimposed for comparison.
Tumor formation in Bard1SF/SF and Bard1KA/KA mice
Homozygous mice with certain hypomorphic Brca1 mutations (e.g., Brca1SF/SF or Brca1tr/tr) are viable, but develop a broad spectrum of tumor types at increased rates relative to their wildtype and heterozygous littermates (Drost and Jonkers, 2009; Ludwig et al., 2001; Shakya et al., 2011). As Brca1SF/SF cells are defective for both HDR and SFP, it is not possible to conclude whether the tumor susceptibility of these Brca1-mutant mice reflects the loss of HDR, SFP, or both. However, since Bard1SF and Bard1KA are separation-of-function alleles that abrogate SFP without affecting HDR, we tested whether HDR alone is sufficient for tumor suppression in the absence of SFP by monitoring cohorts of Bard1SF/SF and Bard1KA/KA mice for tumor formation (Figure 6C). Although some animals developed tumors at an advanced age, the kinetics of tumor formation in Bard1SF/SF and Bard1KA/KA mice was statistically indistinguishable from that of their littermate controls (p=0.9607). In sharp contrast, Brca1SF/SF mice exhibit a markedly increased rate of tumorigenesis relative to controls (p<0.0001) (Shakya et al., 2011). Thus, simultaneous loss of Brca1/Bard1-mediated HDR and SFP, but not loss of Brca1/Bard1-mediated SFP alone, predisposes mice to spontaneous tumor formation.
Stalled fork protection and chromosomal stability are also defective in heterozygous cells harboring BRCT mutations in either Brca1 or Bard1
The analysis of tumor formation in Bard1SF/SF and Bard1KA/KA mice (Figure 6C) indicates that HDR alone is sufficient for tumor suppression in the absence of SFP. However, it does not address whether SFP can itself contribute to tumor suppression, especially in cells that are heterozygous for pathogenic BRCA1/BARD1 mutations. Although these cells retain most BRCA1-mediated functions, including HDR, Pathania et al. observed a defect in stalled fork protection in heterozygous-mutant (BRCA1mut/+) human mammary epithelial cells (Pathania et al., 2014). To determine whether heterozygosity for a Brca1 BRCT mutation also impairs SFP, we conducted DNA fiber analysis of MEFs harboring the Brca1SF allele. Notably, Brca1SF/+ cells displayed a profound defect in SFP (Figure 7A). Likewise, SFP is also defective in heterozygous Bard1KA/+ (Figures 7B) and Bard1SF/+ (data not shown) MEFs.
Figure 7. Heterozygous Bard1SF/+, Bard1KA/+, and Brca1SF/+ cells are haploinsufficient for stalled fork protection and accumulate DNA damage upon replication stress.

A) DNA fiber analysis (as described in Figure 3) of Brca1+/+, Brca1 SF/SF, and Brca1SF/+ MEFs.
B) DNA fiber analysis of Bard1+/+, Bard1KA/KA, and Bard1KA/+ MEFs.
C) Alkaline comet assay (as described in Figure 6) to assess HU-induced DNA damage in isogenic panels of wild type, homozygous-mutant, and heterozygous-mutant MEFs harboring the Brca1SF, Bard1SF, and Bard1KA alleles. Statistical analyses were conducted using one-way ANOVA (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001).
D) DNA fiber analysis of Bard1+/+, Bard1Q552X/+, Bard1co-rec/+, Bard1SF/+, and Bard1KA/+ MEFs.
To ascertain whether cells heterozygous for the Bard1 and Brca1 BRCT mutations accumulate DNA damage in response to replication stress, isogenic MEFs were exposed to HU and evaluated in the alkaline comet assay. As shown in Figure 7C, the heterozygous-mutant clones (Bard1SF/+, Bard1KA/+, and Brca1SF/+) sustained significantly higher levels of DNA damage than their isogenic wildtype controls. Of note, these levels were also lower than those observed in the corresponding homozygous-mutant clones (Bard1SF/SF, Bard1KA/KA, and Brca1SF/SF). Thus, heterozygosity of these mutations is sufficient to confer a defect in SFP, as well as increased susceptibility to DNA damage in the face of replication stress.
It is conceivable that the SFP defects (Figure 7B) and enhanced HU-induced DNA damage (Figure 7C) displayed by heterozygous Bard1SF+ and Bard1KA/+ cells is due to reduced levels of the fully functional wildtype Bard1 protein relative to wildtype Bard1+/+ cells. To evaluate this possibility, we examined cells that are heterozygous for either of two distinct Bard1 alleles that fail to express protein product and are thus effectively null: 1) Bard1co-rec, which is the Cre-recombined product of a conditional-null Bard1co allele that lacks the first coding exon and 2) Bard1Q552X, which mimics a BARD1 nonsense mutation implicated in familial breast cancer (Ratajska et al., 2011) but fails to express a truncated polypeptide (see Experimental Procedures). Although Bard1 protein levels in heterozygous Bard1co-rec/+ and Bard1Q552X/+ cells are significantly reduced relative to those of Bard1+/+ cells (Figure S6C), no impairment in SFP (Figure 7D) or chromosome stability (Figure S6D) is observed in these cells. Thus, reduced expression of wildtype Bard1 protein alone is unlikely to account for the SFP defects or genomic instability of heterozygous Bard1SF/+ and Bard1KA/+ cells.
DISCUSSION
Phospho-recognition by the Brca1 BRCT domain is required for both homology-directed repair (HDR) and stalled fork protection (SFP)
The ability of BRCA1 to promote genome stability is thought to be a central aspect of its tumor suppression activity. While early studies established that BRCA1 and BRCA2 are required for homology-directed repair (HDR) of DSBs, both proteins also protect stalled forks from nucleolytic degradation (Kolinjivadi et al., 2017a; Schlacher et al., 2011; Schlacher et al., 2012; Ying et al., 2012). Moreover, Schlacher et al. identified a BRCA2 mutation that specifically ablates SFP without affecting HDR, and further showed that cells bearing this mutation undergo chromosomal instability upon HU treatment (Schlacher et al., 2011). These findings support two critical notions: first, that SFP and HDR are separable BRCA2 functions and, second, that SFP is itself an important contributor to genome integrity, especially in cells subjected to replication stress. SFP is also dependent on BRCA1, FANCD2, and RAD51, suggesting the existence of a “stalled fork protection” pathway that preserves genome integrity by preventing nucleolytic degradation of stalled forks (Hashimoto et al., 2010; Schlacher et al., 2012). Indeed, multiple components (PTIP, MLL3/4, CHD4, and PARP1) of an opposing pathway that promotes fork degradation by recruiting the MRE11 nuclease to stalled forks have also been identified (Ding et al., 2016; Ray Chaudhuri et al., 2016), and recent work has implicated the four-way reversed fork as the likely substrate for nucleolytic degradation in BRCA1/2-deficient cells (Kolinjivadi et al., 2017b; Lemacon et al., 2017; Mijic et al., 2017; Taglialatela et al., 2017).
Most (>90%) pathogenic BRCA1 lesions implicated in hereditary breast and ovarian cancer disrupt the C-terminal BRCT domain, either by amino acid substitution or, more commonly, by truncation. Here we show that Brca1 BRCT phospho-recognition is required for SFP (Figure 3E). Thus, SFP is likely to depend on the interaction of BRCA1 with one or more of its BRCT phosphoprotein ligands, such as Abraxas, FancJ, or Ctip. Perhaps more importantly, this finding, when combined with our previous evidence that HDR is dependent on Brca1 BRCT phospho-recognition (Shakya et al., 2011), indicates that most (>90%) of the pathogenic BRCA1 mutations responsible for hereditary breast and ovarian cancer abrogate both the HDR and SFP functions of BRCA1. This raises the intriguing possibility that disruption of both pathways is a common, and perhaps requisite, aspect of tumorigenesis in BRCA1 mutation carriers.
Since genome instability can arise due to abrogation of either HDR or SFP (Schlacher et al., 2011), it is important to determine whether the tumor suppression activity of BRCA1 is mediated by its ability to promote HDR, SFP, or both. The BRCT phospho-recognition property of BRCA1 is likely to be required for tumor suppression since a germline missense mutation that ablates this activity (S1655F) has been implicated in hereditary breast cancer and the corresponding mutation of murine Brca1 (S1598F) elicits basal-like triple-negative mammary tumors in mice (Shakya et al., 2011). In addition, homozygous Brca1SF/SF cells are defective for HDR (Shakya et al., 2011), consistent with the fact that the BRCA1 BRCT domain specifically binds at least four phosphoproteins that function in HDR (Abraxas/CCDC98, BACH1/BRIP1/FANCJ, CtIP, and UHRF1). Together, these observations suggest that HDR is a critical component of BRCA1-mediated tumor suppression. However, since Brca1SF/SF cells are also defective for SFP, we cannot discern from the present data whether tumor suppression is dependent on the ability of BRCA1 to promote HDR, SFP, or both. As discussed below, resolving this question will require the identification and analysis of BRCA1/BARD1 separation-of-function mutations that specifically abrogate either HDR or SFP.
Mice defective for phospho-recognition by either the Bard1 (Bard1SF/SF and Bard1KA/KA) or Brca1 (Brca1SF/SF) BRCT domain have distinct phenotypes
BRCA1 exists primarily in a nuclear complex with the BARD1 tumor suppressor (Wu et al., 1996), and the resulting BRCA1/BARD1 heterodimer mediates many of the functions ascribed to BRCA1, including HDR and tumor suppression (Laufer et al., 2007; Shakya et al., 2008; Westermark et al., 2003). Like BRCA1, BARD1 harbors two tandem C-terminal BRCT repeats that form a phosphate binding cleft. Although BRCA1 uses its BRCT domain to recognize particular phosphoproteins, no phosphoprotein ligands have as yet been identified for the BARD1 BRCT domain. Instead, the BARD1 BRCT domain binds in a phospho-dependent manner to poly(ADP-ribose) (PAR), a nucleic acid polymer that is rapidly assembled at sites of DNA breaks and stalled replication forks (Li and Yu, 2013).
