

Abstract
Cancer biology research over recent decades has given ample evidence for the existence of self-renewing and drug-resistant populations within heterogeneous tumors, widely recognized as cancer stem cells (CSCs). However, a lack of clear understanding about the origin, existence, maintenance, and metastatic roles of CSCs limit efforts towards the development of CSC-targeted therapy. In this review, we describe novel avenues of current CSC biology. In addition to cell fusion and horizontal gene transfer, CSCs are originated by mutations in somatic or differentiated cancer cells, resulting in de-differentiation and reprogramming. Recent studies also provided evidence for the existence of distinct or heterogeneous CSC populations within a single heterogeneous tumor. Our analysis of the literature also opens the doors for a novel hypothesis that CSC populations with specific phenotypes, metabolic profiles, and clonogenic potential metastasize to specific organs.
Keywords: Cancer stem cells, metabolic reprogramming, de-differentiation
1. Introduction
Why does a tumor relapse after its initial remission? The quest for an answer to this question led in 1959 to the derivation of the term “tumor stem cells” [1]. Tumors comprise a heterogeneous cell population, with 0.1% to 0.8% of these tumor cells being cancer stem cells (CSCs) [2]. Research on CSCs was launched for the first time in 1994, when Lapidot and colleagues observed in primary human acute myeloid leukemia (AML) that a small subpopulation of cells, CD34+CD38-, initiate tumor in severe combined immune deficient mice (SCID) [3]. In the light of this evidence, following studies from 1994 to date investigated for the presence of CSCs or tumor initiating cells in various cancers and observed that a small population of drug-resistant, tumor-initiating, and stemness-activated CSCs are present in almost all cancer types. Lineage tracing experiments by three independent groups in 2012 further fueled recognition of the existence of CSCs [4–6].
1.1. Relevance of CSCs in cancer initiation
CSCs differentiate into self-renewing cells and differentiated cells that make up the entire bulk of the tumor [7]. According to the CSC hypothesis, “stem cells,” by residing at the top of the cellular hierarchy in each tumor, can self-renew and give rise to heterogeneous cell populations within the tumor. Studies focusing on CSCs demonstrated that implantation of even a small number of CSCs has the ability to form tumors suggesting the significance of CSCs in cancer initiation [8].
This was further confirmed in a study by Driessens et al., wherein using genetic lineage tracing experiments it was demonstrated that a fraction of tumor cells and long term persisting stem-like cells have an increased proliferative potential and produce progeny that occupied a significant part of the tumor in squamous skin cancer [5]. Another study also demonstrate that Lgr5+ stem cells in intestinal adenomas produce the cells of entire adenoma by maintaining Lgr5+ stem cell population [6]. These studies suggest that CSCs are the major culprits for the initiation and progression of cancers.
Four aspects of CSC biology have been investigated in the literature, including origin, manifold existence, maintenance, and metastasis of CSCs (OMMM of CSCs) (Fig. 1). Current evidence suggests that cell fusion, horizontal gene transfer and mutations drive cellular transformation and reprogramming into CSCs. In addition, metabolic shifts from glycolytic to oxidative phosphorylation, or vice versa, also induce cancer stemness [9].
Fig. 1.
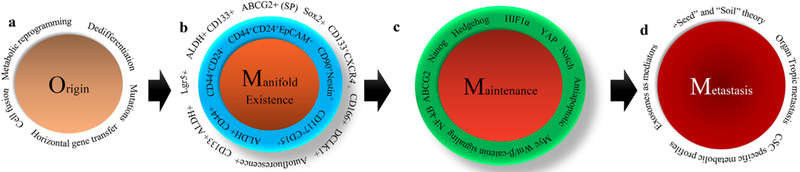
Overall journey of CSCs from origin to metastasis. a) Origin of CSCs. Mutations in adult stem cells (ASCs) or in differentiated somatic cells can lead to CSC origin. Dedifferentiation of somatic differentiated cell in response to various external toxic exposures can give rise to CSC phenotype. Other factors, such as metabolic reprogramming, cell fusion, and horizontal gene transfer can also induce CSCs. b) Multiple CSC populations reside within tumors. CSCs with detoxification systems such as ABCG2-mediated drug efflux mechanism and ALDH-mediated aldehyde toxic substance detoxification systems exist in various tumors. CSCs expressing cell surface markers such as CD44, CD24, and EpCAM together are also the major constituents within various heterogeneous tumors, such as pancreatic tumors. Other CSCs, which express CD133 and CXCR4, also reside within the same tumor. Intestinal tumors consist of Lgr5-expressing CSCs. c) ‘Stemness’ maintenance mechanisms. The stemness in CSCs is largely maintained by specific stemness molecules such as Wnt/β-catenin, Notch and hedgehog, along with other factors such as YAP, HIF1α, NF-kB, PPARγ, and antiapoptotic. d) Role of CSCs in metastasis. The “seed” and “soil” theory, as proposed by Stephen Paget, states that primary site tumor cells (seed) travel to a distant organ (soil), and colonize and initiate the growth of tumor. Based on this theory, it is possible that CSCs from the primary site will travel to distant organs to initiate metastatic tumors. Another hypothetical view suggests that exosomes released by CSCs in the primary site travel to target sites and form the premetastatic niche (PMN) that supports upcoming CSCs or cancer cells. Another view also suggests that distinct CSC population subtypes with subtype-specific metabolic profiles travel to different organs (organ specific metastasis).
A challenge in understanding CSC biology is the lack of consensus about the markers of CSCs. Different studies propose varying markers for CSCs in different cancers. Emerging evidence suggests that tumors consist of heterogeneous CSC subtype populations, and each subtype of CSCs display a unique phenotype and unique clonogenic and metastatic potential. Our understanding of how these heterogeneous CSC subtype populations are maintained and contribute to the cancer biogenesis and aggressiveness after their generation also remains limited.
Studies suggest that CSCs within tumors have the potential to migrate to specific organs [10,11]. Exosomes, small extracellular vesicles released by cells, carry cellular components to distant organs, thereby increasing cellular communications [12]. Recent studies have shown that the exosomes released by cancer cells within a primary tumor travel to distant organs and form a pre-metastatic niche [12,13]. However, it is not known which cell type in a heterogeneous primary tumor releases the exosomes with the capacity to form a pre-metastatic niche in distant organs. In this review, we discussed the concept of CSCs from its origin to metastasis and described the perspective about the role of CSCs in organotrophic metastasis.
2. General features of cancer stem cells
The CSC hypothesis emerged a decade ago; however, it is still unclear what makes these CSCs unique compared to normal cells, other non-cancerous stem cells, or cancer cells. Most cell signaling pathways and cell surface and intracellular markers are similar in normal cell, stem cell, CSC, and cancer cells. For instance, bone marrow-derived mesenchymal stem cells express high levels of the CD90 cell surface marker [14]. However, the same marker has also been proposed as a CSC marker in primary high-grade gliomas and other cancers [15]. Similarly, CD44, an established CSC marker, is also highly expressed in normal endometrial cells and regulates the function of normal endometrium [16]. Here, we explore the features that could potentially derive CSCs.
2.1. Recapitulation of embryonic signature in CSCs
The most prominent and specific feature which can be observed in CSCs is the recapitulation of embryonic pluripotent networks and the overexpression of embryonic genes [17]. During development, TGF-β, FGFR/MAPK or Akt, Wnt, Notch, and sonic hedgehog pathways maintain self-renewal and pluripotency of embryonic stem cells (ESCs). These pathways ultimately activate three major transcription factors: Oct3/4, SOX2, and Nanog. These factors activate ESC-specific genes and maintain the stem cell state of ESCs by inhibiting differentiation genes. During embryo development and organ specification, pluripotent genes are inhibited, and differentiation genes are activated. Thus, in adult tissues, the expression levels of Oct3/4, SOX2, Nanog, and other ESC maintenance genes is very low [18,19]. However, during the initiation and progression of cancers, these ESC genes and networks are activated. The aberrant expression of ESC genes and activation of stemness networks leads to the enrichment of CSCs, which initiate or aggravate tumor.
2.2. Mutations distinguish CSCs from non-CSCs
CSCs also differ from other non-CSC populations in that CSCs show mutations leading to the aberrant regulation of majority of the stemness and proliferation pathways. Even though the same signaling pathways are seen in CSCs and non-CSCs, their enhanced and aberrant activation distinguishes CSCs from non-CSCs. For instance, aberrant activation of the Wnt pathway has been shown to induce CSCs [20,21].
2.3. CSCs are quiescent
CSCs also have another striking specific property; i.e., CSC quiescence. CSCs are a low-cycling quiescent cell population residing within the tumor. These low-cycling CSCs are responsible for the tumor aggressiveness and metastasis [22,23]. Resistance to chemotherapeutic drugs is another specific property of CSCs, and this resistance is usually conferred by ABCG2 and other drug resistance receptors [24].
3. Origin of cancer stem cells
3.1. Cell fusion and stem cells
CSCs originate through cell fusions (Fig. 2), wherein cells acquire extra properties through a process in which two cells fuse together to form hybrids with a higher degree of aneuploidy [25]. The fusion of 5-fluorouracil-(5-FU) resistant cancer cells with methotrexate-resistant cancer cells produces aneuploidy hybrid cells, resistant not only to 5-FU and methotrexate, but also to melphalan. In addition, Rizvi and colleagues showed that the fusion of bone marrow-derived stem cells with intestinal tumor epithelial cells does not produce a resistant CSC population, nor do the new hybrid cells induce cancer [26]. However, polyploid giant cancer cells (PGCCs), which are aroused because of cell fusion have been shown to display drug resistance and tumorigenic properties [27,28]. Overall, poor evidence from literature for the fusion-mediated CSC origin limits this model.
Fig. 2.
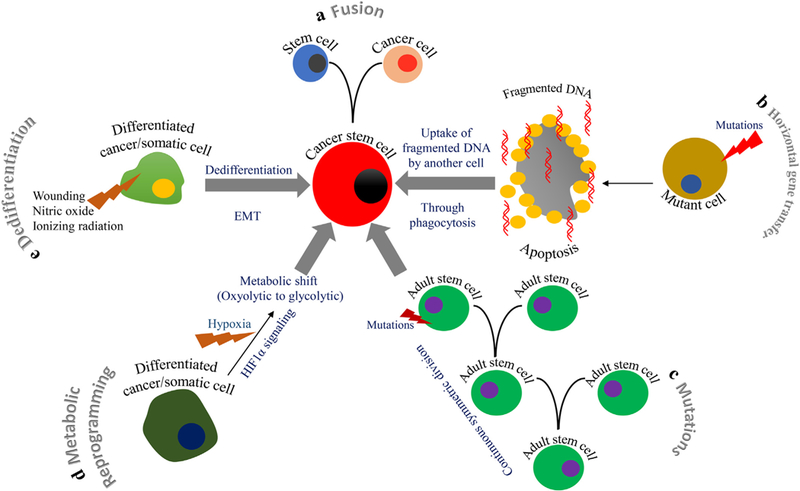
Different modes of CSC origin. a) ‘Cell fusion’ is a process whereby two cells (one stem cell and another cancer cell) fuse together to form CSCs. b) In horizontal gene transfer, mutant fragmented DNA (from a mutant somatic cell that is undergoing apoptosis) is taken up by another somatic or cancer cell, leading to the emergence of CSCs. c) Continuous symmetric divisions in adult stem cells (ASCs) lead to mutation in these cells and give rise to CSCs. d) A metabolic shift in somatic or differentiated cells could reprogram these cells into CSCs. e) Ionizing radiation, wounding, or exposure to toxic chemicals can de-differentiate somatic cells into CSCs.
3.2. Horizontal gene transfer
Another mechanism that could potentially contribute to the origin of CSCs is horizontal gene transfer [29], which is usually an adaptation mechanism found in bacteria and fungi that use this mechanism to acquire resistance to antibiotics [30]. Horizontal transfer is a mechanism whereby DNA from donor cells is delivered into recipient cells, followed by insertion of donor DNA sequences into host genome and expression of inserted gene sequence. The expression of inserted gene sequence in host helps the host cell to acquire resistance to antibiotics. In eukaryotic cells, horizontal transfer occurs between an apoptotic cell (donor) and a recipient cell through endocytosis or phagocytosis. Apoptosis and consequent DNA fragmentation in a somatic cell are the results of mutations. The fragmented DNA is endocytosed by another somatic cell [31] or tumor cell [29], leading to the formation of cells with more aggressive phenotype. This clearly suggests that horizontal gene transfer might play an important role in the generation of resistant CSCs by the transfer of mutated genetic material from apoptotic bodies derived from therapy-sensitive cancer cells to somatic or other therapyresistant cancer cells. However, lack of experimental evidence has limited the reliability of this phenomenon.
