

SUMMARY
Cell adhesion, morphogenesis, mechanosensing, and muscle contraction rely on contractile actomyosin bundles, where the force is produced through sliding of bipolar myosin II filaments along actin filaments. The assembly of contractile actomyosin bundles involves registered alignment of myosin II filaments and their subsequent fusion into large stacks. However, mechanisms underlying the assembly of myosin II stacks, and their physiological functions have remained elusive. Here we identified myosin-18B, an unconventional myosin, as a stable component of contractile stress fibers. Myosin-18B co-localized with myosin II motor domains in stress fibers, and was enriched at the ends of myosin II stacks. Importantly, myosin-18B deletion resulted in drastic defects in the concatenation and persistent association of myosin II filaments with each other, and thus led to severely impaired assembly of myosin II stacks. Consequently, lack of myosin-18B resulted in defective maturation of actomyosin bundles from their precursors in osteosarcoma cells. Moreover, myosin-18B knockout cells displayed abnormal morphogenesis, migration, and ability to exert forces to the environment. These results reveal a critical role for myosin-18B in myosin II stack assembly, and provide evidence that myosin II stacks are important for a variety of vital processes in cells.
Keywords: myosin-18B, actin, stress fiber, actomyosin bundles, myosin II stack
eTOC
Jiu et al. demonstrate that a highly divergent member of the myosin family, myosin-18B, is critical for assembly of myosin II stacks in human osteosarcoma cells. They also provide evidence that these higher-order myosin II -structures are important for generation of functional actomyosin bundles in cells.
Summary statement:
Myosin-18B is critical for assembly and maturation of NMII containing contractile stress fibers.
Graphical Abstract
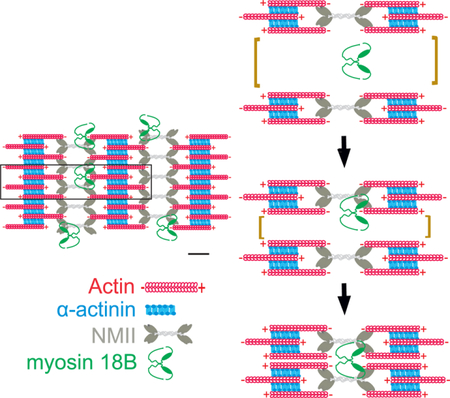
INTRODUCTION
The ability of cells to migrate, change their shape, and exert forces to the environment depends on the actin cytoskeleton. To support these vital processes, actin filaments assemble into protrusive and contractile structures, where force is produced through polymerization of actin filaments against membranes and through sliding of bipolar myosin II filaments along actin filaments, respectively [1]. In muscle cells, highly organized actomyosin bundles called myofibrils provide the force for muscle contraction. Also many non-muscle cell-types harbor related contractile bundles composed of a bipolar array of actin and non-muscle myosin II (NMII) filaments that are collectively called the stress fibers. Although stress fibers are reminiscent of muscle myofibrils, their contractile units are not precisely aligned, and the mechanisms regulating their contractility are different [2, 3].
Stress fibers contribute to adhesion, migration, morphogenesis, and mechanosensing of non-muscle cells [4–9]. Moreover, contractile actomyosin bundles control the subcellular localization and dynamics of cytoplasmic intermediate filaments and propel the transport of T cell receptor microclusters at the immunological synapses [10, 11]. Based on their protein compositions and associations with focal adhesions, stress fibers can be divided into three categories: non-contractile dorsal stress fibers (also called the radial fibers), transverse arcs that are thin, curved actomyosin bundles, and ventral stress fibers that are thick actomyosin bundles directly linked to focal adhesions at both ends. Ventral stress fibers are generated through coalescence of multiple thin transverse arcs during the centripetal flow, and their formation additionally depends on mechanosensitive regulation of actin filament assembly at focal adhesions [7, 12–14].
The fusion of transverse arcs, and consequent generation of ventral stress fibers, are accompanied by an increase in the contractile force [7, 15]. Importantly, recent studies revealed links between arc fusion and formation of larger NMII stacks at the lamellum. Through a combination of super-resolution microscopy approaches, sequential amplification of NMII filaments was identified to occur close to the leading edge. Moreover, formation of larger NMII stacks was shown to take place through expansion of initial NMII filaments and stacks, as well as through concatenation of NMII filaments. These processes are dependent on NMII motor activity and actin filaments, and were proposed to be important for self-organization of arcs into ventral stress fibers [16–18]. Finally, existence of long- range attractive forces was suggested to occur between individual NMII filaments during the stack formation [17, 18]. Thus, arc fusion and NMII stack formation coincide during the formation of contractile stress fibers, but the underlying mechanisms have remained elusive. For example, whether the formation of NMII stacks depends on specific myosin-associated proteins is not known. Moreover, due to lack of specific means for inhibiting stack assembly, the physiological functions of myosin stacks have not been determined.
Beyond actin and NMII, stress fibers are composed of a large array of actin-regulating proteins, including other members of the myosin superfamily [3]. Divergent myosins, which associate with contractile actomyosin bundles, include myosin-18A and myosin-18B. They are composed of myosin II-like head domains followed by a coiled-coil region. In variance to myosin II proteins, myosin-18A harbors an N-terminal extension composed of a lysine/glutamate-rich region and the PDZ domain [19]. Myosin-18B, on the other hand, harbors relatively long N- and C-terminal extensions that do not display detectable homology to known protein domains [20, 21]. At least myosin-18A head domain lacks the actin-activated ATPase activity, and the full-length protein does not form bipolar filaments [22, 23]. Importantly, electron microscopy, co-sedimentation, and single-molecule imaging studies demonstrated that purified myosin-18A co-assembles with NMII in vitro. Moreover, super-resolution microscopy experiments revealed that in cells myosin-18A co-assembles with NMII in mixed bipolar filaments that localize to contractile stress fibers [22].
Myosin-18B was originally identified as a tumor suppressor [20]. Subsequently, mutations in the myosin-18B gene and its altered expression levels were linked to progression of lung, colorectal, and ovarian cancers as well as to cardiomyopathy and muscle weakness [20, 24–27]. Myosin-18B is expressed in cardiac and skeletal muscles, and in some non-muscle tissues [20, 21]. In mice and zebrafish, myosin-18B deficiency results in myocardial defects, and aberrant alignment of thick and thin filaments within sarcomeres [28–30]. However, the molecular functions of myosin-18B have remained elusive.
Here, we demonstrate that myosin-18B exists as compact structures that co-localize with myosin II motor domains in stress fibers of osteosarcoma cells. Deletion of myosin-18B results in defective assembly of large myosin II stacks, leading to severe problems in generation of ventral stress fibers from transverse arcs. Collectively, our study demonstrates that myosin-18B is critical for assembly of myosin II stacks, and that these higher-order myosin-structures are important for generation of functional actomyosin bundles in cells.
RESULTS
Myosin-18B is a stable component of contractile stress fibers in osteosarcoma cells
To identify novel stress fiber components, we performed a proximity dependent biotin identification (BioID) analysis [31] on human osteosarcoma cells (U2OS) using a biotin ligase fused to a central stress fiber component, tropomyosin-1.6 (Tpm1.6). Among the high-confidence interactors was myosin-18B, an unconventional myosin family protein (Figure 1A and 1B; Table S1). Western blot analysis revealed that myosin-18B is highly expressed in cardiomyocytes and U2OS cells, whereas myosin-18A is the predominant myosin-18 family protein in dermal fibroblasts and HeLa cells (Figure 1E).
Figure 1. Myosin-18B is a stable component of contractile stress fibers.
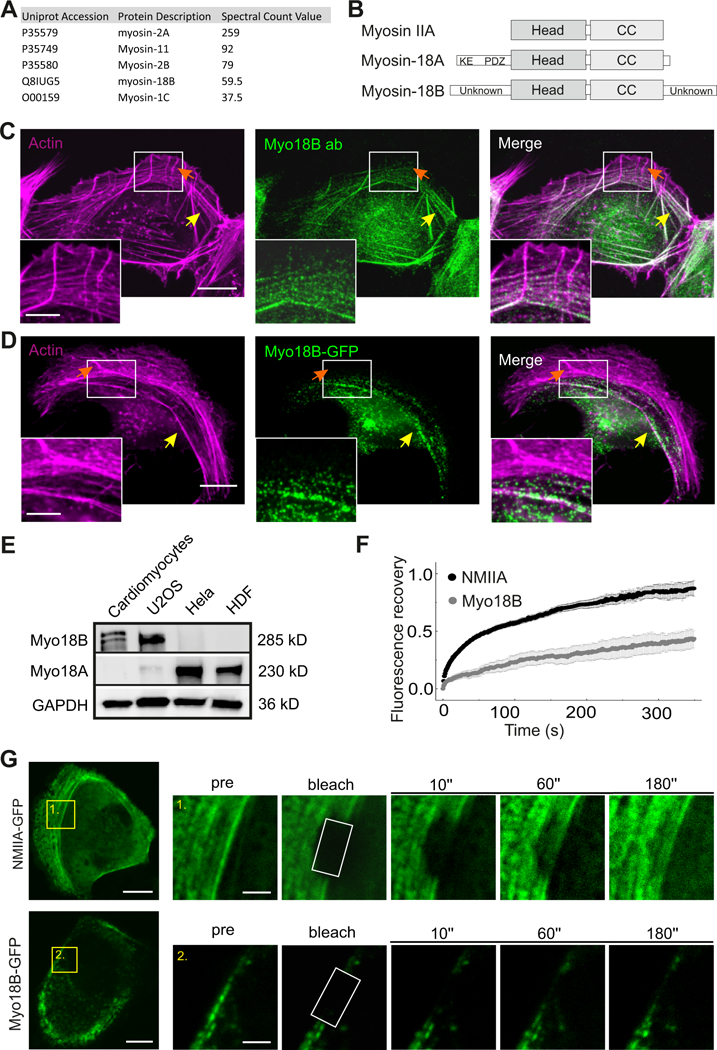
(A) Top myosin family protein hits from the BioID screen for identification of tropomyosin 1.6 (Tpm1.6) -associated proteins in U2OS cells. (B) Domain structures of myosin IIA, myosin-18A and myosin-18B. All three proteins contain a motor-like head domain followed by the coiled-coil (CC) region. Myosin-18A additionally contains a lysine/glutamate (KE) -rich region and the PDZ domain in its N-terminal extension, whereas myosin-18B harbors large N- and C-terminal extensions with unknown domain structures. (C) Immunofluorescence microscopy analysis demonstrating that in U2OS cells endogenous myosin-18B localizes to F-actin -rich (visualized by fluorescent phalloidin) ventral stress fibers (indicated by yellow arrows), but not to non-contractile dorsal stress fibers (indicated by orange arrows). (D) Also myosin- 18B-GFP fusion protein co-localizes with myosin IIB to ventral stress fibers. Bars, 10 µm and 5 µm in the panels C, D and magnified insets, respectively. (E) Western blot analysis of endogenous myosin- 18B and myosin-18A in U2OS, HeLa, and HDF cells. GAPDH was probed for equal sample loading. Please note that the doublet band in the cardiomyocyte extract at ~280 kDa may result from post- translational modification of myosin-18B. (F) Normalized average Fluorescence-recovery-after- photobleaching (FRAP) recovery curves of NMIIA (black line) and myosin-18B (gray line) demonstrating their dynamics on stress fibers. 23 myosin-18B-GFP transfected cells and 22 NMIIA- GFP transfected cells were used for quantification. The data are presented as mean ± s.e.m. (G) FRAP analysis of the dynamics of NMIIA-GFP and myosin-18B-GFP in contractile stress fibers of U2OS cells. Examples of cells expressing NMIIA-GFP and myosin-18B-GFP are shown on the top, and magnified regions on the bottom represent time-lapse images of the bleached regions. Bars, 10 µm (in cell images) and 2 µm (in the magnified time-lapse images). See also Figure S1 and Table S1.
