

Abstract
Mutations in the nuclear localization signal of the RNA binding protein FUS cause both Frontotemporal Dementia (FTD) and Amyotrophic Lateral Sclerosis (ALS). These mutations result in a loss of FUS from the nucleus and the formation of FUS-containing cytoplasmic aggregates in patients. To better understand the role of cytoplasmic FUS mislocalization in the pathogenesis of ALS, we identified a population of cholinergic neurons in Drosophila that recapitulate these pathologic hallmarks. Expression of mutant FUS or the Drosophila homolog, Cabeza (Caz), in class IV dendritic arborization neurons results in cytoplasmic mislocalization and axonal transport to presynaptic terminals. Interestingly, overexpression of FUS or Caz causes the progressive loss of neuronal projections, reduction of synaptic mitochondria, and the appearance of large calcium transients within the synapse. Additionally, we find that overexpression of mutant but not wild type FUS results in a reduction in presynaptic Synaptotagmin, an integral component of the neurotransmitter release machinery, and mutant Caz specifically disrupts axonal transport and induces hyperexcitability. These results suggest that FUS/Caz overexpression disrupts neuronal function through multiple mechanisms, and that ALS-causing mutations impair the transport of synaptic vesicle proteins and induce hyperexcitability.
Keywords: ALS, FUS, Drosophila, neuron, synapse, nuclear transport
1. Introduction
Fused in Sarcoma (FUS) is an integral regulator of multiple aspects of RNA metabolism, including mRNA splicing and export, stress granule formation, and local translation (Lagier-Tourenne et al., 2010; Yang et al., 2010). Mutations in the FUS carboxy-terminal atypical proline-tyrosine nuclear localization signal (PY-NLS) cause ALS, and pathogenesis is thought to be a consequence of the exclusion of FUS from the nucleus and its cytoplasmic aggregation within neurons (Ito et al., 2010; Kwiatkowski et al., 2009; Vance et al., 2009). The precise mechanisms by which mutations in FUS cause neurodegeneration are unknown, but hypotheses include: (1) loss of FUS from the nucleus disrupting mRNA maturation (Zhou et al., 2013); (2) gain of FUS in the cytoplasm altering stress granule dynamics (Li et al., 2013); (3) cytoplasmic FUS transported along axons altering synaptic physiology (Schoen et al., 2015); (4) induction of the DNA damage response (Hicks et al., 2000; Kuroda, 2000; Qiu et al., 2014; Wang et al., 2013), and (5) a combination of these pathomechanisms (Shang and Huang, 2016). One challenge in distinguishing between these possibilities in vivo is that expression of ALS-mutant FUS in Drosophila and mouse motor neurons leads to overexpression in neuronal nuclei in addition to cytoplasmic mislocalization (Lanson et al., 2011; Machamer et al., 2014; Qiu et al., 2014). Animal models expressing ALS-mutant FUS that predominantly localize to the neuronal cytoplasm in order to allow investigation of the cytoplasmic contribution of FUS toxicity have not yet been generated.
Multiple studies in vertebrate models indicate that neuromuscular junction function is impaired during the preclinical stage of ALS (Moloney et al., 2014). For example, overexpression of human FUSP525L or superoxide dismutase 1 (SOD1) with the G93A mutation results in loss of neuromuscular junctions before motor neuron death (Lalancette-Hebert et al., 2016; Rocha et al., 2013). Knockdown or overexpression of mutant FUS results in hyperexcitability and a reduction in the amplitude of synaptic transmission in zebrafish (Armstrong and Drapeau, 2013). The mechanisms by which FUS alters synaptic activity are not well understood, but several lines of evidence indicate that FUS and other RNA binding proteins implicated in ALS including TDP-43 regulate the expression of proteins essential for synaptic transmission. FUS enhances the stability of GluA1 mRNA by binding the 3’ UTR to promote AMPA receptor function (Udagawa et al., 2015). Furthermore, mutations in the Drosophila homolog of TDP-43, tbph, result in reduced levels of the synaptic voltage-gated calcium channel, Cacophony (Chang et al., 2013).
Drosophila has been a useful model for investigating the pathogenic mechanisms underlying ALS caused by mutations in FUS (Lanson et al., 2011; Machamer et al., 2014; Wang et al., 2011). Importantly, FUS is functionally conserved in Drosophila, as human FUS expression in neurons fully rescues lethality and locomotor dysfunction associated with null mutations of the Drosophila homolog Caz (Wang et al., 2011). Overexpression of FUS in Drosophila motor neurons alters neuromuscular junctions , impairs motor activity (Chen et al., 2011), and disrupts axonal transport of mitochondria (Chen et al., 2016). However, interpretation of these studies is confounded by the observation that in these models, ALS-causing mutant FUS still predominantly localizes to the nucleus of motor neurons, with only modest mislocalization into the cytoplasm (Lanson et al., 2011; Machamer et al., 2014). The absence of significant mislocalization in these models underscores the conclusion that we and others have previously come to: toxicity of FUS expression in Drosophila motor neurons is largely independent of ALS-causing mutations, but rather, FUS toxicity correlates most strongly with expression level (Lo Piccolo et al., 2017; Machamer et al., 2014). Furthermore, FUS-associated toxicity in Drosophila overexpression models is dependent on the nuclear rather than cytoplasmic localization of FUS (Jäckel et al., 2015), and overexpression of nuclear FUS is sufficient to induce neuronal death in vitro (Suzuki and Matsuoka, 2015). Thus, identifying a population of neurons in a genetically and experimentally tractable model system that have the hallmark mislocalization of mutant FUS seen in patients may help understand the pathogenesis of ALS caused by FUS mutations.
2. Results
In many FUS ALS models, including Drosophila motor neuron overexpression models, disease-associated mutations in the NLS do not result in the loss of FUS from the nucleus (Machamer et al., 2014; Qiu et al., 2014; Sharma et al., 2016a; Zhou et al., 2013). In an attempt to generate a new model of FUS ALS/FTD that recapitulates the pathophysiology seen in patients, we sought to identify a neuronal population in which NLS mutations cause FUS to be lost from the nucleus and mislocalize to the cytoplasm by overexpressing flag(FL)-tagged FUSP525L with the panneuronal C155-GAL4 driver (Lin and Goodman, 1994). We identified neurons in the larval body wall in which FUSP525L severely mislocalizes to the cytoplasm, and based on the multidendritic appearance and body wall localization, we hypothesized that these cells were class IV dendritic arborization (da) neurons. Indeed, FUSWT localizes to the nucleus of da neurons in third instar larvae when overexpressed with the da-specific ppk-GAL4 driver (Figure 1A) (Ainsley et al., 2003; Zhong et al., 2010) , whereas FUSP525L is predominantly mislocalized to the cytoplasm (Figure 1B). Importantly, these transgenic lines were developed using site-directed insertion to eliminate unequal expression due to position effects seen when transgenic lines are generated by random insertion (Wang et al., 2011). Equivalent expression of mRNA and protein has previously been confirmed for these transgenic lines (Machamer et al., 2014). Thus, the mislocalization of FUSP525L to the cytoplasm is a consequence of the P525L mutation in the NLS and not differential expression levels.
Figure 1: ALS causing mutations in FUS and Caz cause cytoplasmic mislocalization in dendritic arborization neurons.
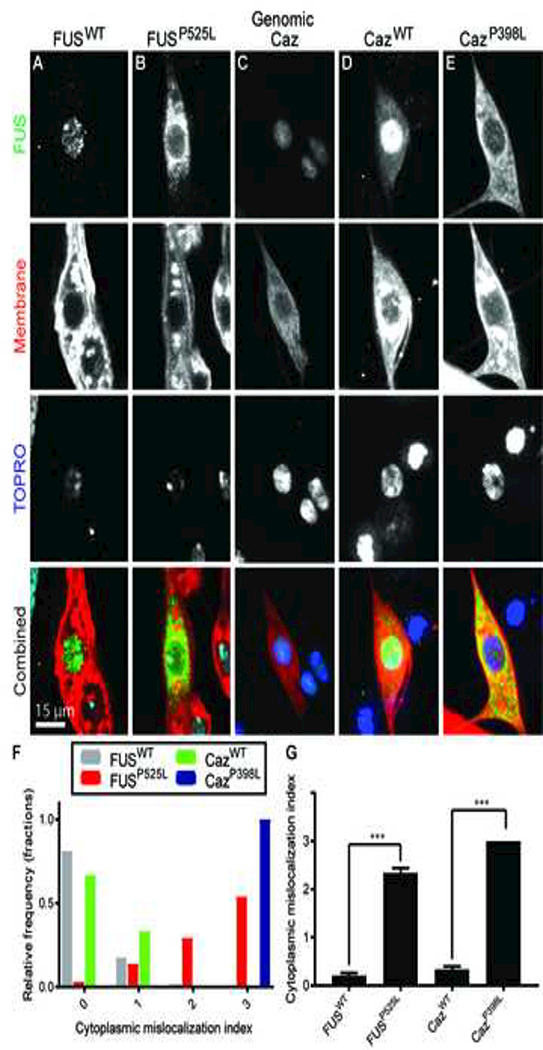
(A) FUSWT or (B) FUSP525L was expressed in da neurons using the ppk-GAL4 driver. Flag (FL)-tagged, wild-type FUS(FUSWT) localizes primarily to the nucleus, whereas, FL-FUSP525L accumulates in the cytoplasm. (C) Endogenous Caz localizes to da neuron nuclei using a transgenic line that expresses FL-Caz under the control of its endogenous enhancer elements. (D) FL-CazWT and (E) FL-CazP398L were expressed using ppk-GAL4, and FL-CazWT localizes primarily to the nucleus whereas FL-CazP398L localizes to the cytoplasm. Transgenic FUS/Caz was visualized with an anti-Flag antibody and neuronal plasma membrane was visualized through transgenic expression of CD8-GFP in (A,B,D, and E) or by staining with anti-HRP antibody (Bashaw, 2010) in (C). The nucleus was visualized with TOPRO staining. (F) The nuclear/cytosplasmic distribution of transgenic FUS/Caz was quantified using the following scale: (0): exclusively nuclear staining, (1): primarily nuclear with faint cytoplasmic staining, (2): strong cytoplasmic staining weaker than nuclear staining, (3): strong cytoplasmic staining greater than nuclear staining and the average values were quantified in (G). N = 60–66 neurons total from 3 animals for each genotype. ***p<0.001. Statistical significance determined by one-way ANOVA with Dunn’s multiple comparison posttest.