3.3. Mutations lead to formation of CSCs
Various conditions, such as radiation treatment, tissue injury, and exposure to toxins (from smoking and the like) induce mutations in certain genes, including p53 (tumor suppressor) and Kras [32]. Fujimori et al. have demonstrated that extracellular stress induces cancerous genes in differentiating embryonic stem cells (ESCs), producing CSC populations [33]. The external harmful signals can also deregulate or enhance certain signaling pathways in normal adult stem cells (ASCs) and can lead to the transformation of these ASCs into CSCs. For instance, arsenic has been shown to induce the transformation of normal prostate epithelial stem cells into CSCs [34]. In addition, mutations in symmetrically dividing normal ASCs transform an ASC into a CSC. Furthermore, ASCs undergo continuous division for a long time, and this increases the chances of accumulating mutations that lead to the transformation of a normal ASC into CSC. On the other hand, mutations in oncogenes and tumor suppressor genes of mesenchymal stem cells (MSCs) induce the transformation of MSCs into CSCs, leading to the initiation and progression of sarcomas [35]. Conditional inactivation of p53, NF1, and Pten tumor suppressors in neural stem/progenitor cells converted these cells into CSCs and initiated brain tumors [36]. A genome-wide mutation pattern analysis in ASCs in human small intestine, colon, and liver tissues showed that mutations accumulate steadily in these tissues at a rate of 40 mutations per year. The mutation spectra of genes associated with carcinogenesis is also like the mutation spectra of tissue-specific ASCs, suggesting that mutations in ASCs can induce carcinogenesis [37].
3.4. De-differentiation of non-CSCs into CSCs
Another phenomenon responsible for the origin of CSCs is cellular de-differentiation. A differentiated cancer cell can de-differentiate into a CSC in response to various factors, including wounding, stress and hypoxia, leading to cancer initiation and progression. A recent study showed that glioma cells could de-differentiate into glioma stem-like cells (GSCs) in response to stress and hypoxia-induced HIF1α signaling. In addition, angiocrine factors such as nitric oxide (NO) have been shown to drive de-differentiation of glioma cells into GSCs [38–40]. Wounding also induces Gata6+ epidermal stem cells into stem-like cells, suggesting that wounding can also de-differentiate a differentiated cell into a stem cell [41]. Ionizing radiation also converted cancer cells into a stem-like phenotype, with increased metastasis [42]. By undergoing EMT, pancreatic ductal cells have been shown to acquire CSC phenotype through de-differentiation [43]. These studies collectively suggest de-differentiation as an important phenomenon involved in generation of CSCs in various cancers.
3.5. Metabolic reprogramming of CSCs
Metabolic adaptation is central in cancer cells, especially regarding glycolysis. In glycolysis, glucose is broken down through a series of steps into pyruvate, producing two molecules of ATP per one molecule of glucose. In the presence of enough oxygen levels, pyruvate enters oxidative phosphorylation (OXPHOS) and produces 36 ATP molecules per one glucose molecule. Thus, OXPHOS is more efficient than glycolysis in generating ATP molecules. However, cancer cells, as hypothesized by Warburg, show increased glycolysis to produce ATP, even in the presence of oxygen, and it converts pyruvate into lactate to create a condition of fermentative metabolism that is highly conducive to the activity of cancer cells. If sufficient levels of glucose are available, aerobic glycolysis can generate more energy more rapidly than can OXPHOS [44].
Otto Heinrich Warburg, a German physiologist, theorized that one of the most important causes of cancer is variation in metabolism. According to his theory, cancer cells generate energy through anaerobic breakdown of glucose even in the presence of oxygen. Hence, these cells are mostly glycolytic. According to this theory, then, cancer is a disease of mitochondrial dysfunction. However, healthy cells produce energy through oxidative phosphorylation, in which the end product of gly-colysis, pyruvate, is oxidized in the mitochondria, and thus these healthy cells are mostly oxyolytic. Very few studies so far have explored the metabolic profiles of CSCs. Several studies suggest that CSCs are mostly glycolytic, while other speculate that these cells are oxyolytic [45,46]. Recently, Folmes and colleagues documented reprogramming of somatic cells into pluripotent cells, and demonstrated that a shift from oxidative phosphorylation to glycolysis in somatic differentiated cells is required for reprogramming of these cells into pluripotent stem cells [47]. Based on this study, it is possible that a non-CSC population is reprogrammed into CSCs during the development of cancer. In support of this, hypoxia has been shown to increase breast CSCs regulated by hypoxia-inducible factor 1 alpha (HIF1α) [9].
To conclude this part of the review, there are two major unknown challenges associated with the elucidation of the origin of CSCs. The preceding discussions conclude that de-differentiation is one of the driving factors for CSC generation, and in this regard, it seems possible that CSCs show plasticity. CSCs typically differentiate and form heterogeneous populations. At the same time, a differentiated non-CSC population, in response to certain conditions including toxic exposure and mutations, may de-differentiate into CSCs and lead to further tumor progression. However, further evidence is necessary to shed light on the complex phenomenon of CSC plasticity, given that tumors contain a heterogeneous population with differing phenotypes and genotypes. Another challenge in the study of CSCs is the identification of differentiated non-CSC populations that undergo transformation and de-differentiation.
The stem cells in a tissue are known to divide and self-renew for long periods, and studies have suggested that these stem cells are more susceptible to mutations. However, no study has been documented in the literature demonstrating the accumulation of mutations in these long-lived dividing stem cell populations. The suggestion that a metabolic shift can reprogram somatic cell into pluripotent stem cells also requires further investigation and in-depth study as such a shift might relate to cancer.
Since CSCs were first identified, their origin has been a pronounced mystery. Toxic exposure, mutations, metabolic shift, cellular plasticity, and de-differentiation have been suggested as contributing factors for the origin of CSC. Finding a link between all these factors and investigating whether these events occur in a sequential manner are challenges.
4. Manifold existence of cancer stem cells and its impact on cancer biogenesis and aggressiveness
So far, a description for the existence of CSCs is controversial, as different studies have shown different CSC markers (Table 1). The lack of a universal CSC marker makes it difficult to characterize CSCs in various kinds of cancer, and much of the evidence suggests that CSCs are heterogeneous. According to this, tumors consist of distinct CSC populations with specific phenotypes and clonogenic potentials. In this part of the review, we will describe the CSC heterogeneity or specific clonogenic populations of CSCs in various cancers and their impact on cancer biogenesis and aggressiveness.
Table 1.
Distinct CSC population in various cancers.
| Tumor type | Type of cancer stem cell existed | |
|---|---|---|
| 1 | Melanoma | CD133+, ABCG2+, CD271+, ABCB5+, ALDH1+, CD20+, PD1+ and CXCR6+ |
| 2 | Bladder cancer | CD44+, CD67LR+, EMA+, ALDH1A1+ and BCMab1+. |
| 3 | Breast cancer | CD44+CD24−, ALDH+, CD44+ALDH+ and CD49f+CD24+ |
| 5 | Leukemia | CD34+CD38−; CD34-Lin+CD38+; CD45RA+; CD34+CD38−CD71−HLA-DR−;CD123+; TIM3+; CXCR4+ |
| 6 | Gastric cancer | Lgr5+, CD26+, CD44+, ALDH1+, CD133+ |
| 7 | Colorectal cancer | CD133+, CD133+CXCR4+, CD44+, CD166+, ALDH+, CD29+, Lgr5+ and CD44+CD24+EpCAM+ |
| 8 | Ovarian cancer | CD44+, CD24+, CD117+ and CD133+ |
| 9 | Head and Neck cancers | CD44+, CD133+, ALDH+, CD98+, CD200+, GRP78+, ALDH+CD44+ and Bmi1+ |
| 10 | Liver cancer | CD133+, ALDH+, CD44+, CD90+, CD13+, OV6+, EpCAM+, ABCG2+ (side population), DLK1+, K19+ and c-Kit+ |
| 11 | Lung cancer | CD44+, CD87+, CD90+, CD117+, CD133+, CD166+, ALDH+, BMI1+, EpCAM+ and Side population. |
| 12 | Pancreatic cancer | CD44+, CD133+, ALDH1+, Side population, CD44+CD24+EpCAM+, autofluorescence+ and CD133+CXCR4+ |
4.1. Cancer stem cell heterogeneity in liquid tumors: establishment of original CSC paradigm
The concept of CSCs was first recognized and analyzed in most detail in liquid tumors such as acute myeloid leukemia (AML), chronic myeloid leukemia (CML) and acute lymphoid leukemia (ALL). Bonnet et al., have identified CD34+CD38− leukemic stem cells in AML for the first time and demonstrated that this particular subset of leukemic stem cell population show self-renewal, proliferative and differentiation potential and induce leukemic transformation in NOD/SCID mice [48]. Later, Cox et al., have showed that cells with CD34+CD19−CD10− phenotype are long term proliferative cells with tumorigenic potential [49]. Later studies in leukemia showed distinct CSC populations including CD34-Lin+CD38+, CD45RA+, CD34+CD38−CD71−HLA-DR−, CD34+CD38− CD123+, T cell immunoglobulin mucin 3 (TIM3+) and CXCR4+ [50–54]. However, CSCs in solid tumors display a completely distinct set of markers as described below.
4.2. Cancer stem cell heterogeneity in solid tumors
CD133+ and ABCG2+ population from surgical biopsy samples of melanoma patients showed an increased tumor initiating property in vivo [55]. In NOD/SCID mice, ALDH+ melanoma cells showed increased tumorigenicity as compared to ALDH− population suggesting the existence of ALDH+ CSCs in melanoma [56]. Circulating melanoma cells showed Nestin expression suggesting this protein as a potential marker for melanoma metastatic stem cells [57]. Transplantation of CD271+ melanoma cells into engrafted human skin produced melanoma tumors, while CD271− population did not, suggesting CD271+ population as a distinct melanoma stem cell population [58]. Fang et al., showed a small proportion of CD20+ population as melanoma stemness population [59].
Furthermore, bladder tumor consists of distinct heterogeneous CSC populations. For example, CD44+BCMab1+ population was identified as CSC population in bladder cancer and they have also shown that this CSC population has an increased GALNT1 expression and Hh signaling leading to increased tumorigenesis [60]. In addition, CD133+ population also showed tumorigenic and stemness potential in bladder cancer [61]. In addition to CD44+ and CD133+ CSCs, glioma tumors consist of distinct other CSC subtype populations expressing MUSASHI1, NESTIN, CD15, L1CAM, and A2B5 [62–65].
A recent emerging study demonstrated that the breast CSCs exist as two distinct populations such as epithelial-to-mesenchymal (EMT) CSCs and mesenchymal-to-epithelial (MET) CSCs [66]. Evidence from this study showed that EMT CSCs (Vimentin+ and EpCAM−) are quiescent; however, MET CSCs (Vimentin− and EpCAM+) are proliferative [66]. Another study demonstrated that a single CD49f+CD24−CD44+ cell derived clones show aggressive phenotype and activation of OCT4/SOX2/lincRNA-RoR signaling suggesting that this subset of population is also stemness population in breast tumors [67].
In contrast, a totally a different set of CSC populations were identified in gastric cancer. Li et al., have showed that invasive intestinal type gastric cancer is originated from Lgr5+ stem cells. In addition, Lgr5+CD54+ cells showed elevated expression of pluripotent and EMT markers in gastric cancer [68]. Apart from these, ALDH+ and CD133+ populations were identified as CSCs in gastric cancer [69]. Similar subset of CSC populations was also identified in colorectal tu-mors. Different subset of CXCR4+Lgr5+ expressing population showed significant tumorigenic activity in colorectal cancer [70]. In addition, CD133+ CSCs showed increased tumorigenicity, and a subset of CD133+CXCR4+ CSCs showed increased metastatic potential along with poor survival in colorectal tumors [71]. DCLK1 has been showed to be associated with increased tumorigenesis, and knockdown of DCLK1 reduced tumor cell pluripotency and prosurvival signaling in colorectal cancer, suggesting the existence of a distinct DCLK1+ subset of CSCs in colorectal tumors [72].
In addition to CD44+, ALDH+ and CD133+ CSC populations, ovarian and head and neck tumors consist of distinct CSC subtype populations. CD117+ CSCs in ovarian cancer is associated with poor survival and increased tumorigenesis. [73]. In contrast, head and neck tumors harbor CD98+, CD200+ and GRP78+ CSC population [74]. Among these, CD200+ CSCs showed increased stemness features and resistance to chemo and radiation treatments [75]. Similarly, liver tumors consist of CD133+, ALDH+ and CD44+ cancer stemness populations [76,77]. In addition, CD90+CD44+ subset of liver cancer cells showed elevated tumorigenicity and metastasis in immunodeficient mice [78]. CSCs in lung tumor is also highly heterogeneous, and for example, CD44+, ALDH+ and EpCAM+ maintain stemness and tumorigenic potential in lung adenocarcinoma [79]. In addition, lung tumor also consists of highly tumorigenic CD166+CD44+ CSC population [80].
Pancreatic tumor harbor several distinct stemness populations such as CD44+, CD133+, ALDH1+, ABCG2+ (SP), EpCAM+, autofluorescence+ (AF) and CXCR4+ CSC populations [81]. Hermann et al., have shown that CD133+ population is highly tumorigenic and a CD133+CXCR4+ subset of population is highly metastatic in pancreatic cancer [11]. Recently, an emerging study demonstrated that intracellular autofluorescence (AF) was used as a CSC marker in pancreatic cancer [82]. Riboflavin-loaded intracellular vesicles expressing ABCG2 shows autofluorescence and maintains stemness potential in pancreatic CSCs [82]. ABCG2 receptors on cell surface confer chemo-resistance to cells, and cells with a greater number of these receptors are considered stem cells or side population (SP). Hence, ABCG2 is also used as CSC marker in various cancers [83,84]. The autofluorescence assay and SP assay are mainly based on ABCG2 expression. However, the autofluorescence population did not overlap with SP cells, further confirming and supporting the non-uniqueness of stemness markers in CSCs.
