

Abstract
The reactivity of 1-hydroxybenzoyl triazole (HOBt) esters with the carboxylate functionality present in peptides is demonstrated in the gas phase with a doubly deprotonated dianion. The reaction forms an anhydride linkage at the carboxylate site. Upon ion trap collisional induced dissociation (CID) of the modified peptide, the resulting spectrum shows a nominal loss of the mass of the reagent and a water molecule. Analogous phenomenology was also noted for model peptide cations that likely contain zwitterionic/salt-bridged motifs in reactions with a negatively charged HOBt ester. Control experiments indicate that a carboxylate group is the likely reactive site, rather than other possible nucleophilic sites present in the peptide. These observations suggest that HOBt ester chemistry may be used as a chemical probe for the presence and location of carboxylate groups in net positively-charged polypeptide ions. As an illustration, deprotonated sulfobenzoyl-HOBt was reacted with the [M+7H]7+ ion of ubiquitin. The ion was shown to react with the reagent and CID of the covalent reaction product yielded an abundant [M+6H-H2O]6+ ion. Comparison of the CID product ion spectrum of this ion with that of the water loss product generated from CID of the unmodified [M+6H]6+ ion revealed the glutamic acid at residue 64 as a reactive site, suggesting that it is present in the deprotonated form.
Keywords: Ion/ion reactions, zwitterion, protonated peptide, carboxylates, triazole ester
Graphical Abstract
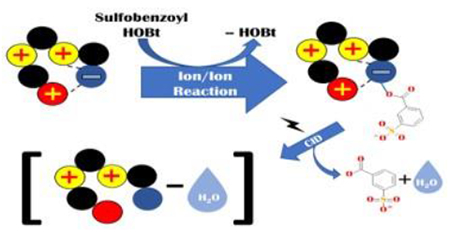
Introduction
Strong electrostatic interactions such as zwitterionic pairing between oppositely charged functional groups offer stabilizing effects to the secondary and tertiary structures of peptides and proteins. Such pairings can be classified as salt bridges (+ − +) or zwitterions (+ −). Determining the existence and location(s) of such interactions in a polypeptide is relevant to its three-dimensional structural characterization. While amino acids in a zwitterionic state are known to exist in solution at neutral pH, their existence in the gas phase has previously been questioned.[1] However, there is growing experimental evidence that indicates that such charge separation is possible in the gas phase for systems as small as two amino acids.[2,3,4] Techniques used to elucidate the location and arrangement of salt bridges in the gas phase include computational studies[5], ion mobility[6], ion spectroscopy [2], and interpretation of fragmentation via mass spectrometry[4,7,8]. Many of the experimental techniques rely upon some form of calculation to provide a precise prediction of zwitterionic structure.
Calculations have been used to characterize the tendency of salt bridge formation involving particular amino acid residues. For instance, it is predicted that zwitterionic states for non-basic residues such as glycine are not favorable in the gas phase;[1] however, both experimental and theoretical research has shown that zwitterionic structures can be favorable for more basic residues such as arginine.[3,9,10,11] Of the residues capable of comprising salt bridges in the gas-phase (arginine, lysine, aspartic acid, and glutamic acid), the arginine side chain is established as the primary positively charged component of zwitterionic conformations.[12] The aspartic acid and glutamic acid side chains and the C-terminus act as negatively charged components.[12] The influence that these residues have on the conformational landscape has been the subject of examination.[13] In the case of small polypeptides, comparisons between theoretical and experimental data can be made. Unfortunately, due to conformational complexity of larger systems, ab initio methods are too computationally expensive to be extended to proteins.
Ion mobility mass spectrometry (IM-MS) provides a direct probe of an ion’s conformation via its drift time through a gas, which can be translated to a collision cross section (CCS). Research using this technique has demonstrated that the cross section of a protein is highly dependent on its charge state.[14] The dependence of CCS on protein charge state between has been attributed in part to conformational differences that arise from the loss of stabilizing electrostatic forces such as salt bridges and hydrogen bonds under the influence of the Coulombic field associated with multiple charging.[15,16] Electrospray ionization (ESI), which tends to lead to multiple charging of polypeptides, is of particular utility for structural studies due to the possibility for the preservation of a least some elements of the native condensed-phase structure when ions are transferred to the gas-phase, provided the solution is not denaturing. [17,18,19,20] IM-MS in conjunction with ESI has been particularly useful in the study of protein and peptide ion higher order structure. However, measurement of the CCS alone can neither provide detailed structural information nor information regarding chemical interactions within an ion.
The fragmentation patterns of proteins and peptides resulting from various activation techniques in tandem mass spectrometry have been reported to be sensitive to the presence of zwitterionic pairing. [21,22,23] For example, unique neutral losses have been attributed to zwitterionic interactions in dipeptides.[3,8] However, such losses have not proved to be conclusive for larger polypeptide ions as other mechanisms might also account for them. Recently, Bonner et al. have shown that photoexcitation of net positively-charged ions that can contain anionic sites, specifically carboxylates, lead to fragmentation analogous to that observed with electron-based dissociation techniques. [7] This technique directly probes for the presence of zwitterionic sites, by photoactivation of the ion. Other light-based activation techniques such as infrared multiple photodissociation spectroscopy also show the ability to directly identify the presence of zwitterionic interactions by revealing unique vibrational signatures of the carboxylate functional group. These approaches that directly activate the carboxylate functionality appear to be useful for determining the presence of zwitterionic interactions.
In this work, we evaluated the possibility that functional-group selective ion/ion reactions might serve as chemical probes for the presence of carboxylate groups in polypeptide cations. Prior work has shown that N-hydroxysuccinimide (NHS) esters can serve as reagents for gas-phase covalent modification of nucleophilic functional groups such as primary amines [24], guanidine [25] and carboxylates.[26] In the reaction between NHS esters and carboxylates, a labile anhydride bond is formed at the carboxylate. Subsequent collisional activation results in the loss of the modification and, nominally, a molecule of water. This reaction does not occur with carboxylic acids (i.e., the protonated form of a carboxylate) as the nucleophilicity of the acidic form is much less than that of its conjugate base. Similar to NHS esters, 1-hydroxy-benzotriazole (HOBt) esters undergo gas phase reactions with amine groups but with increased reactivity, as HOBt is a better leaving group relative to NHS.[27] While HOBt esters have not yet been demonstrated to react with carboxylates in the gas phase, their increased reactivity with primary amines relative to NHS esters suggested to us that they would also be more reactive with carboxylates. We therefore sought to determine if HOBt esters might be reagents that could indicate the presence of carboxylate groups in polypeptide cations. If acidic side-chains or the C-terminus were engaged in a zwitterionic interaction, the nucleophilicity of the site might be sufficient to displace HOBt and form an anhydride. Further interrogation of the modified polypeptide could then provide information regarding sites involved in the zwitterionic interaction.