Although FUS is believed to be expressed in all neurons (Andersson et al., 2008), we confirmed that Drosophila FUS (Caz) is endogenously expressed in da neurons by staining for animals expressing FL-tagged Caz under the control of endogenous Caz enhancer elements (Wang et al., 2011), and find that Caz is expressed in da neurons and localizes to the nucleus (Figure 1C). Next, we tested if mutations in caz homologous to ALS-causing mutations in FUS can also result in the mislocalization of Caz into the cytoplasm. Transgenic CazWT primarily localizes to the nucleus (Figure 1D), whereas CazP398L primarily localizes to the cytoplasm (Figure 1E). We quantified the extent of mislocalization by scoring the localization of FUS using the following scale: (0): exclusively nuclear staining, (1): primarily nuclear with faint cytoplasmic staining, (2): strong cytoplasmic staining weaker than nuclear staining, and (3): strong cytoplasmic staining greater than nuclear staining (Figure 1F-G). Given the dramatic difference in subcellular localization between wild type and mutant FUS and Caz in da neurons, we reasoned that da neurons are a good model to determine the contribution of mislocalized cytoplasmic FUS to neuronal toxicity caused by ALS-associated mutations.
In addition to nuclear and cytoplasmic localization, mutant FUS has been localized to synapses (Belly et al., 2005; Schoen et al., 2015). Thus, we determined whether mutant FUS in the cytoplasm of da neurons was constrained to the cell soma or if it also localizes to neuronal processes. da neurons are nociceptive sensory neurons whose cell bodies and large dendritic fields are located between the cuticle and epithelial cells lining the body wall (Grueber et al., 2002; Im and Galko, 2012). Their dendritic fields tile into non-overlapping domains that correlate with the sensory fields for each neuron. da neurons extend axonal projections to the ventral ganglion where they form synapses that participate in the pain evoked escape responses (Zhong et al., 2010). We find that FUSP525L and CazP398L localize to these synaptic projections (Figure 2A arrows), whereas FUSWT and CazWT are not detectable at the synapse.
Figure 2: Mutant FUS and Caz are transported to synaptic projections.
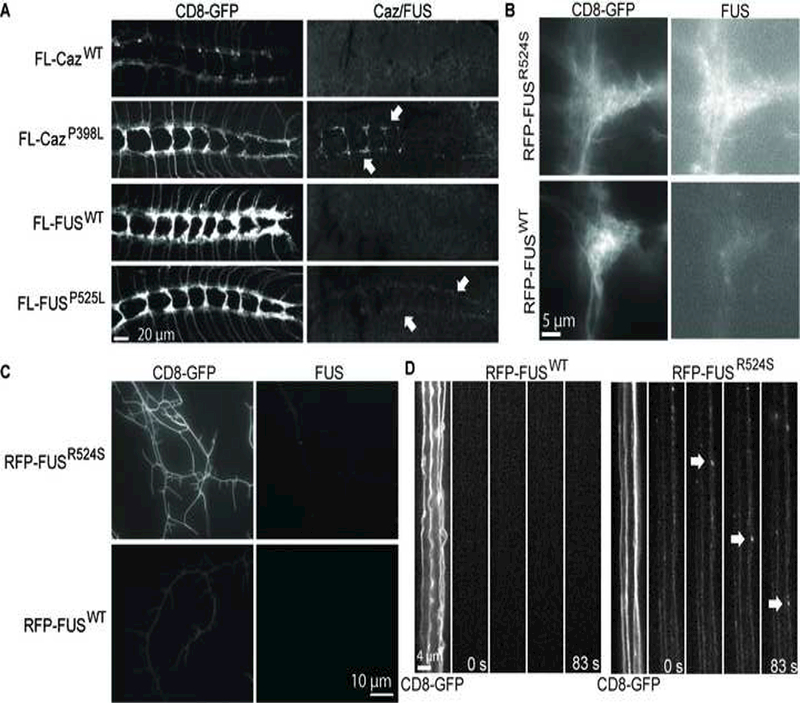
(A) FUSP525L and CazP398L expressed with ppk-GAL4 localize to synaptic projections of da neurons in third instar larvae (arrows), but neither FUSWT nor CazWT are detected within projections. RFP-FUSR524S and RFP-FUSWT were expressed with ppk-GAL4, and RFP-FUSR524S is highly enriched in the synaptic projections of da neurons (B), whereas RFP-FUSWT is barely detectable. RFP-FUSR524S but not RFP-FUSWT localizes to da neuron dendrites (C), and RFP-FUSR524S transports via active axonal transport (D arrows), but RFP-FUSWT is not detectable within axons. The speed of axonal transport was calculated from the average run speed from 5 different vesicles to be 1.45 +/− 0.14 μm/s.
These results suggest that mutant FUS and Caz are actively transported along the axon to the synapse.
To test this, we expressed either RFP-FUSWT or RFP-FUSR524S in da neurons and performed live imaging of the larval body wall. Similar to what is seen with FL-FUSP525L and FL-CazP398L, red fluorescent protein-tagged FUSR524S (RFP-FUSR524S) (Chen et al., 2011) is enriched in synapses (Figure 2B) and dendrites (Figure 2C), whereas wildtype FUS is barely detectable at synaptic projections (Figure 2B) and undetectable in dendrites (Figure 2C). FUSR524S is actively transported to the synaptic projections (Figure 2D and supplemental movie 1) and within dendrites (Supplemental movie 3). The mean run velocity of RFP-FUSR524S undergoing anterograde axonal transport is 1.45 +/− 0.14 μm/s, suggesting a fast axonal transport mechanism. Additionally, we find that similar RFP-FUSR524S punctae, both static and mobile, are localized to synaptic projections (Supplemental movie 2). These data suggest that mutant but not wild type FUS granules are transported to the synapse in da neurons.
To determine the consequences of FUS and Caz overexpression and/or mutant mislocalization on da neuron structure and function, we coexpressed the plasma membrane marker CD8-GFP with FUSWT, FUSP525L, CazWT or CazP398L (Figure 3A) and measured the branching complexity of the large dendritic fields of da neurons using a Sholl analysis (Sholl, 1953) (Figure 3B). Both FUSWT and FUSP525L result in the loss of dendritic branching, but there is no significant difference between FUSWT and FUSP525L. Likewise, the expression of CazWT or CazP398L results a severe reduction in dendritic branching, but no difference is seen between CazWT and CazP398L. These data suggest that FUS-mediated dendrite toxicity in da neurons is independent of the presence of ALS-linked mutations.
Figure 3: Overexpression of FUS and Caz result in simplified dendritic branching.
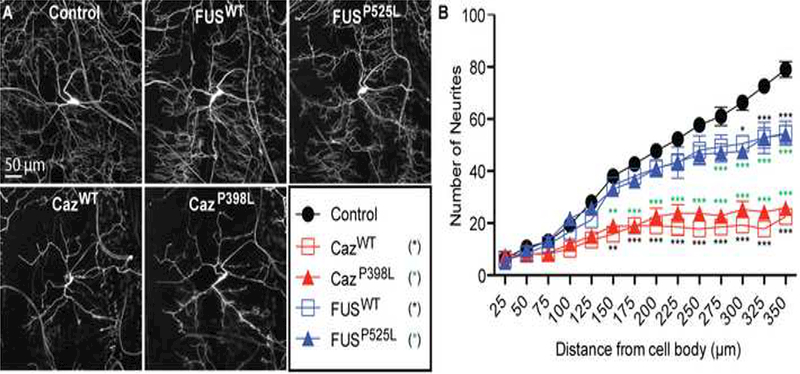
(A-E) Plasma membrane marker CD8-GFP was expressed with FUSWT, FUSP525L, CazWT, CazP398L or alone (control) in da neurons using the ppk-GAL4 driver, and the dendritic branching complexity was measured using Sholl analysis. (F) FUSWT or FUSP525L expression reduces branching complexity of the dendritic network compared to control, with the greatest effect seen on the distal dendritic branches. Expression of CazWT or CazP398L results in a severe loss of dendritic branching complexity. N = 8 animals for each genotype. *p<0.05, **p<0.01, ***p<0.001. Statistical significance determined by two-way ANOVA with Bonferroni multiple comparison posttests.
