

Abstract
The enantioselective construction of carbon–heteroatom and carbon–carbon bonds alpha to ketones forms substructures that are ubiquitous in natural products, pharmaceuticals and agrochemicals. Traditional methods to form such bonds have relied on combining ketone enolates with electrophiles. Reactions with heteroatom-based electrophiles require special reagents in which the heteroatom, which is typically nucleophilic, has been rendered electrophilic by changes to the oxidation state. The resulting products usually require post-synthetic transformations to unveil the functional group in the final desired products. Moreover, different catalytic systems are typically required for the reaction of different electrophiles. Here, we report a strategy for the formal enantioselective α-functionalization of ketones to form products containing a diverse array of substituents at the alpha position with a single catalyst. This strategy involves an unusual reversal of the role of the nucleophile and electrophile to form C–N, C–O, C–S, and C–C bonds from a series of masked ketone electrophiles and a wide range of conventional heteroatom and carbon nucleophiles catalyzed by a metallacyclic iridium catalyst.
Graphical Abstract
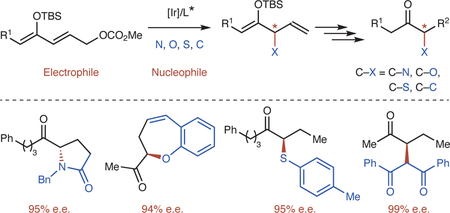
Enantioselective reactions occurring at the alpha positions of ketones are some of the most practiced reactions in organic chemistry. Most often, these reactions join a ketone enolate with an electrophile, especially a carbon electrophile1–2. Reactions of enolates with heteroatom electrophiles to form carbon-heteroatom bonds, however, require special reagents in which the heteroatom position has been made electrophilic by the oxidation state of the heteroatom, and these reagents often do not contain the substituents in the final desired products3–6. For example, the enantioselective formation of C–N bonds at the carbon alpha to a ketone has been achieved with nitrosoarenes, azodicarboxylates, azidoiodinanes and hydroxylamines as electrophiles, but the nitrogen-heteroatom bond and the acyl groups on the nitrogen in the initial product must be cleaved to generate amines3–6. Likewise, enantioselective construction of C–O bonds alpha to ketones has been achieved with electrophilic sources of the heteroatom unit, such as nitrosoarenes7, oxaziridines8, peroxides9, and even oxygen10, but simple formation of a C–O bond between the carbonyl unit and an alkoxide or phenoxide is rare4. The enantioselective formation of C–S bonds occurs with reactions of disulfides, sulfenylamines, and sulfenyl chlorides, but not with simple thiolates4,11.
Many products of these reactions of carbonyl enolates are chiral, and enantioselective bond formation alpha to a carbonyl group also has been a longstanding challenge in organic chemistry. Again, such reactions have most commonly been conducted with enolate nucleophiles and carbon electrophiles with a wide range of catalysts, but special heteroatom electrophiles have been needed for enantioselective formation of carbon-heteroatom bonds, even with chiral amine catalysts12, Lewis acid catalysts13, phase-transfer catalysts14, photoredox catalysts15, or frustrated Lewis acid-Brønsted base pairs16. Although the enantioselective formation of carbon-carbon bonds with a ketone enolate is more common, new strategies are needed to form certain synthetically important classes of products by formation of a carbon-carbon bond at the alpha-position of ketones, such as 1,4-dicarbonyl compounds that do not result from reactions of enolates with alkyl or carbonyl electrophiles and are often synthesized by oxidative coupling of enolates without control of the absolute configuration of the chiral product (Figure 1a)17–19. For these reasons, alternative strategies are needed to enable the enantioselective formation of both carbon-heteroatom bonds and carbon-carbon bonds at the position alpha to a ketone with readily available heteroatom and carbon nucleophiles.
Figure 1. Typical approaches to the α-functionalization of ketones and our design of an alternative strategy involving a reversal of the origin of the nucleophile and electrophile.
