

Abstract
Tailoring nanofibrous matrices ‒ a material with much promise for wound healing applications ‒ to simultaneously mitigate bacterial colonization and stimulate wound closure of infected wounds is highly desirable. To that end, a dual-releasing, multiscale system of biodegradable electrospun nanofibers coated with biocompatible micellar nanocarriers is reported. For wound healing, transforming growth factor (TGF)-β1 was incorporated into polycaprolactone/collagen (PCL/Coll) nanofibers via electrospinning and locally stimulated the myofibroblastic differentiation of human dermal fibroblasts. To prevent infection, biocompatible nanocarriers of polypeptide-based block copolymer micelles (BCMs) were deposited onto the surfaces of PCL/Coll nanofibers using tannic acid as a binding partner. Micelle-modified fibrous scaffolds were favorable for wound healing, not only supporting the attachment and spreading of fibroblasts comparable to those on non-coated nanofibers, but also significantly enhancing fibroblast migration. Micellar coatings could be loaded with gentamicin or clindamycin and exhibited antibacterial activity as measured by Petrifilm and zone of inhibition assays as well as time-dependent reduction of cellular counts of Staphylococcus aureus cultures. Moreover, delivery time of antibiotic dosage was tunable through the application of a novel modular approach. Altogether, this system holds great promise as an infection-mitigating, cell-stimulating, biodegradable skin graft for wound management and tissue engineering.
Keywords: nanofibrous matrices, infection prevention, wound healing, layer-by-layer, dual drug release
Graphical Abstract
Dual-releasing, multiscale system of biodegradable electrospun nanofibers is developed for simultaneous acceleration of wound healing and mitigation of bacterial infection. For wound healing, transforming growth factor (TGF)-β1 was incorporated into polycaprolactone/collagen (PCL/Coll) nanofibers via electrospinning. To provide antibacterial protection, nanofibers were modified with polymer micelles that served as containers for antibiotic loading and provided submicron topography to accelerate wound healing.
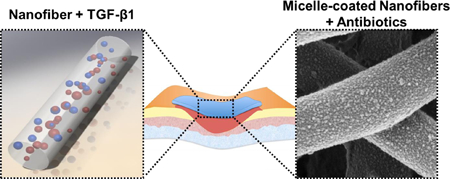
1. Introduction
Chronic wounds are an important medical problem that affect approximately 6.5 million people and cost 6 to 15 billion dollars annually in the United States alone.[1] Chronic wounds display prolonged stages of inflammation and delayed wound healing, due to an imbalance of cytokines and growth factors in the wound site.[2] Many scaffolding materials have been developed to facilitate chronic wound closure,[3] with nanofibrous matrices (NFs) showing much potential and numerous advantages. NFs can morphologically and dimensionally mimic the native extracellular matrix (ECM) fibers[4] due to the tunability of fiber diameter, spatial fiber organization, and interfiber distance (i.e., pore size).[5] Moreover, NFs can create a stimulatory microenvironment that can enhance wound healing through the local release of biomolecules.[6]
NFs can be fabricated from a large variety of polymeric materials (e.g., synthetic, natural, or polyblended) via electrospinning.[7] Polyblended nanofibers of bioresorbable polycaprolactone (PCL) and collagen (Coll) have shown promise for wound healing applications. PCL/Coll NFs support the attachment, spreading, and proliferation of normal human dermal fibroblasts (NHDFs) and the formation of dermal substitutes.[8] Moreover, anisotropically aligned PCL/Coll nanofibers are able to not only enhance cell migration along the oriented nanofibers, but also promote cellular differentiation.[8c] Furthermore, stacking of NHDF-seeded PCL/Coll fibrous matrices can lead to the formation of a 3D tissue construct that closely mimics the layered structure of native dermis within a short culture period.[8b] Considering that compromised wounds have imbalanced growth factors and cytokines, incorporating growth factors into PCL/Coll nanofibers for locally modulating the regenerative capacity of skin cells is an appealing strategy.
To that end, we explore the incorporation of transforming growth factor-β1 (TGF-β1) into PCL/Coll NFs. TGF-β1 is a multifunctional growth factor involved in wound healing by recruiting inflammatory cells and enhancing angiogenesis in wounds while stimulating wound closure.[9] Deficiency in TGF-β1 has shown delayed wound healing in mice.[10] Interestingly, NHDFs cultured on PCL/Coll NFs with TGF-β1 supplemented in media exhibit decreased proliferation, increased migration, and increased expression of α-smooth muscle actin (α-SMA).[11] α-SMA is a well-known marker of myofibroblastic differentiation, and its high-level expression is correlated with high fibroblast contractile activity.[12] Here, we investigate the effects of TGF-β1 incorporated directly into PCL/Coll nanofibers on cellular adhesion, migration, and differentiation.
Healing of wounds may be hampered by bacterial infection, most commonly of Staphylococcus aureus, with a rate of ~6% for skin grafts.[13] Moreover, upon infection with >105 colony forming units (CFU) per gram of tissue, about 80% of skin grafts fail.[14] Post-operative antibiotic treatment has been associated with the survival of skin grafts.[13f, 15] However, systemic delivery of antibiotics can cause undesirable side effects, especially with the most potent antibiotics, which may be mitigated with local delivery. More importantly, local delivery may help avoid overexposure of bacterial strains to antibiotics, which is correlated with increasing antibacterial resistance,[16] as well as the deleterious consequences of systemic administration of antibiotics. In this regard, incorporating antibacterial agents into NFs is an attractive option for local delivery of antibiotics, and has been implemented with varying degrees of success.[17] Water-soluble antibiotics such as tetracycline,[18] gentamicin,[17b] and ciproflaxin[17c, d] among others have been successfully incorporated into NFs via direct addition to electrospinning solution. Although these NFs showed the ability to prevent infection, no evidence indicated they could enhance wound healing.
In another approach to deliver drugs from NFs, large antibiotic-containing polylactic-co-glycolic acid micropatterns (~75 micron) were printed on top of NF membranes for local release.[19] Alternatively, to achieve time-programmed multi-agent release upon dissolution, hydrophilic, biodegradable micelles, loaded with anticancer agents, were included within hydrophilic NFs via electrospinning.[20] However, the aforementioned efforts focused either on bone regeneration or anti-cancer applications and did not involve topographical modification of NF surfaces through micellar attachment. Furthermore, bioactive agents incorporated into NFs via electrospinning are exposed to harsh organic solvents and control over their release kinetics is limited. In recognition of the advantages of micelle-enabled, localized drug release from surface coatings,[21] we explore the decoration of nanofiber surfaces with micelles for release of antibiotics with a particular interest in wound healing. Additionally, immobilization of micellar nanocarriers on NF surfaces introduces secondary topographic features on NFs which could also modulate cellular responses.
To immobilize micellar nanocarriers onto NF surfaces, we use the well-established layer-by-layer (LbL) technique, enabling the construction of stimuli-responsive films for drug delivery.[21–22] In this study, PCL/Coll NFs with large surface area and versatile surface chemistry were used as a 3D structural support to host micelles. Polypeptide-based block copolymer micelles (BCMs) were assembled with tannic acid (TA) to achieve robust films with swelling/deswelling behavior for release of small molecules.[23] TA is an attractive binding partner due to its antibacterial, anti-inflammatory, and pro-wound healing effects.[24] Specifically, TA has been shown to suppress the synthesis of tumor necrosis factor-α cytokine,[25] known for its involvement in chronic inflammation and high levels in chronic wound fluids.[26] Here we show that LbL coatings of BCM/TA on PCL/Coll NFs created a favorable topography for fibroblasts with regard to cellular attachment, spreading, proliferation, and wound gap closure. Moreover, BCM/TA-coated PCL/Coll NFs could be loaded with antibiotic cargo to prevent bacterial infection, with facile control over timing of delivery. Through the use of a modular system, these BCM/TA-coated PCL/Coll NFs can be combined with TGF-β1 loaded PCL/Coll NFs to create a multifunctional platform for simultaneous infection prevention and accelerated wound healing as illustrated schematically in Figure 1.
