

Abstract
Three new bis(2,2′-bipyridine)-heteroleptic Ru(II) dyads incorporating thienyl groups (n=1–3, compounds 1, 2, and 3, respectively) appended to 1,10-phenanthroline were synthesized and characterized to investigate the impact of n on the photophysical and photobiological properties within the series. All three complexes showed unstructured emission near 618 nm from a triplet metal-to-ligand charge transfer (3MLCT) state with a lifetime (τem) of approximately 1 μs. Transient absorption measurements revealed an additional excited state that was nonemissive and long-lived (τTA=43 μs for 2 and 27 μs for 3), assigned as a triplet intraligand (3IL) state that was accessible only in 2 and 3. All three complexes were strong singlet oxygen (1O2) sensitizers, with quantum yields (ΦΔ) for 2 and 3 being the largest (74–78%), and all three were photocytotoxic to cancer cells with visible light activation in the order: 3 > 2 > 1. Cell-free DNA photodamage followed the same trend, where potency increased with decreasing 3IL energy. Compounds 2 and 3 also showed in vitro photobiological effects with red light (625 nm), where their molar absorptivities were <100 M−1 cm−1. These findings highlight that Ru(II) dyads derived from α-oligothiophenes directly appended to 1,10-phenanthroline – namely 2 and 3 – possess low-lying 3IL states that are highly photosensitizing, and they may therefore be of interest for photobiological applications such as photodynamic therapy (PDT).
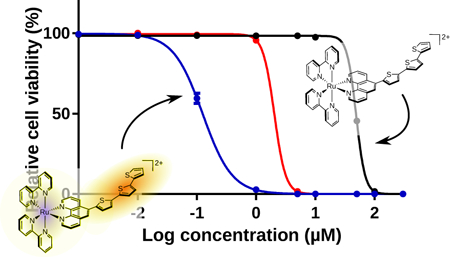
Graphical Abstract
Ru(II) dyads derived from imidazo[4,5-f][1,10]phenanthroline ligands appended with α-oligothienyl groups were investigated for their photophysical and photobiological properties. The compounds differed systematically by the number of thienyl groups, where n=1–3 in 1-3, respectively. All three compounds were effective singlet oxygen generators and were phototoxic to cells with visible light, with potency positively correlated to n. Compounds 2 and 3 were characterized by low-lying intraligand triplet excited states with prolonged lifetimes that give rise to in vitro phototoxic effects with red light, where molar extinction coefficients were vanishingly small.
INTRODUCTION
Ru(II) complexes for photobiological applications
Coordination complexes and organometallic complexes have emerged as attractive alternatives to organic compounds for light-based applications ranging from dye-sensitized solar cells (DSSCs) to photodynamic therapy (PDT). Their modular architectures lend well to systematic structural changes for tuning (photo)chemical, (photo)biological, and (photo)physical properties. An important feature for their use as photosensitizers is the sheer number of excited states that are accessible with visible and near-infrared light (1–4). Metals and ligands can be selected to produce lowest-energy excited states with predictable reactivities that can be fine-tuned with further chemical modification to one or more ligands. This concept has been demonstrated in the widely-studied Ru(II) polypyridyl class of complexes.
Briefly, the excited state dynamics of [Ru(bpy)3]2+ (bpy=2,2′-bipyridine), and many of its derivatives, are governed by a lowest-lying triplet metal-to-ligand charge transfer (3MLCT) excited state that exhibits an intrinsic lifetime of approximately 1 μs in deoxygenated solution at room temperature (1). This state decays primarily through phosphorescence or intersystem crossing (ISC) in the absence of oxygen and other excited state quenchers, but can sensitize singlet oxygen (1O2) in oxygenated solution. While the metal-centered (MC) and intraligand (IL) triplet excited states of [Ru(bpy)3]2+ are higher in energy than the 3MLCT state and largely inaccessible with visible light, judicious changes to the auxiliary ligands have resulted in systems with accessible 3MC or 3IL states that play a defining role in the photophysical dynamics.
Notable strategies include the use of substituted ligands that cause steric clash in the coordination sphere of the metal ion, to populate dissociative 3MC states upon photoexcitation. Photoinduced ligand loss in these strained Ru(II) complexes has been exploited for covalent modification of biomolecules such as DNA as a means of cytotoxicity for photochemotherapy (PCT) (5,6), although PCT has not yet been developed clinically. Alternatively, π-extended ligands have been used to lower the energy of 3IL states and prolong excited state lifetimes for improved 1O2 photosensitization (and to afford additional cytotoxic mechanisms) in the design of better PDT agents (7–12). The term metal-organic dyad has been used to describe these Ru(II) complexes that incorporate π-expanded organic chromophores that are tethered to one of the chelating ligands, and these systems were first studied by groups investigating triplet-triplet energy transfer in bichromophoric systems (13). The predicted utility (at the time) of such supramolecular systems was in the area of optoelectronic and luminescence-based technologies, but more recently, their potential in photobiological applications such as PDT/PCT has been recognized (7,14–20).