We next tested whether ALS-linked mutations in FUS and Caz cause degeneration of synaptic terminals by measuring the volume of bilateral segmental synaptic projections within the ventral ganglion defined by the plasma membrane marker CD8-GFP. In control animals, the synaptic projections of da neurons form a ladder-like pattern with fairly uniform fluorescence intensity (Figure 4A). Expression of FUSP525L and FUSWT minimally reduces synaptic projection volume (Figure 4B, C, F) but leads to synaptic mistargeting defects to regions outside of the synaptic region (arrowheads, Figure 4B). In contrast, expression of both CazWT and CazP398L results in severe disruption of the synaptic architecture in addition to a reduction in projection volume, most significantly in projections from distal segments (A6-A8) (Figure 4A, D, E, F). Many of the proximal projections (A2-A4) in animals expressing CazWT, and to a lesser degree CazP398L, are characterized by the complete loss of normal appearing projections and the appearance of clusters of large round swellings (inset arrowheads). To determine if the effects of FUS and Caz expression are due to early developmental defects or represent degeneration after normal early development, we imaged ventral ganglia from larvae at 72, 96, or 120 hours after egg laying (AEL), corresponding to early, mid, and late third instar larval stages. All animals expressing transgenic FUS and Caz had largely normal synaptic projections at 72 hours AEL, but display a progressive loss of fluorescence at 96 and 120 hours (Supplemental Figure 1). This phenotype is most severe for CazWT and CazP398L expressing animals, which display significant fragmentation of synaptic projections. These data indicate that overexpression of FUS or Caz leads to a progressive loss of both axonal and dendritic neuronal projections.
Figure 4: Overexpression of FUS and Caz result in the loss of synaptic projections.
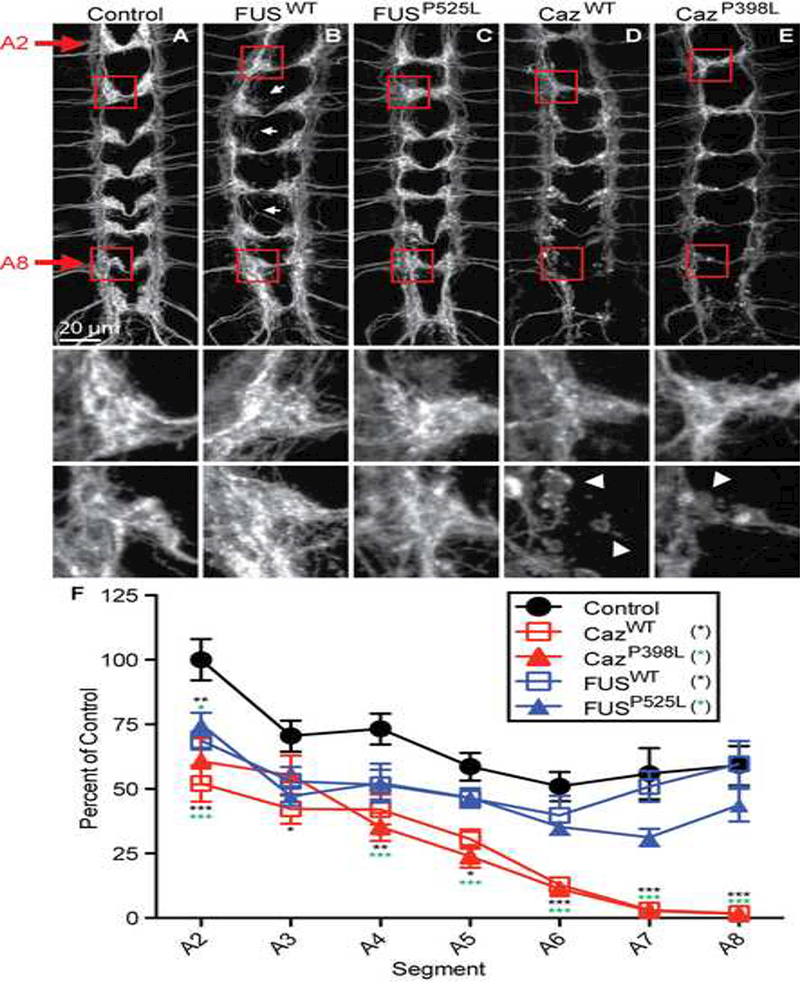
(A) Plasma membrane marker CD8-GFP (A) was coexpressed with (B) FUSWT, (C) FUSP525L, (D) CazWT, or (E) CazP398L with ppk-GAL4, and the synaptic projections of da neurons from the abdominal segments A2-A8 in third instar larvae were imaged. Middle panels are enlargements of proximal segment A2, whereas lower panels are enlargements of distal segment A8. Arrows show aberrant projections. (F) The size of the synaptic projections of da neurons from abdominal segments A2-A8 in third instar larvae were measured using volumetric analysis with Imaris. CazWT or CazP398L expression results in a severe loss of synaptic projections. N = 8 animals for each genotype. *p<0.05, **p<0.01, ***p<0.001. Statistical significance determined by two-way ANOVA with Bonferroni multiple comparison posttests.
To investigate the mechanisms underlying the loss of dendritic arbors and synaptic projections of da neurons, we examined the microtubule cytoskeleton by coexpressing tubulin-GFP with FUSWT, FUSP525L, CazWT or CazP398L. In control animals, the intensity of tubulin-GFP appears mostly uniform throughout distal axons and synaptic termini with periodic punctae (Figure 5A, inset arrow). We find that the microtubule cytoskeleton in FUS-expressing neurons appears largely normal within the terminal projections, though the projections display a less uniform morphology than controls (Figure 5A-C). In contrast, in CazWT and CazP398L expressing neurons, large accumulations of tubulin-GFP are seen in many synaptic termini, with some displaying intense tubulin-GFP signal on the periphery (Figure 5D-F), likely representing microtubule loops known to form within synaptic swellings (Roos et al., 2000). These morphological abnormalities are consistent with the large bulbous swellings seen when Caz is coexpressed with CD8-GFP (Figure 4D-E, arrows). These findings suggest that Caz overexpression disrupts the synaptic terminal cytoskeleton independent of its subcellular localization.
Figure 5: FUS overexpression disrupts the synaptic microtubule cytoskeleton.
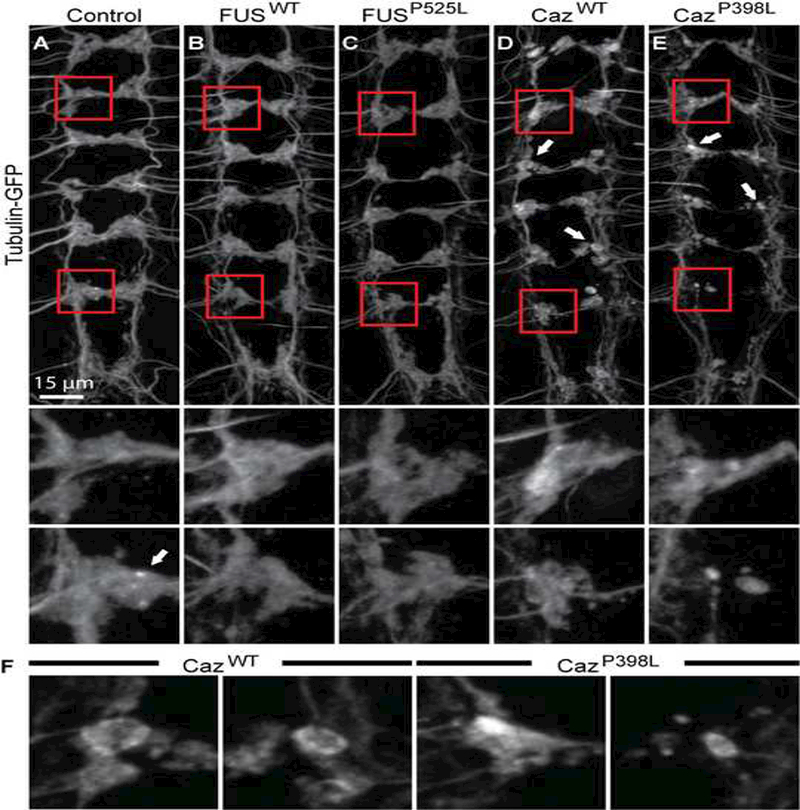
(A) Tubulin-GFP was expressed either alone (A) control, or with (B)FUSWT, (C) FUSP525L, (D) CazWT, or (E) CazP398L using ppk-GAL4 and the synaptic projections of da neurons in third instar larvae were imaged. Middle panels show enlargements of proximal (A2) synaptic projections, and lower panels show distal (A8) projections. In control, FUSWT, and FUSP525L expressing neurons, tubulin-GFP appears homogenously distributed within the projections, with sporadic puncta present (A, arrow). In animals expressing CazWT or CazP398L, tubulin-GFP is abnormally distributed, with instances of locally intense staining around the periphery of round varicosities (D,E arrows). (F) Magnification of projections indicated by arrows in D and E.