To ascertain the functional relevance of BARD1 BRCT phospho-recognition, we generated mice harboring either of two missense mutations predicted to disrupt the BRCT phosphate-binding cleft of Bard1, including one that is structurally equivalent to the Brca1-S1598F mutation. Homozygous Bard1SF/SF and Bard1 KA/KA mice are born at the expected Mendelian ratios and, apart from male infertility, they appear to develop normally. As such, Bard1SF/SF and Bard1KA/KA mice are considerably more robust than Brca1SF/SF mice, which are born at reduced Mendelian ratios (dependent on the genetic background) and exhibit, in addition to male infertility, growth retardation and mild developmental defects (Shakya et al., 2011). Nevertheless, the Bard1 mutations elicit a more severe defect with respect to male germ cell development, resulting in maturation arrest at earlier stages of spermatogenesis in Bard1SF/SF and Bard1KA/KA testes than Brca1SF/SF testes. Together, these observations indicate that the developmental functions of BRCT phospho-recognition by BRCA1 and BARD1 are at least partly distinct.
Phospho-recognition by the Bard1 BRCT domain is essential for SFP but not HDR
Surprisingly, Bard1SF/SF and Bard1KA/KA cells are, unlike Brca1SF/SF cells, competent for HDR as determined by IR-induced Rad51 focus formation and DR-GFP recombination (Figure 2). Nonetheless, since these assays measure HDR of two-ended DSBs, we cannot formally exclude the possibility that Bard1 BRCT phospho-recognition contributes specifically to HDR of one-ended DSBs generated by replication fork collapse, which may have distinct genetic requirements (Willis et al., 2014). Also, while HDR is not dependent on Bard1 BRCT phospho-recognition (Figure 2), it should be noted that HDR is impaired by gross deletion of the entire BARD1 BRCT domain (Laufer et al., 2007) or by mutation of an HP1-binding site that lies near the N-terminal edge of the BRCT domain (Wu et al., 2015b). Thus, it is the phospho-recognition property of the Bard1 BRCT domain in particular that is dispensable for SPF. Nonetheless, DNA fiber analysis of Bard1SF/SF and Bard1KA/KA cells revealed that Bard1 BRCT phospho-recognition is essential for SFP (Figure 3). Although its role in SFP had not been examined previously, it seemed likely that the BARD1 protein would be required given that the stability and nuclear localization of BRCA1 are both dependent on heterodimerization with BARD1. However, in Bard1SF/SF and Bard1KA/KA cells, the mutant Brca1 polypeptides are stably expressed, localize to the nucleus, and readily form Brca1/Bard1 heterodimers (Figure S5). As such, the failure of SFP in these cells cannot be attributed to inappropriate formation of the BRCA1/BARD1 heterodimer. Instead, these data indicate that the phospho-recognition property of the BARD1 BRCT domain is specifically required for stalled fork protection.
BARD1 phospho-recognition is required for the recruitment of BRCA1/BARD1 to stalled forks
Using iPOND technology, Dungrawala et al. demonstrated that BRCA1 and BARD1 levels are highly enriched at HU-stalled DNA replication forks (Dungrawala et al., 2015). Therefore, the ability of the BARD1 BRCT domain to bind poly(ADP-ribose) (PAR) in a phospho-dependent manner (Li and Yu, 2013) suggested a possible mechanism for the SFP defect in Bard1SF/SF and Bard1KA/KA cells. Previous studies have shown that PAR chains are assembled at sites of stalled DNA replication by PAR polymerase 1 (PARP1) and that SFP is dependent on PARP1 activity (Ying et al., 2012). Therefore, to determine whether Bard1 BRCT phospho-recognition is required for recruitment of the Brca1/Bard1 heterodimer to stalled forks, we first examined the ability of Brca1/Bard1 to localize within PCNA-staining replication factories of HU-treated cells (Scully et al., 1997). Notably, the co-localization of Brca1/Bard1 and PCNA in late S phase cells was markedly reduced in HU-treated Bard1SF/SF and Bard1KA/KA cells relative to wildtype cells, as well as in wildtype cells exposed to low doses of the PARP1 inhibitor olaparib (Figure 4). The inability of the Brca1/Bard1 heterodimer of Bard1SF/SF and Bard1KA/KA cells to associate with HU-stalled replication forks was also established biochemically using the iPOND method (Figure 5). These observations indicate that Bard1 BRCT phospho-recognition promotes SFP by mediating PAR-dependent recruitment of Brca1/Bard1 heterodimers to stalled replication forks.
Although Brca1/Bard1 recruitment to stalled forks is dependent on PAR recognition by the Bard1 BRCT domain, we cannot exclude the possibility that the BRCT domain of Bard1 can also mediate other PAR-independent functions by binding one or more as yet unidentified phosphoprotein ligands. For example, the ability of the BARD1 BRCT domain to interact functionally with a phosphoprotein ligand(s) might explain how mutation of its phosphate-binding cleft renders Bard1SF/SF and Bard1KA/KA cells partially hypersensitive to PARP inhibition (Figures 1B and S6B). Interestingly, in this regard, the BRCT domain of NBS1 has the capacity to bind in a phospho-dependent manner with either PAR chains (Li et al., 2013) or CK2-phosphorylated isoforms of the MDC1 protein (Lloyd et al., 2009; Williams et al., 2009).
Loss of SFP alone is sufficient to induce chromosomal instability in Bard1SF/SF and Bard1KA/KA cells
Although BRCA1-mutant cells exhibit chromosomal instability, it is difficult to discern to what degree this instability reflects abrogation of either HDR or SFP individually. Ideally, this information could be attained through analysis of separation-of-function mutations that specifically abrogate either HDR (to generate cells with an HDR–/SFP+ “phenotype”) or SFP (the HDR+/SFP– phenotype). Although no such BRCA1 mutations have as yet been described, the Bard1SF and Bard1KA alleles represent novel separation-of-function mutations that generate the HDR+/SFP– phenotype. Significantly, Bard1SF/SF and Bard1KA/KA cells also display chromosomal instability in response to replication stress (Figure 6), indicating that a defect in SFP alone is sufficient to induce some degree of chromosomal instability.
Loss of SFP does not render Bard1SF/SF and Bard1KA/KA mice prone to tumorigenesis
Since the HDR and SFP functions of BRCA1/BARD1 both contribute to chromosomal stability, it is conceivable that BRCA1/BARD1-mediated tumor suppression activity is dependent on HDR, SFP, or both. To discriminate among these possibilities, it would be helpful to evaluate tumorigenesis in mice harboring Brca1/Bard1 separation-of-function mutations that yield either the HDR–/SFP+ or HDR+/SFP– phenotype. A number of mouse strains exist that harbor Brca1 alleles modeled after pathogenic BRCA1 mutations associated with hereditary breast and ovarian cancer. A subset of these, including the Brca1SF allele, encode polypeptides that retain at least some Brca1 function. Thus, unlike Brca1-null animals, which invariably undergo embryonic lethality, mice that are homozygous for these hypomorphic mutations (e.g., Brca1SF/SF mice) can survive as adults, but develop a wide spectrum of tumor types at increased rates relative to their wildtype and heterozygous littermates (Shakya et al., 2011). However, since Brca1SF/SF cells are deficient for both the HDR and SFP pathways (Figures 2D and3E), this observation does not illuminate the relative contributions of HDR or SFP toBRCA1-mediated tumor suppression. In contrast, Bard1SF/SF and Bard1KA/KA mice, which exhibit the HDR+/SFP– phenotype, do not display increased tumor formation relative to their littermates (Figure 6C), suggesting that abrogation of BRCA1/BARD1-mediated SFP alone is not sufficient to elicit tumor susceptibility. This observation implies that the tumor suppression activity of BRCA1/BARD1 is not dependent on SFP alone, but instead entails either its HDR function alone or a combination of its HDR and SFP functions. Future studies of tumor formation in Brca1/Bard1-mutant animals with an HDR–/SFP+ phenotype should help to distinguish between these alternatives.
SFP is defective in both homozygous and heterozygous Brca1/Bard1-mutant cells
In most families afflicted with BRCA1-linked hereditary breast and ovarian cancer, the tumorprone women are heterozygous mutation carriers with one mutant and one normal BRCA1 allele and, almost invariably, the normal BRCA1 allele is lost or inactivated in the tumors that arise in these women (Foulkes, 2008). Apart from their enhanced tumor susceptibility, BRCA1-mutation carriers are healthy and fertile, as are heterozygous Brca1/Bard1-mutant mice. Indeed, most biological functions attributed to BRCA1, including HDR, appear to be unaffected in cells that are heterozygous for pathogenic BRCA1 mutations (Jiang and Greenberg, 2015; Moynahan and Jasin, 2010; Nagaraju and Scully, 2007; Venkitaraman, 2014). Nonetheless, subtle defects in heterozygous BRCA1-mutant mammary epithelial cells have been reported, including premature senescence, modest chromosomal instability, and aberrant development of mammary epithelial cell populations (Konishi et al., 2011; Lim et al., 2009; Martins et al., 2012; Sedic et al., 2015), suggesting that BRCA1 may be haploinsufficient for at least some of its functions. For example, Pathania et al. showed that while heterozygous-mutant (BRCA1mut/+) human mammary epithelial cells are competent for a number of BRCA1-dependent processes, including HDR, these cells display signs of replication stress and an inability to stabilize HU-stalled DNA replication forks (Pathania et al., 2014). Here we show that SFP is also defective in cells heterozygous for mutations that ablate the BRCT phospho-recognition activities of either Bard1 (Bard1SF/+ and Bard1KA/+) or Brca1 (Brca1SF/+).
How does loss of HDR and SFP contribute to breast cancer formation in BRCA1-mutation carriers?