Ru(II) dyads with low-energy 3IL states and prolonged lifetimes
Ford and Rogers were the first to report on the attachment of organic chromophores to diimine ligands to impart novel photophysical properties to Ru(II) polypyridyl complexes in 1992 (21). In their example, a pyrenyl group appended to 1,10-phenanthroline (phen), spatially isolated from Ru(II), led to an 3IL-3MLCT excited state equilibrium where the 3IL state served as an energy reservoir to prolong the 3MLCT lifetime more than ten-fold (from 0.8 μs to 11.2 μs). Harriman and Ziessel later replaced the flexible hydrocarbon chain of the earlier Ru(II)-pyrenyl metal-organic dyad with an alkynyl linker directly conjugated to bpy, which increased the forward and reverse rates of triplet energy transfer and extended the 3MLCT lifetime to 42 μs (22). Building on this work, Castellano and Ziessel found that including two or three (rather than one) of the pyrenylethynylene-substituted bpy ligands led to pure 3IL states with slightly longer intrinsic lifetimes (57 and 65 μs, respectively) (23).
In 2010 two groups reported separately on Ru(II) dyads incorporating one pyrenylethnylene chromophore appended to phen (PEP), but differing in the substitution position (3-PEP versus 5-PEP) (14,24). These PEP-based Ru(II) dyads also possessed lowest-lying, pure 3IL states with lifetimes of 140 and 240 μs, respectively, that were extremely sensitive to trace oxygen (19). Zhao demonstrated the utility of such systems for luminescent oxygen sensing (24) while McFarland and Thummel demonstrated their potent in vitro PDT effects (19). While pure 3IL states (derived from 3ππ* transitions localized to the organic chromophore) are good producers of cytotoxic 1O2 through Type II energy transfer processes, 3IL states with some amount of charge-transfer character (3ILCT) offer the opportunity to exploit Type I electron transfer reactions as well as potential oxygen-independent pathways for PDT/PCT. Therefore, we had been simultaneously investigating α-oligothienyl-based Ru(II) dyads with accessible 3IL and 3ILCT states.
α-Oligothiophene-based Ru(II) dyads
Thiophene rings linked through the 2,2′ positions form stable, conjugated chains of oligomers and polymers. Oligo- and polythiophene derivatives are generally able to donate and accept electrons at moderate energies without decomposition (25); the interesting photophysical and electrochemical properties of conjugated oligo- and polythiophenes arise from the expansive π-system that can stabilize excited states and distribute positive and negative charges when the number of thiophene rings n is sufficiently large. Optical absorption by the π-extended system leads to symmetry-permitted excited states that can be driven to long-lived charge separation under the influence of a suitable acceptor or donor (26).
The capacity for charge separation and transport is useful in the fields of solar energy conversion and molecular electronics, but even oligomers of smaller n are well suited to such applications. Triplet states are accessible from ISC of a singlet exciton, with an exchange energy of around 0.7 eV in conjugated polymers in general (27) and 1.72 eV for 2,2′:5′,2′′-terthiophene (3T) (28). The “natural” size of the triplet exciton is n=3‒4 thiophene rings (29), but the quantum yield of triplet formation decreases as the number of rings increases, leveling off at around n=5 (30). We were thus interested in oligothiophenes of about n=3–5 for our Ru(II) dyads.
Early work by Scaiano and coworkers showed that relatively short thiophene chains (such as 3T) act as good 1O2 generators (28,31), which led us to focus on 3T as the organic chromophore for certain Ru(II) dyads. The 3ππ* states of 3T have been estimated as 1.72 eV (28) to 1.93 eV (32), lower in energy than the 3MLCT state of most polypyridyl Ru(II) complexes (33) making it possible to capture photons with 1MLCT states and deliver them effectively to spin-forbidden 3IL states on the organic chromophore. The slow ISC rates characteristic of oligothienyl-based 3ππ* states are responsible for the extended triplet lifetimes observed for these Ru(II) dyads, especially those with 3IL states that are sufficiently low in energy that they do not equilibrate with 3MLCT states. The implication is that long intrinsic lifetimes provide ample opportunity for bimolecular reactions with oxygen and other excited state quenchers.