FUS expression has previously been reported to disrupt axonal transport in motor neurons (Chen et al., 2016). The observation that projections from neurons located in distal segments expressing CazWT and CazP398L display more significant morphological alterations than neurons located in proximal segments (Figure 4D-F) and the severe disruption of microtubule cytoskeletons in synaptic projections suggests transgenic FUS expression may also impair axonal transport in da neurons. To determine if the cytoplasmic mislocalization of FUSP525L or CazP398L disrupts axonal transport, we measured the number of “axonal jams” (Lloyd et al., 2012) in da neurons by staining for the synaptic vesicle protein Cysteine string protein (CSP) (Rozas et al., 2012) in animals coexpressing CD8-GFP and FUSWT, FUSP525L, CazWT, or CazP398L. We find that overexpressing CazP398L, but not CazWT, FUSWT or FUSP525L results in the consistent formation of large axonal swellings containing CSP (Figure 6A, B). As FUSP525L and CazP398L undergo aberrant axonal transport, we determined if CazP398L was a component of these axonal jams by coexpressing fluorescently-tagged Synaptotagmin (Syt-GFP) with FUSWT, FUSP525L, CazWT, or CazP398L in da neurons (Figure 6C). We find CazP398L localizes to axonal jams containing Syt-GFP, but no such colocalization is seen with FUSWT, FUSP525L, or CazWT. Together, these data suggest that CazP398L, but not CazWT, disrupts axonal transport of synaptic vesicles.
Figure 6: Mutant FUS disrupts transport of synaptic vesicle proteins to synaptic projections.
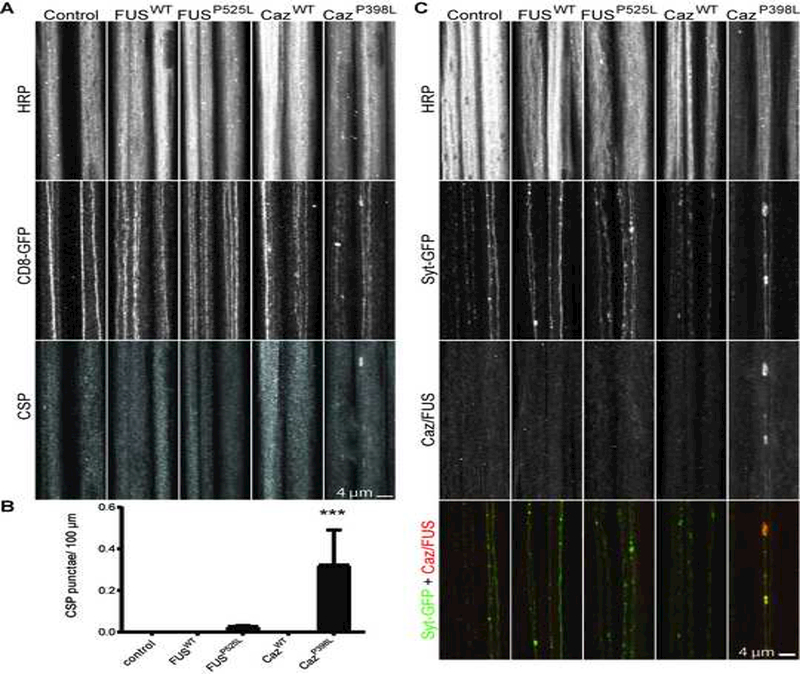
(A) Cysteine string protein (CSP) was stained for segmental nerves of third instar larvae expressing CD8-GFP alone (control), or coexpressed with FUSWT, FUSP525L, CazWT, or CazP398L using ppk-GAL4. The number of axonal swellings containing CSP aggregations was measured (B). CazP398L display significant CSP accumulations in segmental nerves consistent with axonal jams, whereas CazWT, FUSWT, and FUSP525L do not display axonal swellings with CSP accumulations. N = 5–6 animals for each genotype. (C) Syt-GFP was expressed alone (control) or with FUSWT, FUSP525L, CazWT, or CazP398L using ppk-GAL4. Transgenic FUS and Caz were visualized in segmental nerves by staining for the FL epitope tag. CazP398L accumulates with Syt-GFP in axonal swellings, but no swellings are present in FUSWT, FUSP525L, or CazWT expressing animals. ***p<0.001 Statistical significance determined by one-way ANOVA with Bonferroni multiple comparison posttests.
We next measured synaptic Syt-GFP levels in animals coexpressing FUSWT, FUSP525L, CazWT or CazP398L (Figure 7A, B), and we observe a large reduction in Syt in the synapses of da neurons overexpressing either CazWT or CazP398L. Interestingly, we find that the expression of FUSP525L but not FUSWT results in a significant reduction of Syt in da neuron synapses (Figure 7A, B), suggesting that FUSP525L disrupts the delivery of Syt to synapses. These data indicate that ALS-causing mutations in FUS and Caz impair the transport of synaptic vesicle proteins. Additionally, to determine if ALS-causing mutations in FUS also impair the transport of other components of the synapse essential for neuronal function, we coexpressed GFP targeted to mitochondria (mito-GFP) with FUSWT, FUSP525L, CazWT or CazP398L and found that overexpression of CazWT or CazP398L, and to a lesser extent, FUSWT or FUSP525L resulted in a reduction in synaptic mitochondria (Figure 7C, D). Alterations in mitochondria are not restricted to the synaptic terminals as larger mitochondria and/or collections of mitochondria are observed in the axons of animals expressing FUSWT, FUSP525L, CazWT or CazP398L (Supplemental Figure 2). Synaptotagmin is the calcium sensor responsible for synaptic vesicle fusion (Huang et al., 2011), and synaptic mitochondria provide energy for normal synaptic transmission and buffering internal Ca2+ (Vos et al., 2010), suggesting that synaptic transmission may be altered in FUS/Caz-expressing da neurons.
Figure 7: FUS reduces the level of synaptic Synaptotagmin and mitochondria.
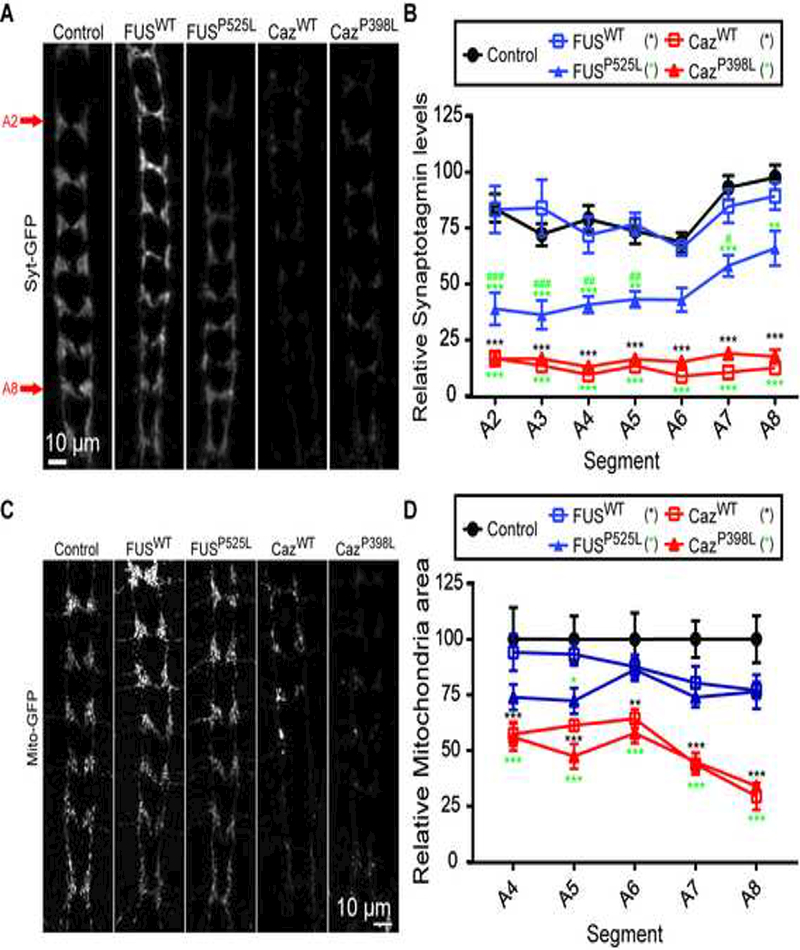
(A) Syt-GFP was expressed alone in controls and with FUSWT, FUSP525L, CazWT, or CazP398L using the ppk-GAL4 driver and the amount of Syt-GFP was quantified in synaptic projections of da neurons in third instar larvae using Imaris volumetric analysis (B). Expression of FUSP525L results in the loss of Syt-GFP from synaptic projections, but expression of FUSWT does not. Expression of either CazWT or CazP398L results in a severe loss of Syt-GFP. N = 4. (C) Mito-GFP was expressed alone (controls) or with FUSWT, FUSP525L, CazWT, or CazP398L using the ppk-GAL4 and the mitochondria in synaptic projections of third instar larvae da neurons were imaged. Expression of either CazWT or CazP398L and to a lesser extent FUSWT or FUSP525L reduces synaptic terminal mitochondria . N = 4–6 animals for each genotype. *p<0.05, **p<0.01, ***p<0.001 vs control. #p<0.05, ##p<0.01, ###p<0.001 vs FUSWT overexpression. Statistical significance determined by two-way ANOVA with Bonferroni multiple comparison posttests.