Experimental
Materials.
Hydroxybenzotriazole hydrate (3-carboxypropyl) trimethylammonium chloride, acetyl chloride, and sodium 3-sulfobenzoate were purchased from Sigma Aldrich (St. Louis, MO, USA) 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) and N, N′-dicyclohexyl carbodiimide (DCC) were purchased from Thermo Fisher Scientific (Rockford, IL, USA). The peptide AADAADAA was custom synthesized by NeoBioLab (Cambridge, MA, USA). The custom peptides YGRAR and YRARG were purchased from GenScript (Piscataway, NJ, USA). Methanol and dimethylformamide (DMF) were purchased from Mallinckrodt (St. Louis, MO, USA). Water (HPLC grade), ammonium hydroxide (ACS grade) and acetonitrile (Optima grade) were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Glacial acetic acid (ACS grade) was purchased from Macron fine chemicals (Center Valley, PA) All peptide solutions were prepared with concentrations of approximately 1 mg/mL and diluted to ~100 μg/mL. YRARG and YGRAR were prepared in 50:50 (v/v) water/methanol. Ac-AADAADAA-Ome solution was prepared in 49.5/49.5/1 (v/v/v) methanol/water/ammonium hydroxide.
Reagent synthesis.
The hydroxybenzotriazole ester of 4-trimethylammonium butyrate (TMAB-HOBt) was prepared from a modified method based on synthesis of [3-(2,5)-dioxopyrrolidin-1-yloxycarbonyl)-propyl] trimethyllammonium.[28] 1.3 mg of (3-carboxypropyl) trimethylammonium chloride, 0.9 mg of HOBt, and 1.7 mg of DCC were dissolved in 1 mL acetonitrile each. 10 μL aliquot of each were combined and diluted 100× in acetonitrile. Sulfobenzoyl HOBt was prepared as previously shown.[27] The group conjugated to HOBt for a particular reagent ion was chosen to attach to the analyte ion polarity of interest. Sulfonate is appropriate for reactions with cations whereas TMAB is appropriate for attachment to anions.
Peptide methyl esterification.
Peptide methyl esterification was performed as described previously.[29] 2 M hydrochloric acid in dry methanol was prepared by combining 40 μL of acetyl chloride and 250 μL of dry methanol. 1 mg of YGRAR was dissolved in 100 μL of this solution and allowed to react for 2 hr. The solution was lyophilized and reconstituted to make a peptide solution as described above.
Carboxyl O18 Labeling.
O18 labeling was performed according to a previously published procedure.[30] Briefly, 1 mg of peptide was dissolved in 200 uL H218O with 1% (v/v) trifluoroacetic acid. The solution was allowed to react at room temperature for approximately 3 days and lyophilized to dryness. The peptide was reconstituted in 50:50 (v/v) water/methanol and diluted to a final concentration of 100 μg/mL.
Mass Spectrometry.
Experiments were performed using a QTRAP 4000 hybrid triple quadrupole/linear ion trap or a TripleTOF 5600 System (SCIEX, Concord, ON, Canada). Both instruments were previously modified to perform ion/ion reactions.[31,32] Anions and cations were sequentially injected via alternatively pulsed nano-electrospray ionization (nESI). First, cations are injected, isolated in Q1, and transferred and stored in q2. Next, reagent ions are injected, isolated in Q1, and transferred to q2.[33] The ions were mutually stored in the q2 reaction cell for 100-1000 ms.[31,34] In the case of experiments performed on the QTRAP, reaction products formed in q2 were transferred to Q3 where they were subjected to MSn using resonance excitation ion-trap CID and analyzed by mass selective axial ejection (MSAE).[35,36] For experiments performed on the TripleTOF 5600, products were subjected to CID in q2. Fragment ions were back transferred to q1 for isolation, sequentially transferred to q2, and mass analyzed via orthogonal time-of-flight (TOF).
Results and Discussion
We begin by demonstrating the reactivity of HOBt esters with carboxylate groups using the same model peptide anion that was used to establish carboxylate reactivity with NHS esters. We then describe results based on the model peptide YGRAR to examine the reactivity of HOBt esters with carboxylate groups that might be engaged in zwitterionic structures in a polypeptide cation. We finish by describing results obtained with a small model protein (i.e., bovine ubiquitin) to provide evidence for the presence and location of a carboxylate in a multiply protonated polypeptide system.
HOBt-TMAB & Ac-ADAADAA-Ome.
The doubly deprotonated peptide Ac-ADAADAA-Ome was reacted with the synthesized TMAB-HOBt reagent cation to demonstrate HOBt reactivity towards carboxylates in the gas phase. The peptide and reagent formed a long-lived electrostatic complex [M-2H+TMAB HOBt]− shown in Figure 1a. When the complex was subjected to CID, neutral HOBt (135 Da) was lost (Figure 1b), analogous to the loss of NHS from reactions with the TMAB-NHS ester cation described previously.[27]The newly formed species [M-2H+TMAB]− species was subjected to collisional activation, which resulted in production of [M-H-H2O]− shown in Figure 1c. The loss of HOBt indicates that a covalent reaction between the HOBt ester and the polypeptide anion takes place. (Note that quaternary ammonium cations react with carboxylates via alkyl cation transfer [37]. The absence of evidence for either proton or alkyl cation transfer indicates that the covalent reaction leading to loss of HOBt is, by far, the dominant process.) Fragmentation of the [M-2H+TMAB]− anion generated from loss of HOBt to yield the [M-H2O]− product is fully consistent with the generation of an anhydride upon reaction with a carboxylate and the TMAB HOBt ester, as illustrated in Scheme 1.