The presence of subtle phenotypic defects in heterozygous BRCA1/BARD1-mutant cells supports emerging models for the development of breast tumors in BRCA1-mutations carriers (Konishi et al., 2011; Lim et al., 2009; Martins et al., 2012; Pathania et al., 2014; Sedic et al., 2015). Here we show that cells heterozygous for mutations that disrupt phospho-recognition by the BRCT domains of Brca1/Bard1 fail to protect stalled replication forks (Figures 7A and7B) and readily accumulate DNA damage in response to replication stress (Figure 7C). However, these abnormalities are unlikely to be caused solely by reduced levels of wildtype Bard1 expression as they are not observed in cells heterozygous for null Bard1 mutations (e.g., Bard1co-rec/+ or Bard1Q552X/+ cells) (Figure 7D and S6D). Thus, it is conceivable that the mutant Brca1/Bard1 proteins of Bard1SF/+, Bard1KA/+, and Brca1SF/+ cells exert a dominant-negative effect that contributes to the observed defects in SPF and genome stability. In any case, women who carry germline mutations that render heterozygous BRCA1mut/+ cells defective for SFP (such as the pathogenic BRCA1SF allele) should experience a “field effect” in which all mammary epithelial cells are prone to replication stress due, at least in part, to their inability to protect stalled replication forks (see Figure S7). The downstream consequences of this stress, especially chromosomal instability, may then allow for the emergence of cells with genetic lesions that circumvent the premature senescence and inviability typically associated with BRCA1-mutant homozygosity (such as p53 or p16 inactivation, which occurs in the vast majority of BRCA1-mutant breast tumors). In these cells or their progeny, subsequent loss of the wildtype BRCA1 allele would yield viable cells that lack HDR activity, and as such would experience more extensive genomic instability at levels sufficient to drive malignant progression (Figure S7). In this scenario, loss of BRCA1-mediated SRF stability would play a critical role in the early stages of tumorigenesis. Though speculative, this model would imply that the SFP and HDR functions of BRCA1 can mediate distinct, but complementary, aspects of tumor suppression, and would also predict that both functions must be abrogated for tumor development in BRCA1-mutation carriers. The identification of a Brca1/Bard1 mutation that yields an HDR–/SFP+ phenotype should provide a critical test of this model, especially if tumor formation is abrogated or reduced in mice bearing this mutation relative to those with Brca1/Bard1 mutations that yield the HDR–/SFP– phenotype (e.g., Brca1SF).
STAR Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Richard Baer (rb670@columbia.edu).
Genetically-engineered mouse models
The mice used in this study were housed in an AAALAC-accredited facility at Columbia University Medical Center. All experiments involving mice were performed according to Columbia University Institutional Animal Care and Use Committee-approved protocols. Six-week-old male mice were used for testes histology. Male and female mice were used for the tumor formation studies and were sacrificed upon detection of a palpable mass or moribund appearance.
To generate mice harboring the Bard1S563F allele, a knock-in targeting vector containing mouse Bard1 genomic DNA was constructed by inserting a neomycin-resistance gene cassette flanked by loxP sites (loxP-PGK-neo-loxP) into intron 7 and the S563F missense mutation into exon 8 (Figure S2B). Likewise, to produce mice with the Bard1K607A allele, a targeting construct was generated by inserting the loxP-PGK-neo-loxP cassette into intron 8 and the K607A missense mutation into exon 9 (Figure S3B). These vectors were then electroporated into KV1 ES cells, properly recombined neomycin-resistant ES clones were identified, and the presence of the desired mutations confirmed by sequence analysis. Two independent clones of both Bard1S563F-neo/+ and Bard1K607A-neo/+ ES cells were injected into C57BL/6J blastocysts for the production of germline-transformed mice. To excise the loxP-PGK-neo-loxP cassette from the targeted alleles (Figures S2C and S3C), chimeric male Bard1S563F-neo/+ and Bard1K607A-neo/+ mice were mated with females carrying a ubiquitously expressed Cre transgene (EIIa-Cre; Tg(EIIa-cre)C5379Lmgd; Jackson Laboratories 003724) to produce offspring with the desired Bard1S563F or Bard1K607A alleles (Figures S2D and S3D). The heterozygous Bard1S563F/+ or Bard1K607A/+ mice were then backcrossed with pure C57Bl/6J mice (Jackson Laboratory) three times to yield animals that were approximately 94% C57BL/6J (N3 backcrossed). All mouse tumor cohorts, mouse embryonic fibroblast (MEF) lines, and embryonic stem (ES) cell lines were generated using mice on this background.
Similar procedures were used to generate mice harboring the Bard1Q552X or Bard1co-rec alleles studied in Figures 7 and S6. The Bard1Q552X allele was modeled after a tumorigenic human BARD1 lesion (c.1690C>T) that generates a nonsense mutation in codon Q564 (Ratajska et al., 2011), and as such may have the potential to encode a truncated polypeptide lacking the C-terminal 214 amino acids of human BARD1. Therefore, ES cells were electroporated with a mouse Bard1 targeting construct containing the equivalent nonsense mutation (Q552X) in exon 7 and the loxP-PGK-neo-loxP cassette in intron 6. Properly recombined neomycin-resistant ES clones were then identified and injected into blastocysts to obtain germline-transformed mouse strains bearing the Bard1Q552X-neo allele. To excise the loxP-PGK-neo-loxP cassette from the targeted alleles, chimeric male Bard1Q552X-neo/+ mice were mated with females carrying a ubiquitously expressed Cre transgene (EIIa-Cre) to produce offspring with the desired Bard1Q552X allele. However, further analysis of these strains revealed that the Bard1Q552X allele is functionally null. First, upon intercrossing heterozygous mutant animals (Bard1Q552X/+), no homozygous mutant pups were obtained (expected Mendelian ratio 25%), indicating that Bard1Q552X/Q552X mice undergo embryonic lethality in a manner reminiscent of Bard1-null (Bard1–/–) mice (McCarthy et al., 2003). Second, Western analysis showed that the steady-state levels of full-length Bard1 protein in Bard1Q552X/+ MEF lines are significantly reduced relative those of wildtype Bard1+/+ MEFs (Figure S6C). Importantly, a truncated Bard1-Q552X polypeptide of the predicted molecular weight (~60 kilodaltons) was not detected in Bard1Q552X/+ MEFs, even after long exposures. Thus, the protein product of the Bard1Q552X allele is not stably expressed, either due to nonsense-mediated mRNA decay or rapid degradation of the Bard1-Q552X polypeptide. In either case, the Bard1Q552X/+ allele would act functionally as a null allele, consistent with the embryonic lethality of homozygous Bard1Q552X/Q552X mice.
To generate a conditional-null Bard1co allele in which the first coding exon is flanked by loxP recombination signals, ES cells were electroporated with a Bard1co-neo targeting construct. Within this targeting construct, a single loxP signal was inserted into an XmnI restriction site located ~900 basepairs upstream of the first Bard1 coding exon, while a second loxP signal was inserted, along with a PGK-neomycin resistance cassette flanked by FRT (Flp recombinase target) signals, into an AgeI restriction site located ~350 basepairs downstream of the first coding exon. Properly recombined neomycin-resistant ES clones were then injected into blastocysts to obtain germline-transformed mouse strains bearing the Bard1co-neo allele. To excise the FRT-PGK-neo-FRT cassette from the targeted allele, chimeric male Bard1co-neo/+ mice were mated with Flpe-expressing (B6;Cg-Tg(ACTFLPe)9205Dym/J, Jackson Laboratories 005703) females to produce offspring with the desired Bard1co allele. The Bard1co-rec allele was then generated by mating Bard1co/+ mice with animals that carry a ubiquitously expressed Cre transgene (EIIa-Cre). Upon intercrossing the Bard1co-rec/+ progeny, wildtype and heterozygous pups were observed at the expected 1:2 ratio, but homozygous Bard1co-rec/co-rec offspring were not obtained (0 of 107 viable pups). Note that the conditional Bard1co allele differs from the previously described conditional Bard1flex1 allele (Shakya et al., 2008) in that the 5’ loxP signal was inserted into the XmnI restriction site (located ~900 basepairs upstream of coding exon 1) instead of the SalI site (~1900 basepairs upstream of coding exon 1). However, since the 3’ loxP signal was inserted into the same downstream AgeI site in both Bard1co and Bard1flex1, Cre-mediated recombination of either allele results in loss of Bard1 coding exon 1 (which encodes the initiator methionine and part of the RING domain).
Cell lines
All mouse embryonic fibroblasts (MEFs) were grown in a sterile 37˚C incubator with a humidified 5% CO2 atmosphere. Primary MEFs were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Cellgro) supplemented with 10% fetal bovine serum (FBS; Cellgro), 100 µg/mL penicillin/streptomycin, 2 mM L-glutamine, 1X nonessential amino acids (Cellgro) 1.25 µg/mL Plasmocin (InvivoGen), and 0.1 mM 2-mercaptoethanol. Immortalized MEFs were cultured in DMEM supplemented with 10% FBS, 100 µg/mL penicillin/streptomycin, and 2 mM L-glutamine.
Embryonic stem cells (ES cells) were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Cellgro) supplemented with 15% Hyclone ES cell screened fetal bovine serum (FBS, Fisher Scientific), 100 µg/mL penicillin/streptomycin, 2 mM L-glutamine, 1X nonessential amino acids (Cellgro), 1.25 µg/mL Plasmocin (InvivoGen), 0.1 mM 2-mercaptoethanol, and 1000 units/mL leukemia inhibitory factor (LIF, Millipore), and housed in a sterile 37˚C incubator with a humidified 5% CO2 atmosphere. To prevent differentiation, ES cells were cultured on a layer of mitotically inactive primary mouse embryonic fibroblasts (MEFs). Primary MEFs were seeded onto 0.2% gelatin coated plates and inactivated when approximately 90% confluent by 2-hour incubation with 5 µg/mL mitomycin C (MMC, Sigma) in primary MEF media (Dulbecco’s Modified Eagle Medium, DMEM; Cellgro) supplemented with 10% fetal bovine serum (FBS; Cellgro), 100 µg/mL penicillin/streptomycin, 2 mM L-glutamine, 1X nonessential amino acids (Cellgro), 1.25 µg/mL Plasmocin (InvivoGen), and 0.1 mM 2-mercaptoethanol). Following MMC inactivation, the primary MEFs were washed twice with 1X PBS and cultured in ES cell media.