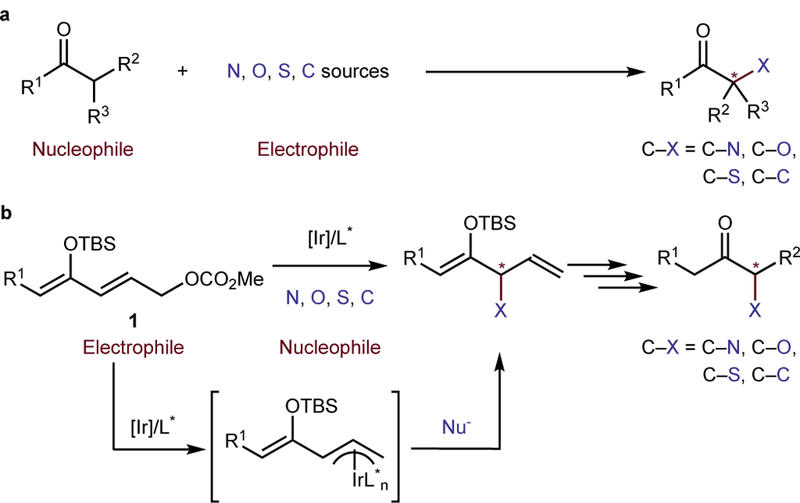
a. Conventional approach to the synthesis of alpha-functionalized ketones. A classical method to prepare ketones bearing substituents at the alpha carbon involves deprotonation of the alpha C–H bond to form an enolate and reaction of this nucleophilic enolate with a suitable electrophile. However, to introduce groups bound to the alpha carbon through a heteroatom, a heteroatom-based electrophile must be used, and preparation of this electrophile usually requires conversion of a heteroatom nucleophile to an electrophilic group by multiple chemical steps. b. Reported strategy with reversal of the nucleophilic and electrophilic components. An alternative strategy involves converting the carbonyl compound to an electrophile for a catalytic reaction with common, commercially available reagents that are nucleophilic at carbon, nitrogen, oxygen or sulfur. We show that the latter strategy allows a broad range of nucleophiles to react with a masked ketone that is electrophilic to form chiral, alpha-functionalized ketones with defined absolute configuration.
We envisioned a strategy shown in Figure 1b in which the reaction between 1 as an allylic electrophile precursor to ketones and a carbon or heteroatom nucleophile in the presence of an iridium catalyst would enantioselectively form protected ketone products containing substituents at the alpha position. Because metallacyclic iridium complexes catalyze the enantioselective formation of carbon-heteroatom and carbon-carbon bonds with a wide range of nucleophiles20–23, these reactions could form protected ketones containing a diverse array of substituents bound to the alpha carbon. Previously, allylic esters containing an α,β-unsaturated ester unit were used as electrophiles for enantioselective reactions of organomagnesium, lithium and zinc reagents catalyzed by copper complexes, but the electrophiles in these reactions were limited to ester derivatives that do not undergo Michael addition and are less acidic at the alpha position than ketones, and the nucleophile was limited to hard main group organometallic reagents24–27. Related strategies with other transition-metal catalysts have led to achiral products from reaction at the terminal position of allylic electrophiles derived from an ester or a ketone28–29.
Here, we report the implementation of a strategy involving reaction of an allylic ester of a ketone dienolate with a wide range of heteroatom and carbon nucleophiles. These nucleophiles include primary amines, secondary amines, alkyl amines, aryl amines, heteroaryl amines, phenoxides, thiolates, malonates, and 1,3-diones, as well as natural products, biologically active amino alcohols, amino acids, and even peptides. Reactions of all of these nucleophiles lead to α-substituted ketone derivatives in high yield and enantioselectivity.
Results and discussion
To test the strategy shown in Figure 1b, we first subjected unprotected γ-carbonato enone 2a in Figure 2 to conditions commonly used for Ir-catalyzed allylic substitution with benzylamine as the nucleophile30. The substitution product was not observed; only the product from Michael addition of the amine to the unsaturated ketone formed (Figure 2a). To prevent competing Michael addition, the ketone was converted to the silyl enol ether. The choice of silyl group proved important for the reaction to occur. Trimethylsilyl enol ether 4a decomposed under the reaction conditions to the corresponding ketone, and the product from Michael addition to the resulting enone was observed (Figure 2b). However, silyl enol ether 1a, bearing a TBS protecting group, was more stable and underwent allylic substitution to provide product 7a in 58% yield, 97% ee and 13:1 branched to linear selectivity (Figure 2c). Further experiments showed that substitution product 7a formed in 82% yield, 99% ee and 12:1 branched to linear ratio with BnNH2 (2.5 equiv) as nucleophile with the combination of [Ir(COD)Cl]2 (2 mol%) and (Sa,S,S)-L (4 mol%) as catalyst precursors in THF (1.0 M) at 40 oC for 12 h.
Figure 2. Importance of the protecting group.