Figure 1:
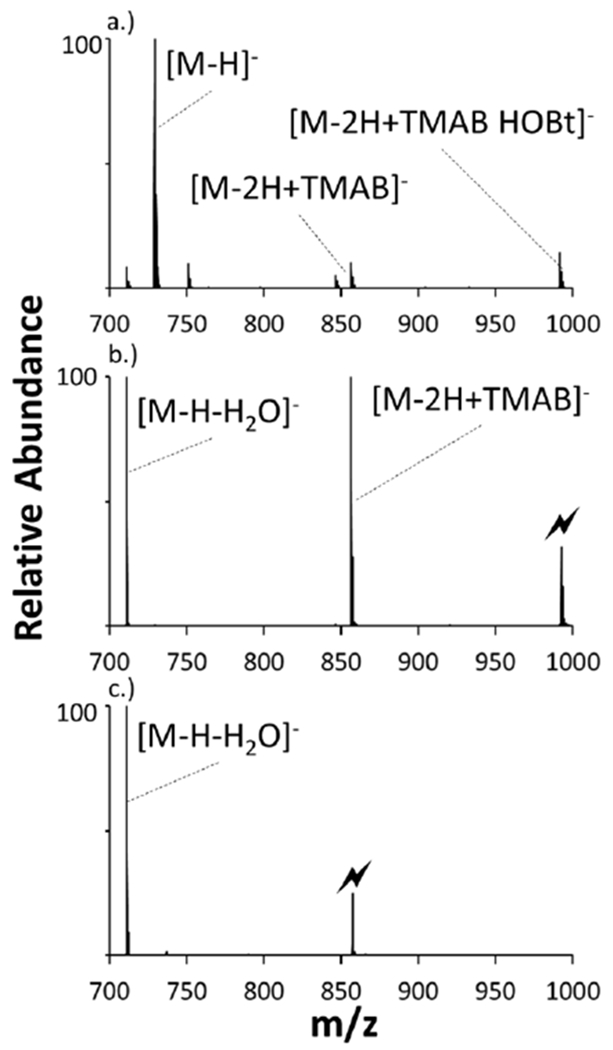
Product ion spectra derived from (a) Ion-ion reaction between [Ac-ADAADAA-Ome-2H]− and [TMAB-HOBt]+ (b) CID of complex [M-2H+(TMAB-HOBt)− (c) CID of [M-2H+TMAB]− Formation of the [M-H-H2O]− (indicated by the blue outline).
Scheme 1:

Reaction between the carboxylate group in a doubly-deprotonated Ac-AADAADAA-Ome with deprotonation at both the aspartic acid side chains with a positively charged TMAB HOBt
Sulfobenzoyl HOBt and ions derived from YGRAR.
The pentapeptide YGRAR was chosen as a model system because it has two arginine residues and a single carboxylic acid group (i.e., the C-terminus) that can engage in the formation of a salt bridge or zwitterion. In a cold-ion UV-IR spectroscopy study that will be described in detail elsewhere, the dominant conformer of singly protonated YGRAR has been shown to lack a carboxylic acid group by virtue of the absence of the signature O-H stretch (free/hydrogen bonded) in the hydride stretch region, the absence of a carboxylic acid C═O stretch in the amide I region, and the presence of the anti-symmetric COO− stretch in the amide II region.[38] This suggests that the C-terminus in this ion is deprotonated. The major conformer of doubly protonated YGRAR, on the other hand, showed the signature absorbances of carboxylic acid. We therefore focused our attention on the reaction of deprotonated sulfobenzoyl HOBt with doubly protonated YGRAR. Attachment of an anion to doubly-protonated YGRAR results in a net singly charged ion population that can be comprised of a combination of structures with different charge partitioning, some of which can contain zwitterionic structures.
The potential nucleophilic sites within a complex comprised of deprotonated sulfobenzoyl HOBt (SB-HOBt) and doubly protonated YGRAR that can react with the HOBt ester include unprotonated basic sites (i.e., the N-terminus or either of the guanidine groups) and a carboxylate group, if present. The ion/ion product ion spectra for the reaction of deprotonated SB-HOBt and all doubly protonated versions of YGRAR (i.e., unmodified, C-terminally 18O-labelled, methyl esterified, and N-terminally acetylated) showed abundant complex formation (e.g., [YGRAR+2H+SB-HOBt]+) as well as single proton transfer to yield the [M+H]+ ion (see Figure S-1 for the post-ion/ion reaction spectra for the four forms of YGRAR mentioned above). Figure 2 compares the CID spectra of the SB-HOBt complexes generated with doubly protonated versions of YGRAR (Figure 2(a)), methyl esterified YGRAR (YGRAR-OMe, Figure 2(b)), C-terminal 18O-labelled YGRAR (indicated as 18O-YGRAR with both C-terminal oxygen atoms exchanged for 18O with isolation of this ion prior to ion/ion reaction shown in Figure S-2, Figure 2(c)), and N-terminally acetylated YGRAR (ac-YGRAR, Figure 2(d)). All four complexes show loss of intact neutral SB-HOBt, which reflects complexes in the activated population that underwent proton transfer, as opposed to covalent reaction. In the cases of YGRAR, 18O-YGRAR, and YGRAR-OMe, however, there is also an abundant product generated by loss of HOBt, which reflects covalent bond formation in the complex. In the case of ac-YGRAR, on the other hand, no evidence for loss of HOBt is apparent. In ac-YGRAR, there are two nominal basic sites (i.e., the two arginine side-chains). In the doubly protonated species, both arginines are expected to be ionized. The sulfonate group of SB-HOBt is expected to form a strong electrostatic interaction with one of these sites. In order for the complex to have a net positive charge, an excess proton must be present on the other arginine and the C-terminus must also be protonated. Hence, there are no available nucleophilic sites to react with the HOBt ester (i.e., the C-terminus and one arginine are protonated and the other arginine is involved with the electrostatic interaction with the sulfonate group). Hence, no nucleophilic displacement of HOBt can occur to give rise to a covalent reaction.