CD44+ and CD133+ cells have been proposed to be CSCs of medullary thyroid carcinoma [85], endometrial cancers [86] and rectal cancer [87]. In addition, choroidal and ciliary body melanoma CSCs express CD117 and CD15 markers [88]. Overall, these studies suggest that tumors of various organs consist of distinct CSC populations with different stemness phenotypes.
4.3. Evidence for the existence of distinct CSC population based on nuclear transcription factors
Studies have demonstrated that certain embryonic genes, including Oct3/4, SOX2, Nanog, KLF4, and PAF1/PD2 are recapitulated during cancer initiation and progression. Most recently, SOX2 was identified as a CSC prognostic marker in pediatric sarcomas and prostate cancer [89,90]. Polycomb group proteins (e.g. EZH2, BMI1) are major drivers of stem cells, and a group of researchers from the University of Queensland have proposed BMI1 and EZH2 as prostate and oral CSC markers, respectively [89,91]. We have identified the novel role of PAF1/PD2 in the maintenance of ovarian and pancreatic CSCs, demonstrating that PAF1/PD2 interacts with Oct3/4 or PHF5A and maintains the stem cell features of ovarian/pancreatic CSCs [92–94]. It is also known that VEGF promotes stemness and has been shown as a breast CSC marker [95].
4.4. Metabolic profiles of CSCs
A previous study demonstrated that self-renewing normal pluripotent stem cells (PSCs) are glycolytic and that during differentiation, these PSCs switch their metabolic profile from glycolytic to OXPHOS [96]. According to this study, even CSCs should be glycolytic, since self-renewal is one of the common properties of normal stem cells and CSCs. Ciavardelli et al. have demonstrated that CD44+ CD24− breast CSCs are glycolytic, and that the inhibition of glycolysis reduced proliferation of this CSC population [45]. In addition, radio and drug resistant populations in nasopharyngeal carcinoma relied on glycolysis for their energy needs. However, upon differentiation, these CSCs shifted their metabolic profile from glycolytic to OXPHOS [97]. In contrast, reduced glycolysis and increased mitochondrial oxidative phosphorylation have been shown to be characteristic features of CSCs. Song and colleagues demonstrated that colon CSCs utilize mitochondrial oxidative phosphorylation for their energy needs [98]. Similarly, glioma and leukemic stem cells have been reported to be oxyolytic [99].
Based on the above studies, it is clear that there is an extensive metabolic variability in CSCs. Recent research suggests the existence of distinct CSC populations, and it may be possible that a specific CSC population subtype shows a unique metabolic profile. Gammon and colleagues showed that there exist two different types of CSC populations in breast tumors, EMT CSCs, which is a CD44+CD24- and quiescent population, and MET CSCs, which comprise ALDH+ and cycling population [50]. The group studied the metabolic profiles of these two CSC subtypes and suggested that EMT CSCs are glycolytic in nature, whereas MET CSCs are oxyolytic [100].
4.5. Plasticity of cancer stem cells
Based on the discussion above, it is clear that CSCs in a tumor are heterogeneous. In addition to this complexity, CSC plasticity is another process, in which there is a transformation between non-stem cells to stem cells or vice versa contribute to the development of heterogeneity within tumor. One of the major processes of CSC plasticity is EMT. As described above, studies in breast cancer showed two distinct CSC populations such as EMT CSCs and MET CSCs, which maintain the plasticity [66]. In pancreatic cancer, metformin treatment induced the transition of oxidative CSCs into drug resistant glycolytic CSCs, further confirming the plasticity of CSCs [8].
In addition to these studies, other reports also demonstrated the transformation of non-CSC into CSCs. For example, JARID1B, a histone demethylase has been shown to regulate tumorigenicity, and however, cells negative for JARID1B also acquired self-renewal suggesting the plasticity of melanoma CSCs [101]. ZEB1, an EMT transcription factor induces the plasticity of CSCs by converting non-CSCs into CSCs in breast cancer [102]. The combined induction of pluripotent transcription factors POU3F2+SOX2+SALL2+OLIG2 reprogram glioblastoma cells into glioma CSCs [103]. Moreover, silencing of PTEN, a tumor suppressor gene induced stemness features in mammary epithelial cells resulting in the development of pre-malignant lesions [104].
To sum up this section, the major current challenge in CSC biology is the lack of a specific CSC marker that can be used to characterize virtually any kind of CSC from any kind of cancer. Self-renewal and cellular quiescence can be exploited to characterize CSCs; however, these properties are common in both normal stem cells and CSCs. To overcome these challenges, the scientific community proposed various biomarkers for CSCs, but great disagreement remains within the scientific community regarding CSC biomarkers. Different studies have proposed different markers, but these markers are not unique to a particular cancer type. Studies have also proposed different markers for CSCs from different cancers. These studies conclude that there is no unique marker for CSCs in different cancers. On the other hand, this discussion can open doors to a novel hypothesis that tumors consist of heterogeneous and distinct CSC populations, and that each CSC subtype shows a unique metabolic profile and clonogenic potential that results in the derivation of heterogenic populations within tumors. Isolation of CSCs from various cancers using various established markers and investigating the common property or marker expression in all these CSC population types may further reveal a single, unique property in CSCs. Such investigation can also address whether different types of CSCs exist, and, if so, describe the function of each type of CSC within tumors.
5. Maintenance of “Cancer stemness”
Controlled signaling networks in normal stem cells maintain tissue homeostasis; however, loss of control in these networks and consequent abnormalities result in elevated self-renewal, increased survival and increased proliferation that lead to CSC generation and enhanced CSC maintenance. Stem cell niche, various signaling molecules and transcription factors maintain stemness homeostasis in normal stem cells. Similarly, CSC niche, mutations and various abnormal extrinsic and intrinsic signals deregulate the pathways leading to the emergence and maintenance of CSCs. In this section of the review, we will discuss the role of tumor microenvironment (TME), specific stemness signaling molecules and transcription factors in CSC maintenance in detail (Fig. 3).
Fig. 3.
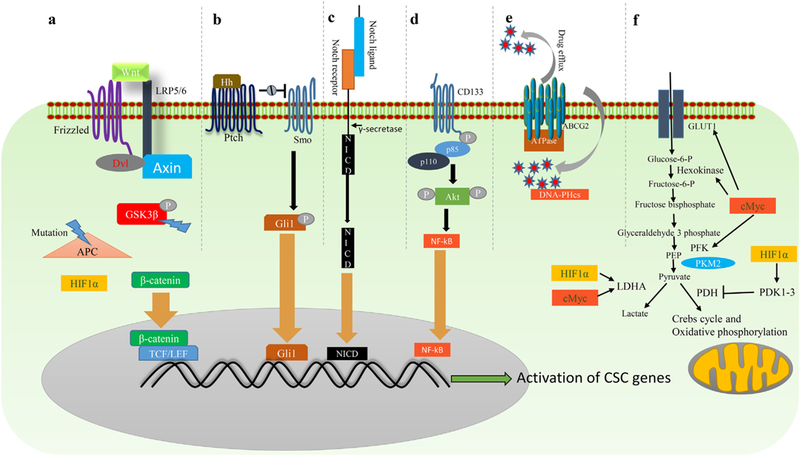
‘Stemness’ maintenance pathways in CSCs. a) Canonical Wnt/β-catenin pathway starts with binding of Wnt ligand to Frizzled receptor (Fzd) and low-density lipoprotein receptor-related protein 5/6 coreceptors (LRP5/6). This results in recruitment of Axin and dishevelled (Dvl) to the plasma membrane and leads to protection of β-catenin from degradation. Free β-catenin in the cytoplasm then translocates to the nucleus and complexes with TCF/LEF to regulate Wnt target stem cell genes. b) Hh signaling pathway is initiated by binding of hedgehog class of ligands to Patched1 (PTCH1) or Patched2 (PTCH2) receptors. The receptor-ligand binding removes Patched receptor mediated inhibition of a G-protein coupled protein, Smoothened (Smo), leading to the nuclear translocation of Gli family transcription factors (a glioma associated oncogene homolog transcription factors). c) Notch signaling is initiated when a notch ligand of a cell binds to the notch receptor of an adjacent cell. This results in release of the intracellular part of the notch, which translocates into the nucleus and acts as a transcriptional co-activator. When the Notch transcriptional co-activators bind to target gene promoter regions, this leads to the activation of its target CSC genes. d) Phosphorylation of the cytoplasmic portion of CD133 leads to the phosphorylation of AKT. Activated AKT activates the NF-kB pathway, resulting in activation of stemness genes. e) ATP-dependent mechanism of drug efflux by ABC transporters. f) HIF1α and cMyc increase glycolysis (or Warburg effect) and inhibit oxidative phosphorylation in CSCs for rapid energy production. HIF1α activates pyruvate dehydrogenase kinases (PDK1–3), which inhibit pyruvate dehydrogenase (PDH), and thus inhibits oxidative phosphorylation. c-Myc and HIF1α also activate lactate dehydrogenase A (LDHA) to favor Warburg effect (aerobic glycolysis). C-Myc also activates the GLUT-1 receptor, hexokinase, and phosphofructokinase (PFK) that favor glycolysis.
5.1. Role of tumor microenvironment in the maintenance of CSCs
The interactions between CSCs and its “niche”, a specific tumor microenvironment (TME) of CSCs maintain “stemness”. Stromal cells, mesenchymal stem cells, hypoxic regions, inflammatory cells, extra cellular matrix (ECM) and angiogenesis are the major components of this specific microenvironment that induce and maintain stem cell features in CSCs. The effect of inflammatory stroma on the dedifferentiation and generation of CSCs was shown recently by Schwitalla et al. in which they showed that increased activation of NF-kB, a transcription factor in the inflammatory TME elevates Wnt signaling and induces dedifferentiation of non-stem cell population into CSC population in intestinal tumors [105]. Another evidence for TME mediated maintenance of CSCs showed that Notch signaling prevents CSCs from differentiation by inhibiting the differentiation signals coming from TME [106]. In addition, mesenchymal stem cells support CSC by secreting a variety of cytokines such as CXCL12 and interleukins that induce stemness through NF-kB pathway [107]. In TME, CSCs escape from NK cell mediated cytotoxic effects and for instance, CD133+ CSCs express low levels of MHC-class 1 molecules leading to the poor recognition by NK cells in glioblastoma [108]. TME hypoxia also promote tumor progression and therapy resistance by preferentially maintaining CSCs in breast cancer [109]. Human renal CSCs release microvesicles (containing miRNAs and mRNAs for VEGF etc.) and induce angiogenesis [110]. These studies collectively suggest that TME is functionally important for the maintenance of CSCs.
5.2. Wnt molecules in the maintenance of self-renewal of cancer stem cells
Wnt proteins are involved in a signaling mechanism that maintains the ability of self-renewal in stem cells. The Wnt ligands bind to different receptors and result in the activation of either canonical or non-canonical signaling pathways that control various cellular functions such as self-renewal, proliferation, survival, and differentiation. In the canonical Wnt signaling pathway, the Wnt ligand binds to the Frizzled receptor (Fzd) and low-density lipoprotein receptor-related protein 5/6 co-receptors (LRP5/6), leading to the formation of the Fzd/LRP5/6 complex. As a result, Axin and dishevelled (Dvl) are recruited to the plasma membrane, leading to the disruption of the β-catenin degradation complex. Free and active forms of β-catenin in the cytoplasm then translocate to the nucleus. In the nucleus, these molecules form complexes with T-cell factor (TCF)/lymphoid enhancer factor (LEF) and lead to the transcriptional activation of Wnt target genes [111]. Various non-canonical Wnt molecules such as Wnt4, Wnt5a, Wnt9b, Wnt10a, and Wnt10b are involved in the fine-tuning of the canonical Wnt signaling mechanism [112]. These non-canonical Wnt molecules bind to the Fzd receptor and Ror1/2 co-receptor, thereby activating non-canonical signaling pathways such as the Wnt/Calcium (Ca2+) and Wnt/ planar cell polarity (PCP) pathways.
Previous studies have shown that Wnt signaling regulates the maintenance of normal intestinal stem cells. The regenerative capacity of intestinal stem cells has been shown to be reduced with the decline in canonical Wnt signaling, suggesting that Wnt signaling plays an important role in the improvement of aging of these cells [113]. Intestinal stem cell markers such as CD44 and Lgr5 are also the direct target genes of the Wnt pathway [114]. It was also shown that Lgr5+ cells are multipotent intestinal stem cells, rapidly cycling and long-lived, with elevated regenerative capacity [114]. Reduced expression of DKK1, an antagonist of the Wnt pathway, increased the stemness of intestinal stem cells, suggesting the importance of this signaling in intestinal stem cells [115]. Previous studies have shown that Wnt signaling maintains the self-renewal of mammary stem cells, and inhibition of Wnt signaling reduced the stemness of mammary stem cells [116]. Protein C receptor, a Wnt target gene, has been shown to maintain mammary stem cells [117]. Another Wnt target gene, Sox9 has been shown to maintain the stemness state of mammary stem cells [118]. Lgr4, a leucine-rich repeat G-protein coupled receptor, is involved in stemness maintenance of intestinal and epidermal stem cells. Lgr4 activates Sox2 through Wnt signaling to maintain stemness in mammary stem cells [119]. In contrast to the stemness maintenance role of Lgr4, a recent study has shown that Lgr4 upregulation also induces the differentiation of prostatestem cells [120]. These studies clearly suggest that Wnt signaling has distinct roles in different organs and importance of Wnt signaling in the maintenance of normal mammary stem cells.