To determine if FUS/Caz expression in da neurons alters synaptic activity, we coexpressed the genetically-encoded calcium sensor GCAMP5 (myr-GCAMP5) (Akerboom et al., 2012) and RFP (CD4-tdTomato) with FUSWT, FUSP525L, CazWT, or CazP398L in da neurons and performed time-lapse imaging of the synaptic projections from segment A5 or A6 to measure changes in local calcium concentration (Figure 8A). Expression of FUSWT, FUSP525L, CazWT, or CazP398L results in large calcium transients not seen in control animals (Figure 8B, D). Local increases in calcium concentration were restrained to subdomains of the synaptic projections and their associated axons (Supplemental Figure 3, arrow), indicating that these transients are likely mediated by voltage-gated calcium channel activation by action potentials formed in the soma or axon initial segment. Additionally, expression of CazP398L, FUSWT or FUSP525L but not CazWT results in a significant increase in the frequency of observed calcium transients in synaptic projections (Figure 8C). We next determined if transgenic expression increased the average Ca2+ concentration within the terminal by measuring the average GCAMP and RFP fluorescence intensity (Figure 8E). We normalized the GCAMP fluorescence to RFP fluorescence to control for any differences in GCAMP expression. We find no significant difference between the average GCAMP fluorescence between the control and animals expressing CazWT, CazP398L, FUSWT or FUSP525L. Together, these data indicate that FUS/Caz expression increases the frequency and/or amplitude of presynaptic calcium transients without changing baseline calcium levels.
Figure 8: FUS alters the frequency and amplitude of synaptic calcium transients.
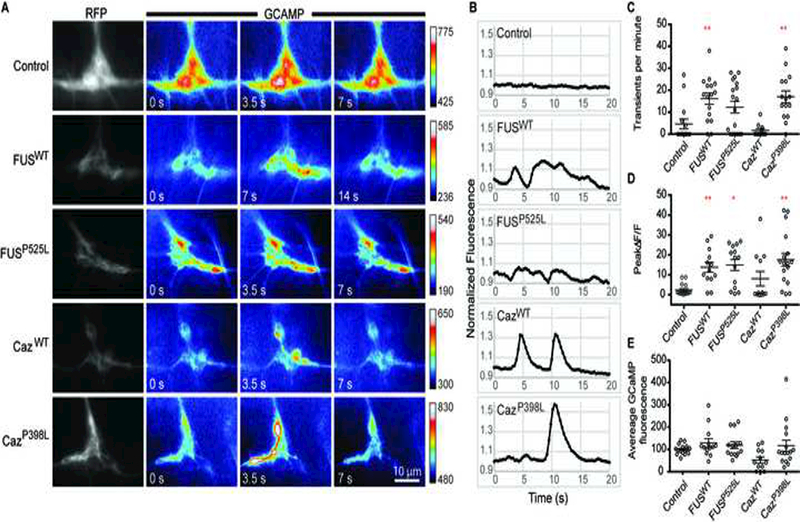
mryGCAMP5 and CD4-tdTomato were expressed in control animals or with FUSWT, FUSP525L, CazWT, or CazP398L using ppk-GAL4, and Ca2+ transients were recorded from single hemisegment projection fields of da neurons from A5 or A6 in third instar larvae (A). Color scales for each image sequence are indicated to the right of the sequence. (B) Representative changes in normalized fluorescence localized to a 2 μm ROI during a 20 s period. Control animals display small fluctuations of GCAMP intensity over the imaging period, whereas large transients are seen in FUSWT, FUSP525L, CazWT, or CazP398L expressing neurons. (C) The frequency of Ca2+ transients was determined by counting the number of GCAMP transients over a 60 second period in projection fields corresponding to hemisegment A5 or A6. Synaptic projections from animals expressing FUSWT or CazP398L have a higher frequency of Ca2+ transients than controls. N = 11–18 animals for each genotype. (D) Fluorescence was measure for a single 2 μm diameter ROI and the largest ΔF/F of a continuous increase in fluorescence over a 105 second period was calculated. Synaptic projections from animals expressing FUSWT, FUSP525L, or CazP398L have significantly larger Ca2+ transients than controls. N = 12–16 animals for each genotype. (E) Average normalized GCAMP fluorescence was calculated by dividing GCAMP fluorescence by RFP fluorescence of synaptic projections calculated from 5–10 images per synapse. Background fluorescence intensity was measured and subtracted from signal intensity for both channels before normalization. N = 12–16 animals for each genotype. *p<0.05, **p<0.01. Statistical significance determined by one-way ANOVA with Bonferroni multiple comparison posttests.
3. Discussion
The majority of ALS and FTD cases show a common feature: the loss of the soluble form of one or more RNA binding proteins (i.e. TDP-43, FUS, hnRNP2A/B, or hnRNP1) from the nucleus and the appearance of aggregates of these proteins in the cytoplasm (Kapeli et al., 2017) . Although in vitro models of FUS-ALS have recapitulated robust mislocalization of FUS to the cytoplasm (Ichiyanagi et al., 2016; Scaramuzzino et al., 2013), in many in vivo models, mutant FUS has failed to significantly mislocalize to the cytoplasm (Huang et al., 2011; Sharma et al., 2016b; Verbeeck et al., 2012). Here, we describe a Drosophila model of FUS-mediated neurodegeneration that reproduces the pathogenic changes in FUS localization seen in the neurons of ALS patients. Drosophila da neurons are cholinergic sensory neurons that have stereotyped development of neuronal projections that have been extensively used to study neuronal development and function (Jan and Jan, 2010) in addition to neurodegeneration (Lee et al., 2011), due to their morphological complexity, experimental accessibility and genetic tractability.
By expressing wild-type or mutant forms of FUS in da neurons, we identified changes in neuronal physiology and function that were mutation-specific as well as those that are mutation-independent. FUS has previously been implicated in modulating synaptic function, but its mechanism of action in this regard is not known (Schoen et al., 2015; Sephton et al., 2014). In our study, mutations in the NLS of FUS and Caz cause their mislocalization to the cytoplasm and transport to synaptic terminals. As a consequence, mutant FUS/Caz impairs axonal transport of synaptic vesicle proteins, thus reducing the level of Synaptotagmin, a key component of the presynaptic release machinery. Disruption of axonal transport has been identified in multiple models of ALS and other neurodegenerative diseases, but whether it’s a causative factor, a participating pathogenic mechanism, or a consequence of neurodegeneration remains unclear (Baldwin et al., 2016; Bilsland et al., 2010; De Vos et al., 2008; Marinkovic et al., 2012). Overexpression of FUS has previously been shown to disrupt transport of mitochondria in Drosophila motor neurons (Chen et al., 2016), with the most severe disruption seen in mutant FUS-expressing animals. Similarly, we see a loss of synaptic mitochondria in da neurons expressing FUS/Caz.
FUS could be impairing synaptic transmission through several mechanisms. Increased transport of FUS into neuronal processes may alter the local concentration of synaptic transcripts and thus disrupt local translation, or increased concentrations of FUS in neuronal process may induce the formation of stress granules, which could alter local translation or sequester proteins essential for normal function.
Alternatively, the changes in synaptic physiology could be downstream of changes in the cell body and dendrites. Axonal hyperexcitability is observed in ALS patients (Bostock et al., 1995; Kanai et al., 2006; Vucic and Kiernan, 2006), and neuronal hyperactivity in ALS patients correlates with disease severity (Kanai et al., 2006). FUS may increase the excitability of neurons by altering the concentration and content of channels required for proper neuronal physiology within the axon or cell body. This is supported by our findings that da neurons overexpressing CazP398L but not CazWT are hyperexcitable as measured by calcium transient frequency.
Although we propose that a genetic model that incorporates ALS-causing mutations is likely to be the most faithful to disease pathogenesis, the effects of wild type FUS/Caz overexpression are likely also relevant. Indeed, rare cases of ALS are caused by increased FUS expression as a result of mutations in 3’ untranslated regulatory elements resulting in aberrant FUS aggregation and stress granule formation (Sabatelli et al., 2013). In our studies, the most striking consequence of FUS/Caz overexpression was the appearance of calcium transients within the axons and synaptic termini of da neurons. Loss of the Drosophila homolog of TDP-43, tbph, reduces the voltage-gated calcium channel cacophony in motor neurons (Chang et al., 2013) suggesting that FUS expression may also regulate Ca2+ levels by controlling calcium channel expression. Alternatively, mitochondria are important for buffering synaptic Ca2+, and the loss of synaptic mitochondria function results in calcium transients of significantly larger amplitudes (Xing and Wu, 2018). Consistent with these studies, Caz expression results in both the loss of synaptic mitochondria and the appearance of large calcium transients. Ca2+ microdomains can regulate calcium sensitive kinases and phosphatases that control cytoskeleton dynamics in growth cones and mature presynaptic and postsynaptic compartments (Bodaleo and Gonzalez-Billault, 2016; Gasperini et al., 2017; Merriam et al., 2013). The loss of synaptic complexity and altered microtubule dynamics suggest a pathological cascade in which FUS expression impairs the function of mitochondria, reducing presynaptic Ca2+ buffering and altering cytoskeletal dynamics leading to disrupted synaptic morphology. It’s intriguing that both nuclear and cytoplasmic FUS/Caz lead to similar degenerative phenotypes, and we postulate that this is due to FUS-mediated sequestration of critical proteins or RNAs that shuttle with FUS through the nuclear pore.