All myosin-18B antibodies tested in this study recognized, in addition to myosin-18B, also other proteins in a Western blot analysis. However, myosin-18B antibody from LSBio (cat # LS-C403352) appeared specific in immunofluorescence microscopy because: i). It displayed specific stress fiber staining in wild-type U2OS cells, whereas only a weak, diffuse signal was detected in myosin-18B knockout cells (Figure S1A), and ii). It faithfully recognized myosin-18B-GFP that was expressed in myosin-18B knockout cells (Figure S1B) and in wild-type cells (Figure S1C). Immunofluorescence microscopy on U2OS cells using this antibody revealed that myosin-18B localizes to contractile ventral stress fibers and to a subset of transverse arcs, but is absent from the non-contractile dorsal stress fibers. In addition to stress fiber localization, myosin-18B appeared to accumulate to the perinuclear region of cells (Figure 1C). Similar localization to ventral stress fibers and perinuclear region was also observed for myosin-18B-GFP expressed in U2OS cells, confirming that myosin-18B is a component of contractile stress fibers (Figure 1D). Myosin-18B-GFP co-localized with Tpm1.6 in ventral stress fibers, but the perinuclear myosin-18B puncta did not clearly accumulate to F-actin rich structures in U2OS cells (Figure S1E).
Live-cell imaging on U2OS cells expressing myosin-18B-GFP and MLC (myosin light chain) - mCherry revealed that the stress fiber -associated population of myosin-18B underwent slow retrograde flow together with NMII, whereas the myosin-18B puncta at the perinuclear region were much more mobile (Figure S1D). Fluorescence-recovery-after-photobleaching (FRAP) was then applied to examine the dynamics of myosin-18B molecules in stress fibers. These experiments revealed that myosin-18B-GFP displays very slow fluorescence recovery on photobleached ventral stress fibers compared to NMIIA-GFP (Figure 1F and 1G). Thus, myosin-18B localizes to the NMII-containing actin stress fibers, and exhibits relatively slow association/dissociation kinetics at these actomyosin bundles.
Myosin-18B co-localizes with NMII motor domains
To examine the association of myosin-18B with stress fibers more precisely, we applied super- resolution structured-illumination microscopy (SIM) on cells expressing the myosin-18B-GFP construct. Consistent with the conventional fluorescence microscopy data, the myosin-18B puncta localized to the perinuclear regions as well as to the F-actin rich stress fibers, and were most prominent in mature ventral stress fibers and in thick arc bundles located close to the cell center (Figure 2A, 2B and S2). The myosin-18B puncta did not co-localize with α-actinin or NMIIA and NMIIB tails in stress fibers (Figure 2B and S2). Instead, most myosin-18B puncta overlapped with NMII motor domains, as visualized by an antibody against the regulatory light chain (RLC) (Figure 2A). Importantly, the myosin-18B puncta were not uniformly distributed along the NMII motor domains in myosin filaments and filament stacks, but were typically enriched at the ends of large filament stacks as well as at the intersections of adjacent NMII stacks (Figure 2A, 2C, and 2D). It is also important to note that many myosin-18B puncta did not stain with an antibody against RLC, suggesting that myosin-18B itself perhaps does not bind RLC.
Figure 2. Myosin-18B forms a compact structure, which co-localizes with the myosin II motor domains typically at the distal regions of myosin bundles and stacks.
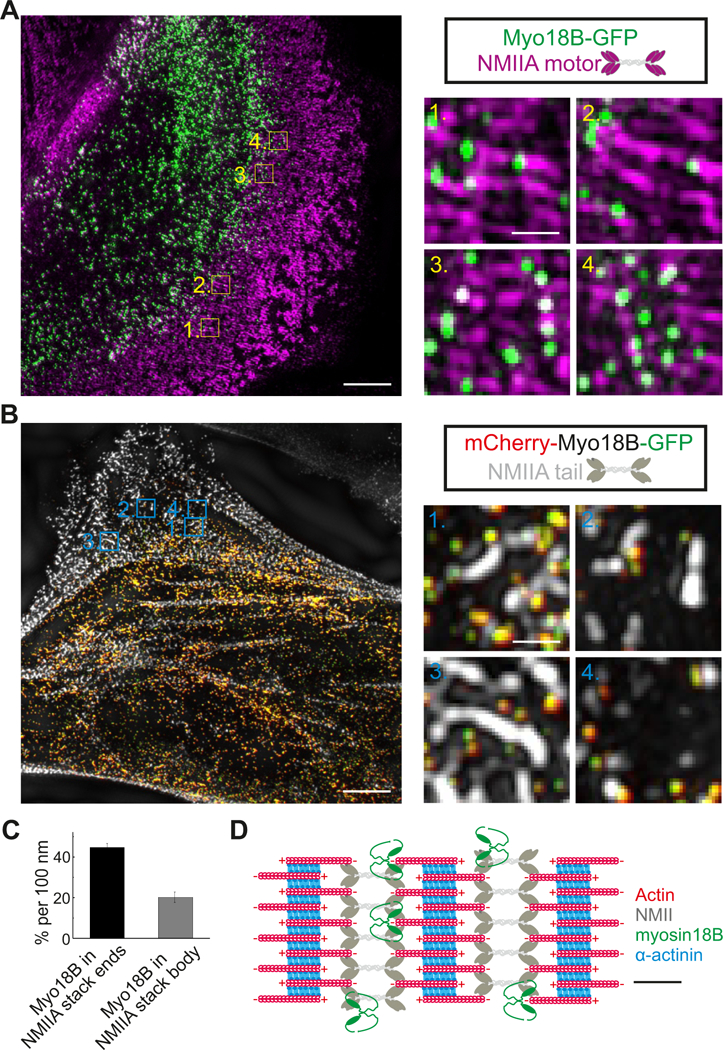
(A) Representative example of a 3D-SIM image obtained from an U2OS cell transfected with a construct expressing myosin-18B- GFP. Myosin II motor domains (magenta) were visualized by an antibody against the regulatory light chain (RLC). Magnified images illustrate that myosin-18B co-localizes with myosin II motor domains at the ends of large myosin stacks (1, 2) and along the short myosin stacks (3, 4). Bars, 5 µm and 0.5 µm in the left and right magnified panels, respectively. (B) Representative example of a 3D-SIM image obtained from a U2OS cell transfected with a construct expressing myosin-18B fused to mCherry and GFP at its N- and C-termini, respectively. Please note that the N- and C-termini of myosin-18B molecules are close (< 0.1 µm) to each other. The NMIIA tail domain (white) was visualized by an antibody against NMIIA C-terminus. Magnified images illustrate localization of myosin-18B molecules adjacent to the NMIIA tails at the ends of myosin stacks (1, 2), in the region flanking the NMIIA tails in the stack (3), and adjacent to NMIIA tails in small myosin structures (4). Bars, 5 µm and 0.5 µm in the left images of cells and right magnified panels, respectively. (C) The percentage of myosin-18B positive segments at the ends and in the central regions of myosin II stacks (i.e. ~45 % of the 100 nm segments at the ends of NMII stacks contained a myosin-18B puncta, whereas myosin-18B occupancy was ~20 % per 100 nm in the central regions of NMII stacks). Please, see ‘Methods’ for details concerning the quantification. n = 8 cells, and data are presented as mean ± s.e.m. (D) Schematic diagram of myosin-18B localization in actomyosin bundles based on the SIM data. Schematic scale bar, 200 nm. See also Figure S2.
To elaborate whether myosin-18B forms extended structures, or exists as more compact structures in cells, we generated a construct where mCherry and GFP were fused at N- and C-termini of myosin-18B, respectively. SIM analysis of U2OS cells expressing this construct revealed that mCherry-myosin-18B- GFP mainly exists as small puncta, where red and green channels exhibit nearly complete overlap with each other (Figure 2B). Thus, the N- and C-termini of the protein are in close proximity to each other, suggesting that myosin-18B is present as fairly compact structures in cells.
Myosin-18B depletion leads to impaired cell migration and defects in maturation of stress fibers
To explore the cellular function of myosin-18B, we generated myosin-18B knockout U2OS cells by the CRISPR/Cas9 approach. Quantitative RT-PCR analysis demonstrated that in wild-type cells myosin- 18B mRNA is much less abundant compared to NMIIA and NMIIB mRNAs, and that the knockout cells do not express detectable levels of myosin-18B mRNA (Figure S3A and B). Moreover, Western blot analysis demonstrated that the obtained knockout cells did not produce detectable myosin-18B protein (Figure 3A). However, because the myosin-18B antibodies were quite unspecific on Western blots (Figure S3C), a Sanger sequencing analysis was performed on the knockout cells. This revealed that the myosin-18B gene in the knockout cell line harbors a single nucleotide deletion in the second exon targeted by sgRNA, resulting in a frame-shift and appearance of a stop codon 16 triplets downstream of the deletion point (Figure S3D). Illumina amplicon next generation sequencing of genomic DNA further revealed a complete absence of wild-type myosin-18B gene in the knockout colonies. Moreover, no compensatory upregulation of myosin-18A protein or mRNA was detected (Figure 3A and S3E).
Figure 3. Myosin-18B knockout cells display defects in migration and stress fiber maturation.