Figure 2:
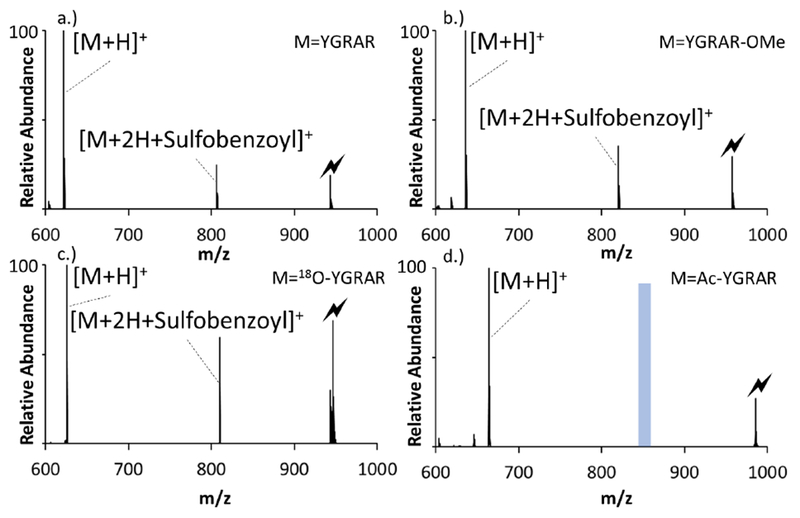
Ion trap CID of the complexes [M+2H+SB-HOBt]+ generated via ion/ion reactions between deprotonated SB-HOBt and doubly protonated (a) YGRAR, (b) YGRAR-OMe,(c) O18-YGRAR, and (d) ac-YGRAR. The lightning bolts indicate the precursor ion subjected to collisional activation and the blue shaded bar in 2d.) indicates the region of the mass scale that the loss of HOBt would be observed if it occurred.
Unmodified YGRAR has three nominal basic sites (i.e., the N-terminus and the two guanidine side-chains of the arginine residues) and one acidic site (i.e., the C-terminus). Upon attachment of a negatively charged sulfobenzoyl reagent to a doubly-charged YGRAR, there are nine ways to partition charge among the acidic and basic sites to result in a singly charged ion. Three conformers involve the protonation of all of the basic sites, with one being associated with the sulfonate group of the reagent and the C-terminus being deprotonated. The six remaining conformers do not involve zwitterion formation (i.e., the C-terminus is neutral, see Scheme S-1). (The same situation prevails for the O18-labelled peptide.) In the case of the methyl-esterified peptide, only the six non-zwitterionic combinations are possible. In all of the non-zwitterionic systems, one unprotonated basic site (i.e., unprotonated N-terminus or an unprotonated arginine residue) is available to react with the ester. In the case of the zwitterionic systems, the only nominal nucleophile available to react is the deprotonated C-terminus. Figure 3 compares the ion trap CID spectra of the covalently modified ions derived from YGRAR (Figure 3(a)) and YGRAR-OMe (Figure 3(b)). The two spectra are similar in that the major product ions are consistent with covalently modified b3 and b4
and b4 ions (the
ions (the  indicates an increase in mass of 183 Da) which suggests covalent modification at either the N-terminus or the central arginine in the peptide. Given the lower proton affinity of the N-terminus relative to the guanidine side-chain of arginine, it is likely that the main reactive site is the N-terminus. Furthermore, acylation of arginine leads to a dominant loss of 35 Da via a concerted loss of ammonia and water [39] when the covalent adduct lacks an alpha-hydrogen, as is the case here. There is a relatively small signal indicating the loss of 35 Da in the spectra of Figure 3. There is also a mechanism for the loss of the adduct plus 42 Da from peptides with acylated arginines [37], which is not observed in these peptides. Hence, relatively little reactivity associated with the arginine residues is apparent in either spectrum of Figure 3. A significant difference between the two spectra is the appearance of a product associated with the loss of 202 Da, corresponding to [M+H-H2O]+, for YGRAR but not for YGRAR-OMe. The analogous experiment performed with modified O18-YGRAR (Figure S-3) showed a loss of 204 Da, which indicates that one of the oxygen atoms associated with the neutral loss arises from the C-terminus. The reaction between SB-HOBt and the carboxylate group of the C-terminus is shown schematically in Scheme 2.
indicates an increase in mass of 183 Da) which suggests covalent modification at either the N-terminus or the central arginine in the peptide. Given the lower proton affinity of the N-terminus relative to the guanidine side-chain of arginine, it is likely that the main reactive site is the N-terminus. Furthermore, acylation of arginine leads to a dominant loss of 35 Da via a concerted loss of ammonia and water [39] when the covalent adduct lacks an alpha-hydrogen, as is the case here. There is a relatively small signal indicating the loss of 35 Da in the spectra of Figure 3. There is also a mechanism for the loss of the adduct plus 42 Da from peptides with acylated arginines [37], which is not observed in these peptides. Hence, relatively little reactivity associated with the arginine residues is apparent in either spectrum of Figure 3. A significant difference between the two spectra is the appearance of a product associated with the loss of 202 Da, corresponding to [M+H-H2O]+, for YGRAR but not for YGRAR-OMe. The analogous experiment performed with modified O18-YGRAR (Figure S-3) showed a loss of 204 Da, which indicates that one of the oxygen atoms associated with the neutral loss arises from the C-terminus. The reaction between SB-HOBt and the carboxylate group of the C-terminus is shown schematically in Scheme 2.
Figure 3:

Ion trap CID of the covalent adducts of (a) YGRAR and (b) YGRAR-OMe. The area shaded in blue indicates where a [M-H2O]+ ion should appear.
Scheme 2:

Reaction between the carboxylate group in a doubly-charged YGRAR with protonation at the N-terminus and both arginine residues with deprotonated SB-HOBt.
The data of Figure 3 are consistent with unique reactivity for YGRAR relative to YGRAR-OMe with SB-HOBt. However, water loss from a peptide ion is hardly unusual and is generally less likely from a methyl-esterified peptide in any case. For this reason, we compared the CID spectrum of the nominal [M+H-H2O]+ ion generated via the ion/ion reaction process associated with Figure 3(a) (designated [M+H-H2O]+i/i rxn ) with that of the [M+H-H2O]+ ion generated from water loss upon CID of singly protonated unmodified YGRAR (see Figure 4b) (designated[M+H-H2O]+CID).