MEF line generation and immortalization
Embryos were harvested under sterile conditions from a pregnant mouse on day E13.5 post-fertilization. After removing the liver and heads of each embryo, the remaining embryonic tissue was placed into a 15 mL tube containing 500 µL of ice-cold 1X trypsin-EDTA (0.25% trypsin/2.21 mM EDTA in Hank’s Balanced Salt Solution without sodium bicarbonate, calcium, and magnesium; Cellgro) inside a sterile tissue culture hood and incubated on ice overnight in a 4°C room. The next morning, excess trypsin was drained from each 15 mL tube before adding 2 µL DNase I (2000 U/mL, New England Biolabs) to the embryo and incubating at 37°C for 5 minutes. Following the incubation, primary MEF media (Dulbecco’s Modified Eagle Medium (DMEM; Cellgro) supplemented with 10% fetal bovine serum (FBS; Cellgro), 100 µg/mL penicillin/streptomycin, 2 mM L-glutamine, 1X nonessential amino acids (Cellgro), 1.25 µg/mL Plasmocin (InvivoGen), and 0.1 mM 2-mercaptoethanol) was used to inactivate the trypsin and break the embryos into a single cell suspension. Finally, the cells were plated on a 10 cm plate coated with 0.2% gelatin.
To immortalize primary MEF, passage two (P2) MEFs at approximately 50% confluence were transfected with SV40 large-T antigen. For transfection, 10 µg of the pMSSVLT plasmid was mixed with 25 µL of Lipofectamine 2000 (Thermo Fisher Scientific) in 1 mL of opti-MEM reduced serum media (Life Technologies) and incubated for 20 minutes. After incubation, the lipofectamine/DNA mix was added dropwise to primary MEFs in a 10-cm dish and returned the incubator. The next morning, the cells were washed with 1x PBS and given fresh primary MEF media. The MEFs were subsequently cultured for 10–12 passages until only immortalized cells remained (approximately 4 weeks in total). The genotype of the immortalized MEFs was confirmed by PCR prior to freezing.
In vitro poly(ADP-ribose)-binding assays
A bacterial expression construct encoding glutathione S-transferase (GST) fused to the C-terminal 389 amino acids of murine Bard1 (GST-Bard1-wt) was generated by inserting a Bard1 cDNA fragment into the pGEX2 vector. Derivatives of this construct encoding polypeptides harboring the S563F (GST-Bard1-S563F) or K607A (GST-Bard1-K607A) missense mutations were then derived by site-directed mutagenesis. The GST-Bard1 fusion proteins, as well as the parental GST protein, were expressed in Escherichia coli, purified by affinity chromatography, and rebound to glutathione resin beads by standard procedures. For each PAR-binding reaction, 3 picomoles of resin-bound GST-Bard1 (or GST only) protein were incubated with 30 picomoles of poly(ADP-ribose) (Trevigen) at 4˚C for 2 hours in NETN-100 buffer (0.5% Nonidet P-40, 2 mM EDTA, 50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM DTT) (Li and Yu, 2013). After washing the beads three times with the same buffer and once with TBS buffer (20 mM Tris-HCl pH 7.6, 0.137 M NaCl, 0.1% 1 mM DTT), the GST-fusion proteins were eluted in TBS containing 10 mM glutathione, and spotted onto a Protran nitrocellulose membrane (Amersham) using a dot blot vacuum apparatus. For the PAR input samples, 30 picomoles of poly(ADP-ribose) (Trevigen) were denatured in 0.4 M NaOH + 10 mM EDTA and spotted onto a Biodyne B membrane (Pall Life Sciences). The membranes were then blocked with 10% milk in TBS-T buffer (20 mM Tris-HCl pH 7.6, 0.137 M NaCl, 0.1% Tween 20) for 30 minutes at room temperature. After blocking, the membranes were incubated with anti-PAR 10H (Millipore) monoclonal antibody diluted 1:300 in 2% milk/TBS-T for two hours and with HRP-conjugated goat anti-mouse (Sigma) secondary antibodies diluted 1:10,000 in 2% milk/TBS-T for one hour. To confirm the presence of the GST-fusion proteins, the Protran nitrocellulose membrane was stripped with Restore Western Blot Stripping Buffer (Pierce) and immunoblotted with anti-GST monoclonal antibody (clone B-14; Santa Cruz Biotechnology).
Subcellular fractionation
To perform cell fractionation, exponentially growing immortalized MEFs seeded 48 hours earlier were harvested and lysed in 5 times the cell pellet volume of buffer A (10 mM Hepes pH 7.9, 10 mM KCl) supplemented with protease inhibitor cocktail (Roche) and incubated for 10 minutes on ice. Following the incubation, 1/16th lysate volume of 10% NP40 was added to the cell lysate and vortexed for 10 seconds. The lysates were then centrifuged at 5,000 rpm for 2 minutes at 4˚C to separate into nuclear (pellet) and cytoplasmic (supernatant) fractions. The resulting supernatant (cytoplasmic fraction) was removed and 0.11 times the total cytoplasmic fraction volume of buffer B (0.3 M Hepes pH 7.9, 1.4 M KCl, 1 mM DTT) was added. The cytoplasmic fraction was then centrifuged at 13,000 rpm for 10 minutes at 4°C and the resulting supernatant collected for protein concentration measurement. To process the nuclear fraction, the cell pellet obtained from the 5,000 rpm centrifugation step was resuspended in 2 times the cell pellet volume of buffer C (20 mM Hepes pH 7.9, 25% v/v glycerol, 0.42 M NaCl, 0.2 mM EDTA, protease inhibitor cocktail, and 1 mM DTT). The nuclear fraction was then vortexed for 10 minutes at 4°C and centrifuged at 13000 rpm for 10 minutes. The resulting supernatant (nuclear extract) was collected for protein concentration measurement. Following electrophoresis, lysates were transferred onto an Amersham Protran 0.45 µm nitrocellulose membrane (GE Healthcare Life Sciences) in western transfer buffer (25 mM Tris-Cl pH 7.6, 190 mM glycine, 20% methanol, 0.04% SDS) at 22 V overnight at room temperature. The following day, the membrane was blocked in 10% milk in TBS-T (20 mM Tris-Cl pH 7.6, 0.137 M NaCl, 0.1% Tween 20) for 30 minutes at room temperature. Membranes were stained with primary antibody diluted in 2% milk/TBS-T for two hours and then incubated with either HRP-conjugated goat anti-mouse (Sigma) or HRP-conjugated donkey anti-rabbit (GE Healthcare Life Sciences) secondary antibodies diluted 1:10,000 in 2% milk/TBS-T for one hour. Anti-Brca1 (1:2000, Shakya et al., 2011), anti-Bard1 (1:2,000, E.E McCarthy et al., 2003), anti-Ctip (1:50, Yu & Baer, 2000), and anti-α-Tubulin (1:10,000, Calbiochem) were used in western blotting experiments.
Co-immunoprecipitation analyses
For Bard1 co-immunoprecipitation experiments, exponentially growing cells seeded 48 hours prior to collection were harvested in low salt lysis buffer (10 mM Hepes pH 7.6, 0.25 M NaCl, 0.1% NP40, 5 mM EDTA, 10% glycerol) supplemented with complete protease inhibitor cocktail, 1 mM dithiothreital (DTT), and 25 mM sodium fluoride. Immunoprecipitation was then performed by incubating 600 µg of protein with mouse Bard1-specific rabbit polyclonal antibody (1:50) at 4˚C on a rotator for 2 hours, adding 50 µL of protein A sepharose CL-4B beads (50% v/v in low salt lysis buffer, GE Healthcare Life Sciences), and incubating on a rotator at 4˚C for an additional 30 minutes. The beads were then washed three times with low salt lysis buffer and resuspended in protein loading dye (0.313 M Tris-Cl pH 6.8, 10% SDS, 50% glycerol, 25% 2-mercaptoethanol, and 0.05% bromophenol blue) before boiling for three minutes to elute bound proteins. For the Mre11 and HP1γ co-immunoprecipitation experiments, chromatin extracts were prepared as described by Wu et al. (2015b). For Mre11 co-immunoprecipitation, 3.0 mg of protein were immunoprecipitated with either the Bard1-specific polyclonal rabbit antiserum or the corresponding pre-immune serum, fractionated by PAGE, and immunoblotted with Mre11-specific monoclonal antibody 18 (Santa Cruz Biotechnology). For HP1γ co-immunoprecipitation, 1.9 mg of protein was immunoprecipitated with an HP1γ-specific monoclonal antibody (clone 42s2; Millipore Sigma) or a “non-specific” monoclonal antibody (Flag-specific clone M2; Sigma-Aldrich), fractionated by PAGE, and immunoblotted with either the Bard1-specific polyclonal antiserum or the HP1γ-specific monoclonal antibody.
Genotoxin sensitivity assays
For all genotoxin sensitivity assays, immortalized MEFs were seeded in 6-well plates at 1000 cells/well for both drug-treated and control plates. Each experimental condition was plated in triplicate (3 wells per condition). At 48 hours after plating, immortalized MEFs were exposed to varying doses of mitomycin C (0 ng/mL, 50 ng/mL, 100 ng/mL, 200 ng/mL, 400 ng/mL, and 800 ng/mL MMC) for 4 hours. After drug treatment, the cells were washed twice with 1x PBS and cultured in fresh media until harvest 5–7 days post treatment. For PARP inhibitor (PARPi) treatment, at 24 hours after plating immortalized MEFs were exposed to various concentrations of olaparib (0 µM, 0.064 µM, 0.16 µM, 0.4 µM, 1.0 µM, 2.5 µM, and 5.0 µM). The media containing olaparib was replaced with fresh media containing olaparib every 48 hours until cell harvest 6–8 days after initial drug treatment. For ionizing radiation (IR) treatment, immortalized MEFs were irradiated 48 hours after plating with varying doses of IR (0 Gy, 2 Gy, 4 Gy, 6 Gy, 8 Gy, and 10 Gy) with an Atomic Energy of Canada Gammacell 40 Cesium unit. After irradiation, the cells were allowed to grow undisturbed until harvest 5–7 days later. In all cases, MEFs were harvested approximately 7 – 9 days after seeding, stained with a 0.5% crystal violet, 50% methanol solution, and surviving colonies (containing > 50 cells) were counted.