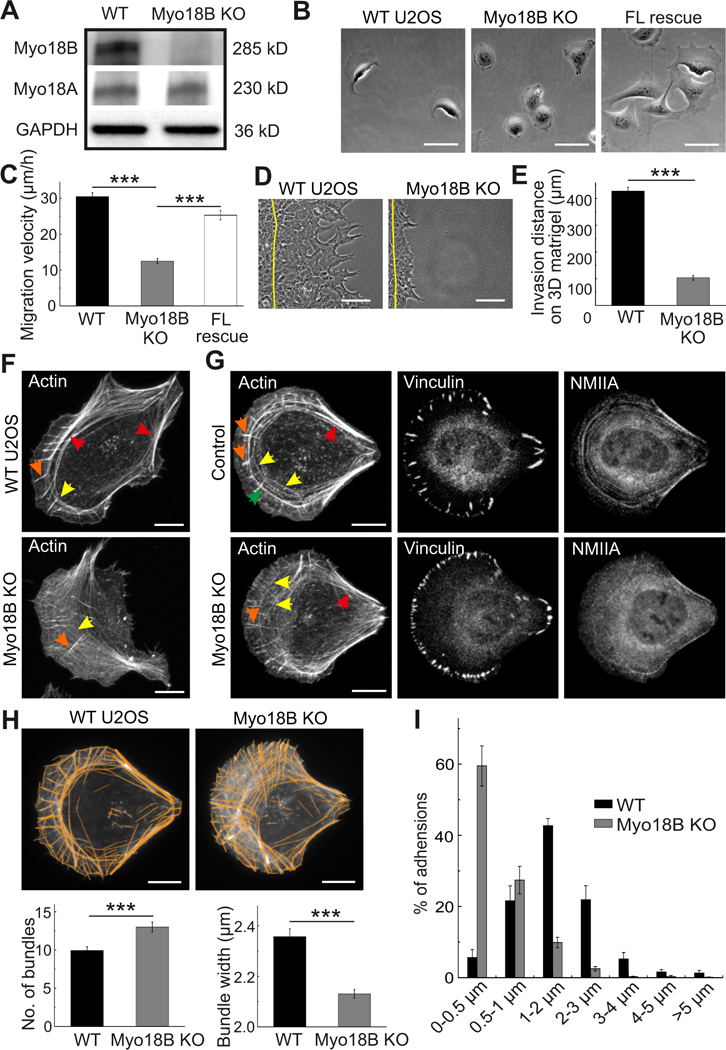
(A) Western blot analysis of endogenous myosin-18B and myosin-18A levels in total cell lysates of wild- type U2OS (WT) and myosin-18B CRISPR/Cas9 knockout (KO) cells. GAPDH was probed for equal sample loading. (B) Representative images from time-lapse videos of wild-type, myosin-18B knockout, and full-length myosin-18B rescue (FL rescue) knockout cells migrating on a fibronectin-coated surface. Bars, 50 µm. (C) Random migration velocities of wild-type, myosin-18B knockout, and full- length myosin-18B rescue knockout cells. Quantification is based on tracking of the displacement of nuclei. The data are presented as average velocity obtained from a 12 hour cell tracking ± s.e.m, ***P = <0.001 (t-test). n= 44 for wild-type, 42 for myosin-18B knockout, and 36 for FL rescue cells. (D) Representative images of wild-type and myosin-18B knockout U2OS cells migrated for 48 hours in a 3D Matrigel matrix. Yellow lines indicate the positions of cells at the beginning of experiment. Bars, 30 µm. (E) Invasion distances of wild-type and myosin-18B knockout cells in 3D Matrigel matrix. Quantification is based on 3 independent experiments. The data are presented as averaged invasion distance after incubation of 48 hours ± s.e.m, ***P=<0.001 (t-test). (F) Representative images of actin filaments visualized by phalloidin in wild-type and myosin-18B knockout cells. Bars, 10 µm. (G) Representative images of actin filaments, focal adhesions and myosin IIA visualized by phalloidin, vinculin and NMIIA antibodies staining, respectively, in wild-type and myosin-18B knockout cells cultured on crossbow shaped fibronectin micro-patterns. Bars, 10 µm. Examples of dorsal stress fibers, thin transverse arcs, thick arcs, and ventral stress fibers in panels F and G are indicated by orange, yellow, green, and red arrows, respectively. (H) Representative examples of cells, and quantification of numbers of actin filament bundles and their widths by software FilamentSensor_0.2.2. Bars, 10 µm. (I) The length distributions of vinculin-positive focal adhesions (FAs). The numbers of adhesions in each size group were divided with the total focal adhesion number of the same cell. n = 774 FAs from 11 cells (wild-type) and 1575 FAs from 10 cells (myosin-18B knockout). Data are presented as mean ± See also Figure S3.
When plated on a fibronectin-coated surface, the myosin-18B knockout cells were slightly smaller and displayed more round morphology compared to wild-type U2OS cells (Figure 3B, S3F and S3G). They also displayed motility defects, as measured from individual cells plated on fibronectin, and this phenotype could be rescued by expressing myosin-18B-GFP in the knockout cells (Figure 3C). The abnormalities in cell shape and motility were further verified from a different myosin-18B knockout clone and by depleting of myosin-18B from U2OS cells by siRNA (Figure S3H and S3I). Moreover, the myosin-18B knockout cells exhibited defects in invasive migration in a 3D environment as examined using Matrigel matrix (Figure 3D and 3E).
Immunofluorescence microscopy revealed that the transverse arcs in myosin-18B deficient cells were typically very thin and appeared to have problems in assembling into thick ventral stress fibers (Figure 3F). To allow more precise analysis of the stress fiber phenotype, cells were plated on crossbow shaped fibronectin micro-patterns, where they obtain nearly identical shapes and display characteristic organization of the stress fiber network [10]. Also on micro-patterns, the arcs were typically thinner compared to control cells (Figure 3G). Similar stress fiber phenotypes were also observed in myosin- 18B siRNA knockdown U2OS cells (Figure S3J and S3K). Moreover, the myosin-18B deficient cells typically displayed abnormally long dorsal stress fibers that is a typical phenotype for U2OS cells lacking thick ventral stress fibers [7].
The stress fiber phenotype was first quantified by ImageJ plugin FilamentSensor [32], which revealed increased number of actin filament bundles in myosin-18B knockout cells, accompanied by decreased average width of the bundles (Figure 3H). Because the FilamentSensor software cannot distinguish between dorsal stress fibers and arcs, we also performed unbiased, blind manual quantification of the numbers of thin and thick arcs in wild-type and myosin-18B knockout cells plated on crossbow micro- patterns. This analysis revealed a dramatic increase in the average number of thin arcs and a decrease in the number of thick arcs in the knockout cells, which could be rescued by wild-type myosin-18B (Figure S4A). Corresponding with the reduced amounts of thick stress fibers, the average sizes of vinculin-positive focal adhesions were diminished in myosin-18B knockout cells (Figure 3I), and they exhibited ∼30% weaker contractile forces to the environment compared to control cells (Figure 4A and 4B). Moreover, the knockout cells displayed wider lamella, increased cell height, and more rearward positioning of the nuclei compared to the wild-type cells (Figure S4B and S4C), which are phenotypes associated with defects in the assembly and contractility of stress fibers [5, 10].
Figure 4. Myosin-18B knockout cells display increased centripetal flow of transverse arcs, and reduced contractility.
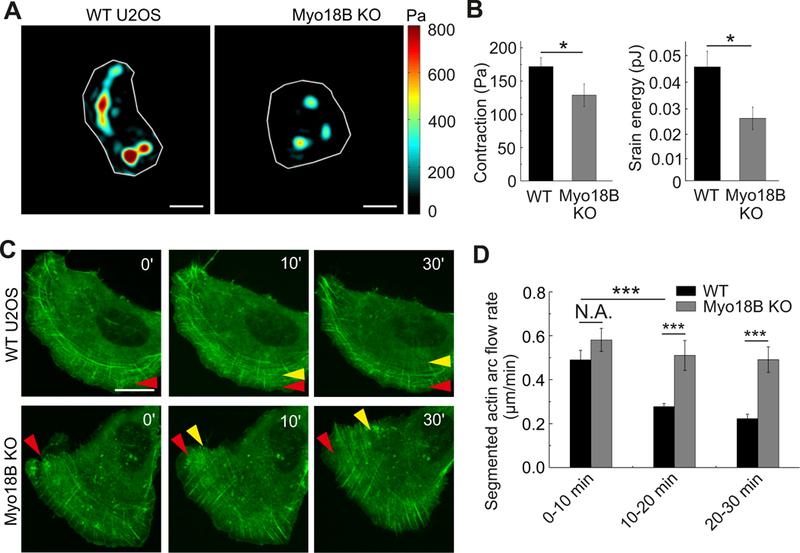
(A) Representative force maps of wild-type and myosin-18B knockout U2OS cells grown on 25 kPa polyacrylamide dishes with fluorescent nanobeads. Bars, 10 µm. (B) Quantification of contraction (root mean square traction) in wild-type (n=68) and myosin-18B knockout cells (n=32) from three independent experiments. The data are presented as mean ± s.e.m. *P=0.033 (non-parametric analysis). (C) Representative examples of transverse arc flow visualized in wild-type and myosin-18B knockout U2OS cells expressing GFP-actin. Red arrows indicate the positions of the observed arcs in the beginning of the movies, and yellow arrows indicate the positions of the same arcs in subsequent time-lapse images. Bars, 20 µm. (D) The time-segmented centripetal flow rates of transverse arcs in wild-type (n=10) and myosin-18B knockout (n=11) cells. Data are represented as mean ± s.e.m. ***P=<0.001 (t-test). See also Figures S4 and S5, and videos S1 and S2.
To elucidate the origin of stress fiber network organization and contractility defects, we examined the dynamics of NMII isoforms. Both NMIIA and NMIIB localized to the remaining thin arcs in myosin- 18B knockout cells (Figure S4D). Moreover, based on FRAP analysis NMIIA-GFP and NMIIB-GFP displayed similar, although slightly increased turnover, on stress fibers of myosin-18B knockout cells compared to control cells (Figure S4E). The moderate increase in NMII dynamics in myosin-18B knockout cells most likely results from the lack of thick ventral stress fibers, because in wild-type cells the dynamics of NMIIA was more rapid in transverse arcs compared to mature ventral stress fibers (Figure S4F). Consistent with earlier studies [33, 34], our FRAP analysis demonstrated that NMIIA displays more rapid dynamics compared to NMIIB.