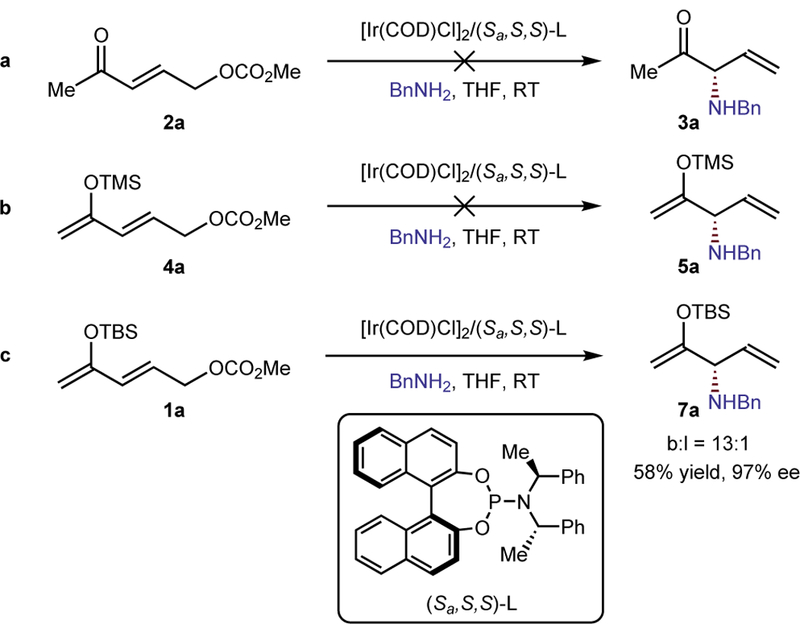
The strategy of forming alpha-functionalized ketones by reaction of an allylic carbonate of a masked ketone required preparation of this reagent with the right silyl group. a. Reaction of an allylic carbonate containing an unmodified ketone did not form the substitution product; instead, the product of Michael addition into the enone occurred. b. Reaction of the trimethylsilyl enol ether derivative of the ketone also did not form the substitution product; in this case, the Si-O bond of the silyl ether cleaved during the reaction to form the ketone, and the product of Michael addition also occurred. c. However, reaction of the silyl enol ether in which the silyl group is the more hindered tert-butyl dimethyl silyl (TBS) group occurred without cleavage of the Si-O bond to give the substitution product in high yield, branched-to-linear selectivity, and enantioselectivity.
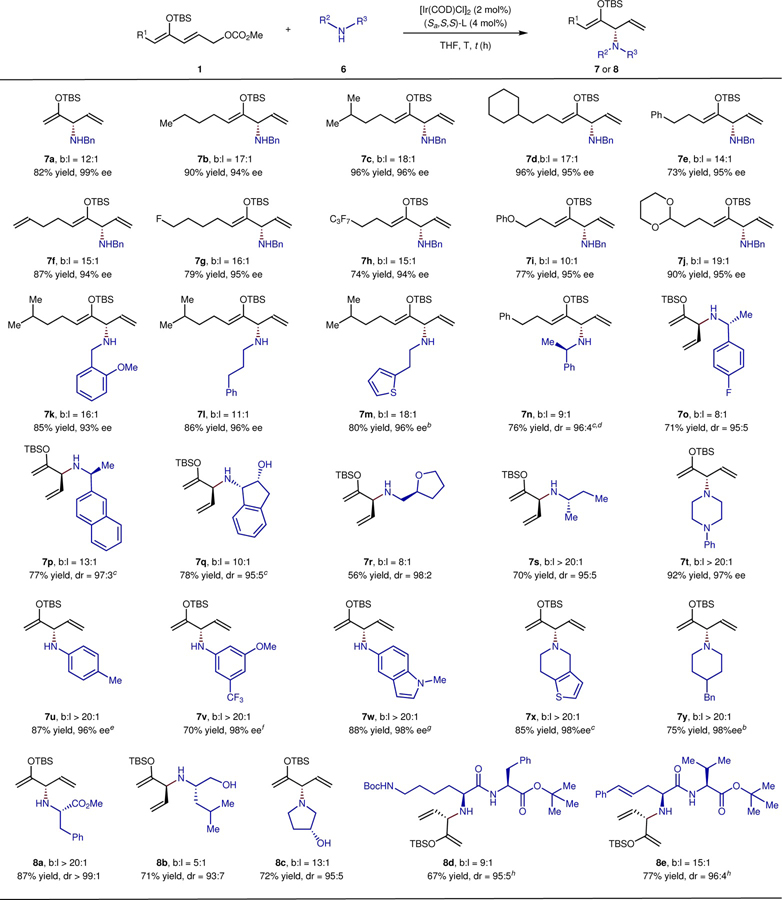
Studies on the scope of the enantioselective α-amination of a series of protected ketones with a variety of nitrogen nucleophiles are shown in Table 1. The scope of ketone derivative that underwent the substitution process is illustrated by examples 7a – 7j. Siloxypentadienyl carbonates containing alkyl, cycloalkyl, phenyl, alkenyl, fluoro, perfluoroalkyl and phenoxyalkyl groups underwent substitution with benzylamine in 73 – 96% yield, 94 – 96% ee, and up to 18:1 b:l selectivity (7b – 7i). The acetal-substituted 1j also reacted in a high 90% yield, 95% ee, and 19:1 b:l selectivity.
Table 1.
Scope of Reaction Partners for the Construction of C–N bondsa
 |
Isolated yields. b:l ratios and dr values were determined by 1H NMR of the crude reaction mixture. Ee values were determined by HPLC or SFC. Reactions were performed under N2 with 1 (0.2 mmol), 6 (0.5 mmol), [Ir(COD)Cl]2 (2 mol%), (Sa,S,S)-L (4 mol%), THF (1 M) at RT – 55 oC for 11 – 24 h. See SI for detailed conditions for each substrate.
Ee values were determined by further hydrogenation of product.
THF (0.5M).
