

SUMMARY
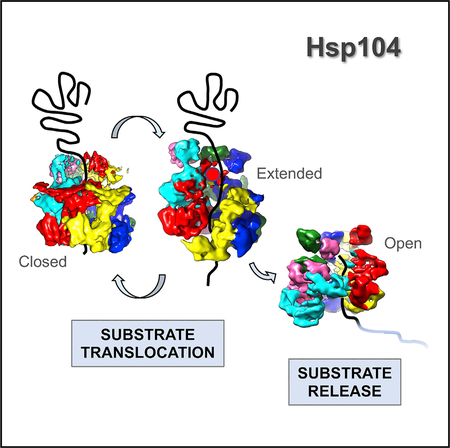
Hsp104 is a ring-forming, ATP-driven molecular machine that recovers functional protein from both stress-denatured and amyloid-forming aggregates. Although Hsp104 shares a common architecture with Clp/Hsp100 protein unfoldases, different and seemingly conflicting 3D structures have been reported. Examining the structure of Hsp104 poses considerable challenges because Hsp104 readily hydrolyzes ATP, whereas ATP analogs can be slowly turned over and are often contaminated with other nucleotide species. Here, we present the single-particle electron cryo-microscopy (cryo-EM) structures of a catalytically inactive Hsp104 variant (Hsp104DWB) in the ATP-bound state determined between 7.7 Å and 9.3 Å resolution. Surprisingly, we observe that the Hsp104DWB hexamer adopts distinct ring conformations (closed, extended, and open) despite being in the same nucleotide state. The latter underscores the structural plasticity of Hsp104 in solution, with different conformations stabilized by nucleotide binding. Our findings suggest that, in addition to ATP hydrolysis-driven conformational changes, Hsp104 uses stochastic motions to translocate unfolded polypeptides.
In Brief
Hsp104 is a ring-forming ATPase that facilitates the disaggregation of amorphous and amyloid-forming protein aggregates. Lee et al. present three distinct cryo-EM structures of a catalytically inactive Hsp104-ATP variant, demonstrating that Hsp104 is a dynamic molecular machine and providing the structural basis for the passive threading of unfolded polypeptides.
Graphical Abstract
INTRODUCTION
Maintaining proteostasis is essential to cell and organismal health. Although molecular chaperones assist protein folding to prevent misfolding and aggregation, the majority of stress-inducible chaperones cannot rescue stress-damaged proteins once aggregation has occurred. Yeast Hsp104 and its bacterial homolog ClpB are ring-forming, ATP-driven protein disaggre-gases that, together with Hsp70 and Hsp40 chaperones, recover functional protein from both amorphous aggregates and amyloid-forming prions (Doyle et al., 2013; Mogk et al., 2015). Hsp104 possesses an N-terminal domain (NTD) and two nucleotide-binding domains (NBD1 and NBD2), which facilitate substrate binding (Gates et al., 2017; Hung and Masison, 2006; Lee et al., 2017b) and abolish Hsp104 function when mutated (Lee et al., 2017a; Lum et al., 2004). A hallmark of Hsp104/ClpB members is the coiled-coil motif within NBD1, which binds Hsp70 (Miot et al., 2011; Rosenzweig et al., 2013; Sielaff and Tsai, 2010) and, in turn, activates the Hsp104 motor for protein disaggregation (Lee et al., 2013; Seyffer et al., 2012). This necessitates the coiled-coil to be exposed on the surface of the Hsp104 hexamer (Gates et al., 2017; Heuck et al., 2016; Lee et al., 2010), where it potentially contacts aggregated proteins.
It is widely presumed that Hsp104 extracts polypeptides from aggregates, followed by threading of the substrate through the axial channel of the barrel-shaped hexamer, resulting in protein unfolding and release of the unfolded polypeptide from the distal end (Doyle et al., 2013; Mogk et al., 2015). Because the ability to disaggregate aggregated proteins requires both ATP binding and hydrolysis (Bösl et al., 2005; Doyle et al., 2007), an Hsp104 variant (Hsp104DWB) featuring mutational alteration of the Walker B motifs in both NBDs is catalytically inactive and functions as a substrate trap (Bösl et al., 2005). Interestingly, adenosine 5′-(β, γ-imido)triphosphate (ADPNP), a non-hydrolyzable ATP analog, does not support substrate binding (Gates et al., 2017) and results in a markedly distinct open-ring structure (Yokom et al., 2016; Yu et al., 2018), arguing for a need to perform structural analyses with physiologically relevant nucleotides.
High-resolution electron cryo-microscopy (cryo-EM) structures of Hsp104/ClpB bound to casein have been reported (Gates et al., 2017; Yu et al., 2018). In the case of Hsp104, two distinct hexamer structures were observed: a near-symmetrical closed ring and an extended ring conformation (Gates et al., 2017), suggesting a ratchet-like mechanism as the structural basis for substrate translocation. Notably, both Hsp104 structures differed from the open spiral conformation observed in the presence of ADPNP (Yokom et al., 2016; Yu et al., 2018). The closed and extended ring structures confirmed the functional role of Hsp104 pore loops in contacting the unfolded polypeptide (Gates et al., 2017). However, no major changes in pore loop conformation or domain organization were observed, as would be expected during the ATP power stroke. Although casein binding is ATP dependent and mimics substrate interaction, casein is an unfolded polypeptide that can be translocated passively in an ATP hydrolysis-independent manner (Li et al., 2015; Nakazaki and Watanabe, 2014). Hence, it is unclear whether the observed Hsp104 conformations are the result of casein binding or induced by different nucleotide species.