Mutations in Wnt signaling molecules are the major drivers of intestinal cancers, including colorectal cancer. A loss of function mutation in APC, a tumor suppressor and a Wnt signaling controller, leads to the deregulation of the Wnt pathway and to the emergence of colorectal CSCs, and consequently to the initiation and progression of colorectal cancer [121]. Mutations in the GSK3β phosphorylation site in β-catenin leads to aberrant Wnt signaling and development of colorectal cancer [122]. Wnt signaling has also been shown to be involved in the maintenance of colon CSCs [123]. Inhibition of Wnt signaling reduces the stemness of breast CSCs and suppresses breast cancer metastasis [124], while the activation of Wnt/β-catenin signaling increased mammosphere formation ability and enriched breast CSCs [125].
5.3. Hedgehog (Hh) in cancer stem cell maintenance
In addition to Wnt, Hh proteins maintain the self-renewal property of stem cells. The Hh signaling pathway begins with the binding of three ligands: Indian hedgehog or desert hedgehog or sonic hedgehog to Patched1 (PTCH1) or Patched2 (PTCH2) receptors. After this binding, the receptor-mediated inhibitory effect on Smoothened (SMO), a G-protein coupled signaling protein, is removed, leading to the activation of a signaling cascade that facilitates the nuclear translocation of GLI family transcription factors (a glioma associated oncogene homolog transcription factors). Once GLI transcription factors enter the nucleus, they bind to DNA and regulate various genes such as Fox, Myc, and Cyclin D [126].
Hh signaling regulates major cellular and molecular events during embryo development and maintains adult tissue homeostasis. It has been shown that Hh signaling regulates the differentiation of ESCs. The signaling is highly activated during the differentiation of ESCs and effects the lineage determination of ESCs [127]. During embryonic chick lung development, the expression patterns of various Hh signaling proteins such as PTCH1 and GLI was also observed [128]. These studies collectively suggest the importance of Hh signaling during embryonic organogenesis and for the regulation of ESCs.
On the other hand, Hh signaling also regulates the stemness of various CSCs. Studies have suggested that multiple myeloma (MM) stem cells, for example, are CD138 negative and that the inhibition of Hh signaling in these cells reduces their clonal ability, suggesting its importance in maintaining MM stem cells [129]. Cyclopamine-mediated inhibition of Hh signaling in GBM neurospheres reduced other stemness markers, such as Nanog, Sox2, and Nestin [130]. The Shh target gene, BMI1, is highly upregulated in CD44+ CD24− breast CSCs and maintains the regulation of self-renewal of breast CSCs [131]. Inhibition of SMO using its antagonist, GDC-0449, and consequent inhibition of Hh signaling, led to apoptosis of pancreatic tumorspheres [132]. These studies clearly implicate Hh signaling in the regulation of stemness in CSCs.
5.4. Notch signaling and self-renewal maintenance
The Notch signaling pathway also governs the self-renewal of stem cells and the development and homeostasis of various tissues. Notch signaling is initiated when two cells, one signal releasing and the other signal receiving, are in contact with each other. The signal-releasing cell produces a ligand, which binds to the receptor on the signal receiving cells and thereby cleaves and translocates the intracellular part of the Notch receptor into the nucleus. Once this enters the nucleus, it acts as a transcriptional co-activator. The binding of the Notch transcriptional co-activators to target gene promoter regions leads to the activation of its target genes, including Myc, p21, and Hes1 [133].
The stemness property of adult normal stem cells in various adult tissues is maintained by Notch signaling. Lgr5+ cells in intestinal tissues are intestinal stem cells, and this Lgr5+ population is responsible for the homeostasis of intestinal tissue. Recent studies have shown that the inhibition of Notch signaling reduces the Lgr5+ intestinal stem cell population [134]. Notch signaling also mainly regulates the maintenance of neural stem cells. Inhibition of Rbpj, a Notch receptor effector molecule, in the adult brain led to the differentiation of neural stem cells into neurons, suggesting that the signaling is crucial for the maintenance of neural stem cells [135].
As in normal stem cells, Notch signaling also plays a crucial role in the regulation of stemness or self-renewal in CSCs, and abnormal activation of the pathway leads to increased self-renewal of CSCs. For instance, Notch signaling controls pancreatic CSCs in pancreatic ductal adenocarcinoma (PDAC), and silencing of Hes1, a Notch pathway target gene, reduced the percentage of pancreatic CSCs [38]. Hes1 upregulation also increases the number of CD133+ cells, side populations, and ability to form tumorspheres in colon cancer. The stemness of renal cell carcinoma CSCs has also been shown to be regulated through the Notch pathway. Blockade of Notch receptors using Numb, an endogenous inhibitor of Notch receptors in CD133+CD24+ renal cell carcinoma CSCs, led to a reduction in self-renewal and drug resistance in these CSCs [136]. Treatment with gamma-secretase inhibitor IX (GSI), a Notch inhibitor, inhibited proliferation and induced apoptosis in CD44+ gastric CSCs [137]. These studies collectively suggest the importance of Notch signaling in the maintenance of stemness in CSCs.
5.5. ABC transporters in the maintenance of drug resistance in CSCs
Drug resistance in CSCs is caused by multiple factors including ABC transporters mediated drug efflux, immune escape due to loss of MHC class 1 molecules, extracellular acidic pH, ALDH enzyme mediated cellular detoxification system and increased DNA repair ability. Among these, ABC transporters mediated drug efflux mechanism is highly activated and unique in CSCs. Previous studies have shown that aberrant regulation of ATP binding cassette (ABC) transporters leads to drug resistance of CSCs [138]. Three major ABC transporters, ABCB1 (MDR1), ABCC1, and ABCG2, have been demonstrated for the drug resistance in CSCs. Multiple signaling mechanisms have been shown to regulate the expression of ABC transporters. MDR1, a multidrug resistant gene, is responsible for the drug resistance of CSCs, and the MDR1 gene is directly regulated by the MYC oncoprotein [139]. In addition, the expression of ABCG2 is regulated by an increase in JNK1 signaling [140]. HIF2α and PPARγ/PTEN/PI3K/Akt have been shown to regulate the ABCG2 gene in breast cancer [141,142]. An increased expression of the estrogen receptor β (ERβ) induces cancer by regulating ABCG2 [143]. NRF2, a redox sensing transcription factor, regulates ABCG2 in lung CSCs [144]. Recently, the YAP pathway (Hippo pathway) has been shown to be a highway for drug resistance in tumors [145]. On the other hand, Notch1 signaling has been shown to regulate the ABCC1 transporter, leading to increased drug resistance in prostate CSCs [146]. In summary, ABCB1, ABCC1 and ABCG2 drug efflux transporters are the major players in the drug resistance of CSCs. Developing therapeutic strategies to target these transporters may be a better strategy to eliminate CSCs from tumor bulk.
5.6. Anti-apoptotic proteins and its importance in CSCs
Anti-apoptotic pathways are primarily responsible for drug resistance in CSCs. Identification of various targets specifically involved in anti-apoptosis of CSCs is crucial for developing CSC-targeted thera-pies. Most studies have repeatedly shown that up-regulation of various anti-apoptotic proteins, such as Bcl-2, Mcl-1, c-FLIP, and survivin, and the down-regulation of pro-apoptotic proteins such as Bid, lead to increased drug resistance in CSCs. Bcl-2, an anti-apoptotic protein, is required for the survival of leukemia stem cells [147]. Glioma stem cells also showed increased expression of Bcl-2, along with another anti-apoptotic protein, Mcl-1 [148,149]. Another significant anti-apoptotic protein, c-FLIP, exerts its function by activating major signaling mole-cules, such as Akt and ERK. c-FLIP is overexpressed in breast CSCs and leads to their increased survival [150]. Survivin, another anti-apoptotic protein, has been shown to be up-regulated in breast CSCs and to lead to therapeutic resistance of breast CSCs [151]. Targeting Bid, a pro-apoptotic protein, in ovarian CSCs reduced the stemness of these cells, suggesting that Bid is responsible for the elevated stemness in ovarian CSCs [152]. Targeting these pro-apoptotic and anti-apoptotic proteins in CSCs may increase sensitivity of CSCs to drug-mediated cell death.
5.7. NF-KB in CSC maintenance
One of the many characteristic features of CSCs is epithelial to mesenchymal transition [50]. Recent studies have suggested that EMT is not only responsible for metastasis, but also for drug resistance EMT is regulated by the TWIST-mediated NF-ƘB pathway in papillary thyroid carcinoma [153]. In addition, SDF-1 activates NF-ƘB, leading to the induction of EMT in breast CSCs [154]. The multidrug resistant gene, MDR1, is regulated by CD133 and the DNA dependent protein kinase (DNA-PK) via the PI3K- or Akt-NF-κB network [155]. Osteo-pontin, an oncoprotein, induces hepatocellular CSCs through an integrin-NF-κB-HIF-1α pathway [156]. Activation of TLR3 was also shown to induce the co-activation of β-catenin and NF-ƘB, leading to the promotion of breast CSCs [156]. In addition to various signaling molecules, miRNAs also induce CSCs by activating NF-κB pathway; for instance, Let7a induces mammosphere formation by activating the Ras/ NF-κB pathway [157].
Given these results, targeting ABC transporters, pro and anti-apoptotic molecules and NF-κB together may be proved to be an efficient therapeutic strategy to eradicate CSC populations from tumor bulk.
5.8. HIF1-mediated metabolic alteration in CSCs
Hypoxia inducible factor 1 (HIF1) is a transcription factor that mediates metabolic configuration under hypoxic conditions. HIF1 consists of two subunits, HIF1α (controlled by oxygen) and HIF1β (expressed constitutively) [158]. HIF1α, under normoxic conditions, is also degraded by oxygen-dependent prolyl-hydroxylases (PHD1–3). Hypoxic conditions or inhibition of PHD enzymes increases the stabilization and nuclear translocation of HIF1α, where it activates its target metabolic genes such as glucose transporters (GLUT1) and pyruvate dehydrogenase kinases (PDK1–3) and results in the switch from mitochondrial oxidative phosphorylation to glycolysis. Increased expression of GLUT1 elevates glucose uptake by cells, and the augmentation of PDK1–3 expression inhibits oxidative phosphorylation by inhibiting pyruvate dehydrogenase (PDH). Furthermore, HIF1α activates pyruvate kinase (PKM2) gene transcription, and this is facilitated by its interaction with the PKM2 gene. PKM2 catalyzes the final step of glycolysis by dephosphorylating phosphoenolpyruvate to pyruvate. Thus, HIF1α plays an important role in increasing glycolysis and decreasing oxidative phosphorylation [158].
A recent study demonstrated that reprogramming of a somatic cell into a pluripotent stem cell requires a metabolic shift from oxidative phosphorylation into glycolysis [158]. In detail, the transfection of dermal fibroblasts with pluripotent factors such as OCT4, SOX2, KLF4, and c-MYC increases expression of HIF1α, which in turn activates its target genes, including PDK1, PDK3, and PKM2. The activation of PDK1 and PDK3 inhibits PDH, leading to a shift from oxidative phosphorylation to glycolysis. These results suggest that a metabolic shift from mitochondrial respiration to glycolysis is mandatory during the reprogramming of somatic cells into stem cells.
In terms of CSCs, HIF1α expression is elevated in prostate CSCs, suggesting that an HIF1α signaling-mediated glycolytic shift may be required for maintenance of CSCs [159]. In addition, HIF1α has been shown to increase β-catenin transcription and to result in the activation of Wnt signaling in leukemia stem cells [160]. Iida et al. showed that HIF1α activates Oct3/4 and Sox2 expression and induces CD133 expression under hypoxic conditions [161]. Despite this evidence, it is not known whether the activation of HIF1α can completely reprogram a cancer cell or a differentiated cell into a CSC through an HIF1α-mediated glycolytic shift.
5.9. Myc and Nanog maintain metabolic alteration in CSCs
The oncoproteins of the Myc family (MYC, MYCN and MYCL) control cell proliferation in various cancers. Apart from this, these oncoproteins have been shown to play a crucial role in metabolic reprogramming under cancerous conditions. MYC activates various metabolic genes, including GLUT1, ENO1, HK1, LDHA and PKM2, and thus regulate the uptake of glucose, glycolysis, and lactic acid production. MYC has a further significant role in the regulation of metabolic pathways in stem cells. The MYC oncoproteins (MYC and MYCN) present in neuronal progenitor cells activate HK2 and LDHA, leading to their increased reliance on glycolysis for energy production. However, during differentiation of these neuronal progenitor cells into neurons, a shift from glycolysis to oxidative phosphorylation was observed that led to a reduction in MYC and MYCN proteins as well as in the glycolytic enzymes [162].