We believe that da neurons can be used to identify important changes to neuronal physiology that are disrupted by FUS mislocalization into the cytoplasm and synapses. da neurons have discrete subcellular compartments, and cell bodies and dendrites are localized in the body wall whereas synapses are localized to the ventral ganglion. These properties make da neurons amenable to novel compartment-specific imaging and biochemical experiments. However, as with any model, there are limitations, such as the fact that da neurons are sensory neurons rather than motor neurons and any larval model is unable to distinguish developmental defects from those resulting from neurodegeneration. Nonethtelss, given their imaging accessibility and genetic tractability, this model is amenable to further studies to identify mechanisms of altered neuronal cell biology and physiology.
4. Experimental Procedure
4.1. Fly Genetics and rearing.
Flies were raised at 25 °C on yeast supplemented standard molasses-based food. Experiments utilized the following transgenes from the indicated stock lines or sources: ppk-GAL4 (BL 32078), UAS-CD8-GFP (BL 31290), UAS-Syt-GFP (BL 6925), UAS-mitoGFP (BL8442), UAS-CD4-tdTomato (BL 35837), UAS-myrGCAMP5 (Mark Wu), UAS-FL-CazWT, UAS-FL-CazP398L, UAS-FL-FUSWT, UAS-FL-FUSP525L (Brian McCabe), UAS-FUSWT-RFP, UAS-FUSP525L-RFP (Jane Wu). Unless otherwise stated, the genetic background of the various transgenes, w1118 (BL 5905) was used for controls.
4.2. Immunohistochemistry
Immunohistochemistry experiments followed previously published protocols (Brent et al., 2009) with a few changes. Briefly, wandering 3rd instar larvae were cut and pinned open in 0 mM Ca2+ HL3, but no internal organs were removed. Animals were immediately fixed in 4% PFA in PBS for 20 minutes. Larvae were then rinsed with PBS 3 times over 5 minutes. While in PBS, the internal organs of the larvae were removed, with the exception of the CNS, segmental nerves, and body wall musculature. Animals were incubated in block solution (PBS, 0.1% Triton-X-100, 5% fetal goat serum) for 30 minutes to 1 hr with rotation at room temperature. Larvae were then incubated with antibodies at the indicated concentration in block solution for 2 hr with rotation at room temperature or overnight at 4 °C. Antibody solution was quickly rinsed off 3 times with PBS-T (PBS, 0.1% Triton-X-100), and then washed with rotation 3 times for 20 minutes each wash. Samples were then incubated in secondary antibody solution at the indicated concentration in block with rotation for 2 hours at room temperature or overnight at 4 °C. Samples were rinsed 3 times with PBS-T and then washed 3 times with rotation for 20 minutes each wash. Samples were prepared on microscope slides using vectashield as a mountant and sealed with nail polish. Antibodies were used at the following concentrations: mouse anti-GFP (Molecular Probes, 1:250), mouse anti-Flag (Sigma: M2, 1:250), goat anti-HRP-Dylight 549 (Jackson Immunoresearch, 1:500), mouse anti-CSP (DSHB, 1:250). Secondary antibodies used were goat anti-mouse Alexafluor 488 (Jackson Immunoresearch, 1:250) and goat anti-mouse Alexafluor 568 (Jackson Immunoresearch, 1:250).
4.3. Confocal microscopy and image analyses.
Imaging of endogenous fluorescence and antibody staining of fixed samples was performed on either a Zeiss LSM 510 or LSM 800 laser scanning confocal microscope using a 20x, 40x, or 63x objective. Sholl analysis of da neuron dendritic branching was performed on animals expressing CD8-GFP under the control of the ppk-GAL4 driver. ImageJ was used to create a mask of concentric circles with radii of 25 to 350 μm increasing in 25 μm increments around the centroid of da type IV neuron cell bodies from segment A4. The number of dendritic branches crossing each concentric circle was counted from 4 animals for the stated genotypes. For quantification of the volume of da neuron synaptic projections, Imaris was used to segment voxels based on signal intensity into three-dimensional surfaces (surface creation tool). For many animals, contiguous volumes were formed consisting of projections from multiple segments. In such cases, projection volumes of neighboring segments were separated using the surface cutting tool. The total volume of projections to each segmental target was then quantified. For Syt quantification, the level of Syt was determined by measuring the total fluorescence within the synaptic projections using surface segmentation. To quantify mitochondria loss from synaptic projections, total mitochondria area was calculated in imageJ by measuring the ROI area produced from thresholding mito-GFP fluorescence of maximum projections images.
4.4. Active transport of FUS.
To characterize the transport of FUS in DA IV neurons, we expressed RFP-FUSWT and RFP-FUSR524S using the ppk-GAL4 driver. Wandering third instar larvae were dissected in 0 mM Ca2+ HL3 on a sylgard cube. The cube was inverted onto a WillCo 35 mM glass bottom dish and excess HL3 was wicked away with a kimwipe. FUS transport was imaged in medial segmental projections (A4-A6) and the corresponding segmental nerves using a Zeiss Axioscope. For axonal transport, images were captured every 2.7 s. The velocity of transporting vesicles was determined in imagej by measuring the average velocity of vesicles during transport runs uninterrupted by pauses or reversals. Values are given in velocity +/− SEM.
4.5. Calcium imaging.
Third instar larvae expressing myrGCAMP5 with the ppk-GAL4 driver were dissected on a sylgard imaging cube in 1.0 mM Ca2+ HL3. The cube was inverted onto a WillCo 35 mM glass bottom dish and imaged using a Zeiss Axioscope inverted epifluorescent microscope. GCAMP fluorescence from presynaptic terminals of da neurons localized to segment A5 or A6 was collected under LED illumination. For representative time-lapse movies and images used for the determination of maximum Ca2+ transient amplitude, RFP (CD4-tdTomato) and GCAMP images were acquired at ~0.75 frames per second.
Measurement of GCAMP transient frequency and transient dynamics were per were performed on time-lapse images sequences acquired at ~3.3 frames per second for 60 s. All images were analyzed in ImageJ. To determine the maximum transient amplitude and to measure transient dynamics, GCAMP fluorescence was measured in an ROI of 2 um diameter placed over synaptic areas that demonstrated large changes in intensity. Transient frequency was counted from a single hemisegment synaptic projection by eye. To compare the relative average calcium concentration between the given genotypes, 5–10 images were acquired of individual hemisegment projections in animals coexpressing myrGCAMP5 and CD4-tdTomato. These images were averaged, background fluorescence was measured from nonsynaptic area and subtracted from each channel. GCAMP fluorescence values were normalized expression level by dividing by RFP fluorescence values. Values calculated from individual animals were normalized to the average of the control values and expressed as percent of control.
4.6. Statistical Analysis.
For figure 1, a one-way ANOVA with Dunn’s multiple comparison posttest was used to determine statistical significance. For figures 3, 4, and 7, two-way ANOVAs with Bonferroni multiple comparison post-tests were used to determine statistical significance. For figure 6 and 8, one-way ANOVAs with Bonferroni multiple comparison posttests were used to determine statistical significance. For all figure except figure 1, the following pairs were tested: control and FL-FUSWT; control and FL-FUSP525L; control and FL-CazWT; control and FL-CazP398L; FL-FUSWT and FL-FUSP525L; FL-CazWT and FL-CazP398L. In two-way ANOVA’s *’s refer to statistical significance versus control and #’s refer to statistical significance versus wildtype overexpression. ***p<0.001, **p<0.01, *p<0.05.
Supplementary Material
Highlights.
A Drosophila model of ALS in cholinergic neurons is described
ALS-mutant FUS and Cabeza are mislocalized to the cytoplasm in da neurons
FUS overexpression impairs axonal transport and alters synaptic structure
FUS overexpression increases amplitude and frequency of calcium transients
Acknowledgements
We thank Brian McCabe, Jane Wu, Mark Wu, and the Bloomington Stock Center for Drosophila reagents. We thank Michele Pucak, and the NINDS Multi-photon Core Facility (MH084020) at JHMI for imaging services.
Funding Sources
T.E.L. is supported by NIH (NINDS R01 NS082563 and NS094239), ALSA, Target ALS, and the Robert Packard Center for ALS. The authors declare no conflicts of interests.