Here, we present the asymmetric cryo-EM structures of an Hsp104DWB hexamer bound to ATP in the absence of substrate. We show that Hsp104 adopts distinct 3D structures in vitreous ice despite being in the same nucleotide state. Three major Hsp104 hexamer conformations converged, which we refer to as the closed ring, the extended ring, and the open ring conformation. Although the closed and extended ring conformations are remarkably similar to cryo-EM structures of Hsp104:casein complexes (Gates et al., 2017), the open ring conformation is virtually identical to the open spiral assembly of Hsp104-ADPNP without casein bound (Yokom et al., 2016). Taken together, our observations reveal a previously underappreciated structural dynamics of Hsp104 in solution and support a model by which Hsp104 uses stochastic motions to translocate unfolded polypeptides.
RESULTS
Cryo-EM Structures of the Hsp104DWB Hexamer Bound to ATP
Hsp104 self-assembles into hexamers that are stabilized by binding of adenine nucleotides (Parsell et al., 1994). Binding of chemically different adenine nucleotides results in distinct 3D ring structures (Gates et al., 2017). Strikingly, the high-resolution cryo-EM structure of Hsp104 bound to ADPNP revealed an unusual left-handed spiral assembly with an unexpected NBD1:NBD2 interface between the first (P1) and sixth subunit (P6) of adjacent protomers (Yokom et al., 2016), deviating from the expected closed ring conformation for Clp/Hsp100 protein unfoldases (Deville et al., 2017; Effantin et al., 2010; Lee et al., 2007). Although the spiral architecture is reminiscent of the helical subunit arrangement observed in crystal structures of Hsp104/ClpB (Heuck et al., 2016; Lee et al., 2003) and by high-speed atomic force microscopy (AFM) (Uchihashi et al., 2018), the functional importance of this NBD1:NBD2 interface remains to be determined. Unlike ATPγS, ADPNP does not support substrate binding (Gates et al., 2017; Lee et al., 2007). Furthermore, high-resolution cryo-EM structures of Hsp104-ATPγS bound to casein (Hsp104:casein) revealed two distinct hexamer conformations (Gates et al., 2017), which are different from the open spiral architecture observed with ADPNP (Yokom et al., 2016). One of those two high-resolution structures (PDB: 5VY9) resembles the previously determined cryo-EM structure of a closed-ring Hsp104DWB hexamer with symmetry imposed (Lee et al., 2010). Because the structure of an ATPγS-bound but casein-free complex was not reported and because Hsp104 slowly hydrolyzes ATPγS (Bösl et al., 2005; Doyle et al., 2007), it remains unclear whether the observed Hsp104:casein structures are the result of casein binding or may represent structures induced by mixtures of ATPγS and other nucleotide species.
Hsp104DWB featuring a Walker B mutation in both NBD1 (Glu285 to Ala) and NBD2 (Glu687 to Ala) is incapable of ATP turnover (Figure 1A). To ensure that loss of ATP hydrolysis is not the result of lack of ATP binding, we determined the nucleotide occupancy of Hsp104DWB using 2′/3′-O-(N-Methyl-anthraniloyl)adenosine 5′-triphosphate (MANT-ATP), a fluorescently labeled ATP analog (Figure 1B), and by UV crosslinking using benzoylbenzoyl-ATP (BzATP) (Figure 1C). As anticipated, nucleotide binding to the Hsp104DWB hexamer is concentration dependent (Figure 1B). At 1 mM nucleotide, 1.89 ± 0.41 molecules of MANT-ATP (Figure 1B) and 1.73 ± 0.22 molecules of BzATP (Figure 1C) are bound per Hsp104DWB monomer (3 μM), demonstrating that at least ten nucleotide binding sites in the Hsp104DWB hexamer are occupied under saturating ATP conditions.
Figure 1. Cryo-EM Imaging of a Catalytically Inactive Hsp104 Variant.

(A) ATPase activities of Hsp104 and Hsp104DWB without and with κ-casein stimulation. Averages of three independent measurements ± SD are shown.
(B and C) Determining the nucleotide occupancy in Hsp104DWB using (B) MANT-ATP and (C) BzATP crosslinking, respectively. Average number of bound nucleotide from at least three independent measurements ± SD is shown.
(D) Representative EM micrograph of Hsp104DWB hexamers embedded in vitreous ice (scale bar, 25 nm).
(E) Images of selected 2D classes from reference-free 2D classification.