Pancreatic CSCs are oxyolytic as the inhibition of MYC contributes to the suppression of mitochondrial respiration in these CSCs [8,163]. Moreover, treatment with metformin, an inhibitor of mitochondrial respiration, induced apoptosis in pancreatic CSCs. However, a very small subset of pancreatic CSCs developed resistance to metformin and displayed intermediate metabolic phenotype. Metformin-induced Myc overexpression also led to a shift from oxidative phosphorylation into glycolysis in these metformin-resistant CSCs.
Nanog is a transcription factor required for the maintenance of pluripotency in stem cells. It binds to the promoter of the genes involved in oxidative phosphorylation, and its overexpression reduces the expression of mitochondrial respiration genes but increases mitochondrial fatty acid oxidation in liver CSCs. The upregulation of genes involved in mitochondrial respiration interestingly reduces self-renewal of liver CSCs [164]. Nanog thus maintains cancer stemness of liver cancer cells by inhibiting mitochondrial oxidative phosphorylation and by inducing fatty acid oxidation.
6. Cancer stemness in metastasis
6.1. “Seed” and “Soil” theory of metastasis
Although there are novel targeted therapies and advanced surgical techniques available to reduce or remove primary tumors, cancer remains one of the leading causes of mortality worldwide with the primary cause of cancer-related deaths being the progressive development of metastasis. Organ-specific metastasis has been a mystery to researchers for over a century. In 1889, Stephen Paget proposed the “seed” and “soil” theory, in which tumor cells (seed) from the primary site travel to a distant organ (soil), which in turn is a favorable environment to support the colonization and growth of the tumor cell. A specific organ, according to this theory, can provide a suitable environment for the growth of a specific tumor cell, resulting in the organ-specific metastasis of given tumor cells [165] (Fig. 4). In support of this notion, it has also been shown that tumor cells create an environment in targeted distant organs that are suitable for the growth of future metastatic cells [166].
Fig. 4.

Hypothetical model showing organ-specific metastasis of CSCs. Distinct CSC populations with differential metabolic profiles (either glycolytic, oxyolytic or intermediate) reside in heterogenous tumors. The exosomes released by a specific CSC subtype population in the primary tumor site may travel to a specific distant organ to form a pre-metastatic niche (PMN), required for survival of upcoming metastatic CSC.
6.2. Impact of cancer stem cell heterogeneity on the establishment of organotropic metastasis
Previous studies have shown that CSCs can not only initiate and aggravate cancer, but that they are also involved in metastasis. However, the role of CSCs in this organ-specific metastasis and in the formation of a pre-metastatic niche has been poorly understood in the literature. A majority of studies suggest that metastatic cells show a CSC phenotype. In pancreatic cancer, the CD133+ cell population has been shown to be highly metastatic. CXCR4 expressing CSCs showed an increased tumorigenic property and elevated metastatic potential. In colon cancer, CD26+ CSCs are known to have metastatic potential. ALDH+ and CD44+CD24-breast CSCs also show high metastatic potential [167]. However, it is not completely understood in the literature whether heterogeneous CSC subtype populations with a specific phenotype and clonogenic potential can metastasize to a specific organ. Gao et al. have shown that CD110-positive CSCs show liver organo-tropism, whereas CDCP1 promotes lung tropism in colorectal cancer. It has been concluded that CD110+ and CDCP1+ metastatic CSCs in colorectal cancer determine organ-specific metastasis [168]. In addi-tion, CXCR4 positive breast CSCs have been shown to metastasize to the lymph node and lung. Among CD44+CD24- breast CSC populations, a subset of CD44v (variant forms of CD44) populations showed enhanced lung metastasis potential [169]. Studies also suggested that distinct CSC populations display differential metabolic profiles. For instance, breast tumors consist of EMT CSCs and MET CSCs, with EMT CSCs being glycolytic but MET CSCs oxyolytic [100]. In pancreatic cancer, Reichert et al. have showed that EMT-MET plasticity is involved in regulating organotropic metastasis. The authors in this study believe that epithelial plasticity, which is regulated by P120CTN is an important factor for the establishment of cancer cell colonization in liver or lung [100,170]. Investigating whether a specific CSC subtype with a specific metabolic phenotype can metastasize to a specific organ remains a challenge (Fig. 4).
6.3. Exosomes in cancer stem cell-mediated metastasis
Of interest, recent studies also showed that exosomes released by certain primary tumor cells travel to specific target organ and mediate the formation of PMN. The exosomes released by primary tumor cells carry integrins α6β4 and αvβ5 to the lung and liver, respectively. The resident cells of liver and lung then receive these integrin-containing exosomes, resulting in the activation of Src phosphorylation and pro-inflammatory S100 gene expression in these target organs, and leading to the formation of PMN [171]. It is unclear whether exosomes released by a specific CSC with a specific phenotype can reach and form a premetastatic niche in a specific distant organ. Cancer cells and CSCs communicate continuously with each other by sharing their protein and nucleic acid contents through exosomes [172]. It has been shown that the exosomes derived from a stem cell can fuse with a non-stem cell and convert this non-stem cell into stem cell. In this process, the stem cell-derived exosome carries stem cell signature proteins, nucleic acids, and other regulatory RNAs, and that they share these contents with the nonstem cell, transforming from a non-stem cell into a stem cell [173]. Based on this finding, it is possible to suggest that the exosomes derived from CSCs with a specific phenotype can reach a specific organ and can form a pre-metastatic niche in the target organ (Fig. 4).
7. Concluding remarks
The CSCs are self-renewing, drug-resistant cells within the tumor bulk. Understanding their origin, existence, and maintenance and their role in metastasis is crucial to develop an efficient CSC-targeted therapy. Apart from cell fusion and horizontal gene transfer, mutations have been shown to contribute to the origin of CSCs. External factors, such as hypoxia and toxic exposure, induce mutations in differentiated cells and lead to the de-differentiation and reprogramming of these cells into CSCs. It is also possible that a metabolic shift can reprogram somatic or differentiated cancer cells into CSCs. Most studies suggest the existence of distinct CSC population in tumors. The Wnt, Hh, and Notch signaling molecules, along with metabolic regulators such as HIF1α, maintain CSC populations within heterogeneous tumors. Recent evidence fuels the concept that a specific CSC subtype with subtype-specific clonogenic potential and metabolic profiles metastasizes to specific organs.
Acknowledgements
The authors in this article were supported, in parts, by the following grants from the National Institutes of Health (P01 CA217798, R01CA183459, R01CA210637, UO1 CA200466, UO1 CA185148, UO1 CA210240 and P50 CA127297), and the Nebraska Department of Health and Human ServicesLB595.
Footnotes
Conflict of interest
SKB is one of co-founders of Sanguine Diagnostics and Therapeutics, Inc. No potential conflicts of interest were disclosed by the other authors.
References
- [1].Makino S, The role of tumor stem-cells in regrowth of the tumor following drastic applications, Acta - Unio Internationalis Contra Cancrum 15 (Suppl. 1) (1959) 196–198. [PubMed] [Google Scholar]
- [2].Konrad CV, Murali R, Varghese BA, Nair R, The role of cancer stem cells in tumor heterogeneity and resistance to therapy, Can. J. Physiol. Pharmacol 95 (2017) 1–15. [DOI] [PubMed] [Google Scholar]
- [3].Lapidot T, Sirard C, Vormoor J, A cell initiating human acute myeloid leukaemia after transplantation into SCID mice, Nature 367 (1994). [DOI] [PubMed] [Google Scholar]
- [4].Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF, A restricted cell population propagates glioblastoma growth following chemotherapy, Nature 488 (2012) 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C, Defining the mode of tumour growth by clonal analysis, Nature 488 (2012) 527–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, Clevers H, Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas, Science (New York, N.Y.) 337 (2012) 730–735. [DOI] [PubMed] [Google Scholar]
- [7].Prasetyanti PR, Medema JP, Intra-tumor heterogeneity from a cancer stem cell perspective, Molecular cancer 16 (2017) 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos-Olivas R, Graña O, Viera Catarina R, Yuneva M, Sainz B, Heeschen C, MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells, Cell Metabolism 22 (2015) 590–605. [DOI] [PubMed] [Google Scholar]
- [9].Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG, Wicha MS, Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 2784–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cioff M, D’Alterio C, Camerlingo R, Tirino V, Consales C, Riccio A, Ierano C, Cecere SC, Losito NS, Greggi S, Pignata S, Pirozzi G, Scala S, Identification of a distinct population of CD133(+)CXCR4(+) cancer stem cells in ovarian cancer, Sci. Rep 5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C, Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer, Cell Stem Cell 1 (2007) 313–323. [DOI] [PubMed] [Google Scholar]
- [12].Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur Basant K., Becker A, Hoshino A, Mark MT, Molina H, Xiang J, Zhang T, Theilen T-M, García-Santos G, Williams C, Ararso Y, Huang Y, Rodrigues G, Shen T-L, Labori KJ, Lothe IMB, Kure EH, Hernandez J, Doussot A, Ebbesen SH, Grandgenett Paul M., Hollingsworth Michael A., Jain M, Mallya K, Batra SK, Jarnagin William R., Schwartz Robert E., Matei I, Peinado H, Stanger BZ, Bromberg J, Lyden D, Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver, Nat. Cell Biol 17 (2015) 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yu Z, Zhao S, Ren L, Wang L, Chen Z, Hoffman RM, Zhou J, Pancreatic cancer-derived exosomes promote tumor metastasis and liver pre-metastatic niche formation, Oncotarget 8 (2017) 63461–63483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mercati F, Pascucci L, Ceccarelli P, Dall’Aglio C, Pedini V, Gargiulo AM, Expression of mesenchymal stem cell marker CD90 on dermal sheath cells of the anagen hair follicle in canine species, Eur. J. Histochem 53 (2009) 159–166. [DOI] [PubMed] [Google Scholar]
- [15].He J, Liu Y, Zhu T, Zhu J, Dimeco F, Vescovi AL, Heth JA, Muraszko KM, Fan X, Lubman DM, CD90 is identified as a candidate marker for cancer stem cells in primary high-grade gliomas using tissue microarrays, Mol. Cell. Proteomics 11 (2012) (M111.010744). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fujita N, Yaegashi N, Ide Y, Sato S, Nakamura M, Ishiwata I, Yajima A, Expression of CD44 in normal human versus tumor endometrial tissues: possible implication of reduced expression of CD44 in lymph-vascular space involvement of cancer cells, Cancer Res 54 (1994) 3922–3928. [PubMed] [Google Scholar]
- [17].Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA, An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors, Nat. Genet 40 (2008) 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dvash T, Mayshar Y, Darr H, McElhaney M, Barker D, Yanuka O, Kotkow KJ, L Rubin L, Benvenisty N, Eiges R, Temporal gene expression during differentiation of human embryonic stem cells and embryoid bodies, Hum. Reprod 19 (2004) 2875–2883. [DOI] [PubMed] [Google Scholar]
- [19].Okita K, Yamanaka S, Intracellular signaling pathways regulating pluripotency of embryonic stem cells, Curr. Stem Cell Res. Ther 1 (2006) 103–111. [DOI] [PubMed] [Google Scholar]
- [20].Moon B-S, Jeong W-J, Park J, Kim TI, Min DS, Choi K-Y, Role of oncogenic K-Ras in cancer stem cell activation by aberrant Wnt/β-catenin signaling, J. Natl. Cancer Inst 106 (2014) djt373. [DOI] [PubMed] [Google Scholar]
- [21].Karamboulas C, Ailles L, Developmental signaling pathways in cancer stem cells of solid tumors, Biochim. Biophys. Acta 1830 (2013) 2481–2495. [DOI] [PubMed] [Google Scholar]