[Ir(COD)Cl]2 (4 mol%), (Sa,S,S)-L (8 mol%).
Additional DABCO (20 mol%) was added.
Additional DABCO (5 mol%) was added.
Additional DABCO (40 mol%) was added.
Reaction condition: 1 (0.2 mmol), peptide (0.1 mmol), [Ir(COD)Cl]2 (4 mol%), (Sa,sS,S)-L (8 mol%), THF (0.5 M) at 50 oC for 2 d.
The scope of amine that underwent this process was also broad. A diverse range of primary alkyl amines including both achiral amines and chiral amines reacted in high yield (56 – 86%) and high ee (93 – 96%) or dr (95:5 – 98:2) (7k – 7s). For example, the reactions of achiral 2-methoxybenzylamine, 3-phenylpropan-1-amine and thienylethylamine underwent the substitution reaction smoothly, affording 7k – 7m in high yield, enantioselectivity and regioselectivity. The reaction of arylethylamines with varying configurations at the α carbon also occurred in high yield, regioselectivity and stereoselectivity (7n – 7p, 71 – 77% yield, 95:5 – 97:3 dr, 8:1 – 13:1 b:l). The (S)-amine reacted with higher selectivity than the (R)-amine with the catalyst derived from (Sa,S,S)-L, but the differences in selectivity were small (7p from (S)-amine: dr = 97:3, b:l = 13:1; 7n and 7o from (R)-amine: dr = 96:4, b:l = 9:1 and dr = 95:5, b:l = 8:1, respectively). In addition, enantioenriched, chiral amines possessing auxiliary functional groups, such as a free hydroxyl group or tetrahydrofuran unit, were compatible with the reaction conditions, forming substitution products in good to excellent yields, diastereoselectivities and regioselectvities (7q – 7r). Finally, the simple, saturated, chiral hydrocarbyl amine 7s reacted in 70% yield, 95:5 dr and > 20:1 b:l.
Secondary alkylamines, arylamines and heteroarylamines (7t – 7y) also reacted in high yield, regioselectivity and enantioselectivity. Reactions of these amines at room temperature or 50 oC afforded the products of substitution in 75 – 92% yield, 96 – 98% ee, and > 20:1 regioselectivity. Cyclic secondary amines, including piperazine, piperidine, and hydrothienopyridine, reacted with 1a in over 97% ee (7t, 7x, 7y) with excellent regioselectivity. The reactions of arylamines required preactivation of the catalyst, as reported previously31, because Ir-catalyzed allylic substitution reactions rely on the formation of a metallacyclic active catalyst by base-assisted cyclometallation, and aromatic amines, are insufficiently basic to induce formation of a catalytically competent metallacycle. Added DABCO as base generated a system that catalyzed reactions of arylamine nucleophiles. Both arylamines and heteroarylamines reacted under these conditions to afford substitution products 7u – 7w in 70 – 88% yield, 96 – 98% ee, and over 20:1 regioselectivity.
Amino acids and amino alcohols are important motifs in biologically active molecules and commercial drugs. Thus, efficient strategies for introduction or modification of these groups would be beneficial to drug discovery. Marsden et al reported iridium-catalyzed allylic substitution reactions of chiral amino acid derivatives with moderate to good diastereoselectivity32. In our case, the amino acid derivative methyl L-phenylalaninate reacted to form optically pure product in 87% yield, > 99:1 diastereoselectivity, and > 20:1 regioselectivity (8a). The reaction of a primary, acyclic amino alcohol formed 8b in high yield and in high dr, but with moderate regioselectivity. A cyclic, secondary amino alcohol reacted to form 8c with yield and diastereoselectivity that were comparable and branched:linear ratio that was higher than those of the reaction to form 8b. Peptides also reacted with the dienyl carbonate. For example, peptides derived from Lys-Phe and styrylAla-Val reacted in high yield and selectivity (8d, 8e), creating potential access to modified biological macromolecules that would otherwise be challenging synthetic targets.
The approach we disclose also provides a method to prepare α-chiral phenoxy ethers, which are common in natural products and biologically active molecules (Table 2A). The reaction of 1a with phenoxide occurred to form 9a in 71% yield, 94% ee, and 13:1 regioselectivity. An ortho-substituted phenoxide reacted more slowly and required a higher temperature than reactions of less hindered phenoxides, but still formed 9b in good yield and with similar selectivity. Oxygen nucleophiles derived from the amino acid tyrosine also reacted with various ketone derivatives, forming substitution products in up to 85% yield, 97:3 dr, and 16:1 regioselectivity (9c – 9f). Even peptides containing tyrosine were suitable oxygen nucleophiles; ether 9g formed in 83% yield, 96:4 diastereoselectivity, and 14:1 b:l ratio. Likewise, the natural product estradiol bearing both a phenoxide and a free hydroxyl group reacted to form aryl ether 9h in high yield without requiring protection of the free hydroxyl group.
Table 2.