To examine the structure of the ATP-bound state, the Hsp104DWB hexamer was isolated by size-exclusion chromatography and the peak fraction was incubated with 5 mM of freshly prepared ATP, representing an ~1,700-fold molar excess of ATP over protein. Imaging of frozen hydrated specimen shows monodispersed particles (Figure 1D). The 2D class averages (Figure 1E) show that Hsp104DWB-ATP hexamers display dominant top views with minor side views in vitreous ice. About 285,000 particles were picked automatically, and after 2D reference-free averaging, ~90% of particles were subjected to initial 3D classification with RELION (Scheres, 2012). Based on 3D map convergence, 76% of pooled particles were further subjected to a second round of classification, resulting in three converged and distinct classes. Three principal structures converged beyond sub-nanometer resolution without symmetry imposed, corresponding to 56,859, 33,666, and 25,582 of particles (Figure 2; see also Figure S1 and Table S1). These structures are hereafter referred to as the closed, extended, and open ring conformations (Figures 3A–3C). The cross- correlation between our 3D reconstructions and the previously reported closed (EMD-8697), extended (EMD-8746), and open conformation (EMD-8267) of Hsp104 with and without casein bound (Gates et al., 2017; Yokom et al., 2016) are 0.9409 (Figure 3D), 0.9338 (Figure 3E), and 0.9380 (Figure 3F). Although the closed ring is 2-fold higher populated than the other two conformations and closely resembles the cryo-EM structure of Hsp104DWB-ATP determined at lower resolution (EMD-1631; Lee et al., 2010), the extended and open-ring conformations are nearly equally populated. All three conformations are biochemically identical and exist in equilibrium. This structural plasticity contrasts previous work with wild-type Hsp104 and ATPγS, showing 80% of hexamers in the closed state, which increases to 100% when casein is also bound (Gates et al., 2017).
Figure 2. Cryo-EM Image Processing and 3D Reconstruction.

Boxed particles were subjected to 2D class averages and 3D classification. After initial 3D classification, the particles in converged maps were pooled and subjected to a second round of 3D classification, yielding three distinct conformations. Three principle structures were refined beyond sub-nanometer resolution, which are referred to as the extended (left), closed (middle), and open ring conformation (right). See also Figure S1 and Table S1.
Figure 3. Three-Dimensional Structures of Hsp104DWB Bound to ATP.

(A–C) Selected 2D averages, cryo-EM maps, and fitted models of Hsp104DWB in the (A) closed, (B) extended, and (C) open ring conformation. Individual subunits are shown in different color together with the fitted models.
(D–F) Superposition of cryo-EM maps in the (D) closed (purple), (E) extended (yellow), and (F) open ring conformation (blue) with the equivalent maps (solid surface; gold) of Hsp104 without (Yokom et al., 2016) and with casein bound (Gates et al., 2017).
See also Video S1.
The Closed Ring Conformation
The closed ring conformation was determined at 7.7Å resolution, enabling the unequivocal assignment of secondary structure elements (Figure 4A), including the coiled-coil motif that can be assigned in three subunits. Strong mass density is observed for the six copies of NBD2 that together with NBD1 make up the barrel-shaped main body. However, the resolution of our 3D reconstruction is insufficient to observe bound nucleotide. There is a flat, open ring on top of the barrel, which can be attributed to the NTDs (Figures 4B and 4C), which are connected to their respective NBD1s via a long and flexible linker that is only partially visible. Interestingly, the gap between neighboring NTDs of P5 and P6 coincides with where the mass densities for the coiled coil (subunits P1 and P6) are most pronounced, with the coiled coil motifs being in direct physical contact (Figure 4C).
Figure 4. Hsp104 in Closed, Extended, and Open Ring Conformation.

(A and B) Surface representation of closed conformation (A) at high contour level (University of California, San Francisco [UCSF] Chimera threshold 0.813) to fit NBD1 and NBD2 and (B) at low contour level (UCSF Chimera threshold 0.437) to fit NTDs and coiled coil motifs.
(C) Top-down view of closed conformation at low contour level. Mass densities for the NTDs (dark pink) and coiled coil motifs (bright pink) are shown in different hues.
(D) Atomic structure fit of the extended conformation model (PDB: 5VYA) to our 3D reconstruction.
(E) Atomic structure fit of the extended conformation model with four NTDs added in our model. The mass density for the NBD1 of P6 is only partially observed, indicating high mobility.
(F) Atomic structure fit of the open conformation model (PDB: 5KNE) with three more NTDs added in our model.
Superposing the 3D reconstruction of the closed ring with the closed conformation of Hsp104:casein (Gates et al., 2017) shows a remarkable structural overlap with similar overall dimensions. Consequently, placing the closed state Hsp104: casein model (PDB: 5VY9) into our reconstruction results in an excellent fit with NBD1 and NBD2 lining the protein translocating channel as previously reported (Gates et al., 2017). However, without casein, the substrate-binding pore loops that contact the unfolded polypeptide were not resolved, indicating mobility.
Unlike the NBDs, the triangular arrangement of the six NTDs observed in the closed state Hsp104:casein model was incompatible with our structure, necessitating manual fitting. In our structure, the NTDs adopt different en bloc conformations, resembling a funnel that gates the entry to the protein translocating channel of the barrel-shaped hexamer (Figure 4C). The coiled-coil motif extends perpendicular from the axial channel, surrounding the main body in a head-to-tail arrangement and forming a belt reminiscent of the repressed state (Heuck et al., 2016; Oguchi et al., 2012; Figure 4B). This conformation is consistent with the notion that Hsp104DWB is catalytically inactive (i.e., repressed) and cannot facilitate the disaggregation of native substrates.