- [22].Jiang YX, Yang SW, Li PA, Luo X, Li ZY, Hao YX, Yu PW, The promotion of the transformation of quiescent gastric cancer stem cells by IL-17 and the under-lying mechanisms, Oncogene 36 (2016) 1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kojima H, Okumura T, Yamaguchi T, Miwa T, Shimada Y, Nagata T, Enhanced cancer stem cell properties of a mitotically quiescent subpopulation of p75NTR-positive cells in esophageal squamous cell carcinoma, Int. J. Oncol 51 (2017) 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Samant MD, Jackson CM, Felix CL, Jones AJ, Goodrich DW, Foster BA, Huss WJ, Multi-drug resistance ABC transporter inhibition enhances murine ventral prostate stem/progenitor cell differentiation, Stem Cells Dev 24 (2015) 1236–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dittmar T, Nagler C, Schwitalla S, Reith G, Niggemann B, Zänker KS, Recurrence cancer stem cells – made by cell fusion? Med. Hypotheses 73 (2009) 542–547. [DOI] [PubMed] [Google Scholar]
- [26].Rizvi AZ, Swain JR, Davies PS, Bailey AS, Decker AD, Willenbring H, Grompe M, Fleming WH, Wong MH, Bone marrow-derived cells fuse with normal and transformed intestinal stem cells, Proc. Natl. Acad. Sci 103 (2006) 6321–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Spies JJ, van Wyk SMC, Cell fusion: a possible mechanism for the origin of polyploidy, South Afr. J. Bot 61 (1995) 60–65. [Google Scholar]
- [28].Zhang S, Mercado-Uribe I, Xing Z, Sun B, Kuang J, Liu J, Generation of cancer stem-like cells through formation of polyploid giant cancer cells, Oncogene 33 (2014), 10.1038/onc.2013.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, Terzis AJA, The origin of the cancer stem cell: current controversies and new insights, Nat. Rev. Cancer 5 (2005) 899–904. [DOI] [PubMed] [Google Scholar]
- [30].Palmer KL, Kos VN, Gilmore MS, Horizontal gene transfer and the genomics of enterococcal antibiotic resistance, Curr. Opin. Microbiol 13 (2010) 632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Holmgren L, Szeles A, Rajnavölgyi E, Folkman J, Klein G, Ernberg I, Falk KI, Horizontal transfer of DNA by the uptake of apoptotic bodies, Blood 93 (1999) 3956–3963. [PubMed] [Google Scholar]
- [32].Gealy R, Zhang L, Siegfried JM, Luketich JD, Keohavong P, Comparison of mutations in the p53 and K-ras genes in lung carcinomas from smoking and nonsmoking women, Cancer Epidemiol. Biomark. Prevent 8 (1999) 297–302. [PubMed] [Google Scholar]
- [33].Fujimori H, Shikanai M, Teraoka H, Masutani M, Yoshioka K, Induction of cancerous stem cells during embryonic stem cell differentiation, J. Biol. Chem 287 (2012) 36777–36791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tokar EJ, Diwan BA, Waalkes MP, Arsenic exposure transforms human epithelial stem/progenitor cells into a cancer stem-like phenotype, Environ. Health Perspect 118 (2010) 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rodriguez R, Rubio R, Menendez P, Modeling sarcomagenesis using multipotent mesenchymal stem cells, Cell Res 22 (2012) 62–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, Alvarez-Buylla A, Parada LF, Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model, Cancer Cell 15 (2009) 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, Nijman IJ, Martincorena I, Mokry M, Wiegerinck CL, Middendorp S, Sato T, Schwank G, Nieuwenhuis EES, Verstegen MMA, van der Laan LJW, de Jonge J, Ijzermans JNM, Vries RG, van de Wetering M, Stratton MR, Clevers H, Cuppen E, van Boxtel R, Tissue-specific mutation accumulation in human adult stem cells during life, Nature 538 (2016) 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Abel EV, Kim EJ, Wu J, Hynes M, Bednar F, Proctor E, Wang L, Dziubinski ML, Simeone DM, The notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer, PLOS One 9 (2014) e91983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lee G, Auffnger B, Guo D, Hasan T, Deheeger M, Tobias AL, Kim JY, Atashi F, Zhang L, Lesniak MS, James CD, Ahmed AU, Dedifferentiation of glioma cells to glioma stem-like cells by therapeutic stress-induced hif signaling in the recurrent GBM model, Mol. Cancer Therapeut 15 (2016) 3064–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang P, Lan C, Xiong S, Zhao X, Shan Y, Hu R, Wan W, Yu S, Liao B, Li G, Wang J, Zou D, Chen B, Feng H, Wu N, HIF1α regulates single differentiated glioma cell dedifferentiation to stem-like cell phenotypes with high tumorigenic potential under hypoxia, Oncotarget 8 (2017) 28074–28092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Donati G, Rognoni E, Hiratsuka T, Liakath-Ali K, Hoste E, Kar G, Kayikci M, Russell R, Kretzschmar K, Mulder KW, Teichmann SA, Watt FM, Wounding induces dedifferentiation of epidermal Gata6+ cells and acquisition of stem cell properties, Nat. Cell Biol 19 (2017) 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lee SY, Jeong EK, Ju MK, Jeon HM, Kim MY, Kim CH, Park HG, Han SI, Kang HS, Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation, Mol. Cancer 16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Miura K, Kimura K, Amano R, Yamazoe S, Ohira G, Nishio K, Kametani N, Hirakawa K, Ohira M, Analysis of the origin of anaplastic pancreatic cancer and the mechanism of its dedifferentiation, J. Hepato-Biliary-Pancreatic Sci 24 (2017) 176–184. [DOI] [PubMed] [Google Scholar]
- [44].Liberti MV, Locasale JW, The Warburg effect: how does it benefit cancer cells?, Trends Biochem. Sci, 41 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ciavardelli D, Rossi C, Barcaroli D, Volpe S, Consalvo A, Zucchelli M, De Cola A, Scavo E, Carollo R, D’Agostino D, Forli F, D’Aguanno S, Todaro M, Stassi G, Di Ilio C, De Laurenzi V, Urbani A, Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment, Cell Death Dis 5 (2014) e1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pasto A, Bellio C, Pilotto G, Ciminale V, Silic-Benussi M, Guzzo G, Rasola A, Frasson C, Nardo G, Zulato E, Nicoletto MO, Manicone M, Indraccolo S, Amadori A, Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation, Oncotarget 5 (2014) 4305–4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Folmes Clifford D.L., Nelson Timothy J., Martinez-Fernandez A, Arrell DK, Lindor Jelena Z., Dzeja Petras P., Ikeda Y, Perez-Terzic C, Terzic A, Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming, Cell Metabolism, 14 264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bonnet D, Dick JE, Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell, Nat. Med 3 (1997) 730–737. [DOI] [PubMed] [Google Scholar]
- [49].Cox CV, Evely RS, Oakhill A, Pamphilon DH, Goulden NJ, Blair A, Characterization of acute lymphoblastic leukemia progenitor cells, Blood 104 (2004) 2919–2925. [DOI] [PubMed] [Google Scholar]
- [50].Tavor S, Petit I, Porozov S, Avigdor A, Dar A, Leider-Trejo L, Shemtov N, Deutsch V, Naparstek E, Nagler A, Lapidot T, CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice, Cancer Res 64 (2004) 2817–2824. [DOI] [PubMed] [Google Scholar]
- [51].Kikushige Y, Miyamoto T, Identification of TIM-3 as a leukemic stem cell surface molecule in primary acute myeloid leukemia, Oncology 89 (Suppl. 1) (2015) 28–32. [DOI] [PubMed] [Google Scholar]
- [52].Al-Mawali A, Gillis D, Lewis I, Immunoprofiling of leukemic stem cells CD34+/ CD38-/CD123+ delineate FLT3/ITD-positive clones, J. Hematol. Oncol 9 (2016) 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kersten B, Valkering M, Wouters R, van Amerongen R, Hanekamp D, Kwidama Z, Valk P, Ossenkoppele G, Zeijlemaker W, Kaspers G, Cloos J, Schuurhuis GJ, CD45RA, a specific marker for leukaemia stem cell sub-popula-tions in acute myeloid leukaemia, Br. J. Haematol 173 (2016) 219–235. [DOI] [PubMed] [Google Scholar]
- [54].Sun Q, So CC, Yip SF, Wan TS, Ma SK, Chan LC, Functional alterations of Lin-CD34+CD38+ cells in chronic myelomonocytic leukemia and on progression to acute leukemia, Leuk. Res 32 (2008) 1374–1381. [DOI] [PubMed] [Google Scholar]
- [55].Monzani E, Facchetti F, Galmozzi E, Corsini E, Benetti A, Cavazzin C, Gritti A, Piccinini A, Porro D, Santinami M, Invernici G, Parati E, Alessandri G, La Porta CA, Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential, Eur. J. Cancer 43 (2007) 935–946. [DOI] [PubMed] [Google Scholar]
- [56].Luo Y, Dallaglio K, Chen Y, Robinson WA, Robinson SE, McCarter MD, Wang J, Gonzalez R, Thompson DC, Norris DA, Roop DR, Vasiliou V, Fujita M, ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets, Stem Cells 30 (2012) 2100–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fusi A, Reichelt U, Busse A, Ochsenreither S, Rietz A, Maisel M, Keilholz U, Expression of the stem cell markers nestin and CD133 on circulating melanoma cells, J. Invest. Dermatol 131 (2011) 487–494. [DOI] [PubMed] [Google Scholar]
- [58].Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271, Nature 466 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, Van Belle PA, Xu X, Elder DE, Herlyn M, A tumorigenic subpopulation with stem cell properties in melanomas, Cancer Res 65 (2005) 9328–9337. [DOI] [PubMed] [Google Scholar]
- [60].Li C, Du Y, Yang Z, He L, Wang Y, Hao L, Ding M, Yan R, Wang J, Fan Z, GALNT1-Mediated Glycosylation and Activation of Sonic Hedgehog Signaling Maintains the Self-Renewal and Tumor-Initiating Capacity of Bladder Cancer Stem Cells, Cancer Res 76 (2016) 1273–1283. [DOI] [PubMed] [Google Scholar]
- [61].Huang P, Watanabe M, Kaku H, Ueki H, Noguchi H, Sugimoto M, Hirata T, Yamada H, Takei K, Zheng S, Xu K, Nasu Y, Fujii Y, Liu C, Kumon H, Cancer stem cell-like characteristics of a CD133(+) subpopulation in the J82 human bladder cancer cell line, Mol. Clin. Oncol (1) (2013) 180–184. [DOI] [PMC free article] [PubMed]
- [62].Kenny HA, Chiang CY, White EA, Schryver EM, Habis M, Romero IL, Ladanyi A, Penicka CV, George J, Matlin K, Montag A, Wroblewski K, Yamada SD, Mazar AP, Bowtell D, Lengyel E, Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion, J. Clin. Invest 124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CLL, Rich JN, Cancer stem cells in glioblastoma, Genes Dev 29 (2015) 1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Muto J, Imai T, Ogawa D, Nishimoto Y, Okada Y, Mabuchi Y, Kawase T, Iwanami A, Mischel PS, Saya H, Yoshida K, Matsuzaki Y, Okano H, RNA-Binding Protein Musashi1 Modulates Glioma Cell Growth through the Post-Transcriptional Regulation of Notch and PI3 Kinase/Akt Signaling Pathways, PLOS ONE 7 (2012) e33431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Read TA, Fogarty MP, Markant SL, McLendon RE, Wei Z, Ellison DW, Febbo PG, Wechsler-Reya RJ, Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma, Cancer Cell 15 (2009) 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, Hong S, Adams A, D’Angelo R, Ginestier C, Charafe-Jauffret E, Clouthier SG, Birnbaum D, Wong ST, Zhan M, Chang JC, Wicha MS, Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts, Stem Cell Rep 2 (2014) 78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Duru N, Gernapudi R, Lo PK, Yao Y, Wolfson B, Zhang Y, Zhou Q, Characterization of the CD49f+/CD44+/CD24- single-cell derived stem cell population in basal-like DCIS cells, Oncotarget 7 (2016) 47511–47525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wang B, Chen Q, Cao Y, Ma X, Yin C, Jia Y, Zang A, Fan W, LGR5 Is a Gastric Cancer Stem Cell Marker Associated with Stemness and the EMT Signature Genes NANOG, NANOGP8, PRRX1, TWIST1, and BMI1, PLoS One 11 (2016) e0168904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Brungs D, Aghmesheh M, Vine KL, Becker TM, Carolan MG, Ranson M, Gastric cancer stem cells: evidence, potential markers, and clinical implications, J. Gastroenterol 51 (2016) 313–326. [DOI] [PubMed] [Google Scholar]
- [70].Wu W, Cao J, Ji Z, Wang J, Jiang T, Ding H, Co-expression of Lgr5 and CXCR4 characterizes cancer stem-like cells of colorectal cancer, Oncotarget 7 (2016) 81144–81155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhang SS, Han ZP, Jing YY, Tao SF, Li TJ, Wang H, Wang Y, Li R, Yang Y, Zhao X, Xu XD, Yu ED, Rui YC, Liu HJ, Zhang L, Wei LX, CD133(+)CXCR4(+) colon cancer cells exhibit metastatic potential and predict poor prognosis of patients, BMC Med 10 (2012) 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chandrakesan P, Yao J, Qu D, May R, Weygant N, Ge Y, Ali N, Sureban SM, Gude M, Vega K, Bannerman-Menson E, Xia L, Bronze M, An G, Houchen CW, Dclk1, a tumor stem cell marker, regulates pro-survival signaling and self-renewal of intestinal tumor cells, Mol. Cancer 16 (2017) 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Conic I, Stanojevic Z, Jankovic Velickovic L, Stojnev S, Ristic Petrovic A, Krstic M, Stanojevic M, Bogdanovic D, Stefanovic V, Epithelial ovarian cancer with CD117 phenotype is highly aggressive and resistant to chemotherapy, The journal of obstetrics and gynaecology research 41 (2015) 1630–1637. [DOI] [PubMed] [Google Scholar]