Analysis of metaphase spreads using telomere fluorescent in situ hybridization (T-FISH)
For T-FISH analysis, metaphase spreads were prepared from passage 3 (P3) or earlier primary MEFS. On the day prior to drug treatment, primary MEFs were plated on 0.2% gelatin-coated plates and allowed to attach overnight. The cells were then treated with either 40 ng/mL mitomycin C (MMC, Sigma) or mock treatment with 1x PBS for 16 hours (overnight). Four hours prior to the end of genotoxin treatment, Karyomax colcemid solution (Thermo Fisher Scientific) was added at a concentration of 0.1 µg/mL. The cells were then harvested, incubated in 0.4% KCl (w/v) solution, fixed in 3:1 methanol/glacial acetic acid solution, and dropped onto glass microscope slides. Telomeres were stained with a Cy3-labled (CCCTAA)3 peptide nucleic acid probe (Biosynthesis, Inc.), and DNA was counterstained with DAPI-containing mounting media (Vectashield; Vector Laboratories). The T-FISH metaphase spreads were imaged on an Axio Imager Z2 fluorescent microscope with Coolcube1 camera (Zeiss). Metafer software version 3.10.6 (Metasystems) was used to automatically locate metaphases at 10x magnification and then automatically capture images at 63x magnification. The metaphases were then analyzed on Isis fluorescent imaging system software (Metasystems). The results shown in Figure 1C represent the data from two independent experiments where, in each experiment, the results from two Bard1+/+ (E and I) and three Bard1SF/SF (A, C, and H) isogenic MEF clones were pooled. Similarly, Figure 1D represents the data from two independent experiments where two Bard1+/+ (I and N) and three Bard1KA/KA (J, L, and N) isogenic MEF clones were pooled.
Rad51 focus formation
Immortalized MEFs were seeded onto poly-L -lysine (Sigma) coated coverslips and exposed to 10 Gy of ionizing radiation 48 hours later using an Atomic Energy of Canada Gammacell 40 Cesium Unit. The cells were harvested one-hour post-IR, fixed with 3.7% paraformaldehyde(PFA)/PBS solution for 20 minutes at room temperature and permeabilized with 1% Triton X-100/PBS for 5 minutes at room temperature. The cells were then blocked in 5% bovine serum albumin (BSA)/PBS for 1 hour at 37°C and incubated with the Rad51 primary antibody (rabbit polyclonal, Millipore AB-1, 1:200 dilution) diluted in 5% BSA/PBS in a humidified chamber for 1 hour 45 minutes at 37°C. Following primary antibody incubation, the cells were incubated with secondary antibody (goat anti-rabbit Alexa 488, Thermo Fisher Scientific, 1:1000 dilution) diluted in 5% BSA/PBS in a humidified chamber for 45 minutes at 37°C. The cells were then mounted onto a glass slide with Vectashield hard set mounting medium with 4’, 6-diamidino-2-phenylindole (DAPI; Vector Laboratories). The cells were imaged on an Axio Imager Z2 fluorescent microscope with Coolcube1 camera (Zeiss) at 40x magnification. Automated Rad51 foci quantification was carried out using the Metafer 4 software (Metasystems). At least 300 cells were counted per trial.
DR-GFP assay
To perform the DR-GFP assay ES cells harboring the DR-GFP reporter at the Pim1 locus growing exponentially on primary MEF feeder cells were harvested by trypsinization and resuspended in ES transfection medium (Dulbecco’s Modified Eagle Medium (DMEM; Cellgro) supplemented with 15% Hyclone ES cell screened fetal bovine serum (FBS, Fisher Scientific), 2 mM L-glutamine, 1X nonessential amino acids (Cellgro), and 0.1 mM 2-mercaptoethanol) at a concentration of 0.8 × 106 cells per mL. While harvesting the cells, the transfection mix was prepared by combining either 0.5 µg of empty vector (pCAGGs), I-SceI expression vector, or GFP expression vector with 1.2 µL of Lipofectamine 2000 (Invitrogen) in 66 µL of opti-MEM reduced serum media (Life Technologies) and incubating for 20 minutes. After incubation, the ES cells from each clone were seeded on gelatin-coated, feeder-less plates and incubated with the appropriate DNA/lipofectamine mix for 6 hours. The transfection mix was then diluted by adding 1 mL of ES culture media (Dulbecco’s Modified Eagle Medium (DMEM; Cellgro) supplemented with 15% Hyclone ES cell screened fetal bovine serum (FBS, Fisher Scientific), 100 µg/mL penicillin/streptomycin, 2 mM L-glutamine, 1X nonessential amino acids (Cellgro), 1.25 µg/mL Plasmocin (InvivoGen), 0.1 mM 2-mercaptoethanol, and 1000 units/mL leukemia inhibitory factor (LIF, Millipore)). The next morning the cells were given fresh ES media and allowed to grow undisturbed for an additional 48 hours before harvest. Flow cytometry was performed on a FACScalibur machine using CellQuest software (BD Biosciences), and analysis of the data was carried out using FlowJo version X software. The efficiency of repair of the I-SceI-induced chromosomal break was measured by the percentage of GFP-positive cells. Gating for ES cells was performed by sorting cells based on side-scatter height vs forward-scatter height and selecting the appropriate sized population of cells. GFP-positive ES cells were then selected by sorting cells by green fluorescence intensity (FL1-H) vs orange fluorescence intensity (FL2-H) and gating for cells that showed a significant increase in green fluorescence intensity compared to orange fluorescent intensity (and therefore were not autofluorescent). At least 25,000 cells were counted per experimental condition. The percent of GFP-positive cells was normalized to transfection efficiency by measuring the percentage of GFP-positive cells in the well transfected with the GFP expression vector and then dividing the percentage of GFP-positive cells in the I-Sce I transfected wells with this value.
DNA fiber assay
Forty-eight hours after seeding in 6-well plates, exponentially growing immortalized MEFs or passage 3 (P3) primary MEFs were pulse labeled with 200 µM 5-iodo-2’-deoxyuridine (IdU, Sigma) for 20 minutes at 37°C. After IdU treatment, the cells were washed three times with warm (37°C) 1X PBS and pulse labeled with 100 µM 5-chloro-2’-deooxyridine (CldU, Sigma) for 20 minutes at 37°C. The cells were again washed with warm 1X PBS three times and then either harvested (untreated control) or treated with 2 mM hydroxyurea (HU, Sigma) for 1.5 hours followed by harvest. To inhibit the Mre11 nuclease, cells were treated with 50 µM mirin (Sigma) during pulse labeling with IdU and CldU, as well as during the subsequent hydroxyurea treatment. The cells were then harvested and resuspended at a concentration of 0.3 × 106 cells/mL in cold 1X PBS. Two microliters of the cell mixture were pipetted onto pre-cleaned glass microscope slides and lysed using pre-warmed spreading buffer at 37°C (0.5% sodium dodecyl sulfate, 20 mM Tris-Cl pH 7.4, and 50 mM EDTA) for 10 minutes in a humidified chamber at room temperature. Spreading of the DNA was then achieved by tilting the slides at a 15° angle relative to horizontal and allowing the cell lysis buffer mixture to run down the slide for 3 minutes. Following spreading, the slides were air dried, fixed by incubating in an ice cold (–20°C) 3:1 methanol:acetic acid mixture at room temperature for 2 minutes, and denatured in a 2.5 M HCl solution for 45 minutes at room temperature. The slides were blocked for 1 hour at room temperature in 3% bovine serum albumin (BSA), 0.1% Triton X-100/PBS. To stain the slides with primary antibody, the slides were incubated with rat anti-BrdU to detect CldU (Abcam ab6326, diluted 1:100) and mouse anti-BrdU to detect IdU (BD biosciences, BD 347580, 1:100 dilution) diluted in blocking solution for 1 hour at room temperature in a humidified chamber. The slides were then stained with anti-mouse Alexa Fluor 488 (Thermo Fisher Scientific, 1:300 dilution) and anti-rat Alexa Fluor 594 (Thermo Fisher Scientific, 1:300 dilution) diluted in blocking solution for 30 min at room temperature in a humidified chamber. Following secondary antibody staining, the slides were mounted in Prolong Gold Antifade (Thermo Fisher Scientific) and kept at 4°C until ready for imaging. Imaging of fibers was carried out on an Eclipse 80i fluorescent microscope (Nikon) with CoolSNAP HQ2 camera (Photometrics) at 40x magnification. Analysis was performed using imageJ software. At least 150 individual fibers were measured per experimental condition. MRE11-dependent fork degradation in BRCA1/2-mutant cells occurs in a directional manner such that the most recently synthesized DNA of the nascent strands is degraded first (Schlacher et al., 2011). Therefore, stalled fork protection (SFP) was assessed by calculating the ratio of the lengths of adjacent IdU and CldU replication tracts. CldU/IdU ratios should approximate unity in cells that are competent for SFP, while significant reductions in CldU/IdU ratios are observed in cells defective for SFP (Schlacher et al., 2011).
PCNA-Bard1 and PCNA-Brca1 staining and co-localization
Immortalized MEFs were seeded onto poly-L-lysine (Sigma) coated coverslips and treated 48 hours later with 2 mM hydroxyurea (HU, Sigma), 100 nM olaparib (PARPi, SelleckChem), and/or PBS for 90 minutes. After treatment, the cells were harvested by washing three times with ice cold CSK buffer (10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), 100 mM NaCl, 300 mM sucrose, and 3 mM MgCl2), permeabilized with ice cold 0.5% Triton/CSK for 5 minutes at 4°C, and fixed by incubating with ice cold 100% methanol at –20˚C for 10 minutes. After fixation, the cells were blocked by incubating the cells in 5% BSA, 0.1% Triton/PBS for 30 minutes. Cells were then stained with the following primary antibodies: PCNA (mouse monoclonal, Santa Cruz Biotechnology PC10, 1:200 dilution) and either Brca1 (rabbit polyclonal 57J, 1:250 dilution) or Bard1 (rabbit polyclonal 1734R, 1:500 dilution) diluted in 1% BSA 0.1% Triton/PBS for 1 hour at room temperature. Cells were then incubated with secondary antibodies (goat anti-rabbit Alexa Fluor 488, Thermo Fisher Scientific, 1:1000 dilution; and goat anti-mouse Alexa Fluor 568, Thermo Fisher Scientific, 1:400 dilution) diluted in 1% BSA, 0.1% Triton/PBS for 30 minutes at room temperature while protected from light. The cells were then mounted onto a glass slide with Vectashield hard set mounting medium with 4’, 6-diamidino-2-phenylindole (DAPI; Vector Laboratories).