Figure 4:

Ion trap product ion spectra obtained from the nominal [M+H-H2O]+i/i rxn a) and b) [M+H-H2O]+CID.
Interestingly, the [M+H-H2O]+i/i rxn species fragments almost exclusively via the loss of 43 Da under ion trap CID conditions, whereas the [M+H-H2O]+CID ion shows somewhat more diverse fragmentation with the dominant process leading to loss of 42 Da. The latter neutral loss species corresponds to carbodiimide (i.e., HN═C═NH). The loss of this species has previously been proposed to arise following water loss from peptide ions with a C-terminal arginine. [40] Scheme 3 reproduces the mechanism proposed for loss of carbodiimide following water loss from a peptide ion with arginine at the C-terminus.
Scheme 3:
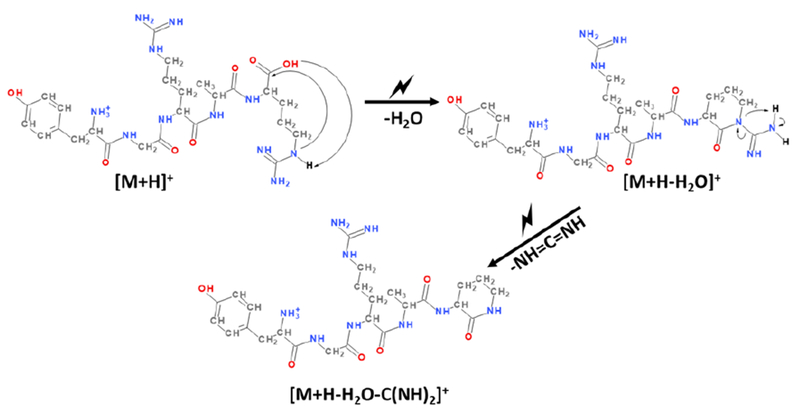
Process leading to loss of carbodiimide following water loss from a protonated peptide with a C-terminal arginine.
The loss of 43 Da from the [M-H2O]+ ion derived from the ion/ion reaction process corresponds to the loss of isocyanic acid (HN═C═O). A prominent loss of isocyanic acid has been reported to occur from peptide ions that have undergone citrullation.[41] In this case, we propose that this loss results from a rearrangement process involving a C-terminal arginine and the anhydride linkage at the C-terminus arising from the ion/ion reaction, as summarized in Scheme 4.
Scheme 4:

Process leading to the formation of the [M-H2O]+ ion from the ion/ion covalent reaction followed by loss of isocyanic acid.
Sulfobenzoyl HOBt & YRARG.
The processes of Schemes 3 and 4 are expected to be most likely when an arginine residue is present at the C-terminus and the oxygen of the water loss comes from the C-terminus. Experiments analogous to those described above for YGRAR were carried out for YRARG to reduce the likelihood for either NH═C═NH or HN═C═O loss. Doubly protonated YRARG was subjected to ion/ion reaction with deprotonated SB-HOBt resulting in proton transfer and in the formation of an electrostatic complex, [YRARG+2H+Sulfobenzoyl HOBt]+, (see Figure S-4(a)). This complex was subjected to CID forming the modified peptide [YRARG+2H+Sulfobenzoyl]+ (see Figure S-4(b)). CID of [YRARG+2H+Sulfobenzoyl]+ complex generated a nominal [M+H-H2O]+ product along with many others, most of which are consistent with a reaction at the N-terminus. The formation of the [M+H-H2O]+ product suggests that at least some of the precursor ion population was composed of a zwitterion and that the carboxylate group reacted with SB-HOBt to generate an anhydride linkage at the C-terminus. Figure 5 compares the CID spectrum of the [M+H-H2O]+ ion derived from the ion/ion reaction process (designated [M+H-H2O]+i/i rxn) (Figure 5(a)) with that of the water loss product derived from CID of the [M+H]+ ion (designated [M+H-H2O]+CID) (Figure 5(b)). In this case, essentially no loss of 43 Da is observed from the ion/ion reaction product, in contrast with the analogous ion derived from YGRAR described above. The spectrum includes an abundant b3 ion and an abundant b4-42 ion, the latter of which is likely to arise from deguanidination of the residue 4 arginine to generate an ornithine side-chain followed by cleavage C-terminal to the ornithine residue [42]. This spectrum is quite distinct from that of the [M+H-H2O]+CID ion. While many of the product ions are held in common, the relative abundances are dramatically different, which indicates that these isomeric ions are of different structure or are comprised of mixtures of structures with different composition. We note that different mechanisms for the generation of [M+H-H2O]+ ions could, in principle, lead to a common structure or mixture of structures. However, at least in the cases of YGRAR and YRARG, the different mechanisms for nominal water loss from the precursor peptide do not lead to the same structure of set of structures.
Figure 5:

Comparison of ion trap CID product ion spectra of the (a) [M+H-H2O]+i/i rxn and (b) [M+H-H2O]+CID.
Sulfobenzoyl HOBt & Ubiquitin [M+7H]7+.