- [74].Bhaijee F, Pepper DJ, Pitman KT, Bell D, Cancer stem cells in head and neck squamous cell carcinoma: a review of current knowledge and future applications, Head & Neck 34 (2012) 894–899. [DOI] [PubMed] [Google Scholar]
- [75].Jung YS, Vermeer PD, Vermeer DW, Lee SJ, Goh AR, Ahn HJ, Lee JH, CD200: association with cancer stem cell features and response to chemoradiation in head and neck squamous cell carcinoma, Head Neck 37 (2015) 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ma S, Chan KW, Lee TK, Tang KH, Wo JY, Zheng BJ, Guan XY, Aldehyde dehydrogenase discriminates the CD133 liver cancer stem cell populations, Mol. Cancer Res 6 (2008) 1146–1153. [DOI] [PubMed] [Google Scholar]
- [77].Rozeik MS, Hammam OA, Ali AI, Magdy M, Khalil H, Anas A, Abo El Hassan AA, Rahim AA, El-Shabasy AI, Evaluation of CD44 and CD133 as markers of liver cancer stem cells in Egyptian patients with HCV-induced chronic liver diseases versus hepatocellular carcinoma, Electr. Phys 9 (2017) 4708–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, Chu PW, Lam CT, Poon RT, Fan ST, Significance of CD90+ cancer stem cells in human liver cancer, Cancer Cell 13 (2008) 153–166. [DOI] [PubMed] [Google Scholar]
- [79].Okudela K, Woo T, Mitsui H, Tajiri M, Masuda M, Ohashi K, Expression of the potential cancer stem cell markers, CD133, CD44, ALDH1, and beta-catenin, in primary lung adenocarcinoma–their prognostic significance, Pathol. Int 62 (2012) 792–801. [DOI] [PubMed] [Google Scholar]
- [80].Zakaria N, Yusoff NM, Zakaria Z, Lim MN, Baharuddin PJ, Fakiruddin KS, Yahaya B, Human non-small cell lung cancer expresses putative cancer stem cell markers and exhibits the transcriptomic profile of multipotent cells, BMC Cancer 15 (2015) 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Vaz AP, Ponnusamy MP, Seshacharyulu P, Batra SK, A concise review on the current understanding of pancreatic cancer stem cells, J. Cancer Stem Cell Res 2 (2014) e1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Miranda-Lorenzo I, Dorado J, Lonardo E, Alcala S, Serrano AG, Clausell-Tormos J, Cioff M, Megias D, Zagorac S, Balic A, Hidalgo M, Erkan M, Kleeff J, Scarpa A, Sainz B Jr., Heeschen C, Intracellular autofluorescence: a biomarker for epithelial cancer stem cells, Nat. Meth 11 (2014) 1161–1169. [DOI] [PubMed] [Google Scholar]
- [83].Kim N, Choung HK, Lee MJ, Khwarg SI, Kim JE, Cancer stem cell markers in eyelid sebaceous gland carcinoma: high expression of ALDH1, CD133, and ABCG2 correlates with poor prognosis, Investig. Ophthalmol. Visual Sci 56 (2015) 1813–1819. [DOI] [PubMed] [Google Scholar]
- [84].Yanamoto S, Yamada S, Takahashi H, Naruse T, Matsushita Y, Ikeda H, Shiraishi T, Seki S, Fujita S, Ikeda T, Asahina I, Umeda M, Expression of the cancer stem cell markers CD44v6 and ABCG2 in tongue cancer: effect of neoadjuvant chemotherapy on local recurrence, Int. J. Oncol 44 (2014) 1153–1162. [DOI] [PubMed] [Google Scholar]
- [85].Bi Y, Meng Y, Wu H, Cui Q, Luo Y, Xue X, Expression of the potential cancer stem cell markers CD133 and CD44 in medullary thyroid carcinoma: a ten-year follow-up and prognostic analysis, J. Surg. Oncol 113 (2016) 144–151. [DOI] [PubMed] [Google Scholar]
- [86].Elbasateeny SS, Salem AA, Abdelsalam WA, Salem RA, Immunohistochemical expression of cancer stem cell related markers CD44 and CD133 in endometrial cancer, Pathol. Res. Pract 212 (2016) 10–16. [DOI] [PubMed] [Google Scholar]
- [87].Nagata T, Sakakura C, Komiyama S, Miyashita A, Nishio M, Murayama Y, Komatsu S, Shiozaki A, Kuriu Y, Ikoma H, Nakanishi M, Ichikawa D, Fujiwara H, Okamoto K, Ochiai T, Kokuba Y, Sonoyama T, Otsuji E, Expression of cancer stem cell markers CD133 and CD44 in locoregional recurrence of rectal cancer, Anticancer Res 31 (2011) 495–500. [PubMed] [Google Scholar]
- [88].Lukenda A, Dotlic S, Vukojevic N, Saric B, Vranic S, Zarkovic K, Expression and prognostic value of putative cancer stem cell markers CD117 and CD15 in choroidal and ciliary body melanoma, J. Clin. Pathol 69 (2016) 234–239. [DOI] [PubMed] [Google Scholar]
- [89].Matsika A, Srinivasan B, Day C, Mader SA, Kiernan DM, Broomfield A, Fu J, Hooper JD, Kench JG, Samaratunga H, Cancer stem cell markers in prostate cancer: an immunohistochemical study of ALDH1, SOX2 and EZH2, Pathology 47 (2015) 622–628. [DOI] [PubMed] [Google Scholar]
- [90].Skoda J, Nunukova A, Loja T, Zambo I, Neradil J, Mudry P, Zitterbart K, Hermanova M, Hampl A, Sterba J, Veselska R, Cancer stem cell markers in pediatric sarcomas: Sox2 is associated with tumorigenicity in immunodeficient mice, Tumour Biol 37 (2016) 9535–9548. [DOI] [PubMed] [Google Scholar]
- [91].Feng JQ, Xu ZY, Shi LJ, Wu L, Liu W, Zhou ZT, Expression of cancer stem cell markers ALDH1 and Bmi1 in oral erythroplakia and the risk of oral cancer, J. Oral Pathol. Med 42 (2013) 148–153. [DOI] [PubMed] [Google Scholar]
- [92].Karmakar S, Seshacharyulu P, Lakshmanan I, Vaz AP, Chugh S, Sheinin YM, Mahapatra S, Batra SK, Ponnusamy MP, hPaf1/PD2 interacts with OCT3/4 to promote self-renewal of ovarian cancer stem cells, Oncotarget 8 (2017) 14806–14820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Vaz AP, Ponnusamy MP, Rachagani S, Dey P, Ganti AK, Batra SK, Novel role of pancreatic differentiation 2 in facilitating self-renewal and drug resistance of pancreatic cancer stem cells, Br. J. Cancer 111 (2014) 486–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Nimmakayala RK, Seshacharyulu P, Lakshmanan I, Rachagani S, Chugh S, Karmakar S, Rauth S, Vengoji R, Atri P, Talmon GA, Lele SM, Smith LM, Thapa I, Bastola D, Ouellette MM, Batra SK, Ponnusamy MP, Cigarette smoke induces stem cell features of pancreatic cancer cells via PAF1, Gastroenterology 155 (2018) 892–908 e896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Wang Z, Shi Q, Wang Z, Gu Y, Shen Y, Sun M, Deng M, Zhang H, Fang J, Zhang S, Xie F, Clinicopathologic correlation of cancer stem cell markers CD44, CD24, VEGF and HIF-1alpha in ductal carcinoma in situ and invasive ductal carcinoma of breast: an immunohistochemistry-based pilot study, Pathol. Res. Pract 207 (2011) 505–513. [DOI] [PubMed] [Google Scholar]
- [96].Zhang J, Nuebel E, Daley GQ, Koehler CM, Teitell MA, Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal, Cell Stem Cell 11 (2012) 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Shen YA, Metabolic reprogramming orchestrates cancer stem cell properties in, 14 (2015), pp. 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Song I-S, Jeong YJ, Han J, Mitochondrial metabolism in cancer stem cells: a therapeutic target for colon cancer, BMB Rep 48 (2015) 539–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, Popescu R, Della Donna L, Evers P, Dekmezian C, Reue K, Christofk H, Mischel PS, Pajonk F, Metabolic state of glioma stem cells and nontumorigenic cells, Proc. Natl. Acad. Sci 108 (2011) 16062–16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Gammon L, Biddle A, Heywood HK, Johannessen AC, Mackenzie IC, Sub-sets of cancer stem cells differ intrinsically in their patterns of oxygen metabolism, PLOS ONE 8 (2013) e62493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, Herlyn M, A temporarily distinct sub-population of slow-cycling melanoma cells is required for continuous tumor growth, Cell 141 (2010) 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, Arendt LM, Kuperwasser C, Bierie B, Weinberg RA, Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state, Proc. Natl. Acad. Sci. U. S. A 108 (2011) 7950–7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Suva ML, Riggi N, Bernstein BE, Epigenetic reprogramming in cancer, Science 339 (2013) 1567–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, Clouthier SG, Wicha MS, Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling, PLoS Biol 7 (2009) e1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, Rupec RA, Gerhard M, Schmid R, Barker N, Clevers H, Lang R, Neumann J, Kirchner T, Taketo MM, van den Brink GR, Sansom OJ, Arkan MC, Greten FR, Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties, Cell 152 (2013) 25–38. [DOI] [PubMed] [Google Scholar]
- [106].Milner LA, Bigas A, Notch as a mediator of cell fate determination in hemato-poiesis: evidence and speculation, Blood 93 (1999) 2431–2448. [PubMed] [Google Scholar]
- [107].Wu XB, Liu Y, Wang GH, Xu X, Cai Y, Wang HY, Li YQ, Meng HF, Dai F, Jin JD, Mesenchymal stem cells promote colorectal cancer progression through AMPK/mTOR-mediated NF-kappaB activation, Sci. Rep 6 (2016) 21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A, Griffero F, Marubbi D, Spaziante R, Bellora F, Moretta L, Moretta A, Corte G, Bottino C, NK cells recognize and kill human glioblastoma cells with stem cell-like properties, J. Immunol 182 (2009) 3530–3539. [DOI] [PubMed] [Google Scholar]
- [109].Kim H, Lin Q, Glazer PM, Yun Z, The hypoxic tumor microenvironment in vivo selects the cancer stem cell fate of breast cancer cells, Breast Cancer Res 20 (2018) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC, Tetta C, Bussolati B, Camussi G, Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche, Cancer Res 71 (2011) 5346–5356. [DOI] [PubMed] [Google Scholar]
- [111].Pohl S-G, Brook N, Agostino M, Arfuso F, Kumar AP, Dharmarajan A, Wnt signaling in triple-negative breast cancer, Oncogenesis 6 (2017) e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Fan J, Wei Q, Liao J, Zou Y, Song D, Xiong D, Ma C, Hu X, Qu X, Chen L, Li L, Yu Y, Yu X, Zhang Z, Zhao C, Zeng Z, Zhang R, Yan S, Wu T, Wu X, Shu Y, Lei J, Li Y, Zhang W, Haydon RC, Luu HH, Huang A, He TC, Tang H, Noncanonical Wnt signaling plays an important role in modulating canonical Wnt-regulated stemness, proliferation and terminal differentiation of hepatic progeni-tors, Oncotarget 8 (2017) 27105–27119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Nalapareddy K, Nattamai KJ, Kumar RS, Karns R, Wikenheiser-Brokamp KA, Sampson LL, Mahe MM, Sundaram N, Yacyshyn M-B, Yacyshyn B, Helmrath MA, Zheng Y, Geiger H, Canonical Wnt signaling ameliorates aging of intestinal stem cells, Cell Rep 18 (2017) 2608–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].de Lau W, Peng WC, Gros P, Clevers H, The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength, Genes Dev 28 (2014) 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Koch S, Nava P, Addis C, Kim W, Denning TL, Li L, Parkos CA, Nusrat A, The Wnt antagonist Dkk1 regulates intestinal epithelial homeostasis and wound repair, Gastroenterology 141 (2011) 259–268 e258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Yu QC, Verheyen EM, Zeng YA, Mammary development and breast cancer: a Wnt perspective, Cancers 8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Wang D, Cai C, Dong X, Yu QC, Zhang X-O, Yang L, Zeng YA, Identification of multipotent mammary stem cells by protein C receptor expression, Nature 517 (2015) 81–84. [DOI] [PubMed] [Google Scholar]
- [118].Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zürrer-Härdi U, Bell G, Tam WL, Mani SA, van Oudenaarden A, Weinberg RA, Slug and Sox9 cooperatively determine the mammary stem cell state, Cell 148 (2012) 1015–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Wang Y, Dong J, Li D, Lai L, Siwko S, Li Y, Liu M, Lgr4 regulates mammary gland development and stem cell activity through the pluripotency transcription factor Sox2, Stem Cells (Dayton, Ohio) 31 (2013) 1921–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Luo W, Rodriguez M, Valdez JM, Zhu X, Tan K, Li D, Siwko S, Xin L, Liu M, Lgr4 is a key regulator of prostate development and prostate stem cell differ-entiation, Stem Cells (Dayton, Ohio) 31 (2013) 2492–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Tanaka N, Mashima T, Mizutani A, Sato A, Aoyama A, Gong B, Yoshida H, Muramatsu Y, Nakata K, Matsuura M, Katayama R, Nagayama S, Fujita N, Sugimoto Y, Seimiya H, APC mutations as a potential biomarker for sensitivity to tankyrase inhibitors in colorectal cancer, Mol. Cancer Therap 16 (2017) 752–762. [DOI] [PubMed] [Google Scholar]