The assembly of thick ventral stress fibers occurs through fusion of arcs during their centripetal flow [7]. Therefore, we applied live-cell imaging on cells expressing GFP-actin to examine if arc fusion was perturbed in myosin-18B knockout cells. In wild-type U2OS cells the arcs first became visible close to the leading edge. During the flow towards cell center, they fused with each other and their centripetal flow rate decreased. In myosin-18B knockout cells, the appearance of arcs at the leading edge and their initial centripetal flow rates were similar to the ones in wild-type cells. However, as the arcs flowed towards the cell center, they failed to fuse with each other and their centripetal flow rates did not decrease as significantly as in the control cells (Figure 4C, 4D and S5A; Videos S1 and S2). These results demonstrate that myosin-18B is critical for the coalescence of thin transverse arcs during their centripetal flow, thus leading to defects in the formation of thick ventral stress fibers in myosin-18B knockout U2OS cells.
Myosin-18B knockout cells display defects in the assembly of large myosin II stacks
Because myosin-18B localizes to non-muscle myosin filaments and filament stacks, we hypothesized that loss of myosin-18B may affect their organization. This was examined by 3D-SIM experiments, where the NMII motor domain was visualized with an antibody against the RLC, the NMIIA tail domain with an antibody against its C-terminal region, and actin filaments by fluorescent phalloidin. Importantly, whereas the morphology and width (longitudinal diameter) of bipolar NMII filaments appeared unaltered in myosin-18B knockout cells, the NMII filament stacks were smaller and less well aligned compared to control cells (Figure 5A and S5B). Quantification of the 3D-SIM data from a region covering 10 µm from the leading edge of the cell demonstrated that the NMII filament density as well as the average lateral stack diameter, were slightly diminished in myosin-18B knockout cells (Figure 5B). Strikingly, the number of large stacks (lateral stack diameter > 500 nm), quantified from 20 µm wide and 900 µm2 regions at the leading edges, was ~10-fold smaller in the myosin-18B knockout cells compared to the wild-type cells (Figure 5C).
Figure 5. Myosin-18B knockout cells display defects in formation of large NMII stacks.
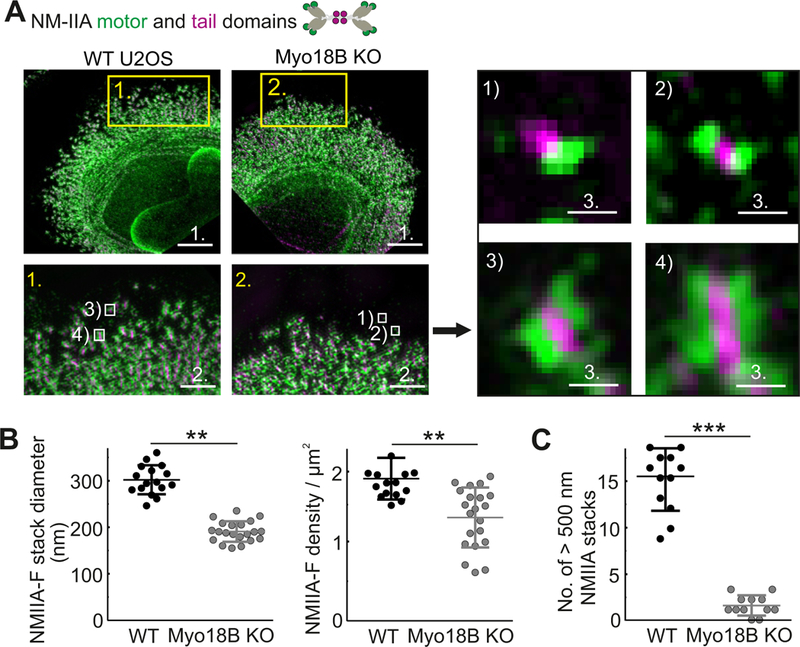
(A) 3D- SIM images of representative wild-type and myosin-18B knockout cells where NMIIA motor and tail domains were visualized by antibodies against regulatory light chain (RLC) and NMIIA C-terminus, respectively. Bars, 5 µm. Magnified images (corresponding to the yellow boxes) display characteristic NMIIA filament distributions in wild-type and myosin-18B knockout cells. Bars, 1 µm. White boxes in magnified regions display examples of: 1) unipolar NMII structures, 2) bipolar NMII filaments, 3) small (lateral diameter < 500 nm) NMII stacks, and 4) large (lateral diameter > 500 nm) NMII stacks, which are displayed on the right. Bars, 200 nm. Bars; 1 (5 µm); 2 (1 µm); 3 (200 nm). (B) The average diameters (nm) and densities (number/µm2) of NMIIA stacks were manually measured. n = 16 cells for wild-type and 21 cells for myosin-18B knockout. Data are represented as mean ± s.e.m. **P=<0.01, ***P=<0.001 (t-test). (C) The numbers of large (lateral diameter >500 nm) NMIIA stacks in the 900 µm2 area within 20 µm wide regions from the edge of cells. n = 12 cells for wild-type and 12 cells for myosin-18B knockout. Data are represented as mean ± s.e.m. ***P=<0.001 (Mann-Whitney u-test). See also Figure S5.
Recent studies demonstrated that large NMII stacks can be generated through parallel alignment and subsequent concatenation of individual bi-polar NMII filaments and small filament stacks [16–18]. To examine whether myosin-18B deficiency leads to problems in this process, we performed live-cell imaging using Zeiss Airyscan microscope on U2OS cells expressing actin-RFP and a construct in which GFP was fused to the N-terminus of the NMIIA motor domain. These experiments revealed relative frequent concatenation (fusion) of NMII filaments and subsequent formation of stable myosin stacks in wild-type U2OS cells (see examples in Figure 6A and S6A; Video S3). In contrast, the NMII filament concatenation events were less frequent in myosin-18B knockout cells (Figure 6C). Instead, the NMII filaments often aligned, but failed to fuse with each other during the observation period or dissociated from each other soon after the fusion event (Figure 6B and S6B; Video S4 and S5). Accordingly, the frequency of myosin II stack splitting was much higher in the myosin-18B knockout cells, despite the fact that they contain significantly lower number of large NMII stacks (Figure 6D). Collectively, these results demonstrate that myosin-18B is critical for proper formation and stability of NMII stacks in U2OS cells.
Figure 6. Myosin-18B knockout cells exhibit decreased stability of NMII stacks.
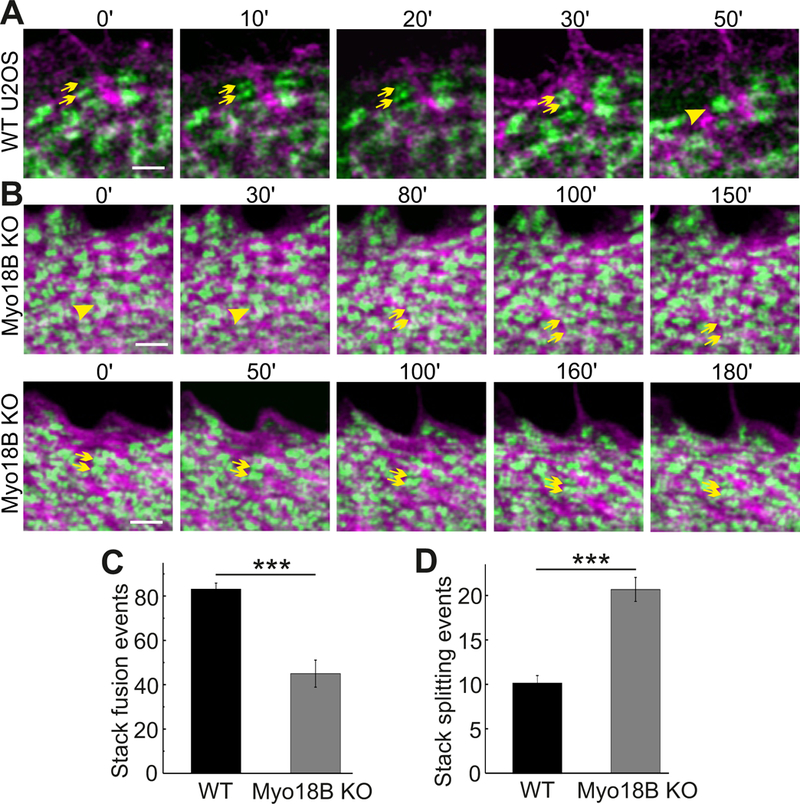
(A and B) Representative time-lapse images recorded with Zeiss Airyscan microscope. Wild-type and myosin- 18B knockout U2OS cells were transfected with plasmids expressing GFP-NMIIA and actin-RFP. An example of NMIIA filament concatenation event is indicated in panel (A) (two NMIIA filaments indicated by yellow arrows fuse with each other to form a NMIIA stack indicated by a yellow arrowhead). An example of NMIIA stack fission event is highlighted in panel (B) upper row (myosin stack indicated by a yellow arrowhead splits into two NMIIA filaments indicated by yellow arrows), and an example of fusion failure of two small NMIIA stacks is highlighted in panel (B) lower row (myosin stacks indicated by yellow arrows). Bars, 1 µm. (C and D) The number of NMIIA filament fusion and splitting events quantified from 900 µm2 areas within 20 µm wide regions from the cell edge in 10 min movies. n = 14 cells for wild-type and 14 cells for myosin-18B knockout. Data are represented as mean ± s.e.m. ***P=<0.001 (Mann-Whitney u-test). See also Figure S6, and videos S3, S4 and S5.
Contribution of myosin-18B domains on its function and dynamics
To explore the contributions of head and coiled-coil domains as well as the N- and C-terminal extensions of myosin-18B for its function, we generated various GFP-tagged deletion constructs of the protein (Figure 7A). The mutant proteins were expressed in U2OS cells (Figure S7A) and tested for their dynamics (Figure 7B and S7B), stress fiber localization (Figure 7C), and ability to rescue the characteristic defects in the size distribution of focal adhesions that result from defects in the assembly of contractile stress fibers in myosin-18B knockout cells (Figure 7D). Only cells expressing intermediate levels of myosin-18B-GFP constructs (as detected by GFP-fluorescence intensity) were selected for further analysis. Based on Western blot and fluorescence imaging experiments (Figure S6C) these cells expressed approximately 10-fold higher levels of GFP-myosin-18B compared to the endogenous myosin-18B levels in U2OS cells.
Figure 7. Localization and dynamics of myosin-18B deletion constructs, and their effects on stress fiber and focal adhesion maturation.