Abbreviations:
- da
dendritic arborization
- CSP
Cysteine String Protein
- Caz
Cabeza
- FL
Flag
- Syt
Synaptotagmin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ainsley JA, Pettus JM, Bosenko D, Gerstein CE, Zinkevich N, Anderson MG, Adams CM, Welsh MJ, Johnson WA, Marder E, Bucher D, Buschges A, Manira AE, Adams CM, Anderson MG, Motto DG, Price MP, Johnson WA, Welsh MJ, Vervoort M, Merritt DJ, Ghysen A, Dambly-Chaudière C, Grueber WB, Graubard K, Truman JW, Grueber WB, Truman JW, Liu L, Yermolaieva O, Johnson WA, Abboud FM, Welsh MJ, Tracey WD, Wilson RI, Laurent G, Benzer S, Grueber WB, Jan LY, Jan YN, Goodman MB, Schwarz EM, Askwith CC, Cheng C, Ikuma M, Benson C, Price MP, Welsh MJ, Lingueglia E, Champigny G, Lazdunski M, Barbry P, Liu L, Leonard AS, Motto DG, Feller MA, Price MP, Johnson WA, Welsh MJ, Waldmann R, Lazdunski M, Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E, Hoshi T, John H, et al. , Grueber WB, Ye B, Moore AW, Jan LY, Jan YN, Ashburner M, Misra S, Roote J, Lewis SE, Blazej R, Davis T, Doyle C, Galle R, George R, Harris N, et al. , Kernan M, Cowan D, Zuker C, Heimbeck G, Bugno V, Gendre N, Haberlin C, Stocker RF, Wang JW, Sylwester AW, Reed D, Wu D-AJ, Soll DR, Wu C-F, Wang JW, Soll DR, Wu C-F, Hannon GJ, Kalidas S, Smith DP, Hong K, Mano I, Driscoll M, Berrigan D, Pepin DJ, Marder E, Calabrese RL, Pearson KG, Friesen WO, Cang J, Martin J-R, Raabe T, Heisenberg M, Strauss R, Varnam CJ, Strauss R, Belle JS, Sokolowski MB, Anderson MG, Chittick P, Perkins GL, Shrigley RJ, Johnson WA, Sharma Y, Cheung U, Larsen EW, Eberl DF, Gloor G, Engels W, Certel SJ, Clyne PJ, Carlson JR, Johnson WA, 2003. Enhanced Locomotion Caused by Loss of the Drosophila DEG/ENaC Protein Pickpocket1. Curr. Biol 13, 1557–1563. 10.1016/S0960-9822(03)00596-7 [DOI] [PubMed] [Google Scholar]
- Akerboom J, Chen T-W, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderon NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S, Shigetomi E, Khakh BS, Baier H, Lagnado L, Wang SS-H, Bargmann CI, Kimmel BE, Jayaraman V, Svoboda K, Kim DS, Schreiter ER, Looger LL, 2012. Optimization of a GCaMP Calcium Indicator for Neural Activity Imaging. J. Neurosci 32, 13819–13840. 10.1523/JNEUROSCI.2601-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, Van Den Berg LH, 2012. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 124, 339–352. 10.1007/s00401-012-1022-4 [DOI] [PubMed] [Google Scholar]
- Andersson MK, Ståhlberg A, Arvidsson Y, Olofsson A, Semb H, Stenman G, Nilsson O, Aman P, 2008. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 9, 37 10.1186/1471-2121-9-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong GAB, Drapeau P, 2013. Loss and gain of FUS function impair neuromuscular synaptic transmission in a genetic model of ALS. Hum. Mol. Genet 22, 4282–4292. 10.1093/hmg/ddt278 [DOI] [PubMed] [Google Scholar]
- Baldwin KR, Godena VK, Hewitt VL, Whitworth AJ, 2016. Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum. Mol. Genet 25, ddw105 10.1093/hmg/ddw105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashaw GJ, 2010. Visualizing Axons in the Drosophila Central Nervous System Using Immunohistochemistry and Immunofluorescence. Cold Spring Harb. Protoc. 2010, pdb.prot5503-prot5503. 10.1101/pdb.prot5503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belly A, Moreau-Gachelin F, Sadoul R, Goldberg Y, 2005. Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: Exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci. Lett 379, 152–157. 10.1016/j.neulet.2004.12.071 [DOI] [PubMed] [Google Scholar]
- Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G, 2010. Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci 107, 20523–20528. 10.1073/pnas.1006869107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodaleo FJ, Gonzalez-Billault C, 2016. The Presynaptic Microtubule Cytoskeleton in Physiological and Pathological Conditions: Lessons from Drosophila Fragile X Syndrome and Hereditary Spastic Paraplegias. Front. Mol. Neurosci 9, 1–16. 10.3389/fnmol.2016.00060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostock H, Sharief MK, Reid G, Murray NM, 1995. Axonal ion channel dysfunction in amyotrophic lateral sclerosis. Brain 217–25. [DOI] [PubMed] [Google Scholar]
- Brent J, Werner K, McCabe BD, 2009. Drosophila larval NMJ immunohistochemistry. J. Vis. Exp 10.3791/1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J-C, Hazelett DJ, Stewart JA, Morton DB, 2013. Motor neuron expression of the voltage-gated calcium channel cacophony restores locomotion defects in a Drosophila, TDP-43 loss of function model of ALS. Brain Res. 10.1016/j.brainres.2013.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Deng J, Wang P, Yang M, Chen X, Zhu L, Liu J, Lu B, Shen Y, Fushimi K, Xu Q, Wu JY, 2016. PINK1 and Parkin are genetic modifiers for FUS-induced neurodegeneration. Hum. Mol. Genet 25, 5059–5068. 10.1093/hmg/ddw310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yang M, Deng J, Chen X, Ye Y, Zhu L, Liu J, Ye H, Shen Y, Li Y, Rao EJ, Fushimi K, Zhou X, Bigio EH, Mesulam M, Xu Q, Wu JY, 2011. Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein Cell 2, 477–486. 10.1007/s13238-011-1065-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CCJ, 2008. Role of Axonal Transport in Neurodegenerative Diseases. Annu. Rev. Neurosci 31, 151–173. 10.1146/annurev.neuro.31.061307.090711 [DOI] [PubMed] [Google Scholar]
- Gasperini RJ, Pavez M, Thompson AC, Mitchell CB, Hardy H, Young KM, Chilton JK, Foa L, 2017. How does calcium interact with the cytoskeleton to regulate growth cone motility during axon pathfinding? Mol. Cell. Neurosci 84, 29–35. 10.1016/j.mcn.2017.07.006 [DOI] [PubMed] [Google Scholar]
- Grueber WB, Jan LY, Jan YN, 2002. Tiling of the Drosophila epidermis by multidendritic sensory neurons. Development 129, 2867–78. [DOI] [PubMed] [Google Scholar]
- Hicks GG, Singh N, Nashabi A, Mai S, Bozek G, Klewes L, Arapovic D, White EK, Koury MJ, Oltz EM, Van Kaer L, Ruley HE, 2000. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat. Genet 24, 175–179. 10.1038/72842 [DOI] [PubMed] [Google Scholar]
- Huang C, Zhou H, Tong J, Chen H, Liu Y-J, Wang D, Wei X, Xia X-G, 2011. FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. PLoS Genet. 7, e1002011 10.1371/journal.pgen.1002011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiyanagi N, Fujimori K, Yano M, Ishihara-Fujisaki C, Sone T, Akiyama T, Okada Y, Akamatsu W, Matsumoto T, Ishikawa M, Nishimoto Y, Ishihara Y, Sakuma T, Yamamoto T, Tsuiji H, Suzuki N, Warita H, Aoki M, Okano H, 2016. Establishment of in Vitro FUS-Associated Familial Amyotrophic Lateral Sclerosis Model Using Human Induced Pluripotent Stem Cells. Stem Cell Reports 6, 496–510. 10.1016/j.stemcr.2016.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SH, Galko MJ, 2012. Pokes, sunburn, and hot sauce: Drosophila as an emerging model for the biology of nociception. Dev. Dyn. 241, 16–26. 10.1002/dvdy.22737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N, 2010. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann. Neurol 69, 152–62. 10.1002/ana.22246 [DOI] [PubMed] [Google Scholar]
- Jäckel S, Summerer AK, Thömmes CM, Pan X, Voigt A, Schulz JB, Rasse TM, Dormann D, Haass C, Kahle PJ, 2015. Nuclear import factor transportin and arginine methyltransferase 1 modify FUS neurotoxicity in Drosophila. Neurobiol. Dis 74, 76–88. 10.1016/j.nbd.2014.11.003 [DOI] [PubMed] [Google Scholar]
- Jan Y-N, Jan LY, 2010. Branching out: mechanismsof dendritic arborization. Nat. Rev. Neurosci 11, 316–328. 10.1038/nrn2836.Branching [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai K, Kuwabara S, Misawa S, Tamura N, Ogawara K, Nakata M, Sawai S, Hattori T, Bostock H, 2006. Altered axonal excitability properties in amyotrophic lateral sclerosis: impaired potassium channel function related to disease stage. Brain 129, 953–962. 10.1093/brain/awl024 [DOI] [PubMed] [Google Scholar]
- Kapeli K, Martinez FJ, Yeo GW, 2017. Genetic mutations in RNA-binding proteins and their roles in ALS. Hum. Genet 136, 1193–1214. 10.1007/s00439-017-1830-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M, 2000. Male sterility and enhanced radiation sensitivity in TLS−/− mice. EMBO J. 19, 453–462. 10.1093/emboj/19.3.453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH, 2009. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. 10.1126/science.1166066 [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Cleveland DW, 2010. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet 19, R46–64. 10.1093/hmg/ddq137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Sharma A, Lyashchenko AK, Shneider NA, 2016. Gamma motor neurons survive and exacerbate alpha motor neuron degeneration in ALS. Proc. Natl. Acad. Sci 113, E8316–E8325. 10.1073/pnas.1605210113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanson NA, Maltare A, King H, Smith R, Kim JH, Taylor JP, Lloyd TE, Pandey UB, 2011. A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum. Mol. Genet 20, 2510–2523. 10.1093/hmg/ddr150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, Bagley JA, Lee HY, Jan LY, Jan Y-N, 2011. Pathogenic polyglutamine proteins cause dendrite defects associated with specific actin cytoskeletal alterations in Drosophila. Proc. Natl. Acad. Sci 108, 16795–16800. 10.1073/pnas.1113573108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YR, King OD, Shorter J, Gitler AD, 2013. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol 201, 361–72. 10.1083/jcb.201302044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DM, Goodman CS, 1994. Ectopic and increased expression of Fasciclin II alters motoneuron growth cone guidance. Neuron 13, 507–23. [DOI] [PubMed] [Google Scholar]