- [122].Kaler P, Augenlicht L, Klampfer L, Activating mutations in β-catenin in colon cancer cells alter their interaction with macrophages; the role of snail, PLOS ONE 7 (2012) e45462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Basu S, Haase G, Ben-Ze’ev A, Wnt signaling in cancer stem cells and colon cancer metastasis [version 1; referees: 3 approved], (2016). [DOI] [PMC free article] [PubMed]
- [124].Jang G-B, Kim J-Y, Cho S-D, Park K-S, Jung J-Y, Lee H-Y, Hong I-S, Nam JS, Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype, Sci. Rep 5 (2015) 12465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Tse CO, Kim S, Park J, Activation of Wnt signaling pathway by AF1q enriches stem-like population and enhance mammosphere formation of breast cells, Biochem. Biophys. Res. Commun 484 (2017) 884–889. [DOI] [PubMed] [Google Scholar]
- [126].Hanna A, Shevde LA, Hedgehog signaling: modulation of cancer properies and tumor mircroenvironment, Mol. Cancer 15 (2016) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Wu SM, Choo ABH, Yap MGS, Chan KK-K, Role of Sonic hedgehog signaling and the expression of its components in human embryonic stem cells, Stem Cell Res 4 (2010) 38–49. [DOI] [PubMed] [Google Scholar]
- [128].Moura RS, Silva-Gonçalves C, Vaz-Cunha P, Correia-Pinto J, Expression analysis of Shh signaling members in early stages of chick lung development, Histochem. Cell Biol 146 (2016) 457–466. [DOI] [PubMed] [Google Scholar]
- [129].Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, Devereux WL, Rhodes JT, Huff CA, Beachy PA, Watkins DN, Matsui W, Hedgehog signaling maintains a tumor stem cell compartment in multiple mye-loma, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 4048–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Clement V, Sanchez P, de Tribolet N, Radovanovic I, Altaba AR, HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal and tumorigenicity, Curr. Biol 17 (2007) 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Liu S, Dontu G, Mantle ID, Patel S, Ahn N, Jackson KW, Suri P, Wicha MS, Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells, Cancer Res 66 (2006) 6063–6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Singh BN, Fu J, Srivastava RK, Shankar S, Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: molecular mechanisms, PLoS One (2011) 6. [DOI] [PMC free article] [PubMed]
- [133].Nowell CS, Radtke F, Notch as a tumour suppressor, Nat. Rev. Cancer 17 (2017) 145–159. [DOI] [PubMed] [Google Scholar]
- [134].Srinivasan T, Than EB, Bu P, Tung K-L, Chen K-Y, Augenlicht L, Lipkin SM, Shen X, Notch signalling regulates asymmetric division and interconversion between lgr5 and bmi1 expressing intestinal stem cells, Sci. Rep 6 (2016) 26069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Imayoshi I, Sakamoto M, Yamaguchi M, Mori K, Kageyama R, Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains, J. Neurosci 30 (2010) 3489–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Xiao W, Gao Z, Duan Y, Yuan W, Ke Y, Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma, J. Exp. Clin. Cancer Res 36 (2017) 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Barat S, Chen X, Cuong Bui K, Bozko P, Götze J, Christgen M, Krech T, Malek NP, Plentz RR, Gamma-Secretase Inhibitor IX (GSI) Impairs Concomitant Activation of Notch and Wnt-Beta-Catenin Pathways in CD44+ Gastric Cancer Stem Cells, STEM CELLS Transl. Med 6 (2017) 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Eckford PDW, Sharom FJ, ABC Efflux Pump-Based Resistance to Chemotherapy Drugs, Chem. Rev 109 (2009) 2989–3011. [DOI] [PubMed] [Google Scholar]
- [139].Chow EKH, Fan L, Chen X, Bishop JM, Oncogene-specific formation of che-moresistant murine hepatic cancer stem cells, Hepatology 56 (2012) 1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Zhu MM, Tong JL, Xu Q, Nie F, Xu XT, Xiao SD, Ran ZH, Increased JNK1 Signaling Pathway Is Responsible for ABCG2-Mediated Multidrug Resistance in Human Colon Cancer, PLOS ONE 7 (2012) e41763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].To KKW, Tomlinson B, Targeting the ABCG2-overexpressing multidrug resistant (MDR) cancer cells by PPARγ agonists, Br. J. Pharmacol 170 (2013) 1137–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Xiang L, Liu Z-H, Huan Q, Su P, Du G-J, Wang Y, Gao P, Zhou G-Y, Hypoxia-inducible factor-2a is associated with ABCG2 expression, histology-grade and Ki67 expression in breast invasive ductal carcinoma, Diagn. Pathol 7 (2012) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Martin CM, Ferdous A, Gallardo T, Humphries C, Sadek H, Caprioli A, Garcia JA, Szweda LI, Garry MG, Garry DJ, Hypoxia-inducible factor-2α transactivates Abcg2 and promotes cytoprotection in cardiac side population cells, Circ. Res 102 (2008). [DOI] [PubMed] [Google Scholar]
- [144].Singh A, Wu H, Zhang P, Happel C, Ma J, Biswal S, Expression of ABCG2 (BCRP), a marker of stem cells, is regulated by Nrf2 in cancer cells that confers side population and chemoresistance phenotype, Mol. Cancer Therap 9 (2010) 2365–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Keren-Paz A, Emmanuel R, Samuels Y, YAP and the drug resistance highway, Nat. Genet 47 (2015) 193–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Liu Z, Xu J, He J, Zheng Y, Li H, Lu Y, Qian J, Lin P, Weber DM, Yang J, Yi Q, A critical role of autocrine sonic hedgehog signaling in human CD138+ myeloma cell survival and drug resistance, Blood 124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM, Liesveld JL, Brookes PS, Becker MW, Jordan CT, BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells, Cell Stem Cell 12 (2013) 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O’Rourke K, Bazan F, Eastham-Anderson J, Yue P, Dornan D, Huang DCS, Dixit VM, Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival, Nature 463 (2010) 103–107. [DOI] [PubMed] [Google Scholar]
- [149].Tagscherer KE, Fassl A, Campos B, Farhadi M, Kraemer A, Bock BC, Macher-Goeppinger S, Radlwimmer B, Wiestler OD, Herold-Mende C, Roth W, Apoptosis-based treatment of glioblastomas with ABT-737, a novel small molecule inhibitor of Bcl-2 family proteins, Oncogene 27 (2008) 6646–6656. [DOI] [PubMed] [Google Scholar]
- [150].Piggott L, Omidvar N, Pérez SM, Eberl M, Clarkson RW, Suppression of apoptosis inhibitor c-FLIP selectively eliminates breast cancer stem cell activity in response to the anti-cancer agent, TRAIL, Breast Cancer Res 13 (2011) R88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Siddharth S, Das S, Nayak A, Kundu CN, SURVIVIN as a marker for quiescent-breast cancer stem cells—An intermediate, adherent, pre-requisite phase of breast cancer metastasis, Clin. Exp. Metastasis 33 (2016) 661–675. [DOI] [PubMed] [Google Scholar]
- [152].Long Q, Yang R, Lu W, Zhu W, Zhou J, Zheng C, Zhou D, Yu L, Wu J, Adenovirus-mediated truncated Bid overexpression induced by the Cre/LoxP system promotes the cell apoptosis of CD133+ ovarian cancer stem cells, Oncol. Rep 37 (2017) 155–162. [DOI] [PubMed] [Google Scholar]
- [153].Lv N, Shan Z, Gao Y, Guan H, Fan C, Wang H, Teng W, Twist1 regulates the epithelial-mesenchymal transition via the NF-kappaB pathway in papillary thyroid carcinoma, Endocrine 51 (2016) 469–477. [DOI] [PubMed] [Google Scholar]
- [154].Kong L, Guo S, Liu C, Zhao Y, Feng C, Liu Y, Wang T, Li C, Overexpression of SDF-1 activates the NF-kappaB pathway to induce epithelial to mesenchymal transition and cancer stem cell-like phenotypes of breast cancer cells, Int. J. Oncol 48 (2016) 1085–1094. [DOI] [PubMed] [Google Scholar]
- [155].Xi G, Hayes E, Lewis R, Ichi S, Mania-Farnell B, Shim K, Takao T, Allender E, Mayanil CS, Tomita T, CD133 and DNA-PK regulate MDR1 via the PI3K- or Akt-NF-kappaB pathway in multidrug-resistant glioblastoma cells in vitro, Oncogene 35 (2016) 241–250. [DOI] [PubMed] [Google Scholar]
- [156].Cao L, Fan X, Jing W, Liang Y, Chen R, Liu Y, Zhu M, Jia R, Wang H, Zhang X, Zhang Y, Zhou X, Zhao J, Guo Y, Osteopontin promotes a cancer stem cell-like phenotype in hepatocellular carcinoma cells via an integrin-NF-kappaB-HIF-1alpha pathway, Oncotarget (6) (2015) 6627–6640. [DOI] [PMC free article] [PubMed]
- [157].Xu C, Sun X, Qin S, Wang H, Zheng Z, Xu S, Luo G, Liu P, Liu J, Du N, Zhang Y, Liu D, Ren H, Let-7a regulates mammosphere formation capacity through Ras/NF-kappaB and Ras/MAPK/ERK pathway in breast cancer stem cells, Cell Cycle 14 (2015) 1686–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Prigione A, Rohwer N, Hoffmann S, Mlody B, Drews K, Bukowiecki R, Blümlein K, Wanker EE, Ralser M, Cramer T, Adjaye J, HIF1α modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1–3 and PKM2, Stem Cells (Dayton, Ohio) 32 (2014) 364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Marhold M, Tomasich E, El-Gazzar A, Heller G, Spittler A, Horvat R, Krainer M, Horak P, HIF1α regulates mTOR Signaling and Viability of Prostate Cancer Stem Cells, Mol. Cancer Res 13 (2015) 556–564. [DOI] [PubMed] [Google Scholar]
- [160].Giambra V, Jenkins CE, Lam SH, Hoofd C, Belmonte M, Wang X, Gusscott S, Gracias D, Weng AP, Leukemia stem cells in T-ALL require active Hif1α and Wnt signaling, Blood 125 (2015) 3917–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Iida H, Suzuki M, Goitsuka R, Ueno H, Hypoxia induces CD133 expression in human lung cancer cells by up-regulation of OCT3/4 and SOX2, Int. J. Oncol, 40 (2012) 71–79. [DOI] [PubMed] [Google Scholar]
- [162].Zheng X, Boyer L, Jin M, Mertens J, Kim Y, Ma L, Ma L, Hamm M, Gage FH, Hunter T, Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation, eLife 5 (2016) e13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Viale A, Giulio F Draetta, Sugar? No Thank You, Just a Deep Breath of Oxygen for Cancer Stem Cells, Cell Metab 22 (2015) 543–545. [DOI] [PubMed] [Google Scholar]
- [164].Chen C-L, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, Hess S, Machida K, NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid meta-bolism, Cell Metab 23 (2016) 206–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Fokas E, Engenhart-Cabillic R, Daniilidis K, Rose F, An H-X, Metastasis: the seed and soil theory gains identity, Cancer Metastasis Rev 26 (2007) 705–715. [DOI] [PubMed] [Google Scholar]
- [166].Psaila B, Lyden D, The Metastatic Niche: adapting the foreign soil, Nat. Rev. Cancer 9 (2009) 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Sampieri K, Fodde R, Cancer stem cells and metastasis, Sem. Cancer Biol 22 (2012) 187–193. [DOI] [PubMed] [Google Scholar]
- [168].Gao W, Chen L, Ma Z, Du Z, Zhao Z, Hu Z, Li Q, Isolation and phenotypic characterization of colorectal cancer stem cells with organ-specific metastatic potential, Gastroenterology 145 (2013) (636–646.e635). [DOI] [PubMed] [Google Scholar]
- [169].Hu J, Li G, Zhang P, Zhuang X, Hu G, A CD44v(+) subpopulation of breast cancer stem-like cells with enhanced lung metastasis capacity, Cell Death Dis 8 (2017) e2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Reichert M, Bakir B, Moreira L, Pitarresi JR, Feldmann K, Simon L, Suzuki K, Maddipati R, Rhim AD, Schlitter AM, Kriegsmann M, Weichert W, Wirth M, Schuck K, Schneider G, Saur D, Reynolds AB, Klein-Szanto AJ, Pehlivanoglu B, Memis B, Adsay NV, Rustgi AK, Regulation of epithelial plasticity determines metastatic organotropism in pancreatic cancer, Dev. Cell, 45 (2018) 696–711.e698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Mark MT, Molina H, Kohsaka S, Di Giannatale A, Ceder S, Singh S, Williams C, Soplop N, Uryu K, Pharmer L, King T, Bojmar L, Davies AE, Ararso Y, Zhang T, Zhang H, Hernandez J, Weiss JM, Dumont-Cole VD, Kramer K, Wexler LH, Narendran A, Schwartz GK, Healey JH, Sandstrom P, Labori KJ, Kure EH, Grandgenett PM, Hollingsworth MA, de Sousa M, Kaur S, Jain M, Mallya K, Batra SK, Jarnagin WR, Brady MS, Fodstad O, Muller V, Pantel K, Minn AJ, Bissell MJ, Garcia BA, Kang Y, Rajasekhar VK, Ghajar CM, Matei I, Peinado H, Bromberg J, Lyden D, Tumour exosome integrins determine organotropic metastasis, Nature 527 (2015) 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Lopatina T, Gai C, Deregibus MC, Kholia S, Camussi G, Cross talk between cancer and mesenchymal stem cells through extracellular vesicles carrying nucleic acids, Front. Oncol 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Fatima F, Nawaz M, Stem cell-derived exosomes: roles in stromal remodeling, tumor progression, and cancer immunotherapy, Chin. J. Cancer 34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]