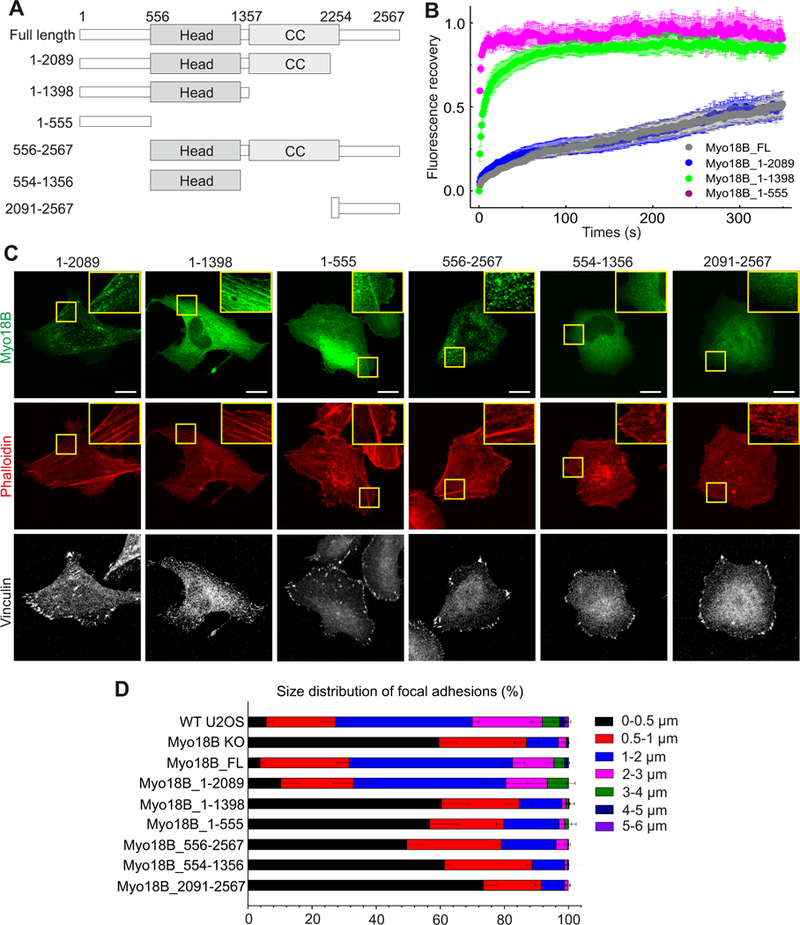
(A) Domain structures of GFP-tagged myosin-18B constructs used in the FRAP and rescue experiments. (B) Normalized average FRAP recovery curves of different myosin-18B-GFP constructs expressed in myosin-18B knockout U2OS cells. 23 myosin-18B-FL-GFP, 18 myosin-18B-1-2089-GFP, 12 myosin-18B-1-1398-GFP, and 13 myosin-18B-1-555-GFP transfected cells were used for quantification. The data are presented as mean ± s.e.m. (C) Representative images of myosin-18B knockout cells transfected with different myosin-18B-GFP constructs shown in panel A. F-actin and focal adhesions were visualized by phalloidin and an antibody against vinculin, respectively. Magnified regions (corresponding to the yellow boxes) highlight localization of the proteins to stress fibers. Bars, 10 µm. (D) The length distributions of vinculin-positive focal adhesions. The number of focal adhesions in each size group was divided with the total adhesion number of the same cell. n = 988 adhesions from 14 cells (wild-type); 2520 adhesions from 16 cells ( myosin-18B knockout,); 979 adhesions from 15 cells (myosin-18B-FL); 1236 adhesions from 12 cells (myosin-18B- 1–2089); 1738 adhesions from 13 cells (myosin-18B-1–1398); 1404 adhesions from 13 cells (myosin18B-1–555 rescue); 2470 adhesions from 13 cells (myosin-18B-556–2567); 1495 adhesions from 13 cells (myosin-18B-554–1356); 1671 adhesions from 13 cells (myosin-18B-2091–2567). Data are represented as mean ± s.e.m. See also Figures S6 and S7.
The protein lacking the C-terminal extension (Myo18B_1–2089) displayed similar stress fiber localization and dynamics to the wild-type protein, and was able to rescue the focal adhesion phenotype nearly as efficiently as the wild-type myosin-18B-GFP. Also the protein lacking the C-terminal extension and the coiled-coil region (Myo18B_1–1398) localized to stress fibers, although more uniformly compared to the full-length protein. However, its dynamics in stress fibers was more rapid compared to full-length myosin-18B, and it was unable to rescue the focal adhesion phenotype. Finally, the N-terminal extension appears to be critical for localization and function of myosin-18B. This is because the isolated N-terminal (Myo18B_1–555) could still localize (although weakly) to stress fibers. Moreover, N-terminal deletions resulted in either punctate (Myo18B_556–2567) or uniform (Myo18B_554–1356, Myo18B_2091–2567) cytoplasmic localization of the protein, and inability to rescue the focal adhesion phenotype (Figure 7B, 7C and 7D). Thus, the N-terminal extension, head domain, and coiled-coil domain are critical for stable stress fiber association and cellular function of myosin-18B, whereas the C-terminal extension has only a minor role in the function of this protein, at least in osteosarcoma cells. Finally, the myosin-18B knockout phenotype could be partially rescued also by over-expressing full-length myosin-18A in U2OS cells, suggesting that the two class XVIII myosins may have partially overlapping functions in cells (Figure S7C).
DISSCUSSION
Assembly of myosin II filaments into large stacks occurs during the formation of contractile actomyosin bundles in interphase cells, and in the contractile ring of dividing cells [5, 16–18]. However, the mechanisms of myosin II stack formation have remained elusive. Here we reveal that: (1) Myosin-18B, a class XVIII unconventional myosin, forms compact structures that display stable association with NMII-rich stress fibers, (2) Myosin-18B is critical for concatenation and persistent association of NMII filaments with each other, and thus promotes the assembly of NMII stacks in cells, and (3) Depletion of myosin-18B leads to defects in the maturation of contractile actomyosin bundles in cultured osteosarcoma cells, and consequently results in abnormal cell morphogenesis and ability to exert forces to the environment. Moreover, our data provide evidence that formation of myosin II stacks is critical for coalescence of transverse arcs during their centripetal flow, and hence for generation of proper, contractile ventral stress fibers.
Several lines of evidence suggest that myosin-18B is directly involved in NMII stack formation. First, myosin-18B predominantly localizes to the ends of large NMII stacks and at the intersections of smaller NMII stacks. Second, myosin-18B depletion results in a dramatic decrease in the density of large (diameter >500 nm) NMIIs stacks in the lamella of osteosarcoma cells. Third, the frequency of NMII filament fusions was decreased, whereas the frequency of NMII stack fissions was increased in myosin-18B knockout cells compared to control cells. Thus, we propose that myosin-18B molecules, which co-localize with the NMII motor domains, enhance association between adjacent NMII filaments with each other to promote myosin stack formation. It is also formally possible that myosin-18B enhances NMII stack formation indirectly, for example by regulating the activity of NMII filaments. However, we consider this as an unlikely scenario, because myosin-18B displays punctuate, non- uniform localization along NMII filaments/filament stacks, and the remaining thin arcs are still contractile in myosin-18B knockout cells as determined by traction force microscopy (although with lower efficiency, most likely due to their failure to assemble into thick ventral stress fibers). Moreover, the thin arcs in myosin-18B knockout cells undergo retrograde flow, a process that is dependent on myosin II activity [14], with similar rates compared to control cells.
Our work provides evidence that coalescence of transverse arcs during their centripetal flow [7] is dependent on myosin-18B -promoted NMII stack formation. This is because myosin-18B appears to directly enhance NMII stack formation, rather than regulating the organization or dynamics of actin filaments in arcs or the activity of NMII molecules. Furthermore, our data reveal that myosin-18B is important for migration of osteosarcoma cells, especially in a 3D matrix, but whether this is due to defects in the formation of thick actomyosin bundles, or due to some other cellular functions of myosin-18B, remains to be elucidated. The effects of myosin-18B depletion on cell migration are also somewhat contradictory to earlier studies, where depletion of NMII enhanced cell migration [35–37]. It is possible that the migration defects of myosin-18B knockout cells arise from their decreased polarity (as measured by circularity index; Figure S3G). The polarity defects may also provide an explanation for why low expression levels of myosin-18B are linked to carcinoma progression [20, 26, 27]. Contractile actomyosin bundles contribute to proper polarity of epithelial cells [38, 39], and thus the lack of functional actomyosin bundles in epithelial cancers is expected to enhance the epithelial- mesenchymal transition (EMT) of carcinoma cells.
Clinical studies have associated mutations in human myosin-18B to cardiomyopathy and nemaline myopathy. A homozygous premature termination codon in the last exon of myosin-18B was discovered in an infant, who suffered with severe cardiomyopathy and died soon after born [25]. Moreover, a non- sense mutation in the last coding exon of myosin-18B, accompanied by a nearly complete loss of myosin-18B transcript, was associated with hypotonia, muscle weakness, Klippel-Feil anomaly, and facial abnormalities in two young patients. Muscle biopsy analysis of one patient displayed abnormalities in thick filaments [24]. These phenotypes are in line with defective sarcomeric organization and function in myosin-18B mutant mice and zebrafish [28–30]. Combined with our work on myosin-18B deficient osteosarcoma cells, these data suggest that myosin-18B is critical for maturation of myofibrils from their precursors, most likely due to its role in assembly (and maintenance) of large, functional myosin II stacks.
Whereas Drosophila expresses only one myosin-18 family protein [21], a gene duplication resulted in the presence of two myosin-18 proteins in vertebrates. Myosin-18B is highly expressed in muscles and in osteosarcoma cells, which are of mesodermal origin. On the other hand, myosin-18A is most prominently expressed in cells of ectodermal origin, such as fibroblasts and HeLa cells (see e.g. Figure 1E). Thus, it is possible that these proteins have similar roles in myosin II stack formation, but function in different cell-types and perhaps through partially different interaction partners (as suggested by differences in their N- and C-terminal extensions). In support of this hypothesis, depletion of myosin- 18A results in defects in the assembly of thick ventral stress fibers in prostate cancer cells [40], and over-expression of myosin-18A could partially rescue the myosin-18B knockout phenotype in U2OS cells (Figure S7C). Moreover, our SIM experiments revealed similar localization pattern for myosin- 18B in the stress fibers of U2OS cells as previously demonstrated for myosin-18A in HeLa and Rat2 cells [22].
What is the precise mechanism by which myosin-18B drives the fusion of NMII filaments? Our results show that myosin-18B forms a relatively compact structure, which displays much slower dynamics at stress fibers compared to the NMIIA heavy chain. Thus, myosin-18B is well-suited for serving as a ‘glue’ to support the lateral association of individual NMII filaments or stacks with each other. Importantly, our SIM data suggest that myosin-18B foci co-localize with NMII motor domains. Thus, it is possible that the head domain of myosin-18B associates with NMII motor domains at the ends of filament stacks, whereas the N-terminal extension or the C-terminal regions of the protein, either directly or through other proteins, promote interactions between adjacent NMII filaments/stacks. This scenario is consistent with myosin-18B deletion analysis demonstrating that the N-terminal extension, head domain and coiled-coil region are required for proper stress fiber localization and function of the protein, whereas at least in osteosarcoma cells, the C-terminal extension has only a minor role. However, the precise molecular mechanism by which myosin-18B incorporates into NMII bundles, and how its different domains together with other potential interacting proteins promote the fusion of NMII filaments into stacks, remain important challenges for future research.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pekka Lappalainen (pekka.lappalainen@helsinki.fi).