The cells were imaged on an Eclipse 80i fluorescent microscope (Nikon) with CoolSNAP HQ2 camera (Photometrics) at 40x magnification. Blue, red, and green channel images were merged and analyzed using imageJ software. At least 200 cells were counted by hand per trial. A ‘Brca1-positive’ cell was defined as a cell containing 5 or more Brca1 foci, while a ‘Bard1-positive’ cell was defined as a cell containing 5 or more Bard1 foci. A ‘PCNApositive’ cell was defined as a cell containing 5 or more PCNA foci. PCNA and Bard1 (or Brca1) were scored as ‘co-localizing’ if over half of the Bard1 (or Brca1) foci overlapped with PCNA foci. Percent co-localization was defined as the number of co-localizing cells over total number of PCNA-positive cells.
iPOND
At 48 hours after seeding, immortalized MEFs were incubated with 10 µM EdU (Thermo Fisher Scientific) for 10 minutes at 37°C and harvested im mediately or after a 90-chase with 2 mM hydroxyurea (HU, Sigma) and/or 100 nM olaparib (PARPi, SelleckChem). The harvested cells were then fixed with 1% formaldehyde/PBS solution for 20 minutes at room temperature. Quenching of the crosslinking reaction was achieved by adding 1.25 M glycine. Cells were then harvested and incubated in permeabilization buffer (0.25% Triton X-100/PBS) at room temperature for 30 minutes. Following permeabilization, cells were washed at 4°C with 0.5% BSA/PBS, and then PBS alone, and then incubated in the “click” (10 mM sodium ascorbate, 2 mM CuSO4, 10 µM biotin-azide, in PBS) or “no-click” (i.e., no biotin-azide) reaction cocktail for one hour at room temperature. After the reaction, cells were resuspended in lysis buffer (1% SDS, 50 mM Tris-Cl pH 8.0) containing protease inhibitor (Roche) and the cell lysates were sonicated five times using a microtip sonicator (Misonix LX) for a 20 second pulse at a power of 15 Watts (with 40 second pauses on ice between pulses). Following sonication, the resulting supernatant was filtered through a 90-micron nylon mesh and diluted 1:1 (v/v) in PBS containing protease inhibitor at 4°C.
The filtered supernatant was incubated for 1 hour at 4°C with 50 µL of packed streptavidin-agarose beads (Thermo Fisher Scientific; pre-washed three times with PBS) supplemented with 50 µL of PBS. The streptavidin-agarose beads containing the captured DNA-protein complexes were then centrifuged for 3 minutes at 1,800g. After washing for 5 minutes each with 1.0 ml of cold lysis buffer, 1.0 ml of 1M NaCl, and twice more with 1.0 ml lysis buffer, the beads were supplemented 1:1 (v/v of packed beads) with SDS sample buffer and incubated at 95˚C for 25 minutes to liberate the proteins. SDS-PAGE fractionation and immunoblotting were then performed as described above.
Alkaline comet assays
Immortalized MEFs were seeded onto 12-well plates and then treated the following day with 2 mM hydroxyurea (HU, Sigma) or PBS mock treatment for 5 hours at 37˚C. Immediately following the drug treatment, the cells were harvested and mixed with 0.5% low melting agarose/PBS and pipetted onto glass slides coated with a double layer of 1% agarose/PBS. Ten µL of the cell/PBS mixture were then added to 75 µL of 0.5% low melting agarose/PBS and pipetted onto the agarose-coated slides. The slides were incubated with pH 10.0 lysis buffer (25 mM NaCl, 100 mM EDTA, 10 mM trizma base, and 1% Triton X-100) at 4°C overnight while protected from light. The following the day, the slides were equilibrated in pre-chilled (4°C) electrophoresis buffer (300 mM NaOH, 1 mM EDTA) for 20 minutes. After equilibration, electrophoresis was performed at 0.6 V/cm in a horizontal chamber (FischerScientific) for 20 minutes. The slides were washed with neutralization buffer (0.4 M Tris-Cl, pH 7.5) and fixed with ice cold 100% ethanol for 20 minutes at room temperature. Following fixation, the comets were stained by pipetting 90 µL of a fluorescent dye (GelRed, Biotium, dilution 1:1000 in H20) onto the slide and placing a coverslip over the dye. The comets were then imaged on an Eclipse 80i fluorescent microscope (Nikon) with CoolSNAP HQ2 camera (Photometrics) at 20x magnification. Comet tail moment values were determined using CometScore Software Version 1.5. At least 75 tails were analyzed per experimental condition. Apoptotic cells (small comet head and very large comet tail) were excluded from the analysis.
Tumor monitoring and histopathology
Mice were monitored for tumor development on a weekly basis. Upon detection of a palpable mass or moribund appearance, the mice were sacrificed and their tissues harvested for histological analysis. The mice were euthanized with CO2 followed by cervical dislocation in accord with the American Veterinary Medical Association Guidelines for the Euthanasia of Animals (2013 Edition). After euthanasia, a piece of the tail was harvested for confirmation of the genotype by PCR or Southern blot. Major organs were collected and fixed overnight in 10% buffered formalin, followed by dehydration with 70% ethanol the next day. Tissues were then embedded in paraffin, sectioned at a thickness of 4 µm, and stained with hematoxylin and eosin for histopathological evaluation.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical differences in the Rad51 foci and T-FISH experiments were determined by unpaired Student’s T test. Statistical differences in the PCNA co-localization experiments and alkaline comet assays were determined by one-way ANOVA. Statistical differences in DNA fiber assays were performed using the Mann-Whitney rank sum test. Statistical differences in the Kaplan-Meier survival curve were determined using the log-rank (Mantel-Cox) test. All statistical analyses were performed using Graphpad Prism software. Details of the statistical analysis specific to each experiment can be found in the figure legends.
KEY RESOURCES TABLE
The table highlights the genetically modified organisms and strains, cell lines, reagents, software, and source data essential to reproduce results presented in the manuscript. Depending on the nature of the study, this may include standard laboratory materials (i.e., food chow for metabolism studies), but the Table is not meant to be comprehensive list of all materials and resources used (e.g., essential chemicals such as SDS, sucrose, or standard culture media don’t need to be listed in the Table). Items in the Table must also be reported in the Method Details section within the context of their use. The number of primers and RNA sequences that may be listed in the Table is restricted to no more than ten each. If there are more than ten primers or RNA sequences to report, please provide this information as a supplementary document and reference this file (e.g., See Table S1 for XX) in the Key Resources Table.
Please note that ALL references cited in the Key Resources Table must be included in the References list. Please report the information as follows:
REAGENT or RESOURCE: Provide full descriptive name of the item so that it can be identified and linked with its description in the manuscript (e.g., provide version number for software, host source for antibody, strain name). In the Experimental Models section, please include all models used in the paper and describe each line/strain as: model organism: name used for strain/line in paper: genotype. (i.e., Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J). In the Biological Samples section, please list all samples obtained from commercial sources or biological repositories. Please note that software mentioned in the Methods Details or Data and Software Availability section needs to be also included in the table. See the sample Table at the end of this document for examples of how to report reagents.
SOURCE: Report the company, manufacturer, or individual that provided the item or where the item can obtained (e.g., stock center or repository). For materials distributed by Addgene, please cite the article describing the plasmid and include “Addgene” as part of the identifier. If an item is from another lab, please include the name of the principal investigator and a citation if it has been previously published. If the material is being reported for the first time in the current paper, please indicate as “this paper.” For software, please provide the company name if it is commercially available or cite the paper in which it has been initially described.
-
IDENTIFIER: Include catalog numbers (entered in the column as “Cat#” followed by the number, e.g., Cat#3879S). Where available, please include unique entities such as RRIDs, Model Organism Database numbers, accession numbers, and PDB or CAS IDs. For antibodies, if applicable and available, please also include the lot number or clone identity. For software or data resources, please include the URL where the resource can be downloaded. Please ensure accuracy of the identifiers, as they are essential for generation of hyperlinks to external sources when available. Please see the Elsevier list of Data Repositories with automated bidirectional linking for details. When listing more than one identifier for the same item, use semicolons to separate them (e.g. Cat#3879S; RRID: AB_2255011). If an identifier is not available, please enter “N/A” in the column.
-
○
A NOTE ABOUT RRIDs: We highly recommend using RRIDs as the identifier (in particular for antibodies and organisms, but also for software tools and databases). For more details on how to obtain or generate an RRID for existing or newly generated resources, please visit the RII or search for RRIDs.
-
○
Please use the empty table that follows to organize the information in the sections defined by the subheading, skipping sections not relevant to your study. Please do not add subheadings. To add a row, place the cursor at the end of the row above where you would like to add the row, just outside the right border of the table. Then press the ENTER key to add the row. Please delete empty rows. Each entry must be on a separate row; do not list multiple items in a single table cell. Please see the sample table at the end of this document for examples of how reagents should be cited.
TABLE FOR AUTHOR TO COMPLETE
Please upload the completed table as a separate document. Please do not add subheadings to the Key Resources Table. If you wish to make an entry that does not fall into one of the subheadings below, please contact your handling editor. (NOTE: For authors publishing in Current Biology, please note that references within the KRT should be in numbered style, rather than Harvard.)