Figure 1.

Schematic representation of a modular system to simultaneously stimulate wound healing and mitigate infection. TGF-β1 was incorporated into PCL/Coll nanofibers to stimulate fibroblast-to-myofibroblast differentiation. Micellar nanocarriers were deposited on the surface of PCL/Coll nanofibers and loaded with antibiotics to prevent infection.
2. Results and Discussion
2.1. TGF-β1-containing NFs Supported Myofibroblastic Differentiation of Fibroblasts
During wound healing, timely closure of wounds with contractile fibroblasts and newly synthesized matrix is essential to accelerate the restoration of the functions of normal skin. TGF-β1 plays a prominent role in directing fibroblast differentiation and the synthesis of new ECM. However, chronic wounds often lack sufficient levels of TGF-β1;[30] therefore, supplementing the wounds with exogenous TGF-β1 could resolve such a challenge. Compared to the addition of free TGF-β1 to the wound bed, local release from PCL/Coll NFs has two-fold advantages: 1) confinement of TGF-β1 to the wound site and 2) better mimicking of the natural secretion of TGF-β1 from ECM. Incorporation of human recombinant TGF-β1 into PCL/Coll NFs was readily done by adding TGF-β1 (4 μg mL−1) into the PCL/Coll (3:1 v/v) spinning solution. Upon electrospinning, TGF-β1-containing NFs were obtained which exhibited similar fiber organization (random) and fiber diameter (216 ± 34 nm vs. 201 ± 31 nm) to those without TGF-β1 (see SI Figure S1). Culturing NHDFs on these NFs with and without TGF-β1 revealed that they both supported the attachment and spreading of NHDFs in a comparable manner (Figure 2). NHDFs displayed spindle-like morphology with obvious focal adhesions formed between the cell membrane and underneath NFs as visualized by vinculin staining (Figure 2A&B). Long, slender spikes of vinculin located to the lamellipodia were seen on both matrices (Insets of Figure 2A&B). Staining for total focal adhesion kinase (FAK), a cytosolic protein involved in cell motility and survival,[31] showed comparable intracellular expression (Figure 2C&D), implying the presence of TGF-β1 may have limited effect on NHDF migration. Indeed, measurements of NHDF migration rate and time-lapse motility on both NFs showed no noted difference (Figure 2 E&F).
Figure 2.

PCL/Coll nanofibers with TGF-β1 support the attachment and fibroblast-to-myofibroblast differentiation of NHDFs. Immunofluorescent staining of focal adhesion protein vinculin (green) and nuclei with DAPI (blue) (A,B), and immunofluorescent staining of FAK (green) (C,D), and α-SMA (red) (E, F) after 7 days culture of 2×104 cells/sample. (G) Cell migration tracks, with individual cell tracks displayed in different colors. Each track start was equalized to the center of the plot. Plot is shown from −150 to 150 μm. (H) Mean displacement for the NHDFs (n=10) tracked at 60-min lapse interval. (I) Quantification of α-SMA positive cells normalized against the cell nuclei stained with DAPI. Data presented as mean ± SD, n=3, p-values are calculated using an unpaired student t-test, **p <0.01.
In contrast, the majority of NHDFs cultured on TGF-β1 containing NFs for 7 days were positive for α-SMA, a representative marker of myofibroblasts[32] (Figure 2G&H). This was markedly different from that of TGF-β1-free NFs, on which only a limited number of cells were α-SMA positive (Figure 2I). This observation is consistent with previous findings that while TGF-β1 strongly induces fibroblast-to-myofibroblast differentiation, it has less potent effects on cellular migration.[33] Furthermore, this result also indicates the preservation of the bioactivity of TGF-β1 incorporated into PCL/Coll NFs via electrospinning, suggesting the utility of this method for the inclusion of other biomolecules.
2.2. Comparing BCM/TA Coatings on Flat and Nanofibrous Substrates
Polyvinylpyrrolidone-b-polyureido(ornithine-co-lysine) (PVP-b-PUOL), a block copolymer that exhibits upper critical solution temperature (UCST) behavior, forms BCMs above 33 °C.[23] Previously, we reported on the assembly of these BCMs with TA on flat substrates. Here, we explore the possibility of depositing this micellar system onto the surfaces of electrospun PCL/Coll NFs. In contrast to planar surfaces, assembly onto 3D substrates is highly challenging due to the intrinsically uneven topography. After successful deposition, the ability of such BCM-coated NFs to uptake antibiotics and efficacy of antibiotic-loaded BCM/TA-coated NFs to simultaneously prevent bacterial colonization and control cell migration was explored.
To deposit micelles onto planar surfaces (Si wafers) and 3D substrates (PCL/Coll NFs), a priming layer of branched polyethylenimine (BPEI) was used. BCMs (0.5 mg mL−1, pH 7.4) were then deposited followed by a top coat of TA (0.2 mg mL−1, pH 7.4). TA hydrogen bonds with the outer shell of the micelles (composed of PVP), which enables TA to secure the micelles onto the surface. We have previously shown that deposition of a top coat over stimuli-responsive micelles prevents the desorption of micelles while enabling retention of their morphology and responsive behavior.[22e, f]
We first investigated whether the chemistry of the nanofibers and their surface curvature had any effects on micellar deposition. The PCL/Coll NFs in the electrospun matrices were randomly organized with an average fiber diameter of 482 ± 6 nm. SI Figure S2 shows the surface morphology of bare or BCM/TA-modified PCL/Coll NFs as well as surface morphology of BCM/TA coatings on Si wafers. When BCM/TA coatings were deposited at the surface of Si wafers, the average micelle diameter was 36 ± 14 nm as quantified by ImageJ analysis of scanning electron microscopy (SEM) images.[23] Depositing the same micellar system on PCL/Coll NFs resulted in a similar average micelle diameter of 34 ± 19 nm. Thus, curvature of the substrate had no effect on micellar flattening upon adhesion to the surface.
Figure 3A–C shows that surface coverage of BCMs on PCL/Coll NFs could be controlled by deposition time, with increasing time leading to an increase in coverage. The micelle coverage was 23 ± 4%, 42 ± 3% and 78 ± 2% for 5, 10, and 30 min deposition times, respectively, as determined by ImageJ software threshold analysis (five randomly selected areas per image were analyzed, the total surface area was approximately 400 µm2). Similarly, the micellar coverage of planar Si wafer substrate was also quantified, reaching 67 ± 4% after 30-min deposition. The increased micellar coverage on NFs may partly come from the easy access of BCMs to nanofiber surfaces as a result of the open interfiber space. Since electrospun matrices generally have a wide distribution of fiber diameters, it was interesting to see whether such variation of fiber diameter influenced the micellar coating. Quantification of the micellar coverage on individual fibers with different diameters (482 ± 6 nm vs. 259 ± 13 nm) revealed a slightly higher occupancy of micelles on larger fibers (78 ± 2%) than smaller ones (62 ± 8%). Such a noted difference suggests to a certain degree that fiber diameter influences micellar adhesion. Theoretically, all the fiber diameters are large enough for BCMs to attach, however, finer fibers have a higher curvature, which could impede BCM adhesion and lead to lower coverage.
Figure 3.

Surface coverage of BCM on PCL/Coll NFs depends on deposition time while average micellar size does not. Morphology of PCL/Coll NFs after 1.5 bilayers of BCM/TA deposited for 5 min (A) or 30 min (B). Surface coverage and average micellar size as a function of deposition time (C). Data presented as mean ± SD, n=2.