The preceding results indicate that an ion/ion reaction can be sensitive to the presence of a carboxylate group in a net positively charged polypeptide ion. For the small model systems, however, the attachment of a sulfonate group to the cation could play a major role in determining the charge distribution within the ion/ion electrostatic complex. As the polypeptide ion increases in size and charge, the perturbation associated with attachment of the reagent anion is expected to decrease in relative importance. We therefore examined SB-HOBt chemistry with multiply-protonated ubiquitin ions and summarize here the results derived from examination of the [M+7H]7+ ion. The reaction of singly deprotonated SB-HOBt with the ubiquitin [M+7H]7+ ion resulted in the attachment of the reagent anion to the protein ion as the dominant process (Figure S-6(a)) and CID of the electrostatic complex predominantly gave rise to HOBt loss to give [M+7H+sulfobenzoyl]6+ with a minor degree of proton transfer to give [M+6H]6+ (Figure S-6(b)). Ion trap CID of the covalently modified protein (i.e., the [M+7H+sulfobenzoyl]6+ ion) yielded a variety of b- and y-type ions with and without the covalent adduct, as expected for modifications at neutral basic sites (Figure S-7). However, the base product ion peak corresponded to [M+6H-H2O]6+ with very little formation of [M+6H]6+. Figure S-8 provides a full ‘butterfly’ plot of the ion trap CID product ion spectra of the [M+6H-H2O]6+ ion derived from the ion/ion reaction process (positive abundance axis designated [M+6H-H2O]6+i/i rxn) and the [M+6H-H2O]6+ ion derived from ion trap CID (negative abundance axis designated [M+6H-H2O]6+CID). While both ion populations are comprised of a mixture of structures, there are many similarities in the spectra and a few notable differences. The identities and relative abundances of most of the product ions in the two spectra are very similar. The differences between the two spectra are highlighted in Figure 6, which displays a subset of the data displayed in Figure S-8. By examining complementary ion pairs, it is possible to narrow down the origins of the sites of water loss. For example, in the case of the [M+6H-H2O]6+CID the complementary ion pair from cleavage of the D39-Q40 amide linkage is in the form of y37-H2O/b39, as opposed to y37/b39-H2O. For the complementary pair from D58-Y59 amide bond cleavage, on the other hand, the dominant combination is y18/b58-H2O rather than y18-H2O/b58. This places the origin of the water loss to be largely arising from the region spanned by residues Q40-D58 (see Scheme 5). Comparable abundances of b52 and b52-H2O ions are observed, which is consistent with water loss taking place from more than one site but largely within the residue Q40-D58 span. The results for the [M+6H-H2O]6+i/i rxn is fully consistent with little water loss up to residue 40. However, comparable contributions from y18/b58-H2O and y18-H2O/b58 indicate that there is a water loss site beyond residue D58 in many of the ions derived from ion/ion reaction. Of particular note is the presence of y12 ions (with very little y12-H2O) in both plots and a y13-H2O ion in the ion/ion reaction product data that is absent in the data from CID of the water loss peak derived directly from the [M+6H]6+ precursor. These results suggest that some of the ions lost a water molecule from residue 64, a glutamic acid. The findings described above for the model systems taken collectively with the comparison of Figure 6 lead us to hypothesize that the glutamic acid of residue 64 is reactive with SB-HOBt, at least for some of the structures in the precursor ion population. For the reactive structure(s), the glutamic acid at position 64 is thus likely to exist with a negative charge possessing sufficient nucleophilicity to lead to covalent reaction. We note that there may be other carboxylates within the ion that are not accessible to the reactive site of the reagent due to the restricted number and locations of reagent attachment sites.
Figure 6:

Comparison of ion trap CID of [M+6H-H2O]6+i/i rxn (top) and ion trap CID of the [M+6H-H2O]6+CID (bottom). (M = Ubiquitin) Water loss ions with major differences in relative abundances in the two plots are labeled in blue font. All other water loss ions are labeled in red font.
Scheme 5:
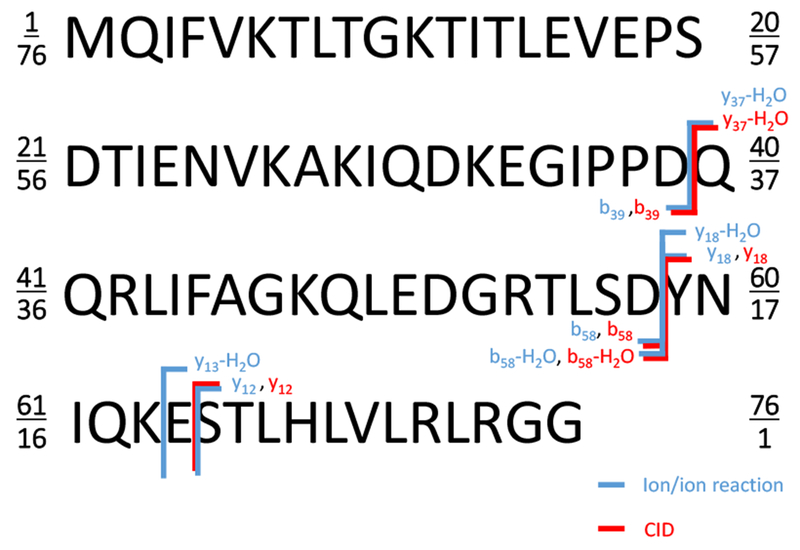
Ubiquitin sequence showing key products from CID of the [M+6H-H2O]6+ ion generated via the ion/ion reaction (blue font) and [M+6H-H2O]6+ ions formed from CID of the [M+6H]6+ precursor ion (red font).
Conclusions
The carboxylate group has been shown to react with triazole esters in the gas phase, in analogy with N-hydroxysuccinimide esters, via the reaction of the TMAB-HOBt cation with dianions of Ac-ADAADAA-Ome. A labile anhydride linkage is formed between the carboxylate and the triazole ester. When a triazole ester is linked to a readily ionized group, it can be used as a reagent to covalently modify a carboxylate-containing ion of opposite charge. In this work, we explored the possibility that a triazole ester ion might react with a cation that contains a carboxylate group. Such a scenario nominally prevails in zwitterionic/salt bridged polypeptide cations. The nucleophilicity of a carboxylate group engaged in an interaction with one or more cations, however, may be significantly lower than that of a carboxylate in a net negative ion. Nevertheless, we demonstrate here that there is some reactivity using the dications of a series of modified and unmodified ions derived from the peptide YGRAR and deprotonated SB-HOBt. Upon CID of a peptide cation that is covalently modified at a carboxylate group, the anhydride linkage cleaves to generate a nominal [M+nH-H2O]n+ ion. The structure or mixture of structures that comprise this ion population may very well differ from the structure or mixture of water loss structures generated via CID of the corresponding [M+nH]n+ ion because the mechanisms of formation differ. The CID spectrum of [M+nH-H2O]n+ peak can be used to localize the site(s) of modification. The reaction of deprotonated SB-HOBt with the [M+7H]7+ ion of ubiquitin and subsequent CID of the [M+6H-H2O]6+i/i rxn ion led to the conclusion of carboxylate location at the glutamic acid at residue 64. The results indicate the potential for triazole esters as chemical probes that can be used to complement other approaches for the study of gas-phase zwitterion/salt bridge structures.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (NIH) under Grant GM R37-45372.