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| Rabbit polyclonal anti-Bard1 | Richard Baer (E.E. McCarthy et al., 2003) | ||
| Rabbit polyclonal anti-Brca1 | Richard Baer (Shakya et al., 2011) | ||
| Mouse monoclonal anti-Brca1 | Andre Nussenzweig (Fernandez-Capetillo & Nussenzweig, 2013) |
||
| Mouse monoclonal anti-BrdU | BD biosciences | Cat#347480 RRID: AB_400326 |
|
| Rat monoclonal anti-BrdU | Abcam | Cat#ab6326 RRID: AB_305426 |
|
| Mouse monoclonal anti-CtIP | Richard Baer (Yu & Baer, 2000) | ||
| Rabbit polyclonal anti-Ctip | Richard Baer (Reczek et al., 2013) | ||
| Mouse monoclonal anti-PCNA | Santa Cruz Biotechnology | Cat# sc-56; RRID:AB_628110 |
|
| Rabbit polyclonal anti-PCNA | Santa Cruz Biotechnology | Cat# sc-7907 RRID:AB_2160375 |
|
| Rabbit polyclonal anti-Rad51 | Millipore | Cat# PC130; RRID: AB_2238184 |
|
| Mouse monoclonal anti-α-Tubulin | Millipore | Cat# CP06 RRID:AB_2617116 |
|
| HRP-conjugated goat polyclonal anti-mouse IgG | Sigma-Aldrich | Cat# A5278 RRID:AB_258232 |
|
| HRP-conjugated donkey polyclonal anti-rabbit IgG | GE healthcare | Cat# NA934 RRID:AB_772211 |
|
| Goat polyclonal anti-rabbit Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11008 RRID:AB_143165 |
|
| Goat polyclonal anti-mouse Alexa Fluor 488 | Thermo Fisher Scientific |
Cat# A-11001 RRID:RRID:AB_253 4069 |
|
| Goat polycolonal anti-rat Alexa Fluor 594 | Thermo Fisher Scientific |
Cat# A-11007 RRID:AB_141374 |
|
| Goat polyclonal anti-mouse IgG2a Alexa Fluor 594 | Thermo Fisher Scientific |
Cat# A-21135 RRID:AB_2535774 |
|
| Rabbit polyclonal anti-Mre11 |
Cell Signaling Technology |
Cat# 4895 RRID:AB_2145100 |
|
| Mouse monoclonal anti-Mre11 | Santa Cruz Biotechnology |
Cat# sc-135992 RRID:AB_2145244 |
|
| Rabbit polyclonal anti-Histone H4 |
Abcam | Cat# ab10158 RRID:AB_296888 |
|
| Mouse monoclonal anti-PAR | Millipore | Cat# AM80–100UG RRID:AB_212695 |
|
| Mouse monoclonal anti-GST | Santa Cruz Biotechnology |
Cat# sc-138 RRID:AB_627677 |
|
| Mouse monoclonal anti-HP1γ (clone 42s2) | Millipore | Cat# 05–690 RRID:AB_309910 |
|
| Mouse monoclonal anti-FLAG M2 | Sigma-Aldrich | Cat# F3165 RRID:AB_259529 |
|
| Chemicals, Peptides, and Recombinant Proteins | |||
| Transfection reagent: Lipofectamine 2000 | Thermo Fisher Scientific |
Cat# 11668019 | |
| Protease inhibitor cocktail | Roche | Cat#11697498001 | |
| Protein A sepharose CL-4B | Ge healthcare | Cat# 17078001 | |
| Mitomycin C | Sigma-Aldrich | Cat# m0503 | |
| Olaparib | SelleckChem | Cat# AZD2281 | |
| KaryoMax Colcemid | Thermo Fisher Scientific |
Cat# 15212012 | |
| VECTASHIELD Anti-fade mounting medium with DAPI | Vector laboratories | Cat# H-1200 | |
| Leukemia inhibitory factor | Millipore | Cat# ESG1107 | |
| IdU | Sigma-Aldrich | Cat# I7125 | |
| CldU | Sigma-Aldrich | Cat# C6891 | |
| Mirin | Sigma-Aldrich | Cat# M9948 | |
| Hydroxyurea | Sigma-Aldrich | Cat# H8627 | |
| Prolong Gold Anti-fade | Thermo Fisher Scientific | Cat# P36930 | |
| Biotin azide | Thermo Fisher Scientific |
Cat# B10184 | |
| EdU | Thermo Fisher Scientific | Cat# A10044 | |
| Streptavidin-agarose | Thermo Fisher Scientific | Cat# SA10004 | |
| GelRed | Biotium | Cat# 41002 | |
| Poly(ADP-Ribose) | Trevigen | Cat# 4336–100-01 | |
| Protran nitrocellulose membrane | Sigma-Aldrich | Cat# GE10600002 | |
| Biodyne B nylon transfer membrane | Pall Life Sciences | Cat# 60201 | |
| GST-Bard1-wt | This paper | N/A | |
| GST-Bard1-S563F | This paper | N/A | |
| GST-Bard1-K607A | This paper | N/A | |
| Deposited Data | |||
| Raw images | Mendeley Data | http://dx.doi.org/10.17632/shfsd2z354.1 | |
| Experimental Models: Cell Lines | |||
| Bard1-S563F MEFs & ES cells | This paper | N/A | |
| Bard1-K607A MEFs & ES cells | This paper | N/A | |
| Bard1-Q552X MEFs | This paper | N/A | |
| Bard1-co-rec MEFs | This paper | N/A | |
| Brca1-S1598F MEFs | This paper | N/A | |
| Brca1-S1598F ES cells | Shakya et al., 2011 | N/A | |
| Experimental Models: Organisms/Strains | |||
| Mouse: Bard1S563F | This paper | N/A | |
| Mouse: Bard1K607A | This paper | N/A | |
| Recombinant DNA | |||
| Plasmid: pMSSVLT | Schuermann M. (1990) Nucl. Acids Res. 18: 4945 |
||
| Plasmid: pCAGGS | Westermark et al., 2003 |
||
| Plasmid: pCAGGS I-SCEI | |||
| Plasmid: pCAGGS GFP | |||
| Software and Algorithms | |||
| Metafer 4 scanning and imaging platform | Metasystems | https://metasystems-international.com/us/products/metafer/ | |
| Isis fluorescent imaging platform | https://metasystems-international.com/us/products/isis/ | https://metasystems-international.com/us/products/isis/ | |
| BD CellQuest Pro | BD Biosciences | https://metasystems-international.com/us/products/isis/ | |
| CometScore software version 1.5 | TriTek | http://autocomet.com/index.php?id=cometscore | |
| FlowJo version 10 | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo | |
| ImageJ | National Institutes of Health |
https://imagej.nih.gov/ij | |
| Prism | GraphPad | https://www.graphpadcom/scientific-software/prism | |
| Other | |||
| Gammacell 40 cesium irradiator | Atomic energy of Canada |
N/A | |
| Axio Imager Z2 fluorescent microscope | Zeiss | N/A | |
| BD FACSCalibur cell analyzer | BD Biosciences | N/A | |
| Eclipse 80i fluorescent microscope | Nikon | N/A | |
| Microtip sonicator | Misonix | N/A | |
TABLE WITH EXAMPLES FOR AUTHOR REFERENCE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat# 3879S; RRID: AB_2255011 |
|
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat# T9026; RRID: AB_477593 |
|
| Rabbit polyclonal anti-BMAL1 | This paper | N/A | |
| Bacterial and Virus Strains | |||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Krashes et al., 2011 | Addgene AAV5; 44361-AAV5 |
|
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP | Hope Center Viral Vectors Core |
N/A | |
| Cowpox virus Brighton Red | BEI Resources | NR-88 | |
| Zika-SMGC-1, GENBANK: KX266255 | Isolated from patient (Wang et al., 2016) |
N/A | |
| Staphylococcus aureus | ATCC | ATCC 29213 | |
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC 700294 | |
| Biological Samples | |||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ |
Cat# UMB1455 | |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ | |
| Patient-derived xenografts (PDX) | Children’s Oncology Group Cell Culture and Xenograft Repository |
http://cogcell.org/ | |
| Chemicals, Peptides, and Recombinant Proteins | |||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350–13-2 |
|
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433–59-5 (free base) |
|
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124- 87–8 |
|
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
|
| Activated S6K1 | Millipore | Cat# 14–486 | |
| GST-BMAL1 | Novus | Cat# H00000406- P01 |
|
| Critical Commercial Assays | |||
| EasyTag EXPRESS 35S Protein Labeling Kit | Perkin-Elmer | NEG772014MC | |
| CaspaseGlo 3/7 | Promega | G8090 | |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202–1012 | |
| Deposited Data | |||
| Raw and analyzed data | This paper | GEO: GSE63473 | |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 | |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium |
http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ | |
| Nanog STILT inference | This paper; Mendeley Data |
http://dx.doi.org/10.17632/wx6s4mj7s8.2 | |
| Affinity-based mass spectrometry performed with 57 genes |
This paper; and Mendeley Data |
Table S8; http://dx.doi.org/10.17632/5hvpvspw82.1 |
|
| Experimental Models: Cell Lines | |||
| Hamster: CHO cells | ATCC | CRL-11268 | |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon |
FlyBase: FBtc0000181 |
|
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility |
N/A | |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09–0021) |
HSCI iPS Core | hES Cell Line: HUES- 8 |
|
| Experimental Models: Organisms/Strains | |||
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center |
WB Strain: BC4011; WormBase: WBVar00241916 |
|
| D. melanogaster: RNAi of Sxl: y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
|
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 | |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 | |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
|
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein- Volhard, 2000 |
ZFIN: ZDB-GENO- 060207–1 |
|
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al., 2012 | N/A | |
|
Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 |
NASC | NASC ID: N70450 | |
| Oligonucleotides | |||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA |
This paper | N/A | |
| Primers for XX, see Table SX | This paper | N/A | |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC |
This paper | N/A | |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT |
Gene Tools | ZFIN: ZDB- MRPHLNO-061106- 5 |
|
| ACTB (hs01060665_g1) | Life Technologies | Cat# 4331182 | |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper |
N/A | |
| Recombinant DNA | |||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat# 632162 | |
| Plasmid: GFP-Nito | This paper | N/A | |
| cDNA GH111110 | Drosophila Genomics Resource Center |
DGRC:5666; FlyBase:FBcl013041 5 |
|
| AAV2/1-hsyn-GCaMP6- WPRE |
Chen et al., 2013 |
N/A | |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al., 2009 | Addgene Plasmid #21339 |
|
| Software and Algorithms | |||
| Bowtie2 | Langmead and Salzberg, 2012 |
http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ | |
| Weighted Maximal Information Component Analysis v0.9 |
Rau et al., 2013 | https://github.com/ChristophRau/wMICA | |
| ICS algorithm | This paper; Mendeley Data |
http://dx.doi.org/10.17632/5hvpvspw82.1 | |
| Other | |||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal. |
This paper | http://aml31.genomewustl.edu | |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite | |
Highlights.