Experimental Model and Subject Details
Cell culture and transfections
Human osteosarcoma (U2OS) cells (derived from a fifteen-year-old human female suffering from osteosarcoma), HeLa cells (derived from cervical cancer cells taken from a human female), and Human dermal fibroblast (HDF) cells (derived from the dermis of normal male neonatal foreskin) were maintained in high glucose (4.5 g/L) Dulbecco’s modified Eagle’s medium (DMEM) (BE12–614F, Lonza) supplemented with 10% fetal bovine serum (10500–064 Gibco), 10 U/ml Penicillin, 10 µg/ml streptomycin and 20 mM L-Glutamine (from 100X concentrate, Gibco) (later referred as complete DMEM) at 37 °C in humidified atmosphere with 5 % CO2. Transient transfections were performed with Fugene HD (Promega) according to manufacturer’s instructions using 3.5:1 Fugene to DNA ratio and 24 hour incubation prior fixation with 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS). 48 hours incubation before fixation was used for mutants rescue experiments. For live-cell and 3D-SIM imaging, cells were detached 24 h post-transfection with (0.25 % w/v) Trypsin-EDTA and plated onto glass-bottomed 35 mm dishes (Greiner bio-one) coated with 10 µg/ml fibronectin (Roche) diluted in PBS. siRNA experiments were performed with Lipofectamin RNAiMAX (Invitrogen) according to manufacturer’s instructions using 40 nM On-target siRNA pool of myosin-18B (Dhmacon) (Target sequence 5’ GCAGAAAGGCCUCGGAUAC 3’, 5’ AAUCAGAGAAGUUGCGGAA 3’, 5’ GAGAACAUGACGCGGAACA 3’, 5’ CGGCAAAAGTACUGTCAUU 3’), myosin-18A (Dhmacon) (Target sequence 5’ CCAAGAAACACGGGCGUAA 3’, 5’ AGGAAGACAUGAACGAAUU 3’, 5’ CAAUGGAGGUGGAGAUCGA 3’, 5’ UGAAAGGACGCAAGUGAA 3’) or 40 nM AllStars negative control siRNA (Qiagen). 72h incubation period was used to efficiently deplete target protein.
Method Details
BioID screen
BioID analysis for tropomyosin 1.6 was performed as described in [10, 31]. To obtain a plasmid for expressing BirA fused to the N-terminus of Tpm1.6, human Tpm1.6 cDNA was amplified and subcloned into the pcDNA3.1 mycBioID vector (Addgene). The fusion protein displayed similar stress fiber localization compared to the endogenous tropomyosin1.6 when expressed in U2OS cells. For proteomics analysis, U2OS cells growing on five 15 cm tissue culture plates were transfected with the plasmid using FuGENE HD transfection reagent (Promega) according to the manufacturer’s instructions. Backbone vector pmycBioID-C1 was transfected as control. Cells were grown for 24 h without biotin and another 24 h in the presence of 50 µM biotin. The cells were harvested in PBS and stored at −80 Co. Single-step affinity purifications of the biotinylated proteins, as well as liquid chromatography mass spectrometry sample preparation, and mass spectrometry were performed as in [10]. To obtain a list of high-confidence protein interactions for Tpm1.6 the data were filtered against our in-house BioID contaminant database.
CRISPR knockout cell line generation
Guide sequences targeting exon 1 and exon 2 of human myosin-18B were selected based on CRISPR Design Tool (crispr.mit.edu) with quality scores of 83 and 92, respectively [41]. Oligonucleotides for cloning guide RNA into pSpCas9 (BB)-2A-GFP vector (a gift from Feng Zhang, Addgene # 48138) were designed as described previously [42]. Forward primer: 5’ CACCGCTCATCACGCCTCGCCCTGT 3’ and reverse primer: 5’ AAACACAGGGCGAGGCGTGATGAGC 3’ (guide sequence underlined) for targeting exon 1. Forward primer: 5’ CACCGTCATTAAGCAACTGGTCCGG 3’ and reverse primer: 5’ AAACCCGGACCAGTTGCTTAATGAC 3’ for targeting exon 2. Transfected cells were detached 24 h post-transfection, suspended into complete DMEM with 20 mM HEPES and sorted with FACS Aria II (BD Biosciences), using on low intensity GFP-expression pass gating, as single cells onto 96-well plate supplemented DMEM containing 20 % FBS and 10 mM HEPES. CRISPR clones were cultivated for two weeks prior selecting clones with no discernible myosin-18B protein expression. Out of total eight surviving clones screened with identical phenotype and no myosin-18B protein expression, one which targeted to exon 2 was selected to use in this study. Cell migration assay presented in supplementary Fig. 3H was performed by CRISPR cell line targeted to exon 1 (Myo18B KO T2 in the figure).
In order to confirm the CRISPR knockout, the region for sequencing was amplified using 5’ – ATCTCATGTGCTGCGTGTGTC – 3’ and 5’ - CTCGCTCTCCTTGCCCAGAAT - 3’ or 5’ – GCCTCTTTCAGCTCTGTGGTC – 3’. The obtained PCR products were sanger-sequenced with the following primer 5’ – ATTCGGGAAGAGGACAAGAGC – 3’. Additional validation was performed using Illumina next generation sequencing (NGS). The genomic region containing second exon was amplified using the oligonucleotides 5’ – CTTGGTCATGTATGTCTCCTC – 3’ and 5’ – GGATGACAAATGACTCAGAGG – 3’, both annealing to the introns surrounding the 2nd exon. More than 300 000 reads were obtained for wild-type and knockout cells, and in both cases 100 % corresponded to wild-type and mutant myosin-18B sequences, respectively.
Plasmids
Plasmids for expressing full length human myosin-18B-GFP (1–2567), myosin-18B-GFP (1–555), myosin-18B-GFP (556–2567), myosin-18B-GFP (1–2089) and myosin-18B-GFP (554–1356) were kind gifts from Jun Yokota (National Cancer Center Research Institute, Tokyo, Japan) and sequenced for verification. All the other myosin-18B constructs were cloned using NEBuilder kit (NEB, #E5520S). The backbone and insert were amplified using the primers 5’ - GACTCAGATCTCGAGCTATGGCCATCTCATCACGCCTCGCC - 3’ and 5’ - GTATGGCTGATTATGATCAGTTATCTACTTGTACAGCTCGTCCATGCCGAGAGTG - 3’ for mCherry-myosin-18B-GFP, 5’ – CGGACTCAGATCTATGGCCATCTCATCACGCCTCGCC - 3’ and 5’ – ATTATGATCAGTTAGGTGGCACTAAGTAGAGGCTGGAGGGAAC - 3’ for myosin- 18B-GFP (1–1398) and 5’ – CGGACTCAGATCTCCTCTACTTAGTGCCACCATTGGAACTG - 3’ and 5’ - ATTATGATCAGTTACTTCTGGAGGTATTTCTTCATTATGCTCGCAAC - 3’ for myosin- 18B-GFP (2091–2567). Then mixed with NEBuilder Master Mix according to manufacture protocol. Other plasmids used in this study are listed in the Table S2.
Immunofluorescence (IF) microscopy
Immunofluorescence experiments were performed as previously described [10]. Briefly, U2OS cells were fixed with 4 % PFA-PBS for 20 min at room temperature (RT), washed three times with 0.2% BSA in Dulbecco’s phosphate buffered saline and permeabilized with 0.1% Triton X-100 in PBS for 4 min. Both primary and secondary antibodies were applied onto cells and incubated in RT for 1 hour. Alexa-conjugated phalloidin was added together with primary antibody solution onto cells. All IF data were obtained either with a wide-field fluorescence microscope (Leica DM6000) with a HCXPL APO 63×1.40–0.60 NA oil objective or by Leica TCS SP5 laser scanning confocal microscope with a 63×1.3 NA glycerol objective. Cells were plated on CYTOOchips™ prior to fixation as described [10]. 3D- SIM imaging and processing was performed on a GE Healthcare DeltaVision OMX equipped with a 60× 1.42 NA Oil objective and sCMOS camera as described previously [17]. The following myosin- 18B antibodies were tested: Sigma (cat # HPA000953), LSBio (cat # LS-C403352), Novus (cat # NBP1–89967), Bioss (cat # bs-2965R), Sdix (cat # 3681.00.02) and Abcam (cat # ab121125). We used the antibody from LSBio for immunofluorescence microscopy on U2OS cells and antibody from Sigma for Western Blots. The following primary antibodies were used for immunofluorescence: myosin-18B rabbit polyclonal antibody (1:50 dilution; LS-C403352, LSBio); myosin-18B rabbit polyclonal antibody (1:50 dilution; HPA000953, Sigma); vinculin mouse monoclonal antibody (1:100 dilution; V9131, Sigma); NMIIA rabbit polyclonal antibody binding to C-terminal tail residues 1948–1960 (1:1000 dilution; 909801, BioLegend); NMIIA RLC mouse polyclonal antibody (1:100 dilution; M4401, Sigma); NMIIB rabbit polyclonal antibody binding to C-terminal tail residues 1965–1976 (1:100 dilution; 909901, BioLegend); α-actinin-1 mouse monoclonal antibody (1:200 dilution; A 5044, Sigma). F-actin was visualized with Alexa Fluor 488-, 568- or 647 conjugated to phalloidin (dilution 1:200; Invitrogen). Secondary antibodies were conjugated to Alexa Fluor 488, 568 or 647 (Invitrogen). The numbers and width of actin filaments bundles from cells plated on crossbow micro-patterns were analysed by the FilamentSensor software as described before [32]. The numbers of thin arcs and thick arcs were manually counted from leading edge of the cells plated on micro-pattern as previously described [42]. The manual quantification in this assay, as well as in the one presented in Figure 5 for the number of myosin II stacks, was blinded in the way that the person analysing the images was provided a randomized set of images for quantification.