- Lloyd TE, Machamer J, O’Hara K, Kim JH, Collins SE, Wong MY, Sahin B, Imlach W, Yang Y, Levitan ES, McCabe BD, Kolodkin AL, 2012. The p150(Glued) CAP-Gly domain regulates initiation of retrograde transport at synaptic termini. Neuron 74, 344–360. 10.1016/j.neuron.2012.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Piccolo L, Jantrapirom S, Nagai Y, Yamaguchi M, 2017. FUS toxicity is rescued by the modulation of lncRNA hsrω expression in Drosophila melanogaster. Sci. Rep 7, 1–17. 10.1038/s41598-017-15944-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machamer JB, Collins SE, Lloyd TE, 2014. The ALS gene FUS regulates synaptic transmission at the Drosophila neuromuscular junction. Hum. Mol. Genet 23, 3810–22. 10.1093/hmg/ddu094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinkovic P, Reuter MS, Brill MS, Godinho L, Kerschensteiner M, Misgeld T, 2012. Axonal transport deficits and degeneration can evolve independently in mouse models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U. S. A 109, 4296–301. 10.1073/pnas.1200658109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merriam EB, Millette M, Lumbard DC, Saengsawang W, Fothergill T, Hu X, Ferhat L, Dent EW, 2013. Synaptic Regulation of Microtubule Dynamics in Dendritic Spines by Calcium, F-Actin, and Drebrin. J. Neurosci 33, 16471–16482. 10.1523/JNEUROSCI.0661-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney EB, de Winter F, Verhaagen J, 2014. ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci 8, 252 10.3389/fnins.2014.00252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Lee S, Shang Y, Wang W-Y, Au KF, Kamiya S, Barmada SJ, Finkbeiner S, Lui H, Carlton CE, Tang AA, Oldham MC, Wang H, Shorter J, Filiano AJ, Roberson ED, Tourtellotte WG, Chen B, Tsai L-H, Huang EJ, 2014a. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Invest 124, 981–99. 10.1172/JCI72723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Lee S, Shang Y, Wang W-YY, Au KF, Kamiya S, Barmada SJ, Finkbeiner S, Lui H, Carlton CE, Tang AA, Oldham MC, Wang H, Shorter J, Filiano AJ, Roberson ED, Tourtellotte WG, Chen B, Tsai L-HH, Huang EJ, 2014b. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Invest 124, 981–999. 10.1172/JCI72723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha MC, Pousinha PA, Correia AM, Sebastião AM, Ribeiro JA, 2013. Early Changes of Neuromuscular Transmission in the SOD1(G93A) Mice Model of ALS Start Long before Motor Symptoms Onset. PLoS One 8, e73846 10.1371/journal.pone.0073846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos J, Hummel T, Ng N, Klämbt C, Davis GW, 2000. Drosophila Futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron 26, 371–382. 10.1016/S0896-6273(00)81170-8 [DOI] [PubMed] [Google Scholar]
- Rozas JL, Gómez-Sánchez L, Mircheski J, Linares-Clemente P, Nieto-González JL, Vázquez ME, Luján R, Fernández-Chacón R, Fernández-Chacón R, Südhof TC, et al. 2012. Motorneurons require cysteine string protein-α to maintain the readily releasable vesicular pool and synaptic vesicle recycling. Neuron 74, 151–65. 10.1016/j.neuron.2012.02.019 [DOI] [PubMed] [Google Scholar]
- Sabatelli M, Moncada A, Conte A, Lattante S, Marangi G, Luigetti M, Lucchini M, Mirabella M, Romano A, Del Grande A, Bisogni G, Doronzio PN, Rossini PM, Zollino M, 2013. Mutations in the 3’ untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 4748–4755. 10.1093/hmg/ddt328 [DOI] [PubMed] [Google Scholar]
- Scaramuzzino C, Monaghan J, Milioto C, Lanson NA, Maltare A, Aggarwal T, Casci I, Fackelmayer FO, Pennuto M, Pandey UB, 2013. Protein Arginine Methyltransferase 1 and 8 Interact with FUS to Modify Its Sub-Cellular Distribution and Toxicity In Vitro and In Vivo. PLoS One 8, e61576 10.1371/journal.pone.0061576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoen M, Reichel JM, Demestre M, Putz S, Deshpande D, Proepper C, Liebau S, Schmeisser MJ, Ludolph AC, Michaelis J, Boeckers TM, 2015. Super-Resolution Microscopy Reveals Presynaptic Localization of the ALS/FTD Related Protein FUS in Hippocampal Neurons. Front. Cell. Neurosci 9, 496 10.3389/fncel.2015.00496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Tang AA, Kulkarni A, West J, Brooks M, Stubblefield JJ, Liu Y, Zhang MQ, Green CB, Huber KM, Huang EJ, Herz J, Yu G, 2014. Activity-dependent FUS dysregulation disrupts synaptic homeostasis. Proc. Natl. Acad. Sci 111, E4769–E4778. 10.1073/pnas.1406162111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Huang EJ, 2016. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 1647, 65–78. 10.1016/j.brainres.2016.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, Shneider NA, 2016a. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun 7, 10465 10.1038/ncomms10465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, Shneider NA, 2016b. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun 7, 10465 10.1038/ncomms10465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHOLL DA, 1953. Dendritic organization in the neurons of the visual and motor cortices of the cat. J. Anat 87, 387–406. [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Matsuoka M, 2015. Overexpression of nuclear FUS induces neuronal cell death. Neuroscience 287, 113–124. 10.1016/j.neuroscience.2014.12.007 [DOI] [PubMed] [Google Scholar]
- Udagawa T, Fujioka Y, Tanaka M, Honda D, Yokoi S, Riku Y, Ibi D, Nagai T, Yamada K, Watanabe H, Katsuno M, Inada T, Ohno K, Sokabe M, Okado H, Ishigaki S, Sobue G, 2015. FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat. Commun 6, 1–13. 10.1038/ncomms8098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo J-M, Miller CC, Shaw CE, 2009. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. 10.1126/science.1165942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeeck C, Deng Q, DeJesus-Hernandez M, Taylor G, Ceballos-Diaz C, Kocerha J, Golde T, Das P, Rademakers R, Dickson DW, Kukar T, 2012. Expression of Fused in sarcoma mutations in mice recapitulates the neuropathology of FUS proteinopathies and provides insight into disease pathogenesis. Mol. Neurodegener 7, 53 10.1186/1750-1326-7-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M, Lauwers E, Verstreken P, 2010. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front. Synaptic Neurosci 2, 1–10. 10.3389/fnsyn.2010.00139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic S, Kiernan MC, 2006. Axonal excitability properties in amyotrophic lateral sclerosis. Clin. Neurophysiol 117, 1458–1466. 10.1016/j.clinph.2006.04.016 [DOI] [PubMed] [Google Scholar]
- Wang J-W, Brent JR, Tomlinson A, Shneider NA, McCabe BD, 2011. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J. Clin. Invest 121, 4118–4126. 10.1172/JCI57883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W-Y, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IRA, Huang EJ, Tsai L-H, 2013. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci 16, 1383–91. 10.1038/nn.3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing X, Wu C-F, 2018. Unraveling Synaptic GCaMP Signals: Differential Excitability and Clearance Mechanisms Underlying Distinct Ca2+ Dynamics in Tonic and Phasic Excitatory, and Aminergic Modulatory Motor Terminals in Drosophila. eNeuro ENEURO.0362–17.2018. 10.1523/ENEURO.0362-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Warraich ST, Nicholson GA, Blair IP, 2010. Fused in sarcoma/translocated in liposarcoma: a multifunctional DNA/RNA binding protein. Int. J. Biochem. Cell Biol 42, 1408–1411. 10.1016/j.biocel.2010.06.003 [DOI] [PubMed] [Google Scholar]
- Zhong L, Hwang RY, Tracey WD, 2010. Pickpocket is a DEG/ENaC protein required for mechanical nociception in Drosophila larvae. Curr. Biol 20, 429–34. 10.1016/j.cub.2009.12.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liu S, Liu G, Oztürk A, Hicks GG, 2013. ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 9, e1003895 10.1371/journal.pgen.1003895 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.