The Extended Ring Conformation
The extended ring conformation was determined at 9.3 Å resolution and resembles the overall structure of the closed ring hexamer but with a major gap between the P1 and P6 subunits that deviate from ring planarity. The mass density is well defined for the main body, enabling assignment of NBD1 and NBD2, with additional, albeit weak, mass density at one end corresponding to the NTD. However, no mass density is observed for the coiled coil motif, indicating mobility. We find that the overall shape and dimensions of the extended Hsp104DWB-ATP hexamer closely resembles the extended conformation of Hsp104:casein (Gates et al., 2017), despite absence of bound polypeptide. Not surprisingly, placing the atomic model for the extended Hsp104:casein conformation, a structure that lacks the NTD and coiled coil motif (PDB: 5VYA), resulted in an excellent fit (Figure 4D). The NTDs for subunits P2, P3, P4, and P5 were fitted manually, which follow the right-handed helical assembly of the main body and line the entry to the protein-translocating channel (Figure 4E).
In the extended conformation, the P2–P5 subunits are arranged similarly to those in the closed ring conformation (root-mean-square deviation [RMSD] 1.44 Å over 2,077 Cα atoms). The NBD1 of P6 rotates by 20° and moves upward by 10 Å, and the NBD1 of P1 undergoes a 7 Å downward shift concomitant with a 14° rotation and the NBD2 of P1 a downward shift of 14 Å and a 29° rotation (Figures 4D and 4E). We note that the mass density for NBD1 of the P6 subunit is the least well defined among NBDs, followed by the NBD1 of P1. In the casein-bound structure, NBD1 of P6 makes the initial contact with the unfolded polypeptide at the pore opening (Gates et al., 2017). In the absence of bound polypeptide, the NBD1 of P6 is presumably mobile and therefore less well defined.
The Open Ring Conformation
The open ring conformation was determined at 9.3 Å resolution and resembles the lock-washer assembly of Hsp104 with ADPNP (Yokom et al., 2016). Mass densities for the NTD, NBD1, and NBD2 are well defined. Superposition of the two independent 3D reconstructions of our open ring conformation and the Hsp104-ADPNP hexamer showed a near perfect correlation in spite of the different nucleotide-bound state (Figure 4F). Consequently, placing the atomic model for the Hsp104-ADPNP hexamer (PDB: 5KNE), featuring the NTDs of P2, P3, and P4, resulted in an excellent fit, confirming that the lock-washer assembly represents an on-pathway state. The remaining NTDs were fitted manually (Figure 4F). Because Hsp104DWB is incapable of ATP hydrolysis, we excluded the possibility that an ADP state was observed. The most striking structural feature is the unusual NBD1:NBD2 interface between P1 and P6 as previously observed in Hsp104/ClpB structures with ADPNP (Yokom et al., 2016; Yu et al., 2018), which do not support casein binding (Gates et al., 2017; Lee et al., 2007). Although the domain arrangements and inter-subunit interfaces for the other four subunits (P2–P5) are similar to the closed and extended ring conformations, their overall arrangement deviates in the open conformation. Nevertheless, each protomer interface is similar when compared pairwise. This is consistent with a ring expansion, resulting in a wider protein translocation channel in the open conformation.
DISCUSSION
Hsp104 is a ring-forming ATP-driven protein machine that facilitates the recovery of functional protein from aggregates. How Hsp104 harnesses metabolic energy to generate the ATP power stroke remains unclear. For one, Hsp104 possesses an innate ATPase activity, making it difficult to pursue structural studies under physiological conditions with ATP. To address this issue, we examined the structure of a catalytically inactive Hsp104 variant and determined the single-particle cryo-EM structures of the Hsp104DWB-ATP hexamer at resolution ranging from7.7 Å to 9.3 Å (Figure S1). Our work uncovered a previously underappreciated structural plasticity with Hsp104 adopting different 3D structures in solution despite being in the same nucleotide state (Figure 3; see also Video S1). We observed three principal hexamer conformations (closed, extended, and open), which exist in equilibrium with only a 2-fold preference for the closed ring. Because Hsp104DWB is incapable of ATP hydrolysis, we ruled out the possibility that our observed conformations are the result of different nucleotide species. However, it is likely that other, less populated conformations also exist because some 2D classes were discarded because of low convergence.
Our observed structures are consistent with recent high speed-AFM work (Uchihashi et al., 2018) and virtually identical to high-resolution cryo-EM structures of Hsp104 without and with casein bound (Gates et al., 2017; Yokom et al., 2016) but with one major difference that all of these conformations are observed in the same, ATP-bound state. In the absence of casein, we did not observe mass densities for pore loops, supporting a role of the channel-facing pore loops in contacting the translocating polypeptide that in turn stabilizes pore loop conformation. Although casein stimulates the ATPase activity of Hsp104/ClpB, casein is an unfolded polypeptide that is threaded passively (Li et al., 2015; Nakazaki and Watanabe, 2014) and does not feature the physico-chemical properties of bona fide substrates. Consistent with this notion, no major structural differences or domain motions between the casein-bound (Gates et al., 2017) and unbound Hsp104 hexamer (this work) in the closed and extended conformation were observed, as would be expected for a true substrate-bound complex, indicating that the observed structures are the result of spontaneous conformational switching as opposed to ATP-driven changes.
What is the structural basis for the observed Hsp104 plasticity? We found that the three principal hexamer conformations (closed, extended, and open) exist in equilibrium and are biochemically indistinguishable. Because we prepared our cryo-EM sample under saturating conditions using a vast excess of ATP, exceeding even those used in our binding experiments (Figures 1B and 1C), we expect that 10–12 nucleotide binding sites are occupied with ATP. This is consistent with the high-resolution cryo-EM structures of Hsp104/ClpB:casein complexes that clearly resolved the bound nucleotide (Gates et al., 2017; Yu et al., 2018). Because we did not observe bound ATP, we cannot rule out that the distinct Hsp104 conformations are the result of different nucleotide occupancies. However, in the case of yeast Hsp104-ADPNP (Yokom et al., 2016) and Hsp104:casein (Gates et al., 2017), whose structures are virtually identical to those observed here, all twelve nucleotide binding sites are occupied in all three hexamer conformations with only one or two sites showing partial occupancy.