FRAP
Wild-type or myosin-18B knockout cells were transfected with NMIIA-GFP, NMIIB-GFP, myosin- 18B-GFP as well as various deletions of myosin-18B-GFP and incubated for 24 hours. Confocal images were acquired with a 3I Marianas imaging system (3I Intelligent Imaging Innovations), consisting of an inverted spinning disk confocal microscope Zeiss Axio Observer Z1 (Zeiss), a Yokogawa CSU-X1 M1 confocal scanner and 63x/1.2 WC-Apochromat Corr WD=0.28 M27 objective (Zeiss). Heated sample environment (+37 °C) and CO2 control were used. SlideBook 6.0 software (3I Intelligent Imaging Innovations) was used for the image acquirement. Five pre-bleach images were acquired followed by bleaching scans with 100 % intensity laser lines over the region of interest. Recovery of fluorescence was monitored 50 times every 200 ms and 300 times every 1s. The intensity of the bleached area was normalized to a neighbouring non-bleached area. Mean scatter plots were calculated from different FRAP experiments and the means and standard deviations were calculated.
Western blotting
U2OS cell lysates (WT and KO) were prepared by washing cells once with PBS and scraping into lysis buffer (50 mM Tris-HCl pH 7.5 150 mM NaCl, 1 mM EDTA, 10 % Glycerol, 1 % Triton X-100) supplemented with 1 mM PMSF, 10 mM DTT, 40 µg/ml DNase I and 1 µg/ml of leupeptin, pepstatin A and aprotinin. All preparations were conducted at 4 °C. Protein concentrations were determined with Bradford reagent (#500–0006, Bio-Rad) and equal amount of the total cell lysates mixed and boiled with Laemmli Sample Buffer (LSB), were loaded and run on 4–20 % gradient SDS-PAGE gels (#4561096, Bio-Rad). Proteins were transferred to nitrocellulose membrane with Trans-blot Turbo transfer system (Bio-Rad) using Mini TGX gel transfer protocol. Membrane was blocked in 5 % milk- TBS with 0.1 % Tween20 (TBS-T) for one hour at RT. Primary and secondary antibodies were diluted into fresh blocking buffer with overnight at 4 °C and one hour at RT, respectively. Proteins were detected from the membrane with Western Lightning ECL Pro substrate (PerkinElmer). All quantifications of band intensities from WB data were conducted with ImageJ densitometry analysis and normalized to GAPDH protein level. For Western blot of hESc H9 cells -derived cardiac embryoid bodies (EBs), the cell lysates of differentiated EBs were prepared in the same manner as described for U2OS cells. The following primary antibodies were used: myosin-18B rabbit polyclonal antibody (1:500 dilution; HPA000953, Sigma); myosin-18A mouse monoclonal antibody (1:1000 dilution; H-10, Santa Cruz); NMIIA rabbit polyclonal antibody (1:1000 dilution; 909801, BioLegend); NMIIB rabbit polyclonal antibody (1:1000 dilution; 909901, BioLegend); GFP rabbit polyclonal antibody (1:2000 dilution; G1544, Sigma); anti-sarcomeric-α-actinin mouse monoclonal antibody (1:500 dilution; clone EA 53, Sigma-Aldrich), cardiac myosin heavy chain (MHC) mouse monoclonal (1:500 dilution; clone 3–48, Thermoscientific), GAPDH mouse polyclonal antibody (1:1000 dilution; G8795, Sigma).
Cell migration assay
Phase contrast time-lapse imaging of migrating U2OS cells were conducted in continuous cell culturing platform Cell-IQ (CM Technologies). Twelve-well plates (Greiner) were coated with fibronectin and the plate lid was switched to Cell-Secure (CM Technologies) enabling insertion of CO2 input and output valves. Cells were allowed to attach for two hours, washed twice with PBS and replaced with DMEM containing 10 mM HEPES prior starting the imaging. 5 % CO2-flow was set cycling between 8 min on, 20 min off. Average migration velocity of wild-type and myosin-18B knockout cells was quantified by tracking the nucleus movement in between 10 min imaging cycles for 10 hours with Cell- IQ analyser (CM Technologies). Only cells that did not collide with one another were selected for measurements. Cell circularity was measured by ImageJ and calculated with equation circularity index = perimeter2/(4π × area).
3D matrigel invasion assay
3D-like circular invasion assay was performed as previously [43], where cell-free space was created using cell stoppers (#81176, Ibidi). After removing the stopper, cells were covered with a thin layer of Matrigel (4 mg/ml) to create a matrix barrier (0.8 mm high) against the cellular surface and allowed to polymerize for 2 hours prior to adding growth medium on the top of the Matrigel. Cell migration was then recorded by Cell-IQ (CM Technologies).
Traction force microscopy
To measure the contractile forces that cell exert upon their substrate, also called traction, we used traction force microscopy, as previously described [42]. In brief, both wild-type and myosin-18B knockout cells were cultured for 3–8 hours on custom made 35 mm dishes (Matrigen) with fibronectin- coated polyacrylamide gel (Young’s modulus = 25 kPa). 200 nm YG fluorescent (505/515) microspheres were immobilized to the surface of the gel. Images of the cells and of the fluorescent microspheres directly underneath the cells were acquired during the experiments and after cell detachment with trypsin. We computed the traction force fields using the approach of constrained fourier-transform traction cytometry and reported the results as the root mean squared (RMS) values.
Live cell imaging
After transient transfection, the cells were incubated for 24 hours and re-plated prior to imaging on 10 µg/ml fibronectin-coated glass-bottomed dishes (MatTek Corporation). The time lapse images of actin- GFP for centripetal arc flow were acquired with 3I Marianas imaging system (3I intelligent Imaging Innovations), consisting of an inverted spinning disk confocal microscope Zeiss Axio Observer Z1 (Zeiss) and a Yokogawa CSU-X1 M1 confocal scanner. 63x/1.2 WC-Apochromat Corr WD=0.28 M27 objective (Zeiss), appropriate filters, heated sample environment (+37°C), and CO2 control were used. SlideBook 6.0 software (3I intelligent Imaging Innovations) was used for the image acquirement. Neo sCMOS (Andor) camera was used for image recording. The time-lapse live cell imaging of GFP- NMIIA and actin-RFP for NMII stack formation were acquired with Zeiss LSM 880 confocal microscope combined with Airyscan detector. ZEN software (Zeiss), 63x magnified plan-apochromat oil immersive objective with NA=1.40, heated sample environment (+37°C), and CO2 control were used for the image acquirement. The recording setting is every 5 s for 10 min. One focal plane was recorded for all time lapse videos. The total events of NMII stack fusion and splitting were measured by manually blind quantification from 10 min time lapse videos in constant area of 20 µm wide and 900 µm2 at the leading edge of the cell.
Focal adhesion data quantification
Focal adhesion (FA) lengths were manually quantified with ImageJ from untransfected wild-type, and myosin-18B knockout cells, as well as from knockout cells expressing myosin-18B-GFP deletion constructs. Cells adhered to several neighbouring cells were discarded from the analysis, and only the cells that displayed low or moderate GFP intensity were selected for analysis. Adhesions sizes were classified to seven size groups and percentual ratio of FA in each class was obtained by dividing the FA number on individual size class with the total detected FAs for the same cell.
Real-time quantitative PCR
Total mRNA was extracted with GeneJET RNA purification kit (#K0731, Thermo Scientific) and single stranded cDNA was synthetized (#K1671, Thermo Scientific) from 500 ng of extracted mRNA. The following primers were used: forward myosin-18B 5’ ATGGCCATCTCATCACGCCTC 3’, reverse myosin-18B 5’ GCCTCTTTCAGCTCTGTGGTC 3’, forward myosin-18A 5’ GGACATGGTGACAAAGTATCAGAA 3’, reverse myosin-18A 5’ TTTGACAACCAGGACTTGACC 3’, forward GAPDH 5’ GAAGGTGAAGGTCGGAGTC 3’, reverse GAPDH 5’ GAAGATGGTGATGGGATTTC 3’. Quantitative PCR reactions were carried out with Maxima SYBR Green/ROX (#K0221, Thermo Scientific) in Bio-Rad CFX96 (Bio-Rad). Changes in expression were calculated with 2^∆Ct method, normalized to GAPDH (∆Ct) and WT expression levels respectively.
Quantification and Statistical Analysis
Statistics were performed with Excel (Microsoft) and SigmaPlot (Systat Software Inc). Sample sizes and the numbers of replications are included in the images. For data following normal distribution, Student’s two-sample unpaired t-test was used. If data did not follow normal distribution, Mann- Whitney u-test for two independent samples was conducted. To quantify myosin-18B localization in 3D-SIM images, NMII stacks were divided to 100 nm segments. The percentage of myosin-18B positive segments in the ends and in the middle sections of the NMII stacks were quantified (from 50 stacks, randomly chosen from each cell). Please note that, instead of 100 nm, the length of the ‘last segment’ of the middle section varied between 51 and 149 nm. Statistical differences in RMS traction between the WT and myosin-18B KO groups were assessed by using the non-parametric Mann– Whitney–Wilcoxon rank-sum (MWW) test. For all image analyses, no raw data were excluded with the exception of cells that were not in focus or the ones that were overexposed in the obtained images. Geneious (Biomatters Limited) analysis tool was used to construct sequence alignments in Figure S3D.
Supplementary Material
The time-lapse movie was obtained for 30 minutes with 10 second intervals using 3I Marianas microscope. The display rate is 24 frames per second.
The time-lapse movie was obtained for 30 minutes with 10 second intervals using 3I Marianas microscope. The display rate is 24 frames per second.
The time-lapse movie was obtained with 5 second intervals using Zeiss Airyscan confocal microscope. The display rate is 5 frames per second.
The time-lapse movies were obtained with 5 second intervals using Zeiss Airyscan confocal microscope. The display rate is 5 frames per second.
HIGHLIGHTS.
Myosin-18B is a stable component of stress fibers in osteosarcoma cells.
Stress fiber maturation from their precursors requires myosin-18B.
Myosin-18B promotes the assembly of myosin II stacks.
Myosin II stacks are important for cell morphogenesis and migration.