In recognition of the effect of nanofiber curvature on micellar deposition, it is necessary to determine whether such curvature could potentially alter the responsive function of micelles. Temperature-responsive release of a model drug (pyrene) into phosphate buffered saline (PBS, pH 7.4) from BCMs immobilized on either Si wafer or PCL/Coll NFs was determined via measuring the fluorescence intensity at 371 nm of the collected PBS samples. To achieve on-demand, pulsated release, the temperature of the release solution was switched between 5 °C and 40 °C, which were below and above the micellar UCST, respectively. SI Figure S3 shows the cumulative continuous release of pyrene from BCM/TA films on Si wafers and PCL/Coll NFs. For both substrates, an on-off pyrene release was observed, with the release occurring exclusively at 40 °C. These results suggest that the curvature of the NFs exhibits minimal effects on micellar response.
The attachment of BCMs onto the fiber surface relied exclusively on non-covalent hydrogen bonding interactions; however, the micellar coatings were robust and did not decompose or desorb from the fiber surface even after 21 days of exposure to physiological conditions (37 °C, PBS, pH 7.4, with 250 rpm shaking) as can be seen in SI Figure S4A, top row. Moreover, the binding of TA with micellar corona enabled the retention of micellar structure even after exposure to PBS at temperatures higher than UCST (SI Figure S4A, top row). Considering the primary purpose of this study was to create multifunctional wound dressings, it was of paramount importance that the BCM/TA-coated NFs could survive the enzymes commonly found in chronic wounds, such as collagenase.[34] Based on image analysis, micellar coatings showed stability upon exposure to collagenase (more details can be found in the Supporting Information). Ability to absorb water is also an important parameter for wound dressings.[35] As shown in SI Figure S5, deposition of BCM/TA coatings on PCL/Coll NFs drastically improved the wettability of the substrates. In contrast to the limited wettability of uncoated PCL/Coll NFs (i.e., a contact angle of 77 ± 13°), BCM/TA-coated NFs showed complete wetting by PBS (pH 7.4). The complete wetting capacity of BCM/TA-coated NFs is most likely due to the high hydrophilicity of BCM/TA coatings, which were demonstrated in our previous study to have high swelling ratios in PBS[23] and displayed complete surface wetting when on Si wafers (data not shown).
2.3. Cytocompatibility and Cell Migration on BCM/TA-modified NFs
Upon modification with BCM/TA coatings, the surface of PCL/Coll NFs became topographically rougher with secondary nanosized attributes (i.e., micelles) (see SI Figure S2) despite overall morphologic similarity. While it was not possible to measure the surface roughness of BCM/TA-coated NFs, a root-mean-square roughness of 4.2 ± 1.5 was observed for 2.5-bilayer micellar films on planar substrates.[23] Such nanofeatured surfaces differentially regulate cellular responses, especially those related to wound healing.[36] Thus, bare and BCM/TA-modified PCL/Coll NFs were compared side-by-side for their influence on the attachment, proliferation and migration of NHDFs. After 24 h culture, NHDFs were stained for viability or processed for SEM examination. As shown in Figure 4A, NHDFs attached to bare and BCM/TA-modified NFs in a similar fashion, that is, developing strong cell-fiber connections and exhibiting a spindle-like shape. Staining the cultured NHDFs with a live/dead kit showed negligible cell death on both NFs (Figure 4B), suggesting the cytocompatibility of BCM/TA coatings with NHDFs. Interestingly, although BCM/TA-coated NFs retained a random orientation, NHDFs on such modified matrices somehow displayed a more aligned, elongated morphology, which was typically observed only on anisotropically aligned PCL/Coll NFs.[29] Further staining the cells for vinculin and FAK affirmed such observation (Figure 4C&D), in which focal adhesion complex (vinculin) distributed primarily on the elongated filopodia. While the cause of such cell orientation is still under investigation, clearly the BCM/TA coating plays a significant role. Proliferation of NHDFs on both PCL/Coll NFs was determined by DNA assay for up to 7 days (SI Figure S6). Significant stimulation of cell proliferation was observed on BCM/TA-modified NFs, especially for prolonged culture duration (day 4 and 7).
Figure 4.

Bare and BCM/TA-coated PCL/Coll NFs support the growth and attachment of NHDFs. Attachment of NHDFs as imaged by SEM imaging after 24 h culture of 5.0×104 cells (A). (B) Live/dead staining of NHDFs after 24 h culture of 5.0×104 cells/sample. Immunofluorescent staining of focal adhesion protein vinculin (green) and nuclei with DAPI (blue) (C), and FAK (green) (D) with DAPI (blue) after 48 h culture of 2×104 cells/sample.
In recognition of the elongated cell morphology ‒ normally associated with facilitated cell migration ‒ an in vitro wound-healing assay was performed to evaluate the migratory activity of NHDFs on bare and BCM/TA-coated PCL/Coll NFs. Staining the culture after 24 and 72 h with methylene blue revealed that the ability of NHDFs to bridge the model wound gap greatly depended on the substrate. On bare PCL/Coll NFs, the migration of NHDFs into the gap was moderate and only achieved approximately 20% and 50% closure by 24 h and 72 h, respectively (Figure 5). In contrast, on the BCM/TA-coated PCL/Coll NFs, NHDF migration into the gap was significantly enhanced and reached approximately 60% and 80% closure by 24 h and 72 h, respectively (Figure 5). This trend also agrees with the increased cell proliferation on BCM/TA-coated PCL/Coll NFs (SI Figure S6). Therefore, we believe that the combination of unique surface topography of BCM/TA coating and increased cell number account for the differences in migration rates. Taken together, BCM/TA-coated PCL/Coll NFs create a favorable environment for NDHFs to migrate and proliferate, two necessary requirements for wound-healing.
Figure 5.

BCM/TA coatings enhance NHDFs migration rate and proliferation on PCL/Coll NFs. (A) NHDFs (2×105 per matrix) were seeded on either bare or BCM/TA coated PCL/Coll NFs with an insert in the middle. After 24 h, the insert was removed to generate a 0.9-mm wound gap. Cells were allowed to migrate into the wound gap, and visualized after 24 and 72 h using methylene blue staining. (B) Quantification of the distance between the front lines of migrating NDHFs. Data presented as mean ± SD, n=3, p-values are calculated using an unpaired student t-test, *p <0.05.
2.4. Antibacterial Performance of Antibiotic-loaded BCM/TA Coatings
Ability of wound healing materials to prevent bacterial infection is of paramount importance. Therefore, we aimed to explore the capability of BCM/TA-coated NFs to act as cargo holders for biomolecules using two model antibiotics: gentamicin, a bactericidal aminoglycoside, and clindamycin, a bacteriostatic antibiotic. Gentamicin works by binding to the 30S ribosomal subunit, thus preventing bacterial protein synthesis,[37] while clindamycin works by binding to the 50S ribosomal subunit, causing dissociation of peptidyl-tRNAs from the ribosome.[38] Clindamycin is particularly attractive because it is widely used in clinics for its effectiveness against some strains of methicillin-resistant S. aureus (MRSA), which is the most common cause of skin infections.[39]
To determine the efficiency of assembled micellar films in loading and release of antibiotics, 3.5-bilayer films of BCM/TA were built on Si wafers and loaded with gentamicin (1 mg mL−1, pH 7.5 aqueous solution) at 4 °C for 20 h. This concentration of gentamicin was reported to have no effect on NHDF growth.[40] Gentamicin-loaded and unloaded films were challenged with 103, 105, and 107 CFU cm−2 of S. aureus ATCC 12600, and the growth of bacteria was enumerated using Petrifilm plates (Figure 6) after 48 h. The Si wafers coated with unloaded films did not demonstrate significant prevention of bacterial growth, while wafers with gentamicin-loaded BCM/TA films were able to kill >99.99% of S. aureus for a relatively high number of bacteria, 105 CFU cm−2. A control group with polymeric layers (3.5 bilayers of PVP/TA) not containing micelles showed negligible antibiotic uptake and no impact on bacterial CFUs. These results indicate that micellar films can be loaded with antibiotics at concentrations above the minimum inhibitory concentration (MIC) (12.5 μg mL−1) and that micellar cores can act as cargo holders for antibiotics. Because LbL deposition was used for micellar coating construction, the amount of antibiotics included in the film can be easily increased by increasing the number of layers in the film.[28, 41]
Figure 6.