Our three principal hexamer conformations (closed, extended, and open) are populated in a near 2:1:1 ratio, although other conformations likely exist. Because Hsp104DWB is deficient in ATP hydrolysis, we propose that changes between distinct Hsp104DWB-ATP hexamer conformations are the result of “breathing” and not caused by ATP hydrolysis as previously suggested (Gates et al., 2017; Uchihashi et al., 2018). Breathing is compatible with conformational switching as well as the “passive” diffusion of casein (Li et al., 2015; Nakazaki and Watanabe, 2014) but distinct from the ATP power stroke (Figure 5).
Figure 5. Mechanistic Model of Protein Disaggregation by Hsp104.

Proposed model of the ATP power stroke that facilitates protein disaggregation followed by substrate threading of the unfolded polypeptide through the Hsp104 hexamer. Initial substrate binding is mediated by the NTD and NBD1 pore loop, enabling unidirectional substrate translocation in an active or passive manner.
We propose a model in which Hsp104 cycles between a closed and extended conformation, going from an extended ring to a closed ring to an open spiral conformation, consistent with binding, threading, and releasing of the unfolded polypep-tide (Figure 5). Hsp104 structures of the pre- and post-power stroke required for protein disaggregation remain unresolved. Contacts between substrate and the NTD and NBD1 pore loop provide directionality for threading of both native and unfolded polypeptides, as previously suggested (Doyle et al., 2012; Lee et al., 2017a). Hence, in addition to ATP-driven changes required for the disaggregation of amorphous and amyloid-forming aggregates, we propose that Hsp104 uses stochastic motions to drive the translocation of unfolded polypeptides. The conformational changes associated with the ATP power stroke are currently unknown and require further investigation.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Francis T.F. Tsai (ftsai@bcm.edu).
METHOD DETAILS
Protein production and purification
Saccharomyces cerevisiae Hsp104 and Hsp104E285A/E687A (Hsp104DWB) featuring a TEV-cleavable N-terminal His6-tag were overex-pressed in Escherichia coli BL21-CodonPlus (DE3)-RIL cells by inducing the bacterial cultures with 0.5 mM IPTG for 4 h at 25°C (Lee et al., 2010). Bacterial cells were pelleted, resuspended in 25 mM Tris pH 7.5, 300 mM NaCl, 30 mM imidazole, 5% glycerol and 5 mM β-mercaptoethanol, and lysed using a microfluidizer. Insoluble materials were cleared by centrifugation at 45,000 rpm for 1 h at 4°C. Hsp104 and Hsp104DWB were purified from the soluble lysate on a Ni-NTA agarose affinity column. The His-tag was cleaved off with His6-TEV protease and reapplied to the Ni-NTA agarose affinity column to remove the liberated His-tag, the TEV protease, and any uncleaved His-tagged protein. Hsp104 and Hsp104DWB were further purified by Q-Sepharose Fast Flow column chromatography. Hexamers were isolated by size exclusion chromatography on a Superdex 200 10/300 GL column pre-equilibrated in 25 mM Tris-HCl pH 7.5, 150 mM NaCl, 5% glycerol and 1 mM DTT.
ATP binding and activity assays
ATPase activities of Hsp104 and Hsp104DWB (0.5 μM monomer) were determined at 22°C in 25 mM HEPES pH 7.5, 150 mM potassium acetate, 10 mM magnesium acetate, 10 mM DTT, and 2 mM ATP without and with k-casein by measuring the amount of inorganic phosphate released after 15 min using the malachite green assay. The malachite green assay was performed by mixing 5 μl of sample with 80 μl of malachite green assay solution (0.03% malachite green, 1.05% ammonium molybdate, 1 N HCl, 0.02% Triton X-100). After incubating the mixture for 1 min at 22°C, 10 μl of 34% citric acid was added to stop the reaction. After 30 min at 22°C, the amount of inorganic phosphate was quantified by measuring the absorbance at 660 nm. Specific ATPase activity of Hsp104 and Hsp104DWB was calculated by subtracting the amount of inorganic phosphate present in the absence of protein.
To determine the stoichiometry of nucleotide binding using fluorescently labeled ATP, 0.3 mg/ml Hsp104DWB (3 μM monomer) was incubated with 10 μM, 100 μM, or 1 mM of MANT-ATP for 10 min at 22μC in 50 mM MOPS pH 7.5, 2 mM DTT and 10 mM MgCl2. Mixtures were kept on ice for 30 min before removing unbound MANT-ATP using a Zeba Spin desalting column. Then, 2 mM ATP was added to the flow-through to compete off MANT-ATP. The fluorescence signal of MANT-ATP in the sample was measured using a LS-55 fluorescence spectrophotometer (Perkin Elmer, Waltham, MA) at 360 nm (excitation) and 440 nm (emission) with slit size of 5 nm, and quantified using a standard curve.