ACKNOWLEDGEMENTS
This study was supported by grants from Academy of Finland (274565 to Y.J), from Jane and Aatos Erkko Foundation and Sigrid Juselius Foundation (to P.L), and from the National Institute of Health (R21HL123522 to R.K and N.S; R35 GM125028 to D.T.B; F31 HL136081 to A.M.F). We thank Dr. Carina Wollnik for helping with the FilamentSensor analyzes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Pollard TD, and Cooper JA (2009). Actin, a central player in cell shape and movement. Science 326, 1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Livne A, and Geiger B (2016). The inner workings of stress fibers - from contractile machinery to focal adhesions and back. J. Cell Sci 129, 1293–1304. [DOI] [PubMed] [Google Scholar]
- 3.Tojkander S, Gateva G, and Lappalainen P (2012). Actin stress fibers--assembly, dynamics and biological roles. J. Cell Sci 125, 1855–1864. [DOI] [PubMed] [Google Scholar]
- 4.Burridge K, and Wittchen ES (2013). The tension mounts: stress fibers as force-generating mechanotransducers. J. Cell Biol 200, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burnette DT, Shao L, Ott C, Pasapera AM, Fischer RS, Baird MA, Der Loughian C, Delanoe-Ayari H, Paszek MJ, Davidson MW, et al. (2014). A contractile and counterbalancing adhesion system controls the 3D shape of crawling cells. J. Cell Biol 205, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanger JW, Kang S, Siebrands CC, Freeman N, Du A, Wang J, Stout AL, and Sanger JM (2005). How to build a myofibril. J. Muscle Res Cell Motil 26, 343–354. [DOI] [PubMed] [Google Scholar]
- 7.Tojkander S, Gateva G, Husain A, Krishnan R, and Lappalainen P (2015). Generation of contractile actomyosin bundles depends on mechanosensitive actin filament assembly and disassembly. eLife 4, e06126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong AJ, Pollard TD, and Herman IM (1983). Actin filament stress fibers in vascular endothelial cells in vivo. Science 219, 867–869. [DOI] [PubMed] [Google Scholar]
- 9.Lee S, and Kumar S (2016). Actomyosin dtress fiber mechanosensing in 2D and 3D. F1000Res 5, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiu Y, Lehtimaki J, Tojkander S, Cheng F, Jäälinoja H, Liu X, Varjosalo M, Eriksson JE, and Lappalainen P (2015). Bidirectional Interplay between Vimentin Intermediate Filaments and Contractile Actin Stress Fibers. Cell Rep 11, 1511–1518. [DOI] [PubMed] [Google Scholar]
- 11.Murugesan S, Hong J, Yi J, Li D, Beach JR, Shao L, Meinhardt J, Madison G, Wu X, Betzig E, et al. (2016). Formin-generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse. J. Cell Biol 215, 383–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burnette DT, Manley S, Sengupta P, Sougrat R, Davidson MW, Kachar B, and Lippincott-Schwartz J (2011). A role for actin arcs in the leading-edge advance of migrating cells. Nat. Cell Biol 13, 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hotulainen P, and Lappalainen P (2006). Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J. Cell Biol 173, 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tee YH, Shemesh T, Thiagarajan V, Hariadi RF, Anderson KL, Page C, Volkmann N, Hanein D, Sivaramakrishnan S, Kozlov MM, et al. (2015). Cellular chirality arising from the self-organization of the actin cytoskeleton. Nat. Cell Biol 17, 445–457. [DOI] [PubMed] [Google Scholar]
- 15.Soine JR, Brand CA, Stricker J, Oakes PW, Gardel ML, and Schwarz US (2015) Model-based traction force microscopy reveals differential Tension in Cellular Actin Bundles. PLoS Comput Biol 11, e1004076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beach JR, Bruun KS, Shao L, Li D, Swider Z, Remmert K, Zhang Y, Conti MA, Adelstein RS, Rusan NM, et al. (2017). Actin dynamics and competition for myosin monomer govern the sequential amplification of myosin filaments. Nat. Cell Biol 19, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fenix AM, Taneja N, Buttler CA, Lewis J, Van Engelenburg SB, Ohi R, and Burnette DT (2016). Expansion and concatenation of non-muscle myosin IIA filaments drive cellular contractile system formation during interphase and mitosis. Mol. Biol. Cell 27, 1465–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu S, Dasbiswas K, Guo Z, Tee YH, Thiagarajan V, Hersen P, Chew TL, Safran SA, Zaidel-Bar R, and Bershadsky AD (2017). Long-range self-organization of cytoskeletal myosin II filament stacks. Nat. Cell Biol 19, 133–141. [DOI] [PubMed] [Google Scholar]
- 19.Furusawa T, Ikawa S, Yanai N, and Obinata M (2000). Isolation of a novel PDZ- containing myosin from hematopoietic supportive bone marrow stromal cell lines. Biochem Biophys Res Commun 270, 67–75. [DOI] [PubMed] [Google Scholar]
- 20.Nishioka M, Kohno T, Tani M, Yanaihara N, Tomizawa Y, Otsuka A, Sasaki S, Kobayashi K, Niki T, Maeshima A, et al. (2002). MYO18B, a candidate tumor suppressor gene at chromosome 22q12.1, deleted, mutated, and methylated in human lung cancer. Proc. Natl Acad. Sci. USA 99, 12269–12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salamon M, Millino C, Raffaello A, Mongillo M, Sandri C, Bean C, Negrisolo E, Pallavicini A, Valle G, Zaccolo M, et al. (2003). Human MYO18B, a novel unconventional myosin heavy chain expressed in striated muscles moves into the myonuclei upon differentiation. J. Mol. Biol 326, 137–149. [DOI] [PubMed] [Google Scholar]
- 22.Billington N, Beach JR, Heissler SM, Remmert K, Guzik-Lendrum S, Nagy A, Takagi Y, Shao L, Li D, Yang Y, et al. (2015). Myosin 18A coassembles with nonmuscle myosin 2 to form mixed bipolar filaments. Curr. Biol 25, 942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guzik-Lendrum S, Heissler SM, Billington N, Takagi Y, Yang Y, Knight PJ, Homsher E, and Sellers JR (2013). Mammalian myosin-18A, a highly divergent myosin. J. Biol. Chem 288, 9532–9548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alazami AM, Kentab AY, Faqeih E, Mohamed JY, Alkhalidi H, Hijazi H, and Alkuraya FS (2015). A novel syndrome of Klippel-Feil anomaly, myopathy, and characteristic facies is linked to a null mutation in MYO18B. J. Med. Genet 52, 400–404. [DOI] [PubMed] [Google Scholar]
- 25.Malfatti E, Böhm J, Lacène E, Romero N, and Laporte J (2015). A Premature Stop Codon in MYO18B is Associated with Severe Nemaline Myopathy with Cardiomyopathy. J. Neuromuscul. Dis 2, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakano T, Tani M, Nishioka M, Kohno T, Otsuka A, Ohwada S, and Yokota J (2005). Genetic and epigenetic alterations of the candidate tumor-suppressor gene MYO18B, on chromosome arm 22q, in colorectal cancer. Gene. Chromosome Canc 43, 162–171. [DOI] [PubMed] [Google Scholar]
- 27.Yanaihara N, Nishioka M, Kohno T, Otsuka A, Okamoto A, Ochiai K, Tanaka T, and Yokota J (2004). Reduced expression of MYO18B, a candidate tumor-suppressor gene on chromosome arm 22q, in ovarian cancer. Int. J. Cancer 112, 150–154. [DOI] [PubMed] [Google Scholar]
- 28.Ajima R et al. Deficiency of Myo18B in mice results in embryonic lethality with cardiac myofibrillar aberrations. Genes Cells 13, 987–999 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Berger J, Berger S, Li M & Currie PD Myo18b is essential for sarcomere assembly in fast skeletal muscle. Hum. Mol. Genet 26, 1146–1156 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Gurung R, Akazawa H, Kodama M, Takeshita F, Otsuka A, Kohno T, Komuro I, Ochiya T, and Yokota J (2017). A Zebrafish Model for a Human Myopathy Associated with Mutation of the Unconventional Myosin MYO18B. Genetics 205, 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eltzner B, Wollnik C, Gottschlich C, Huckemann S, and Rehfeldt F (2015). The filament sensor for near real-time detection of cytoskeletal fiber structures. PloS One 10, e0126346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vicente-Manzanares M, Koach MA, Whitmore L, Lamers ML, and Horwitz AF (2008). Segregation and activation of myosin IIB creates a rear in migrating cells. J. Cell Biol 183, 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shutova MS, Asokan SB, Talwar S, Assoian RK, Bear JE, and Svitkina TM (2017). Self-sorting of nonmuscle myosins IIA and IIB polarizes the cytoskeleton and modulates cell motility. J. Cell Biol 216, 2877–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jorrisch MH, Shih W, and Yamada S (2013). Myosin IIA deficient cells migrate efficiently despite reduced traction forces at cell periphery. Biol. Open 2, 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shih W, and Yamada S (2010). Myosin IIA dependent retrograde flow drives 3D cell migration. Biophys. J 98, L29–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehtimäki J, Fenix AM, Kotila TM, Balistreri G, Paavolainen L, Varjosalo M, Burnette DT, Lappalainen P (2017). UNC-45a promotes myosin folding and stress fiber assembly. J. Cell Biol 12, 4053–4072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lecuit T, Lenne PF, and Munro E (2011). Force generation, transmission, and integration during cell and tissue morphogenesis. Annu. Rev. Cell. Dev. Biol 27, 157–184. [DOI] [PubMed] [Google Scholar]
- 39.Zihni C, Vlassaks E, Terry S, Carlton J, Leung TKC, Olson M, Pichaud F, Balda MS, Matter K (2017). An apical MRCK-driven morphogenetic pathway controls epithelial polarity. Nat. Cell Biol 19, 1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makowska KA, Hughes RE, White KJ, Wells CM, and Peckham M (2015). Specific Myosins Control Actin Organization, Cell Morphology, and Migration in Prostate Cancer Cells. Cell Rep 13, 2118–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ran FA, Hsu PD, Lin C, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiu Y, Peränen J, Schaible N, Cheng F, Eriksson JE, Krishnan R, Lappalainen P (2017). Vimentin intermediate filaments control actin stress fiber assembly through GEF-H1 and RhoA. J. Cell Sci 130, 892–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu X, and Machesky LM (2012). Cells assemble invadopodia-like structures and invade into matrigel in a matrix metalloprotease dependent manner in the circular invasion assay. PloS One 7, e30605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gateva G, Kremneva E, Reindl T, Kotila T, Kogan K, Gressin L, Gunning PW, Manstein DJ, Michelot A, and Lappalainen P (2017). Tropomyosin Isoforms Specify Functionally Distinct Actin Filament Populations In Vitro. Curr Biol 27, 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei Q, and Adelstein RS (2000) Conditional expression of a truncated fragment of nonmuscle myosin II-A alters cell shape but not cytokinesis in HeLa cells. Mol Biol Cell 11, 3617–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The time-lapse movie was obtained for 30 minutes with 10 second intervals using 3I Marianas microscope. The display rate is 24 frames per second.
The time-lapse movie was obtained for 30 minutes with 10 second intervals using 3I Marianas microscope. The display rate is 24 frames per second.
The time-lapse movie was obtained with 5 second intervals using Zeiss Airyscan confocal microscope. The display rate is 5 frames per second.
The time-lapse movies were obtained with 5 second intervals using Zeiss Airyscan confocal microscope. The display rate is 5 frames per second.