BCM/TA coatings on Si wafers can be loaded with gentamicin and prevent bacterial growth. (A) 3.5 bilayer PVP/TA or BCM/TA coatings loaded and unloaded with gentamicin were inoculated with 103, 105, 107 CFU cm−2 of S. aureus ATCC 12600 and the growth of bacteria enumerated using Petrifilm plates (B).
We then tested the antibacterial efficacy of the BCM/TA coatings deposited on PCL/Coll NFs. Hypothetical release of antimicrobials into tissues was mimicked in vitro using zone of inhibition (ZOI) assays (Figure 7). Prior to bacterial assays, bare PCL/Coll NFs and BCM/TA-coated PCL/Coll NFs were sterilized via UV irradiation and loaded with clindamycin hydrochloride (1 mg mL−1, pH 7.5 aqueous solution) at 4 °C for 20 h. Clindamycin was measured to have an MIC of 0.25 μg mL−1 (SI Figure S8). Figure 7 A–C show that bare NFs had a 28% smaller (p < 0.02) ZOI than BCM/TA-coated NFs (ZOI diameters of ~23 ± 9 mm and ~32 ± 6 mm, respectively). By estimating the mass ratios of micelles to nanofibers, micelles were calculated to be approximately 10 times more efficient in absorbing antibiotics than bare NFs. Ability of wound dressings to be fabricated and stored for long periods of time before use is of paramount importance for application in clinics. BCM/TA-coated NFs displayed such an ability as demonstrated by ZOI diameters produced by clindamycin-loaded coatings after 20 weeks of storage of similar size to those observed with fresh BCM/TA-coated NFs (Figure 7A). This suggests high robustness of BCM/TA-coated NFs as a platform for antibiotic delivery and a high level of potential for clinical application.
Figure 7.

BCM/TA coatings on PCL/Coll NFs inhibit growth of S. aureus on agar plates and in TSB solution. ZOI diameter in mm (A) and images of ZOIs from bare (B) and BCM/TA-coated (C) PCL/Coll NFs loaded with clindamycin. ZOIs for BCM/TA-coated PCL/Coll NFs loaded with clindamycin are shown for freshly prepared samples (red) as well as samples after 20-weeks of storage at 5 °C (blue). OD of bacterial culture (D) and CFU per mL (E) over time show that clindamycin-loaded BCM/TA coatings on NFs significantly reduce OD and CFU. Data presented as mean ± SD, n=12 for A, n = 2 for D and E, p-values are calculated using an unpaired student t-test, *p <0.02.
To better mimic bacterial contamination in real wounds, S. aureus ATCC 12600 was cultured in large volumes of growth media (tryptic soy broth, TSB) with shaking in the presence of BCM/TA-coated NFs loaded with clindamycin. As controls, clindamycin-free fibers and free clindamycin solutions (MIC, 0.25 μg mL−1) were used. A separate study to evaluate the correlation of clindamycin concentrations with NHDF growth showed that clindamycin at and above the MIC had minimal effect on NHDF growth (see SI Figure S7). As can be seen in Figure 7D, all control experiments result in an increase of optical density (OD) of the culture along with an increase in bacterial cellular counts, suggesting that S. aureus grew normally in the presence of micellar scaffolds without antibiotics. On the other hand, adding clindamycin to the solution or adding the BCM/TA-coated NFs loaded with clindamycin minimized increases in OD and cellular counts. Specifically, over a culturing period of 5 h, a two-log reduction in bacterial counts was observed for clindamycin-loaded, BCM/TA-coated NFs and was similar to that observed with free clindamycin in solution. This finding suggests that BCM/TA coatings on PCL/Coll NFs could uptake at least 12.5 μg of clindamycin, considering the culturing volume of 50 mL and the clindamycin MIC of 0.25 μg mL−1. It is possible that the actual amount of loaded antibiotic was higher, since a further reduction in OD and cellular counts could not be observed for higher concentrations of clindamycin because of its bacteriostatic nature (SI Figure S9).
2.5. Functionality of Stacking Multiple NFs
In response to the need to deliver multiple biomolecules into chronic wounds, multi-modular matrices containing stacked NFs were designed. This approach takes advantage of the surface functionalization of NFs, and combines them with other types of matrices to create multifunctional matrices as illustrated in Figure 8, top. Combination of multiple matrices is advantageous for two reasons. First, this strategy enables control over the delivery rate of bioactive molecules through introduction of spacer layers (n; n=2 is shown in Figure 8, top). Second, multiple matrices can be fabricated and functionalized separately to enable dual-agent or multiple-agent release capability.
Figure 8.

Highly tunable inhibition of S. aureus from clindamycin-loaded coatings on PCL/Coll NFs via a modular approach. (A) Images of zones of inhibition from clindamycin-loaded coatings on NFs with 0, 2 and 4 spacer layers at time points, 15 min, 1 and 3 h. (B) Quantification of zones of inhibition from clindamycin-loaded coatings on NFs with 0, 2 and 4 spacer layers. Data presented as mean ± SD, n=2.
Here, we demonstrated the feasibility of the first strategy in such a system, in which spacer layers were introduced to enable control over timing of antibiotic delivery. Specifically, 2 and 4 spacer layers (n=2 and n=4) of Whatman filter paper were added between the antibiotic-loaded NFs and agar, which equates to approximately a thickness of 0.5 mm and 1 mm, respectively. Clindamycin-loaded NF samples with and without spacers were exposed to preheated (at 37 °C) agar for various times (15 min, or 1, 3 or 8 h). After this time, the samples were removed and S. aureus streaked onto the agar plate from a ~105 CFU mL−1 TSB culture as described in the experimental section. Without the addition of spacers, one antibiotic-loaded layer appears to completely empty its contents within <1 h, as the ZOI remained the same even for the longest time point (8 h). On the other hand, samples with 2 or 4 spacers (which are ‘empty’ and do not contain antibiotic) demonstrated pronounced delays in the antibiotic delivery rate to a several-hour scale, as seen by smaller ZOIs at shorter time points (Figure 8). Assuming that the antibiotic diffusion coefficient is ~10−6 cm2 s−1 (i.e. similar to that of a small fluorescent dye[42]), it is expected to take ~42 min and ~2.8 h for clindamycin, released from the coatings to diffuse through 0.5 and 1 mm, respectively. The observed results match closely with expectations. Importantly, the final zone size after 3 and 8 h, for both the 2 and 4 spacer systems, was the same as the zone generated by a sample without spacers, which suggests no loss of antibiotics into the spacer layers. These results suggest that by strategically assembling NFs together, the delivery time and the effective concentration delivered can be easily tuned. Such a strategy can be readily applied to NFs infiltrated with other biomolecules such as TGF-β1 (see 2.1.). This approach also can be utilized to develop multifunctional constructs, e.g. by combining TGF-β1-infilitrated PCL/Coll NFs with antibiotic-loaded BCM-modified PCL/Coll NFs.
3. Conclusion
We report direct coating of micellar nanocarriers onto electrospun nanofibers to achieve a hierarchical structure in which the micellar compartments provide significant benefits for controlling interactions with bacteria and mammalian cells. TGF-β1 was directly incorporated into PCL/Coll NFs via electrospinning and stimulated the fibroblast-to-myofibroblast differentiation of normal human dermal fibroblasts seeded on top of NFs. At the same time, LbL-enabled micellar coatings on NFs provided several advantageous features. First, the nanostructured landscape of the fiber surfaces, achieved through attachment of micelles, promoted strong fibroblast adhesion, spreading, proliferation, and enhanced migration even in the absence of a cell-stimulating agent. Second, surface-attached micelles could be used as nanocontainers for the incorporation of small bioactive molecules after fabrication of NFs as shown with antibiotics. These nanoscale-thin micellar coatings on nanofibers efficiently mitigated the infection challenge of S. aureus. Moreover, the feasibility of tuning the delivery rate of bioactive molecules via a modular approach that involved stacking functionalized NFs has been demonstrated. This highly versatile, flexible approach provides a promising platform that might enable employment and independent control over the delivery rate of multiple bioactive agents.