To determine the stoichiometry of nucleotide binding using BzATP, 0.3 mg/ml Hsp104DWB (3 μM monomer) was incubated with 1 mM BzATP for 10 min at 22°C in 50 mM MOPS pH 7.5, 2 mM DTT and 2 mM MgCl2. UV crosslinking was done at 366 nm for 10 min on ice with a hand-held UV illuminator. Unbound BzATP was removed using a PD-10 desalting column pre-equilibrated in 25 mM HEPES pH 7.5, 150 mM NaCl and 2 mM DTT. Crosslinked Hsp104DWB-BzATP was concentrated and denatured in 6 M guanidine hydrochloride and 20 mM sodium phosphate buffer pH 6.5 to measure the absorbance at 260 nm (A260). The concentration of BzATP in the crosslinked sample was quantified by subtracting the A260 signal of Hsp104DWB and calculated using a molar extinction coefficient of 32,500 M−1cm−1 for BzATP.
Specimen preparation and image acquisition
The peak fraction corresponding to the Hsp104DWB hexamer was incubated at 22°C for 10 min with 5 mM ATP in buffer consisting of 50 mM MOPS pH 7.5, 2 mM DTT, and 10 mM MgCl2, and kept on ice for another 30 min. 3.0 μl of sample (0.3 mg/ml) was applied to glow discharged copper grids (Quantifoil, Groβlöbichau, Germany), blotted, and frozen in liquid ethane. Frozen hydrated grids were imaged using a 300-kV Polara G2 electron microscope (FEI, Hillsboro, OR) equipped with a field emission gun. Images were recorded on an electron counting K2 Summit direct detection camera (Gatan Inc., Pleasanton, CA) operated in super-resolution mode at 23,000× nominal magnification corresponding to 1.68 Å/pixel. Dose fractionated imaging was performed by semi-automated collection methods using SerialEM (Mastronarde, 2005). 7.6 s exposures were collected at 200 msec/frame, with a total electron dose of 38 e− per micrograph in 38 frames.
Data processing and 3D reconstruction
Each movie stack was initially binned by two and then corrected for drift and radiation damage using UNBLUR (Grant and Grigorieff, 2015) for total frames. From an initial dataset, ~2,000 particles from 20 images were automatically picked by EMAN2 (Tang et al., 2007) and the contrast transfer function (CTF) parameters were estimated internally based on the boxed particles (e2ctf.py). Extracted particles were subjected to reference-free 2D classification and the resulting 2D class averages were used to generate an initial 3D model with C1 symmetry in EMAN2. For all images, the CTF was determined with CTFFIND4 (Rohou and Grigorieff, 2015). A total of ~285,000 particles were automatically selected from 1,394 micrographs based on 2D references generated in EMAN2 using RELION 1.4 (Scheres, 2012). After 2D class averaging, bad classes were discarded and the remaining 256,378 particles were subjected to 3D classification into eight groups (K = 8). After 3D classification, 24% of total particles in poor classes were discarded because of preferred orientation with particles displaying dominant end-on views in ice. We pooled 76% particles from five classes together and subjected them to a second round of 3D classification (K = 5). Three resulting classes displayed convergence beyond 10 Å resolution, and each of the three classes was subjected to 3D auto-refinement in RELION. The resolution of the final map is 7.7 Å for the closed ring, 9.3 Å for the extended ring, and 9.3 Å for the open ring conformation according to the gold-standard protocol implemented in RELION.
Model building
Structure of the closed conformation. The structure was generated by placing the hexamer model of the casein-bound closed conformation (PDB: 5VY9) into the 3D reconstruction. The initial fit was done manually and then optimized using the “fit-in-map” routine in UCSF Chimera (Pettersen et al., 2004). Individual subunits were fitted as rigid bodies, resulting in a good fit for the main body. However, the orientation of the six NTDs was incompatible with our structure and had to be rotated manually by ~60° degrees around the 6-fold axis. After rotation, the fit for each NTD was further optimized manually using chimera resulting in the NTD conformation of the P5 and P6 subunits to deviate from the pseudo 3-fold symmetry observed in 5VY9. The mass densities for the NTDs of subunit P5 and P6 are weaker compared to the other four NTDs, indicating greater mobility. The coiled-coil motif was also fitted as a rigid body, maintaining only those of subunits P1, P2, and P6 intact based on existing densities.
Structure of the extended ring conformation. We manually placed the extended Hsp104:casein structure (PDB: 5VYA) into our image reconstruction. Then each subunit was fitted as a rigid body using the “fit-in-map” routine in UCSF Chimera. Unlike the template structure, the mass density corresponding to the NBD1 large domain of subunit P1 and the NBD1 large and small domains of subunit P6 are very weak, indicating high mobility in the absence of casein. The extended ring conformation deviates from planarity with a large gap between the P6 and P1 subunits resembling a split lock-washer assembly. NTD densities were observed in our reconstruction for subunits P2, P3, P4, and P5. Added NTD conformations are similar to those observed in the closed conformation (PDB: 5VY9). We were unable to fit the NTDs of subunits P1 and P6.
Structure of the open conformation. The hexamer model in the open conformation (PDB: 5KNE) was fitted into our 3D reconstruction using UCSF Chimera (Pettersen et al., 2004). We observed additional mass densities accounting for the missing NTDs of subunits P1, P5, and P6, which were fitted when possible. The coiled-coil motif was less well ordered and was removed from subunits P3, P5, and P6, keeping only the coiled-coil motif of subunit P4.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data shown in Figure 1 represent the average ± standard deviation (SD) from at least three technical replicates. Statistical analyses were performed in Microsoft Excel. Quantification, statistical analysis, and validation are implemented in the software packages used for 3D reconstruction and model refinement.