References
- 1.Ding Y, Krogh-Jespersen K: The glycine zwitterion does not exist in the gas phase: results from a detailed ab initio electronic structure study. Chemical Physics Letters. 199, 261–266 (1992). doi: 10.1016/0009-2614(92)80116-S [DOI] [Google Scholar]
- 2.Prell JS, O’Brien JT, Steill JD, Oomens J, Williams ER: Structures of Protonated Dipeptides: The Role of Arginine in Stabilizing Salt Bridges. J. Am. Chem. Soc. 131, 11442–11449 (2009). doi: 10.1021/ja901870d [DOI] [PubMed] [Google Scholar]
- 3.Mertens LA, Marzluff EM: Gas Phase Hydrogen/Deuterium Exchange of Arginine and Arginine Dipeptides Complexed with Alkali Metals. J. Phys. Chem. A. 115, 9180–9187 (2011). doi: 10.1021/jp204896z [DOI] [PubMed] [Google Scholar]
- 4.Hiserodt RD, Brown SM, Swijter DFH, Hawkins N, Mussinan CJ: A Study of b1+H2O and b1-Ions in the Product Ion Spectra of Dipeptides Containing N-Terminal Basic Amino Acid Residues. Journal of the American Society for Mass Spectrometry. 18, 1414–1422 (2007). doi: 10.1016/j.jasms.2007.04.018 [DOI] [PubMed] [Google Scholar]
- 5.Popa V, Trecroce DA, McAllister RG, Konermann L: Collision-Induced Dissociation of Electrosprayed Protein Complexes: An All-Atom Molecular Dynamics Model with Mobile Protons. J. Phys. Chem. B. 120, 5114–5124 (2016). doi: 10.1021/acs.jpcb.6b03035 [DOI] [PubMed] [Google Scholar]
- 6.Jenner M, Ellis J, Huang W-C, Lloyd Raven E, Roberts GCK, Oldham NJ: Detection of a Protein Conformational Equilibrium by Electrospray Ionisation- Ion Mobility- Mass Spectrometry. Angewandte Chemie International Edition. 50, 8291–8294 (2011). doi: 10.1002/anie.201101077 [DOI] [PubMed] [Google Scholar]
- 7.Bonner J, Lyon YA, Nellessen C, Julian RR: Photoelectron Transfer Dissociation Reveals Surprising Favorability of Zwitterionic States in Large Gaseous Peptides and Proteins. J. Am. Chem. Soc. 139, 10286–10293 (2017). doi: 10.1021/jacs.7b02428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kjeldsen F, Silivra OA, Zubarev RA: Zwitterionic States in Gas-Phase Polypeptide Ions Revealed by 157-nm Ultra-Violet Photodissociation. Chem. Eur. J. 12, 7920–7928 (2006). doi: 10.1002/chem.200600248 [DOI] [PubMed] [Google Scholar]
- 9.Jockusch RA, Price WD, Williams ER: Structure of Cationized Arginine (Arg M+, M = H, Li, Na, K, Rb, and Cs) in the Gas Phase: Further Evidence for Zwitterionic Arginine. J. Phys. Chem. A. 103, 9266–9274 (1999). doi: 10.1021/jp9931307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Price WD, Jockusch RA, Williams ER: Is Arginine a Zwitterion in the Gas Phase? J Am Chem Soc. 119, 11988–11989(1997). doi: 10.1021/ja9711627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ling S, Yu W, Huang Z, Lin Z, Harañczyk M, Gutowski M: Gaseous Arginine Conformers and Their Unique Intramolecular Interactions. J. Phys. Chem. A. 110, 12282–12291 (2006). doi: 10.1021/jp0645115 [DOI] [PubMed] [Google Scholar]
- 12.Marchese R, Grandori R, Carloni P, Raugei S: On the Zwitterionic Nature of Gas-Phase Peptides and Protein Ions. PLOS Computational Biology. 6, el000775 (2010). doi: 10.1371/journal.pcbi.1000775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaeqx S, Oomens J, Rijs AM: Gas-phase salt bridge interactions between glutamic acid and arginine. Phys. Chem. Chem. Phys. 15, 16341–16352 (2013). doi: 10.1039/C3CP52508B [DOI] [PubMed] [Google Scholar]
- 14.Scarff CA, Thalassinos K, Hilton GR, Scrivens JH: Travelling wave ion mobility mass spectrometry studies of protein structure: biological significance and comparison with X-ray crystallography and nuclear magnetic resonance spectroscopy measurements. Rapid Commun. Mass Spectrom. 22, 3297–3304 (2008). doi: 10.1002/rcm.3737 [DOI] [PubMed] [Google Scholar]
- 15.Covey T, Douglas DJ: Collision cross sections for protein ions. J Am Soc Mass Spectrom. 4, 616–623 (1993). doi: 10.1016/1044-0305(93)85025-S [DOI] [PubMed] [Google Scholar]
- 16.Clemmer DE, Hudgins RR, Jarrold MF: Naked Protein Conformations: Cytochrome c in the Gas Phase. J. Am. Chem. Soc. 117, 10141–10142 (1995). doi: 10.1021/ja00145a037 [DOI] [Google Scholar]
- 17.Wyttenbach T, Bowers MT: Structural Stability from Solution to the Gas Phase: Native Solution Structure of Ubiquitin Survives Analysis in a Solvent-Free Ion Mobility-Mass Spectrometry Environment. J. Phys. Chem. B. 115, 12266–12275 (2011). doi: 10.1021/jp206867a [DOI] [PubMed] [Google Scholar]
- 18.Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT: Collision Cross Sections of Proteins and Their Complexes: A Calibration Framework and Database for Gas-Phase Structural Biology. Anal. Chem. 82, 9557–9565 (2010). doi: 10.1021/acl022953 [DOI] [PubMed] [Google Scholar]
- 19.Silveira JA, Fort KL, Kim D, Servage KA, Pierson NA, Clemmer DE, Russell DH: From Solution to the Gas Phase: Stepwise Dehydration and Kinetic Trapping of Substance P Reveals the Origin of Peptide Conformations. J. Am. Chem. Soc. 135, 19147–19153 (2013). doi: 10.1021/ja4114193 [DOI] [PubMed] [Google Scholar]
- 20.Pagel K, Natan E, Hall Z, Fersht AR, Robinson CV: Intrinsically Disordered p53 and Its Complexes Populate Compact Conformations in the Gas Phase. Angew. Chem. Int. Ed. 52, 361–365 (2013). doi: 10.1002/anie.201203047 [DOI] [PubMed] [Google Scholar]