Scope of the Formation of C–O, C–S, and C–C bondsa
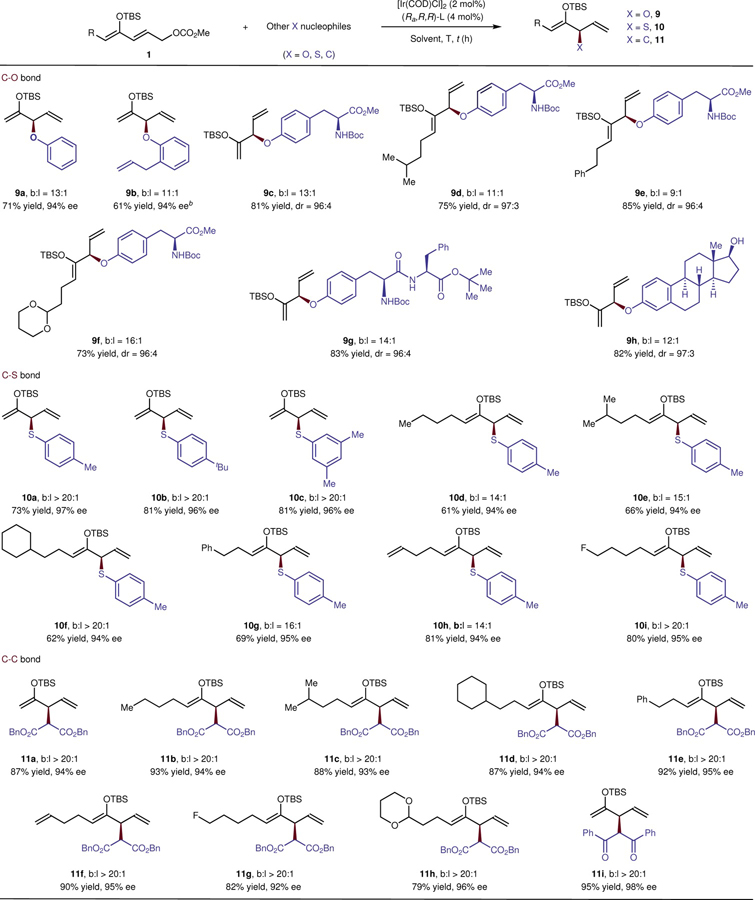 |
Isolated yields. b:l ratios and dr values were determined by 1H NMR of the crude reaction mixture. Ee values were determined by HPLC. Reaction conditions: for 9a – 9h, reactions were performed under N2 with 1 (0.2 mmol), ROLi (0.1 mmol), [Ir(COD)Cl]2 (4 mol%), (Ra,R,R)-L (8 mol%), THF (0.5 M) at 5 oC for 20 h; for 10a – 10i, reactions were performed under N2 with 1 (0.2 mmol), RSNa (0.1 mmol), [Ir(COD)Cl]2 (5 mol%), (Ra,R,R)-L (10 mol%), LiCl (0.3 mmol), DCM (0.1 M) at RT – 35 oC for 14 – 24 h; for 11a – 11i, reactions were performed under N2 with 1 (0.1 mmol), NaCHR2 (0.2 mmol), [Ir(COD)Cl]2 (2 mol%), (Ra,R,R)-L (4 mol%), LiCl (0.1 mmol), THF (0.5 M) at RT for 12 – 20 h. See SI for detailed reaction conditions for each substrate.
The reaction was conducted at 50 oC for 2 d.
α-Sulfenylated carbonyl compounds serve as versatile synthetic intermediates and valuable subunits in natural products and drugs11,33. In contrast to conventional methods to form C–S bonds alpha to a carbonyl, in which sulfur is an electrophilic site, the enantioselective formation of C–S bonds in our system occurred with sodium arenethiolates as nucleophiles (Table 2B). Although thiols can poison transition-metal catalysts34, the reaction of several arenethiolates occurs in high yield, high enantioselectivity and high regioselectivity with ketone derivative 1a (10a – 10c, 73 – 81% yield, 96 – 97% ee, > 20:1 b:l). A broad range of ketone derivatives also underwent substitution with 4-methylbenzenethiolate in high yield (61 – 81% yield) with high regioselectivity (b:l > 14:1) and enantioselectivity (94 – 95% ee, 10d – 10i).
Finally, malonates and 1,3-diones reacted with ketone-derived electrophiles to form products containing new C–C bonds (Table 2C). A variety of ketone derivatives underwent substitution with sodium dibenzyl malonate in 79 – 93% yield and 92 – 96% ee, forming a single constitutional isomer (11a – 11h). Reaction of the anion of dibenzoylmethane as the nucleophile occurred in 95% yield and with 98% ee (11i). This process serves as a new strategy for the asymmetric coupling of two carbonyl compounds to form masked 1,4-dicarbonyl products, as well as a novel route to enantioselective alkylation of ketones2.