DATA AND SOFTWARE AVAILABILITY
Cryo-EM maps and atomic coordinates of Hsp104DWB have been deposited in the EMDB and PDB under ID codes EMD-0376 and PDB: 6N8V (open ring), EMD-0375 and PDB: 6N8T (closed ring), and EMD-0377 and PDB: 6N8Z (extended ring).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli BL21-CodonPlus (DE3)-RIL | Stratagene | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ATP | Roche | Cat.#10127531001; CAS: 51963-61-2 |
| MANT-ATP | Jena Bioscience | Cat.#NU-202; CAS: 151481-86-6 |
| BzATP | Sigma-Aldrich | Cat.#B6396; CAS: 112898-15-4 |
| Malachite green | Sigma-Aldrich | Cat.#M9636; CAS: 123333-61-9 |
| Ammonium molybdate | Sigma-Aldrich | Cat.#A1343; CAS: 12054-85-2 |
| Citric acid | Sigma-Aldrich | Cat.#C7129; CAS: 5949-29-1 |
| Ni-NTA agarose | QIAGEN | Cat.#30230 |
| Q-Sepharose Fast Flow | GE Healthcare | Cat.#17051001 |
| Superdex 200 10/300 GL column | GE Healthcare | Cat.#17108801 |
| Zeba Spin desalting column | Thermo Fisher | Cat.#89882 |
| PD-10 column | GE Healthcare | Cat.#17085101 |
| κ-casein | Sigma-Aldrich | Cat.#C0406; CAS: 9000-71-9 |
| Deposited Data | ||
| Hsp104DWB (Open) | This paper | PDB: 6N8V; EMD-0376 |
| Hsp104DWB (Closed) | This paper | PDB: 6N8T; EMD-0375 |
| Hsp104DWB (Extended) | This paper | PDB: 6N8Z; EMD-0377 |
| Recombinant DNA | ||
| Plasmid expressing Hsp104 | Lee et. al, 2010 | pHsp104 |
| Plasmid expressing Hsp104DWB | Lee et. al, 2010 | pTRAP |
| Software and Algorithms | ||
| Microsoft Excel | Microsoft | https://products.office.com/en-us/excel |
| SerialEM | Mastronarde, 2005 | http://bio3d.colorado.edu/SerialEM/ |
| UNBLUR | Grant and Grigorieff, 2015 | http://grigoriefflab.janelia.org/unblur |
| EMAN2 | Bell etal., 2016 | https://blake.bcm.edu/emanwiki/EMAN2 |
| RELION 1.4 | Scheres, 2012 | http://www2.mrc-lmb.cam.ac.uk/relion |
| Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera |
| Other | ||
| Holey carbon grid, Cu 400 mesh | Quantifoil Micro Tools GmbH | Quantifoil R 1.2/1.3 |
Highlights.
Cryo-EM analysis shows that the Hsp104-ATP hexamer adopts distinct conformations
Observed Hsp104-ATP structures are chemically identical and exist in equilibrium
Changes between closed, extended, and open hexamers are the result of “breathing”
Breathing motions enable the passive threading of unfolded polypeptides
ACKNOWLEDGMENTS
We thank W. Chiu for access to the cryo-EM facility of the NCMI, which is supported by NIH grant number P41GM103832. This work was supported by the NIH under award numbers R01GM104980, R01GM111084, R01GM107629, and R01GM110243, the Welch Foundation (Q-1530), and research resettlement funds for new faculty of Seoul National University.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes one figure, one table, and one video and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.037.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following reference appears in the Supplemental Information: Kucukelbir et al. (2014).