4. Experimental Section
4.1. Materials
Tannic acid (TA) was purchased from Alfa Aesar chemicals (Tewksbury, MA). Clindamycin hydrochloride was obtained from TCI chemicals (Portland, OR). Collagenase was obtained from Worthington Biomedical Corporation (Lakewood, NJ). Tryptic soy broth (TSB) powder was obtained from MP biomedicals (Solon, OH). Difco™ Technical Agar was obtained from BD Biosciences (San Jose, CA). Branched polyethylenimine (BPEI, weight-average molecular weight Mw of 750,000 g mol−1), sodium phosphate monobasic dihydrate, poly(epsilon-caprolactone) (PCL, Mw = 80,000 g mol−1), and trypsin were purchased from Sigma-Aldrich (Allentown, PA). 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) was obtained from Oakwood products Inc. (West Columbia, SC). Type I collagen (Coll) was obtained from Elastin Products Inc. (Owensville, MO). All other solvents were obtained in anhydrous form and used without further purification. Boron-doped silicon (Si) wafers were purchased from University Wafer, Inc. Recombinant human transforming growth factor-β1 (TGF-β1) was purchased from Invitrogen (Invitrogen, Carlsbad, CA).
4.2. Fabrication of Nanofibers
NFs of PCL and Coll at a weight ratio of 3:1 were fabricated using electrospinning as previously described.[27] Briefly, 8% (w/v) solution of 3:1 PCL/Coll was prepared by dissolving PCL and Coll in HFIP. Then, the solution was loaded into a 3-mL syringe with a 20-gauge stainless steel blunt-tip needle and electrospun at 10 kV using a custom electrospinning apparatus. The polymer solution was dispensed using a syringe pump (Kdscientific, Holliston, MA) at 10 μL min−1. PCL/Coll NFs for micellar modification were electrospun onto 12-well-plate-sized paper rings (filter paper, inner diameter 0.5” and outer diameter 0.75”) or metal rings (diameter 0.6”) with a collecting surface of aluminum foil. Before NF spinning, rings were pre-coated with BPEI (0.2 mg mL−1, pH 9) for 20 min. PCL/Coll NFs with TGF-β1 were prepared by electrospinning PCL/Coll solution mixed with reconstituted TGF-β1 at a final concentration of 500 ng mL−1 onto glass coverslips. The estimated amount of TGF-β1 in each sample was 5 ng. The use of an organic solvent during fabrication of TGF-β1-incorporated nanofibers may affect TGF-β1 bioactivity. However, based on our previous findings with fibronectin-incorporated nanofibers, [43] and consistent with the bioactivity results of this work (page 7, Figure 2), the effect was small. Once sufficient NFs were collected, the NFs were cut along the edges of the coverslip to assure full coverage of the entire coverslip surface.
4.3. Preparation of Biocompatible Micellar Nanocontainers and Their Deposition on Substrate Surfaces
A block copolymer of polyvinylpyrrolidone-b-polyureido(ornithine-co-lysine) (PVP-b-PUOL) was synthesized via ring opening polymerization followed by post-polymerization functionalization with ureido groups as reported earlier.[23] The final product of PVP162-b-PUOL513 had the number-average molecular weight Mn of 233,000 g mol−1 and a polydispersity index of 1.17. This polymer forms spherical BCMs in solutions at temperatures lower than ambient.[23] Temperature of 10 °C was used for construction of LbL films of BCM and TA using the dip-deposition technique. In order to deposit BCM/TA films onto surfaces, first a priming layer of BPEI was deposited at pH 9 for 30 min from 0.2 mg mL−1 solution. Afterwards, alternating layers of TA (pH 7.4, 0.2 mg mL−1) and BCM (pH 7.4, 0.5 mg mL−1) were deposited for 30 min at 10 °C so that the final coating had BPEI, TA, BCM, TA (1.5 bilayers) coated on top. Control coatings of PVP and TA were deposited in an analogous manner with PVP solution (pH 7.4, 0.5 mg mL−1) replacing the BCM solution. For ease of characterization of the micellar coatings, 3.5-bilayer films (capped with TA) were also deposited on flat substrates (Si wafers). For surface coverage studies, samples were prepared with varying deposition times of the BCM layer but constant deposition times for the other layers. To monitor coating deposition, SEM performed with a JEOL JSM-7500F instrument was used for imaging, after sputter coating samples with 3 nm Pt/Pd alloy. Static contact angle measurements were taken using an Optical Contact Angle and Surface Tension Meter CAM 101 (KVS Instrument Inc.) with pH 7.4 PBS. Prior to bacterial and cell culture, samples were sterilized with UV irradiation for 15 min on each side for NF substrates or 30 min for Si wafer substrates. A control test on Si wafers showed that film swelling (determined with a spectroscopic ellipsometry (M-2000, J.A. Woollam) as described in next section) before and after UV sterilization remained constant, suggesting negligible effects of UV on film function.
4.4. Degradation Studies on Flat and Structured Substrates
4.4.1. Flat Substrates
Two 3.5-bilayer films of BCM/TA on Si wafers were exposed to 5 mL of collagenase (0.1%) or trypsin (0.25%) in PBS at 37 °C. At designated time points, films were removed from solution, washed with pH 7.4 PBS, and dried with nitrogen gas. Dry thicknesses of coatings on Si wafers were measured with spectroscopic ellipsometry (M-2000, J.A. Woollam) at angles of incidence 45, 55, 65 and 75°. A Cauchy layer model was used for analysis with the wavelength-independent fitting parameter set to 1.5, which allowed for automatic calculation of the best fit thickness. After measurements, samples were returned to and kept in fresh solutions of collagenase or trypsin until the next measurement.
4.4.2. Nanofiber Substrates
To perform degradation studies, 1.5-bilayer BCM/TA coated nanofibers (together with the paper rings) were soaked in 1 mL of sterilized PBS containing trypsin-EDTA (0.25%) in a 12-well plate and incubated at 37 °C under shaking (40 ± 1 rpm). The solution was refreshed every three days. All samples were vigorously washed with deionized water twice and dried at room temperature. For SEM imaging, the samples were examined with a LEO 982 FEG SEM.
4.5. Bacterial Studies
4.5.1. Antibiotic Loading
3.5-bilayer films of BCM/TA and control polymeric layers without micellar cores (PVP/TA) were loaded with gentamicin (1 mg mL−1, pH 7.5 aqueous solution) at 4 °C for 20 h. Bare PCL/Coll NFs and BCM/TA-coated PCL/Coll NFs were loaded with clindamycin hydrochloride (1 mg mL−1, pH 7.5 aqueous solution) at 4 °C for 20 h after UV irradiation. All samples were washed with pH 7.5 aqueous solution to remove excess antibiotic and then dried (with nitrogen gas for silicon wafers or air dried for nanofiber samples).
4.5.2. Bacterial Strain and MIC Assays
For all bacterial studies, Staphylococcus aureus (S. aureus, ATCC 12600) was streaked on TSB agar plates from a frozen glycerol stock, incubated at 37 °C for 16 h to obtain single colonies, and stored at 4 °C before bacterial cultures. All cultures were started with colonies from plates of less than a week old. The MIC of clindamycin hydrochloride for S. aureus was determined by the antibiotic concentration at which no growth in the main streak was observed upon streaking TSB agar plates with different antibiotic concentrations. The MIC of gentamicin was 12.5 μg mL−1 as previously measured by us.[28]
4.5.3. Petrifilm Assay
For Petrifilm (3M Petrifilm Aerobic Count Plates, Nelson Jameson, Marshfield, WI, USA) assays, a single colony from the agar plate was inoculated in 2 mL of TSB and grown overnight at 37 °C with shaking (250 rpm). Afterwards, the optical density at 600 nm (OD600) of the culture was measured and used as a guideline to prepare cultures containing 109, 107 and 105 CFU mL−1 via dilution with TSB. Petrifilm plates were treated with 1 mL of sterilized DI water for 30 min to hydrate the plate. Si wafers coated with 3.5-bilayer BCM/TA films were placed face up on the hydrated Petrifilms and 10 µL of bacterial suspension was placed onto each sample and incubated at 37 °C for 48 h. After 48 h, the bacterial colonies appearing on top of the samples were counted.