Transformations of several products of these substitution processes revealed the potential applications of our strategy to the enantioselective construction of carbon-heteroatom and carbon-carbon bonds alpha to a ketone (Figure 3). Compound 7e was prepared on a half-gram scale from the reaction of 1e with benzylamine in the same yield, regioselectivity and enantioselectivity as the reaction on smaller scale and was used for preparation of derivatives. Hydrogenation of 7e, followed by the cleavage of the TBS protecting group by Et3N·3HF afforded chiral α-amino ketone 12 in 85% yield and 94% ee.
Figure 3. Selected transformations of the allylic substitution products.
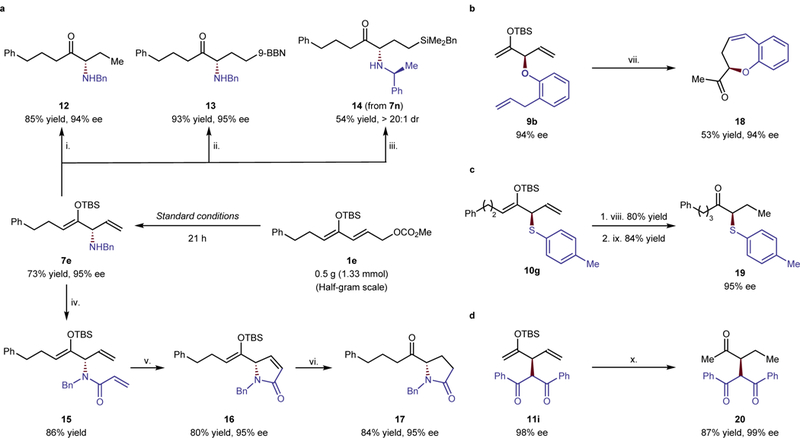
The ketone can be unmasked after common transformations of the olefin. A series of transformations of the olefin, followed by cleavage of the silyl ether with fluoride are shown. a. Derivatives of the product from formation of a C–N bond. i. (1) PtO2 (25 mol%), H2 in MeOH at RT for 7 h; (2) Et3N·3HF (2.0 equiv) in THF at −78 to 0 oC for 1 h. ii. 9-BBN (1.2 equiv) in THF at −78 to 35 oC for 18 h, then Et3N·3HF (2.0 equiv) at −78 oC to RT for 6 h. iii. (1) RhCl(PPh3)3 (10 mol%), BnMe2SiH (3.0 equiv), in toluene at 50 oC for 16 h; (2) Et3N·3HF (2.0 equiv) in THF at 0 oC to RT for 3 h. iv. Acryloyl chloride (1.5 equiv), DIPEA (2.0 equiv) in DCM at 0 oC to RT for 6 h. v. Zhan Catalyst-1B (5 mol%) in DCM at 45 oC for 12 h. vi. (1) 10 wt% Pd/C (5 mol%), H2 in EtOAc at 0 oC to RT+ for 4 h; (2) Et3N·3HF (2.0 equiv) in THF at 0 oC to RT for 3 h. b. Derivatives of the product from formation of a C–O bond. vii (1) Zhan Catalyst-1B (5 mol%) in DCM at RT for 12 h; (2) Et3N·3HF (3.0 equiv) in THF at 0 oC to RT for 11 h. c. Derivatives of the product from formation of a C–S bond. viii. NBSH (2.0 equiv), Et3N (4.0 equiv), THF/iPrOH (1/1), RT, 7.5 h. ix. Et3N·3HF (8.0 equiv) in THF at 0 oC to RT for 20 h. d. Derivatives of the product from formation of a C–C bond. x. (1) Lindlar catalyst (3.0 equiv), H2 in EtOAc at RT for 12 h; (2) Et3N·3HF (4.0 equiv) in THF at RT for 12 h. NBSH: o-nitrobenzenesulfonylhydrazide.
After checking a series of reagent for deprotection of the TBS group, we found that cleavage of this group with Et3N·3HF occurred with the least racemization and occurred in all cases without erosion of enantioselectivity. Cleavage of the silyl ether with KHF2 (2.0 equiv) at 0 oC for 1 h also occurred, but compound 12 was obtained with an enantioselectivity of 93%, which is slightly lower than the 95% ee of the product of the substitution reaction. Cleavage with TBAF (1.5 equiv) at - 78 oC for 1 h occurred, but product 15 was obtained with an enantioselectivity of 70%, which is much lower than the 94% ee of the product of the substitution reaction. The other reagents HF·Pyridine, KF, LiF, LiBF4, and CsF only gave trace or no product.