The BRCT domain of BARD1 recruits BRCA1/BARD1 to stalled replication forks
Stalled fork protection (SFP), but not HDR, requires BARD1 BRCT phospho-recognition
SFP and HDR both require BRCA1 BRCT phospho-recognition
HDR is sufficient for tumor suppression in the absence of SFP
ACKNOWLEDGEMENTS
We are especially grateful to Jean Gautier and all members of the Baer and Ciccia laboratories for advice and critical comments. This work was supported by NIH/NCI grants to R.B. (R01-CA172272 and P01-CA174653), A.C. (R01-CA197774), and S.Z. (P01-CA174653), as well as by an OCRFA Liz Tilberis Award 368964 and Susan G. Komen CCR16377030 (A.C.). D.B. was supported by an NIH training grant (T32-GM007367) and an NCI fellowship (F31-CA200181). M.H. was supported by the JSPS Overseas Research Fellowships (JSPS-2016180) and the Uehara Memorial Foundation Research Fellowship (201740041). G.L. was supported by Italian Association for Cancer Research (AICF) and Italian Association for Cancer Research (AIRC) post-doctoral research fellowships, and W.J. was supported by NIH/NCI grant P01-CA174653.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain
REFERENCES
- Ayi TC, Tsan JT, Hwang LY, Bowcock AM, and Baer R (1998). Conservation of function and primary structure in the BRCA1-associated RING domain (BARD1) protein. Oncogene 17, 2143–2148. [DOI] [PubMed] [Google Scholar]
- Birrane G, Varma AK, Soni A, and Ladias JA (2007). Crystal structure of the BARD1 BRCT domains. Biochemistry 46, 7706–7712. [DOI] [PubMed] [Google Scholar]
- Botuyan MV, Nomine Y, Yu X, Juranic N, Macura S, Chen J, and Mer G (2004). Structural basis of BACH1 phosphopeptide recognition by BRCA1 tandem BRCT domains. Structure 12, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapperton JA, Manke IA, Lowery DM, Ho T, Haire LF, Yaffe MB, and Smerdon SJ (2004). Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nature Struct. Mol. Biol 11, 512–518. [DOI] [PubMed] [Google Scholar]
- De Brakeleer S, De Greve J, Desmedt C, Joris S, Sotiriou C, Piccart M, Pauwels I, and Teugels E (2016). Frequent incidence of BARD1-truncating mutations in germline DNA from triple-negative breast cancer patients. Clin. Genet 89, 336–340. [DOI] [PubMed] [Google Scholar]
- Ding X, Ray Chaudhuri A, Callen E, Pang Y, Biswas K, Klarmann KD, Martin BK, Burkett S, Cleveland L, Stauffer S, et al. (2016). Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat. Commun 7, 12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost RM, and Jonkers J (2009). Preclinical mouse models for BRCA1-associated breast cancer. Br. J. Cancer 101, 1651–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, and Cortez D (2015). The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 59, 998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, et al. (2008). A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol 4, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RA, Lee MS, Tsutakawa SE, Williams RS, Nazeer I, Kleiman FE, Tainer JA, and Glover JN (2008). The BARD1 C-terminal domain structure and interactions with polyadenylation factor CstF-50. Biochemistry 47, 11446–11456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulkes WD (2008). Inherited susceptibility to common cancers. New Eng. J. Med 359, 2143–2153. [DOI] [PubMed] [Google Scholar]
- Glover JN, Williams RS, and Lee MS (2004). Interactions between BRCT repeats and phosphoproteins: tangled up in two. Trends Biochem. Sci 29, 579–585. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Ray Chaudhuri A, Lopes M, and Costanzo V (2010). Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nature Struct. Mol. Biol 17, 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, and Greenberg RA (2015). Deciphering the BRCA1 Tumor Suppressor Network. J. Biol. Chem 290, 17724–17732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Xu XL, Yang M-CW, Wei F, Ayi T-C, Bowcock AM, and Baer R (1997). Cell-cycle dependent colocalization of BARD1 and BRCA1 in discrete nuclear domains. Proc. Natl. Acad. Sci. USA 94, 12075–12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, de Antoni A, Techer H, Baldi G, and Costanzo V (2017a). Moonlighting at replication forks - a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett [DOI] [PubMed]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017b). Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 67, 867–881.e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi H, Mohseni M, Tamaki A, Garay JP, Croessmann S, Karnan S, Ota A, Wong HY, Konishi Y, Karakas B, et al. (2011). Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc. Natl. Acad. Sci. USA 108, 17773–17778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufer M, Nandula SV, Modi AP, Wang S, Jasin M, Murty VVVS, Ludwig T, and Baer R (2007). Structural requirements for the BARD1 tumor suppressor in chromosomal stability and homology-directed DNA repair. J. Biol. Chem 282, 34325–34333. [DOI] [PubMed] [Google Scholar]
- Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, et al. (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun 8, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Lu L-Y, Yang C-Y, Wang S, and Yu X (2013). The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes & Dev . 27, 1752–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, and Yu X (2013). Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 23, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat ML, Gyorki DE, Ward T, Partanen A, et al. (2009). Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nature Med 15, 907–913. [DOI] [PubMed] [Google Scholar]
- Lloyd J, Chapman JR, Clapperton JA, Haire LF, Hartsuiker E, Li J, Carr AM, Jackson SP, and Smerdon SJ (2009). A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell 139, 100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig T, Fisher P, Ganesan S, and Efstratiadis A (2001). Tumorigenesis in mice carrying a truncating Brca1 mutation. Genes & Dev 15, 1188–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manke IA, Lowery DM, Nguyen A, and Yaffe MB (2003). BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302, 636–639. [DOI] [PubMed] [Google Scholar]
- Martins FC, De S, Almendro V, Gonen M, Park SY, Blum JL, Herlihy W, Ethington G, Schnitt SJ, Tung N, et al. (2012). Evolutionary pathways in BRCA1-associated breast tumors. Cancer Discov 2, 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy EE, Celebi JT, Baer R, and Ludwig T (2003). Loss of Bard1, the heterodimeric partner of the Brca1 tumor suppressor, results in early embryonic lethality and chromosomal instability. Mol. Cell. Biol 23, 5056–5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, et al. (2017). Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun 8, 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, and Jasin M (1999). Brca1 controls homology-directed DNA repair. Mol. Cell 4, 511–518. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, and Jasin M (2010). Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell. Biol 11, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraju G, and Scully R (2007). Minding the gap: The underground functions of BRCA1 and BRCA2 at stalled replication forks. DNA Repair 6, 1018–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania S, Bade S, Le Guillou M, Burke K, Reed R, Bowman-Colin C, Su Y, Ting DT, Polyak K, Richardson AL, et al. (2014). BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat. Commun 5, 5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Hu P, Han M, Ellis N, and Jasin M (2001). Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes & Dev 15, 3237–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajska M, Antoszewska E, Piskorz A, Brozek I, Borg A, Kusmierek H, Biernat W, and Limon J (2011). Cancer predisposing BARD1 mutations in breast-ovarian cancer families. Breast Cancer Res. Treat [DOI] [PubMed]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, and Jasin M (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, and Livingston DM (1997). Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 90, 425–435. [DOI] [PubMed] [Google Scholar]
- Sedic M, Skibinski A, Brown N, Gallardo M, Mulligan P, Martinez P, Keller PJ, Glover E, Richardson AL, Cowan J, et al. (2015). Haploinsufficiency for BRCA1 leads to cell-type-specific genomic instability and premature senescence. Nat. Commun 6, 7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin C-S, deRooij DG, Hirsch S, Kandasamy R, Hicks JB, et al. (2011). BRCT phosphoprotein recognition, but not E3 ligase activity, is essential for BRCA1 tumor suppression. Science 334, 525–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya R, Szabolcs M, McCarthy EE, Ospina E, Basso K, Nandula SV, Murty VV, Baer R, and Ludwig T (2008). The basal-like mammary carcinomas induced by Brca1 or Bard1 inactivation implicate the BRCA1/BARD1 heterodimer in tumor suppression. Proc. Natl. Acad. Sci. USA 105, 7040–7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozaki EN, Gu L, Yan N, and Shi Y (2004). Structure of the BRCT repeats of BRCA1 bound to a BACH1 phosphopeptide: implications for signaling. Mol. Cell 14, 405–412. [DOI] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, and Cortez D (2012). Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nature Protocols 7, 594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang J-W, Madubata C, Anand R, Levy B, Rabadan R, et al. (2017). Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 68, 414–430.e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma AK, Brown RS, Birrane G, and Ladias JA (2005). Structural basis for cell cycle checkpoint control by the BRCA1-CtIP complex. Biochemistry 44 , 10941–10946. [DOI] [PubMed] [Google Scholar]
- Venkitaraman AR (2014). Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science 343, 1470–1475. [DOI] [PubMed] [Google Scholar]
- Westermark UK, Reyngold M, Olshen AB, Baer R, Jasin M, and Moynahan ME (2003). BARD1 participates with BRCA1 in homology-directed repair of chromosome breaks. Mol. Cell. Biol 23, 7926–7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, Classen S, Glover JNM, Iwasaki H, Russell P, et al. (2009). Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 139, 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Lee MS, Hau DD, and Glover JN (2004). Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nature Struct. Mol. Biol 11, 519–525. [DOI] [PubMed] [Google Scholar]
- Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, and Scully R (2014). BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510, 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, Yang M-CW, Hwang L-Y, Bowcock AM, and Baer R (1996). Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nature Genet 14, 430–440. [DOI] [PubMed] [Google Scholar]
- Wu Q, Jubb H, and Blundell TL (2015a). Phosphopeptide interactions with BRCA1 BRCT domains: More than just a motif. Prog. Biophys. Mol. Biol 117, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Nishikawa H, Fukuda T, Vittal V, Asano M, Miyoshi Y, Klevit RE, and Ohta T (2015b). Interaction of BARD1 and HP1 Is Required for BRCA1 Retention at Sites of DNA Damage. Cancer Res 75, 1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Hamdy FC, and Helleday T (2012). Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 72, 2814–2821. [DOI] [PubMed] [Google Scholar]
- Yu X, Chini CC, He M, Mer G, and Chen J (2003). The BRCT domain is a phospho-protein binding domain. Science 302, 639–642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.