4.5.4. ZOI Assay
To perform ZOI assays, 4 mL of TSB was inoculated with 5 single colonies from the TSB agar plate and then grown for 17 h. The OD600 of the culture was measured and diluted in TSB to ~105 CFU mL−1. For ZOI agar plates, 20 mL of TSB was dried overnight, pre-heated at 37 °C for 2 h, and then exposed to bare or BCM-coated PCL/Coll NFs (with and without clindamycin loading) for 1 h (unless otherwise noted). Afterwards, the NF samples were removed, and the agar plate was streaked three times with a sterile cotton swab from a 105 CFU mL−1 culture, rotating the plate 60° between each streak. Plates were allowed to dry for 5 min and incubated at 37 °C for 24 h and used for zone of inhibition measurements.
4.5.5. Optical Density and CFU mL−1 Assay
For OD600 measurements as a function of time, 5 single colonies in 4 mL of TSB were grown for 17 h at 37 °C with shaking (250 rpm). 50 mL of TSB was inoculated with 80 μL of the overnight culture at 37 °C with shaking (250 rpm). Cultures were grown for 3 h and then NF samples placed into the culturing flask. At each time point, 0.5 mL of the culture was collected and used for OD600 measurements and plated at various dilutions in duplicate to measure cellular counts in solution. Plates were grown for 20 h and colonies were counted manually.
4.6. Human Cell Studies
For human cell studies, PCL/Coll NFs with or without TGF-β1 on glass coverslips and 1.5 bilayer BCM/TA-coated or bare PCL/Coll NFs on paper rings were used.
4.6.1. Cell Seeding and Culture
Primary normal human skin fibroblasts (NHDFs) from newborn foreskin (R2F) were a gift from Dr. James G. Rheinwald of the Harvard NIH Skin Disease Research Center. The cells were grown in monolayer culture in Dulbecco’s modified Eagle media (DMEM, low glucose) supplemented with 10% FBS and 1% penicillin and streptomycin (Sigma) at 37 °C in a humidified 5% CO2 atmosphere. Cells were routinely subcultured and used at passages 7–10. For each experiment, 500 μL of cell suspension (4×104 cells/mL) was carefully added onto the coverslips coated with nanofibers to avoid any spillage over the edge of the coverslips. The seeded cells were then incubated for 60 min prior to the addition of fully supplemented media. Cells were cultured up to 7 days and media was refreshed every 2–3 days.
4.6.2. Cell Proliferation
Cell proliferation was determined using the CyQUANT® Cell Proliferation Assay Kit (Molecular Probes, Inc., Eugene, OR) following the manufacturer’s manual. Briefly, cell culture samples (in triplicate, n=3) were harvested on day 1, 4 and 7, respectively. After removal of media and rinsing with pH 7.4 PBS, the samples were snap-frozen and stored at −80 °C. All the samples were lysed in CyQUANT® cell- lysis buffer for 1 h at room temperature and then 200 µL of CyQUANT® GR dye/cell-lysis buffer was added to each sample and incubated for 2–5 min at room temperature in the dark. The fluorescence intensity of cell lysates was measured using the multi-mode BioTek microplate reader (Synergy™ HT, BioTek Instruments Inc., Winooski, VT) at 480 nm ex./520 nm em.
4.6.3. Live/Dead Cell Staining
Viability of NHDFs on NFs was determined by fluorescent staining with a Live-Dead Assay Cytotoxicity Kit (Thermo Fisher Sceintific). Specifically, NHDF-seeded NFs after culture for 1 day were incubated with 4 mM calceinacetoxymethyl ester-AM (stains the cytoplasm of live cells green) and 4 mM ethidiumhomodimer (stains the nuclei of dead cells red) for 5 min at room temperature. After rinsing with PBS, the stained samples were visualized with a Nikon Eclipse 80i fluorescence microscope.
4.6.4. Immunofluorescent Staining
Immunofluorescent staining of cells was performed as previously described.[29] Briefly, cultured cells were fixed in 4% paraformaldehyde for 10 min and then permeabilized with 0.2% Triton X-100 in PBS. Primary antibodies used were: phalloidin-FITC (Sigma, 1:500), anti-α-smooth muscle actin, anti-vinculin (Sigma, 1:200), and anti-focal adhesion kinase (Sigma 1:1000). The cells were further incubated with goat anti-mouse or goat anti-rabbit TRITC conjugated IgG secondary antibody (Caltag, 1:400). Cell nuclei were stained with DAPI (Sigma, 1:1000). The staining was examined under a Nikon Eclipse 80i fluorescence microscope.
4.6.5. Cell Migration
Migration of fibroblasts on PCL/Coll NFs with and without TGF-β1 was monitored using time-lapse microscopy. Briefly, the culture was placed in a climate-controlled incubator (temperature- and CO2-controlled chamber) under the eclipse Ti Nikon fluorescence microscope. Each sample was imaged at 10-min intervals for 5 h. Acquired images were analyzed by ImageJ to track at least 10 cells per condition and calculate the mean displacement per 10-min time lapse interval for each cell. To evaluate the migratory activity of NHDFs in relation to wound healing, in vitro CytoSelect™ 24-Well Wound Healing Assay Kit (Cell Biolabs Inc., San Diego, CA) was used. Briefly, 2×105 NHDFs suspended in the culture media were seeded onto the surfaces of NFs with the insert in place. After 24 h, the insert was removed to generate a consistent 0.9-mm-wound gap among the cells. Cells were allowed to migrate into the wound gap for 24 and 72 h. After staining the cells with 0.1% methylene blue, images of the wound gap were taken to analyze the gap distance (n=3).
4.6.6. Cell Susceptibility to Clindamycin Hydrochloride in Solution
Cell susceptibility to clindamycin hydrochloride was determined via MTT assay after 24-h culture in a 6-well plate with a series of clindamycin hydrochloride dilutions (0.0625, 0.125, 0.25, 0.5, and 1 μg mL−1) added to the culture media, respectively. Briefly, the culture was incubated with thiazolyl blue tetrazolium bromide (MTT; Sigma, St. Louis, MO) solution (0.5 mg mL−1 in culture media) at 37 °C in the dark for 4 h. Upon removal of the nonreacted dye, the formazan product was extracted with DMSO and 100 μL of the extract was transferred to a 96-well plate for absorbance measurement. The absorbance was measured at a wavelength of 570 nm with a Synergy HT Multi-Detection Microplate Reader (BioTek Instruments, Winooski, VT).
4.7. Statistical Analysis
All data are expressed as the mean ± standard deviation (SD). Each experiment was repeated at least 3 times on different days. For bacterial studies, each experiment was repeated at least twice on different days with data collected in at least duplicate for each group. Statistical evaluation was performed using an unpaired student t-test. A value of p < 0.05 was considered statistically significant.
The statistical analysis was performed by using Minitab 18 software.