- 21.Skinner OS; McLafferty FW; Breuker K: How ubiquitin unfolds after transfer into the gas phase. J. Am. Soc. Mass Spectrom, 23 (2012) 1011–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z; Browne SJ; Vachet RW: Exploring Salt Bridge Structures of Gas-Phase Protein Ions using Multiple Stages of Electron Transfer and Collision Induced Dissociation. J. Am. Soc. Mass Spectrom, 25 (2014) 604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z; Vachet RW: Gas-phase protein salt bridge stabilities from collisional activation and electron transfer dissociation. Int. J. Mass Spectrom, 420 (2017) 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mentinova M; McLuckey SA: Covalent Modification of Gaseous Peptide Ions with N-Hydroxysuccinimide Ester Reagent Ions. J. Am. Chem. Soc, 132 (2010) 18248–18257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGee WM; Mentinova M; McLuckey SA: Gas-Phase Conjugation to Arginine Residues in Polypeptide Ions via N-Hydroxysuccinimide Ester-based Reagent Ions. J. Am. Chem. Soc, 134(2012) 11412–11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng Z, McGee WM, Bu J, Barefoot NZ, McLuckey SA: Gas Phase Reactivity of Carboxylates with N-Hydroxysuccinimide Esters. J. Am. Soc. Mass Spectrom. 26, 174–180 (2015). doi: 10.1007/sl3361-014-1002-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bu J, Peng Z, Zhao F, McLuckey SA: Enhanced Reactivity in Nucleophilic Acyl Substitution Ion/Ion Reactions Using Triazole-Ester Reagents. J. Am. Soc. Mass Spectrom. 28, 1254–1261 (2017). doi: 10.1007/sl3361-017-1613-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mirzaei H, Regnier F: Enhancing Electrospray Ionization Efficiency of Peptides by Derivatization. Anal. Chem. 78, 4175–4183 (2006). doi: 10.1021/ac0602266 [DOI] [PubMed] [Google Scholar]
- 29.Peng Z, Pilo AL, Luongo CA, McLuckey SA: Gas-Phase Amidation of Carboxylic Acids with Woodward’s Reagent K Ions. J. Am. Soc. Mass Spectrom. 26, 1686–1694 (2015). doi: 10.1007/sl3361-015-1209-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niles R, Witkowska HE, Allen S, Hall SC, Fisher SJ, Hardt M: Acid-Catalyzed Oxygen-18 Labeling of Peptides. Anal. Chem. 81, 2804–2809 (2009). doi: 10.1021/ac802484d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hager JW: A new linear ion trap mass spectrometer. Rapid Commun. Mass Spectrom. 16, 512–526 (2002). doi: 10.1002/rcm.607 [DOI] [PubMed] [Google Scholar]
- 32.Xia Y, Chrisman PA, Erickson DE, Liu J, Liang X, Londry FA, Yang MJ, McLuckey SA: Implementation of Ion/Ion Reactions in a Quadrupole/Time-of-Flight Tandem Mass Spectrometer. Anal. Chem. 78, 4146–4154 (2006). doi: 10.1021/ac0606296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xia Y, Liang X, McLuckey SA: Pulsed dual electrospray ionization for In/In reactions. J Am Soc Mass Spectrom. 16, 1750–1756 (2005). doi: 10.1016/j.jasms.2005.07.013 [DOI] [PubMed] [Google Scholar]
- 34.Xia Y, Wu J, McLuckey SA, Londry FA, Hager JW: Mutual storage mode ion/ion reactions in a hybrid linear ion trap. J Am Soc Mass Spectrom. 16, 71–81 (2005). doi: 10.1016/j.jasms.2004.09.017 [DOI] [PubMed] [Google Scholar]
- 35.Louris JN, Cooks RG, Syka JEP, Kelley PE, Stafford GC, Todd JFJ: Instrumentation, applications, and energy deposition in quadrupole ion-trap tandem mass-spectrometry. Anal. Chem. 59, 1677–1685 (1987). [Google Scholar]
- 36.Londry FA, Hager JW: Mass selective axial ion ejection from a linear quadrupole ion trap. J Am Soc Mass Spectrom. 14, 1130–1147 (2003). doi: 10.1016/S1044-0305(03)00446-X [DOI] [PubMed] [Google Scholar]
- 37.Gilbert JD, Prentice BM, McLuckey SA: Ion/Ion Reactions with “onium” Reagents: An Approach for the Gas-phase Transfer of Organic Cations to Multiply-Charged Anions. J. Am. Soc. Mass Spectrom, 29, 818–825 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harrilal Christopher. “Investigating Electronic and Structural Changes Imposed by Zwitterionic Pairing in Model Peptides Systems Using IR-UV Double Resonance Spectroscopy.” 66th ASMS Conference on Mass Spectrometry and Allied Topics, American Society for Mass Spectrometry. San Diego Convention Center, CA, June, 2018. [Google Scholar]
- 39.McGee WM, McLuckey SA: Gas Phase Dissociation Behavior of Acyl-Arginine Peptides. Int. J. Mass Spectrom, 354–355, 181–187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deery MJ; Summerfield SG; Buzy A; Jennings KR: A Mechanism for the Loss of 60 u from Peptides Containing an Arginine Residue at the C-Terminus. J. Am. Soc. Mass Spectrom. 8, 253–261(2009). [Google Scholar]
- 41.Hao G, Wang D, Gu J, Shen Q, Gross SS, Wang Y: Neutral loss of isocyanic acid in peptide CID spectra: a novel diagnostic marker for mass spectrometric identification of protein citrullination. J Am Soc Mass Spectrom. 20, 723–727 (2009). doi: 10.1016/j.jasms.2008.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGee WM; S.A. McLuckey SA: “The Ornithine Effect in Peptide Cation Dissociation.” J. Mass Spectrom, 48, 856–861 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.