Other enantioenriched α-amino ketones, including 13 and 14, were synthesized from 7e or 7n in high yield by functionalizations of the terminal olefins prior to unmasking of the ketone. A sequential intramolecular metathesis of 9b, which was prepared from the reaction of 1a with the ortho-substituted phenoxide, and removal of the TBS group, afforded product 18 containing an oxepane fused to an aryl ring in moderate yield and high enantioselectivity. Oxepanes fused to aryl rings can be found in natural products, such as (+)-Heliannuol B & D35. The α-sulfenylated ketone 19 was prepared from 10g by simple hydrogenation and deprotection in high yield without erosion of the enantiomeric excess obtained from the allylic substitution. A similar strategy was applied to the synthesis of the product 20 from coupling of two different ketones. Hydrogenation and deprotection occurred in 87% yield to give the 1,4-dicarbonyl compound in 99% ee. Finally, acylation, ring-closing metathesis, reduction and deprotection formed the enantioenriched pyrrolidinone 17 in excellent yield without reduction of the enantiomeric excess of the silyl enol ether 7e. This sequence demonstrates the ability to use the alkene functionality of the initial product to construct structural units that could not be obtained directly by substitution at the position alpha to a ketone.
In conclusion, we report a new, broadly applicable strategy for the enantioselective construction of C–N, C–S, C–O and C–C bonds alpha to the carbonyl group in ketones by iridium-catalyzed allylic substitution. In contrast to conventional methods for α-functionalization of ketones with electrophilic reagents, our approach employs common reagents containing nucleophilic nitrogen, oxygen, sulfur, and carbon atoms that react directly without the requirement for conversion to an electrophile. This requirement has limited the scope of heteroatom-based reagents that can be introduced into the alpha position and has required subsequent, often harsh, reactions to generate the desired product. Our allylic substitution occurs with primary amines, secondary amines, alkylamines, arylamines, heteroarylamines, amino acids, amino alcohols, peptides, phenoxides, thiolates, malonates, and 1,3-diones in uniformly high yield, enantioselectivity and regioselectivity without the need to identify or develop catalytic systems for each class of reagent. The common deprotection of a TBS group from the product after derivatization of the alkene releases the ketone unit in the final product.
Supplementary Material
Acknowledgements
Financial support for this work was provided by the NIH (GM-55382). Z.-T.H. thanks a joint postdoc fellowship from Pharmaron and Shanghai Institute of Organic Chemistry (SIOC). We thank Sophie I. Arlow for assistance in the preparation of this manuscript.
Footnotes
Data availability. All relevant data are available with the manuscript, in the Supplementary Information files.
Competing interests
The authors declare no competing interests.
References
- 1.Bellina F, Rossi R Transition metal-catalyzed direct arylation of substrates with activated sp3-hybridized C–H bonds and some of their synthetic equivalents with aryl halides and pseudohalides. Chem. Rev 110, 1082–1146 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Cano R, Zakarian A, McGlacken GP Direct asymmetric alkylation of ketones: still unconquered. Angew. Chem., Int. Ed 56, 9278−9290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janey JM Recent advances in catalytic, enantioselective α aminations and α oxygenations of carbonyl compounds. Angew. Chem. Int. Ed 44, 4292–4300 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Smith AMR & Hii KK Transition metal catalyzed enantioselective α heterofunctionalization of carbonyl compounds. Chem. Rev 111, 1637–1656 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Zhou F, Liao F-M, Yu J-S & Zhou J Catalytic asymmetric electrophilic amination reactions to form nitrogen-bearing tetrasubstituted carbon stereocenters. Synthesis 46, 2983–3003 (2014). [Google Scholar]
- 6.Maji B & Yamamoto H Use of in situ generated nitrosocarbonyl compounds in catalytic asymmetric α-hydroxylation and α-amination reactions. Bull. Chem. Soc. Jpn 88, 753–762 (2015). [Google Scholar]
- 7.Bøgevig A, Sundén H, Córdova A Direct catalytic enantioselective α-aminoxylation of ketones: a stereoselective synthesis of α-hydroxy and α,α’-dihydroxy ketones. Angew. Chem., Int. Ed 43, 1109–1112 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Zhou L, Wang B, Mu H, Zhang H, Song Y & Qu J Development of tartaric acid derived chiral guanidines and their application to catalytic enantioselective α-hydroxylation of β-dicarbonyl compounds. Org. Lett 15, 3106–3109 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Yao H, Lian M, Li Z, Wang Y & Meng Q Asymmetric direct α-hydroxylation of β-oxo esters catalyzed by chiral quaternary ammonium salts derived from cinchona alkaloids. J. Org. Chem 77, 9601−9608 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Yin H, Tang X, Wu Y, Meng Q & Gao Z A series of cinchona-derived N-oxide phase-transfer catalysts: application to the photo-organocatalytic enantioselective α-hydroxylation of β-dicarbonyl compounds. J. Org. Chem 81, 7042−7050 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Yu J-S, Huang H-M, Ding P-G, Hu X-S, Zhou F & Zhou J Catalytic enantioselective construction of sulfur-containing tetrasubstituted carbon stereocenters. ACS Catal 6, 5319−5344 (2016). [Google Scholar]