Supplementary Material
Acknowledgements
Use of the TAMU Materials Characterization Facility is acknowledged. We acknowledge financial support from Texas A&M University/Association of Former Students Graduate Merit Fellowship, Texas A&M Engineering Experiment Station (TEES) and partial support by NSF-DMR award number 1610725, NIAMS award number 1R01AR067859, NSF-DMR award number 1508511. V.A., M.X., S.S., and H.W. prepared the manuscript. M.X. electrospun TGF-β1 containing nanofibers and performed all the cell studies. A.P. synthesized BCMs, deposited LbL coatings, did SEM imaging of coatings, and performed degradation studies on flat substrates. V.A., A.J., and S.S. designed bacterial experiments. V.A. performed release studies and antibacterial experiments. J.C. electrospun nanofibers. M.S. prepared nanofiber substrates. B.Z. performed degradation studies on nanofibers. All authors have given approval of the final version of the manuscript.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Morphology and average diameter of PCL/Coll NFs with and without TGF-β1. Morphology of bare and BCM/TA-coated PCL/Coll NFs as well as BCM/TA-coated Si wafer. Temperature-triggered release of pyrene from BCM/TA coatings on Si wafers or PCL/Coll NFs. Degradability of BCM/TA films on planar and NF substrates. Wettability of PCL/Coll NFs and BCM/TA coated NFs. Fibroblast proliferation on bare or BCM/TA-coated PCL/Coll NFs up to seven days as measured by DNA assay. Fibroblast growth in the presence of clindamycin. MIC determination of clindamycin hydrochloride for S. aureus. OD and cellular counts for S. aureus cultures containing various clindamycin hydrochloride concentrations. Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Victoria Albright, Department of Materials Science and Engineering, Texas A&M University, 575 Ross Street, College Station, TX 77843, USA.
Meng Xu, Department of Chemistry and Chemical Biology, Stevens Institute of Technology, 1 Castle Point on the Hudson, Hoboken, NJ 07030, USA.
Anbazhagan Palanisamy, Department of Materials Science and Engineering, Texas A&M University, 575 Ross Street, College Station, TX 77843, USA.
Jun Cheng, Department of Chemistry and Chemical Biology, Stevens Institute of Technology, 1 Castle Point on the Hudson, Hoboken, NJ 07030, USA.
Mary Stack, Department of Biomedical Engineering, Stevens Institute of Technology, 1 Castle Point on the Hudson, Hoboken, NJ 07030, USA.
Beilu Zhang, Department of Chemistry and Chemical Biology, Stevens Institute of Technology, 1 Castle Point on the Hudson, Hoboken, NJ 07030, USA.
Prof. Arul Jayaraman, Department of Chemical Engineering, Department of Biomedical Engineering, Texas A&M University
Prof. Svetlana A. Sukhishvili, Department of Materials Science and Engineering, Texas A&M University, 575 Ross Street, College Station, TX 77843, USA, svetlana@tamu.edu
Prof. Hongjun Wang, Department of Chemistry and Chemical Biology, Stevens Institute of Technology, 1 Castle Point on the Hudson, Hoboken, NJ 07030, USA, Hongjun.Wang@stevens.edu
References
- [1].a)Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK, Gottrup F, Gurtner GC, Longaker MT, Wound Repair Regen 2009, 17, 763. [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Markova A, Mostow EN, Dermatol. Clin 2012, 30, 107. [DOI] [PubMed] [Google Scholar]
- [2].a)Frykberg RG, Banks J, Adv. Wound Care 2015, 4, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Panuncialman J, Falanga V, Clin. Plast. Surg 2007, 34, 621. [DOI] [PubMed] [Google Scholar]
- [3].a)Dickinson LE, Gerecht S, Front. Physiol 2016, 7, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Zhong SP, Zhang YZ, Lim CT, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol 2010, 2, 510. [DOI] [PubMed] [Google Scholar]
- [4].Hinderer S, Layland SL, Schenke-Layland K, Adv. Drug Delivery Rev 2016, 97, 260. [DOI] [PubMed] [Google Scholar]
- [5].Wang K, Xu M, Zhu M, Su H, Wang H, Kong D, Wang L, J Biomed. Mater. Res. A 2013, 101, 3474. [DOI] [PubMed] [Google Scholar]
- [6].a)Chou S-F, Carson D, Woodrow KA, J. Controlled Release 2015, 220, 584. [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Wang Z, Qian Y, Li L, Pan L, Njunge LW, Dong L, Yang L, J. Biomater. Appl 2016, 30, 686. [DOI] [PubMed] [Google Scholar]
- [7].a)Liu M, Duan X-P, Li Y-M, Yang D-P, Long Y-Z, Mater. Sci. Eng. C 2017, 76, 1413. [DOI] [PubMed] [Google Scholar]; b)Chen S, Liu B, Carlson MA, Gombart AF, Reilly DA, Xie J, Nanomedicine (London, U.K.) 2017, 12, 1335. [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Nwachukwu CC, Einstein GP, Tulp OL, FASEB J 2017, 31, 333.2.28049156 [Google Scholar]
- [8].a)Venugopal JR, Zhang Y, Ramakrishna S, Artif. Organs 2006, 30, 440. [DOI] [PubMed] [Google Scholar]; b)Yang X, Shah JD, Wang H, Tissue Eng. Part A 2009, 15, 945. [DOI] [PubMed] [Google Scholar]; c)Chen X, Fu X, Shi J.-g., Wang H, Nanomedicine (N. Y., NY, U. S.) 2013, 9, 1283. [DOI] [PubMed] [Google Scholar]
- [9].a)Pakyari M, Farrokhi A, Maharlooei MK, Ghahary A, Adv. Wound Care 2013, 2, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Diegelmann RF, Evans MC, Front. Biosci 2004, 9, 283. [DOI] [PubMed] [Google Scholar]
- [10].Crowe MJ, Doetschman T, Greenhalgh DG, J. Invest. Dermatol 2000, 115, 3. [DOI] [PubMed] [Google Scholar]
- [11].Fu X, Xu M, Jia C, Xie W, Wang L, Kong D, Wang H, J. Mater. Chem. B 2016, 4, 5246. [DOI] [PubMed] [Google Scholar]
- [12].a)Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C, Mol. Biol. Cell 2001, 12, 2730. [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G, J. Cell Biol 1993, 122, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Ronnov-Jessen L, Petersen OW, Lab. Invest 1993, 68, 696. [PubMed] [Google Scholar]
- [13].a)Wukich DK, Ahn J, Raspovic KM, Gottschalk FA, La Fontaine J, Lavery LA, Foot Ankle Int 2017, 38, 388. [DOI] [PubMed] [Google Scholar]; b)Sánchez-Sánchez M, Cruz-Pulido WL, Bladinieres-Cámara E, Alcalá-Durán R, Rivera-Sánchez G, Bocanegra-García V, Int. J. of Lower Extremity Wounds 2017, 16, 129. [DOI] [PubMed] [Google Scholar]; c)Maruccia M, Onesti MG, Sorvillo V, Albano A, Dessy LA, Carlesimo B, Tarallo M, Marcasciano M, Giudice G, Cigna E, Ribuffo D, BioMed Res. Int 2017, 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]; d)Rahim K, Saleha S, Zhu X, Huo L, Basit A, Franco OL, Microb. Ecol 2017, 73, 710. [DOI] [PubMed] [Google Scholar]; e)Gjødsbøl K, Christensen JJ, Karlsmark T, Jørgensen B, Klein BM, Krogfelt KA, Int. Wound J.l 2006, 3, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]; f)Alexander JW, MacMillan BG, Law EJ, Krummel R, Trauma J Acute Care Surg 1982, 22, 687. [DOI] [PubMed] [Google Scholar]