REFERENCES
- Bell JM, Chen M, Baldwin PR, and Ludtke SJ (2016). High resolution single particle refinement in EMAN2.1. Methods 100, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bösl B, Grimminger V, and Walter S (2005). Substrate binding to the molecular chaperone Hsp104 and its regulation by nucleotides. J. Biol. Chem 280, 38170–38176. [DOI] [PubMed] [Google Scholar]
- Deville C, Carroni M, Franke KB, Topf M, Bukau B, Mogk A, and Saibil HR (2017). Structural pathway of regulated substrate transfer and threading through an Hsp100 disaggregase. Sci. Adv 3, e1701726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle SM, Shorter J, Zolkiewski M, Hoskins JR, Lindquist S, and Wick-ner S (2007). Asymmetric deceleration of ClpB or Hsp104 ATPase activity unleashes protein-remodeling activity. Nat. Struct. Mol. Biol 14, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle SM, Hoskins JR, and Wickner S (2012). DnaK chaperone-dependent disaggregation by caseinolytic peptidase B (ClpB) mutants reveals functional overlap in the N-terminal domain and nucleotide-binding domain-1 pore tyrosine. J. Biol. Chem 287, 28470–28479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle SM, Genest O, and Wickner S (2013). Protein rescue from aggregates by powerful molecular chaperone machines. Nat. Rev. Mol. Cell Biol 14, 617–629. [DOI] [PubMed] [Google Scholar]
- Effantin G, Ishikawa T, De Donatis GM, Maurizi MR, and Steven AC (2010). Local and global mobility in the ClpA AAA+ chaperone detected by cryo-electron microscopy: functional connotations. Structure 18, 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates SN, Yokom AL, Lin J, Jackrel ME, Rizo AN, Kendsersky NM, Buell CE, Sweeny EA, Mack KL, Chuang E, et al. (2017). Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 357, 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, and Grigorieff N (2015). Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife 4, e06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuck A, Schitter-Sollner S, Suskiewicz MJ, Kurzbauer R, Kley J, Schleiffer A, Rombaut P, Herzog F, and Clausen T (2016). Structural basis for the disaggregase activity and regulation of Hsp104. eLife 5, e21516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung GC, and Masison DC (2006). N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics 173, 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, and Tagare HD (2014). Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sowa ME, Watanabe YH, Sigler PB, Chiu W, Yoshida M, and Tsai FTF (2003). The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell 115, 229–240. [DOI] [PubMed] [Google Scholar]
- Lee S, Choi JM, and Tsai FTF (2007). Visualizing the ATPase cycle in a protein disaggregating machine: structural basis for substrate binding by ClpB. Mol. Cell 25, 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sielaff B, Lee J, and Tsai FTF (2010). CryoEM structure of Hsp104 and its mechanistic implication for protein disaggregation. Proc. Natl. Acad. Sci. USA 107, 8135–8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim J-H, Biter AB, Sielaff B, Lee S, and Tsai FTF (2013). Heat shock protein (Hsp) 70 is an activator of the Hsp104 motor. Proc. Natl. Acad. Sci. USA 110, 8513–8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Sung N, Mercado JM, Hryc CF, Chang C, Lee S, and Tsai FTF (2017a). Overlapping and specific functions of the Hsp104 N domain define its role in protein disaggregation. Sci. Rep 7, 11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Sung N, Yeo L, Chang C, Lee S, and Tsai FTF (2017b). Structural determinants for protein unfolding and translocation by the Hsp104 protein disaggregase. Biosci. Rep 37, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Weaver CL, Lin J, Duran EC, Miller JM, and Lucius AL (2015). Escherichia coli ClpB is a non-processive polypeptide translocase. Biochem. J 470, 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum R, Tkach JM, Vierling E, and Glover JR (2004). Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J. Biol. Chem 279, 29139–29146. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Miot M, Reidy M, Doyle SM, Hoskins JR, Johnston DM, Genest O, Vitery MC, Masison DC, and Wickner S (2011). Species-specific collaboration of heat shock proteins (Hsp) 70 and 100 in thermotolerance and protein disaggregation. Proc. Natl. Acad. Sci. USA 108, 6915–6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A, Kummer E, and Bukau B (2015). Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation. Front. Mol. Biosci 2, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazaki Y, and Watanabe YH (2014). ClpB chaperone passively threads soluble denatured proteins through its central pore. Genes Cells 19, 891–900. [DOI] [PubMed] [Google Scholar]
- Oguchi Y, Kummer E, Seyffer F, Berynskyy M, Anstett B, Zahn R, Wade RC, Mogk A, and Bukau B (2012). A tightly regulated molecular toggle controls AAA+ disaggregase. Nat. Struct. Mol. Biol 19, 1338–1346. [DOI] [PubMed] [Google Scholar]
- Parsell DA, Kowal AS, and Lindquist S (1994). Saccharomyces cerevisiae Hsp104 protein. Purification and characterization of ATP-induced structural changes. J. Biol. Chem 269, 4480–4487. [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig R, Moradi S, Zarrine-Afsar A, Glover JR, and Kay LE (2013). Unraveling the mechanism of protein disaggregation through a ClpBDnaK interaction. Science 339, 1080–1083. [DOI] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyffer F, Kummer E, Oguchi Y, Winkler J, Kumar M, Zahn R, Sourjik V, Bukau B, and Mogk A (2012). Hsp70 proteins bind Hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces. Nat. Struct. Mol. Biol 19, 1347–1355. [DOI] [PubMed] [Google Scholar]
- Sielaff B, and Tsai FTF (2010). The M-domain controls Hsp104 protein remodeling activity in an Hsp70/Hsp40-dependent manner. J. Mol. Biol 402, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ (2007). EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol 157, 38–46. [DOI] [PubMed] [Google Scholar]
- Uchihashi T, Watanabe YH, Nakazaki Y, Yamasaki T, Watanabe H, Maruno T, Ishii K, Uchiyama S, Song C, Murata K, et al. (2018). Dynamic structural states of ClpB involved in its disaggregation function. Nat. Commun 9, 2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokom AL, Gates SN, Jackrel ME, Mack KL, Su M, Shorter J, and Southworth DR (2016). Spiral architecture of the Hsp104 disaggregase reveals the basis for polypeptide translocation. Nat. Struct. Mol. Biol 23, 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lupoli TJ, Kovach A, Meng X, Zhao G, Nathan CF, and Li H (2018). ATP hydrolysis-coupled peptide translocation mechanism of Myco-bacterium tuberculosis ClpB. Proc. Natl. Acad. Sci. USA 115, E9560–E9569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM maps and atomic coordinates of Hsp104DWB have been deposited in the EMDB and PDB under ID codes EMD-0376 and PDB: 6N8V (open ring), EMD-0375 and PDB: 6N8T (closed ring), and EMD-0377 and PDB: 6N8Z (extended ring).