- 12.Kumaragurubaran N, Juhl K, Zhuang W, Bøgevig A, Jørgensen KA Direct L-proline-catalyzed asymmetric α-amination of ketones. J. Am. Chem. Soc 124, 6254–6255 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Yang X & Toste FD Direct asymmetric amination of α-branched cyclic ketones catalyzed by a chiral phosphoric acid. J. Am. Chem. Soc 137, 3205−3208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohmatsu K, Ando Y, Nakashima T & Ooi T A modular strategy for the direct catalytic asymmetric α-amination of carbonyl compounds. Chem 1, 802–810 (2016). [Google Scholar]
- 15.Huang X, Webster RD, Harms K & Meggers E Asymmetric catalysis with organic azides and diazo compounds initiated by photoinduced electron transfer. J. Am. Chem. Soc 138, 12636–12642 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Shang M, Wang X, Koo SM, Youn J, Chan JZ, Yao W, Hastings BT & Wasa M Frustrated Lewis acid/Brønsted base catalysts for direct enantioselective α-amination of carbonyl compounds. J. Am. Chem. Soc 139, 95–98 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Guo F, Clift MD & Thomson RJ Oxidative coupling of enolates, enol silanes, and enamines: methods and natural product synthesis. Eur. J. Org. Chem 2012, 4881–4896 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Y, Zhang L & Luo S Asymmetric α-photoalkylation of β-ketocarbonyls by primary amine catalysis: facile access to acyclic all-carbon quaternary stereocenters. J. Am. Chem. Soc 136, 14642−14645 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Jang H-Y, Hong J-B & MacMillan DWC Enantioselective organocatalytic singly occupied molecular orbital activation: the enantioselective α-enolation of aldehydes. J. Am. Chem. Soc 129, 7004–7005 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Hartwig JF & Stanley LM Mechanistically driven development of Iridium catalysts for asymmetric allylic substitution. Acc. Chem. Res 43, 1461–1475 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu W-B, Xia J-B & You S-L Iridium-catalyzed asymmetric allylic substitutions. Top. Organomet. Chem 38, 155–208 (2012). [Google Scholar]
- 22.Hethcox JC, Shockley SE & Stoltz BM Iridium-catalyzed diastereo-, enantio-, and regioselective allylic alkylation with prochiral enolates. ACS Catal 6, 6207–6213 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qu J, Helmchen G Applications of iridium-catalyzed asymmetric allylic substitution reactions in target-oriented synthesis. Acc. Chem. Res 50, 2539–2555 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Murphy KE & Hoveyda AH Enantioselective synthesis of α-alkyl-β,γ-unsaturated esters through efficient Cu-catalyzed allylic alkylations. J. Am. Chem. Soc 125, 4690–4691 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Lee Y, Hoveyda AH Lewis base activation of Grignard reagents with N-heterocyclic carbenes, Cu-free catalytic enantioselective additions to γ-Chloro-α,β-unsaturated esters. J. Am. Chem. Soc 128, 15604–15605 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Gao F, Lee Y, Mandai K, Hoveyda AH Quaternary carbon stereogenic centers through copper-catalyzed enantioselective allylic substitutions with readily accessible aryl- or heteroaryllithium reagents and aluminum chlorides. Angew. Chem., Int. Ed 49, 8370−8374 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Den Hartog T, Maciá B, Minaard A, Feringa BL Copper-catalyzed asymmetric allylic alkylation of halocrotonates: efficient synthesis of versatile chiral multifunctional building blocks. Adv. Synth. Catal, 352, 999−1013 (2010). [Google Scholar]
- 28.Ashfeld BL, Miller KA, Martin SF Direct, stereoselective substitution in [Rh(CO)2Cl]2-catalyzed allylic alkylations of unsymmetrical substrates. Org. Lett 6, 1321−1324 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Ohmatsu K, Ito M, Kunieda T, Ooi T Exploiting the modularity of ion-paired chiral ligands for palladium-catalyzed enantioselective allylation of benzofuran-2(3H)-ones. J. Am. Chem. Soc 135, 590–593 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Leitner A, Shu C & Hartwig JF Effects of catalyst activation and ligand steric properties on the enantioselective allylation of amines and phenoxides. Org. Lett 7, 1093–1096 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Shu C, Leitner A & Hartwig JF Enantioselective allylation of aromatic amines after in situ generation of an activated cyclometalated iridium catalyst. Angew. Chem., Int. Ed 43, 4797–4800 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Tosatti P, Horn J, Campbell AJ, House D, Nelson A & Marsden SP Iridium-catalyzed asymmetric allylic amination with polar amines: access to building blocks with lead-like molecular properties. Adv. Synth. Catal 352, 3153–3157 (2010). For most cases, the dr values of products are less than 89:11. [Google Scholar]
- 33.Trost BM α-Sulfenylated carbonyl compounds in organic synthesis. Chem. Rev 78, 363−382 (1978). [Google Scholar]
- 34.Hegedus LL; McCabe RW Catalyst Poisoning; Marcel Dekker: New York, 1984. [Google Scholar]
- 35.Manabe Y, Kanematsu M, Hiromasa Y, Yoshida M, Shishido K Concise total syntheses of heliannuols B and D. Tetrahedron 70, 742–748 (2014). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.