- [14].Robson MC, Surg. Clin. North Am 1997, 77, 637. [DOI] [PubMed] [Google Scholar]
- [15].Kuijpers DIM, Smeets NWJ, Lapière K, Thissen M, Krekels GAM, Neumann HAM, J. Eur. Acad. Dermatol. Venereol 2006, 20, 1296. [DOI] [PubMed] [Google Scholar]
- [16].Taubes G, Science 2008, 321, 356. [DOI] [PubMed] [Google Scholar]
- [17].a)Gao Y, Bach Truong Y, Zhu Y, Louis Kyratzis I, J. Appl. Polym. Sci 2014, 131; b)Dave R, Jayaraj P, Ajikumar PK, Joshi H, Mathews T, Venugopalan VP, J. Biomater. Sci., Polym. Ed 2013, 24, 1305. [DOI] [PubMed] [Google Scholar]; c)Bakhsheshi-Rad HR, Hadisi Z, Hamzah E, Ismail AF, Aziz M, Kashefian M, Mater. Lett 2017, 207, 179 [Google Scholar]; d)Zupančič Š, Sinha-Ray S, Sinha-Ray S, Kristl J, Yarin AL, Mol. Pharmaceutics 2016, 13, 1393. [DOI] [PubMed] [Google Scholar]
- [18].a)Kenawy -R, Bowlin GL, Mansfield K, Layman J, Simpson DG, Sanders EH, Wnek GE, J. Controlled Release 2002, 81, 57. [DOI] [PubMed] [Google Scholar]; b)Xu X, Zhong W, Zhou S, Trajtman A, Alfa M, J. Appl. Polym. Sci 2010, 118, 588. [Google Scholar]
- [19].Chen XN, Gu YX, Lee JH, Lee WY, Wang HJ, Eur. Cells Mater 2012, 24, 237. [DOI] [PubMed] [Google Scholar]
- [20].a)Yang G, Wang J, Li L, Ding S, Zhou S, Macromo.l Biosci 2014, 14, 965. [DOI] [PubMed] [Google Scholar]; b)Yang G, Wang J, Wang Y, Li L, Guo X, Zhou S, ACS Nano 2015, 9, 1161. [DOI] [PubMed] [Google Scholar]
- [21].Zhu Z, Gao N, Wang H, Sukhishvili SA, J. Controlled Release 2013, 171, 73. [DOI] [PubMed] [Google Scholar]
- [22].a)Zhu Z, Sukhishvili SA, J. Mater. Chem 2012, 22, 7667 [Google Scholar]; b)Xu L, Zhu Z, Sukhishvili SA, Langmuir 2011, 27, 409. [DOI] [PubMed] [Google Scholar]; c)Tan WS, Zhu Z, Sukhishvili SA, Rubner MF, Cohen RE, Macromolecules 2011, 44, 7767 [Google Scholar]; d)Xu L, Zhu Z, Sukhishvili SA, Langmuir 2010, 27, 409. [DOI] [PubMed] [Google Scholar]; e)Zhu Z, Sukhishvili SA, ACS Nano 2009, 3, 3595. [DOI] [PubMed] [Google Scholar]; f)Erel I, Zhu Z, Zhuk A, Sukhishvili SA, J. Colloid Interface Sci 2011, 355, 61. [DOI] [PubMed] [Google Scholar]; g)Kim B-S, Park SW, Hammond PT, ACS Nano 2008, 2, 386. [DOI] [PubMed] [Google Scholar]; h)Kim B-S, Lee H.-i., Min Y, Poon Z, Hammond PT, Chem. Commun 2009, 4194. [DOI] [PMC free article] [PubMed]; i)Sukhishvili SA, Curr. Opin. Colloid Interface Sci 2005, 10, 37 [Google Scholar]; j)Kharlampieva E, Kozlovskaya V, Sukhishvili SA, Adv. Mater 2009, 21, 3053 [Google Scholar]; k)Decher G, Hong JD, presented at Proceedings of the Annual Conference on Engineering in Med. and Biol 1990; l)Bertrand P, Jonas A, Laschewsky A, Legras R, Macromol. Rapid Commun 2000, 21, 319 [Google Scholar]; m)Hammond PT, Adv. Mater 2004, 16, 1271 [Google Scholar]; n)Tang Z, Wang Y, Podsiadlo P, Kotov NA, Adv. Mater 2006, 18, 3203 [Google Scholar]; o)Richardson JJ, Cui J, Björnmalm M, Braunger JA, Ejima H, Caruso F, Chem. Rev 2016, 116, 14828. [DOI] [PubMed] [Google Scholar]
- [23].Palanisamy A, Albright V, Sukhishvili SA, Chem. Mater 2017, 29, 9084. [Google Scholar]
- [24].Ninan N, Forget A, Shastri VP, Voelcker NH, Blencowe A, ACS Appl. Mater. & Interfaces 2016, 8, 28511. [DOI] [PubMed] [Google Scholar]
- [25].Kozlovskaya V, Xue B, Lei W, Padgett LE, Tse HM, Kharlampieva E, Adv. Healthcare Mater 2015, 4, 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tarnuzzer RW, Schultz GS, Wound Repair and Regen 1996, 4, 321. [DOI] [PubMed] [Google Scholar]
- [27].Zhang YZ, Venugopal J, Huang ZM, Lim CT, Ramakrishna S, Biomacromolecules 2005, 6, 2583. [DOI] [PubMed] [Google Scholar]
- [28].Albright V, Zhuk I, Wang Y, Selin V, van de Belt-Gritter B, Busscher HJ, van der Mei HC, Sukhishvili SA, Acta Biomater 2017, 61, 66. [DOI] [PubMed] [Google Scholar]
- [29].Huang C, Fu X, Liu J, Qi Y, Li S, Wang H, Biomaterials 2012, 33, 1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M, Wound Repair and Regen 2008, 16, 585. [DOI] [PubMed] [Google Scholar]
- [31].Parsons JT, J. Cell Sci 2003, 116, 1409. [DOI] [PubMed] [Google Scholar]
- [32].Rao K B, Malathi N, Narashiman S, Rajan ST, J. Clin. Diagn. Res 2014, 8, ZC14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hinz B, Gabbiani G, Thromb. Haemostasis 2003, 90, 993. [DOI] [PubMed] [Google Scholar]
- [34].a)Schultz GS, Sibbald RG, Falanga V, Ayello EA, Dowsett C, Harding K, Romanelli M, Stacey MC, Teot L, Vanscheidt W, Wound Repair and Regen 2003, 11, S1. [DOI] [PubMed] [Google Scholar]; b)Saarialho-Kere UK, Kovacs SO, Pentland AP, Olerud JE, Welgus HG, Parks WC, J. Clin. Invest 1993, 92, 2858. [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Schiffer D, Blokhuis-Arkes M, van der Palen J, Sigl E, Heinzle A, Guebitz GM, Br. J. Dermatol 2015, 173, 1529. [DOI] [PubMed] [Google Scholar]
- [35].Ousey K, Cutting KF, Rogers AA, Rippon MG, J. Wound Care 2016, 25, 122. [DOI] [PubMed] [Google Scholar]
- [36].Yim EKF, Leong KW, Nanomedicine (N. Y., NY, U. S.) 2005, 1, 10. [DOI] [PubMed] [Google Scholar]
- [37].McCoy LS, Xie Y, Tor Y, Wiley Interdiscip. Rev.: RNA 2011, 2, 209. [DOI] [PubMed] [Google Scholar]
- [38].Tenson T, Lovmar M, Ehrenberg M, J. Mol. Biol 2003, 330, 1005. [DOI] [PubMed] [Google Scholar]
- [39].King MD, Humphrey BJ, Wang YF, Kourbatova EV, Ray SM, Blumberg HM, Ann. Intern. Med 2006, 144, 309. [DOI] [PubMed] [Google Scholar]
- [40].Ingeburg EG, Randi M, Catherine JW, In Vitro 1979, 15, 114.457178 [Google Scholar]
- [41].a)Pavlukhina S, Zhuk I, Mentbayeva A, Rautenberg E, Chang W, Yu X, van de Belt-Gritter B, Busscher HJ, van der Mei HC, Sukhishvili SA, NPG Asia Mater 2014, 6, e121 [Google Scholar]; b)Pavlukhina SV, Kaplan JB, Xu L, Chang W, Yu X, Madhyastha S, Yakandawala N, Mentbayeva A, Khan B, Sukhishvili SA, ACS Appl. Mater. Interfaces 2012, 4, 4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pristinski D, Kozlovskaya V, Sukhishvili SA, J. Chem. Phys 2005, 122, 14907. [DOI] [PubMed] [Google Scholar]
- [43].Yang X, Ogbolu KR, Wang H, J. Exp. Nanosci 2008, 3, 329. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.