In addition, Ru(II) dyads having oligothienyl-substituted ligands with n as small as 2 exhibit long-lived charge separation via 3ILCT states formed by reductive quenching of the initially populated MLCT state as demonstrated by Wolf and MacDonnell (33–35) These 3ILCT states can participate in electron transfer reactions with oxygen and also undergo autoionization steps to propagate chain reactions. This Type I/II dual reactivity has been explored computationally for Ru(II) dyads containing the oligothienyl-appended imidazo[4,5-f][1,10]phenanthroline (IP-nT) ligand, where n=1–4 (36), to explain the potent in vitro PDT effects observed with certain members of this family (7). One example that serves to illustrate the potential of such systems in photobiological applications is [Ru(dmb)2(IP-3T)]Cl2 (dmb=4,4′-dimethyl-2,2′-bipyridine), also known as TLD1433, a Ru(II) dyad that we developed that is the first Ru(II)-based photosensitizer to enter a human clinical trial and is being investigated for treating nonmuscle invasive bladder cancer with PDT (ClinicalTrials.gov Identifier: NCT03053635).
In the present investigation, we explore a new family of Ru(II) dyads with thiophene units (n=1–3) directly attached to phen at C5 (Chart 1). These structures are reminiscent of the analogous pyrenyl-containing system first reported by Castellano that exhibited a 24 μs lifetime (37). This study arose out of our ongoing interest in exploring the photophysical and photobiological properties of oligothienyl-based chromophores in a variety of Ru(II) dyad constructs.
MATERIALS AND METHODS
Synthesis and characterization.
1,10-phenanthroline monohydrate, ammonium acetate, thiophene, n-butyllithium, tributyltinchloride, tetrakis(triphenylphosphine)-palladium(0), thiophene (1T), 2,2′-bithiophene (2T), 2,2′:5′,2′′-terthiophene (3T), were purchased from Sigma-Aldrich and used without further purification. [Ru(bpy)2Cl2]∙2H20 was prepared by an established procedure (38). Microwave reactions were performed in a CEM Discover microwave reactor. NMR spectra were collected using Bruker AVANCE 500 (Dalhousie University Nuclear Magnetic Resonance Research Resource) or 300 (Acadia Centre for Microstructureal Analysis) MHz spectrometers, and ESI mass spectra were obtained using a Bruker microTOF focus mass spectrometer (Dalhousie University Mass Spectrometry Laboratory). HPLC analysis was carried out using a Hypersil GOLD C18 reversed-phase column with an A-B gradient (98% → 40% A; A=0.1% formic acid in H2O, B=0.1% formic acid in MeOH). Reported retention times are correct to within ± 0.1 min.
The metal complexes for this investigation were isolated and purified as PF6− salts and subsequently subjected to anion metathesis on Amberlite IRA-410 with MeOH in order to yield more water-soluble Cl− salts for biological analysis. 1H NMR and electrospray ionization ESI (+ve) mass spectra were collected on PF6− salts in CD3CN and CH3CN, respectively.
5-bromo-1,10-phenanthroline.
5-bromo-1,10-phenanthroline was prepared according to an adapted literature procedure (39,40). 1,10-Phenanthroline monohydrate (4.02 g) was dried in a vacuum oven (50 °C) for 3 days before use, resulting in a final weight of approximately 3.67 g. A dry heavy-walled pressure tube was charged with dry 1,10-phenanthroline (3.6 g, 20 mmol) and then cooled in an ice bath. Oleum (sulphuric acid fuming 20%, 12 mL) was added and the tube was stirred for 10 min, followed by dropwise addition of bromine (0.6 mL, 11.6 mmol) dropwise using a dry 2.5 mL gastight syringe. The pressure tube was tightly capped with a PTFE stopper, and heated slowly to 135 °C. Two days later, the color changed from a dark brown to a clear orange/red. The reaction mixture was cooled, then slowly and carefully poured into a beaker containing ice water and swirled, where the color changed to a brown/orange color. The mixture was neutralized with stirring by adding NH4OH dropwise, changing the pH from 4 to pH 10. The mixture became a yellow/orange cloudy color, with a dark red oil separated. The mixture was transferred to a separatory funnel and two chloroform extractions (~15 mL) were performed. The chloroform layer, a clear, light yellow color, was introduced to a beaker and a scoopful of activated charcoal was added and stirred for 15–20 min. The mixture was then poured into an Erlenmeyer flask containing anhydrous MgSO4, then gravity filtered through Whatman #1 filter paper into a round-bottom flask, and concentrated under vacuum. The light pink/purple solid was suspended in warm ether (300–400 mL), resulting in a light purple solution with precipitate, and this was filtered over a bed of Celite by applying light vacuum. The filtrate was then concentrated under reduced pressure and dried to yield the final product as a colorless solid (2.448 g, 47%). Rf = 0.38 (alumina, 5% MeOH/DCM). 1H NMR (300 MHz, CDCl3): δ 9.26 – 9.17 (m, 2H; a, h), 8.67 (d, J = 6.6 Hz, 1H; f), 8.21 – 8.11 (m, 2H; c,d), 7.81 – 7.59 (m, 2H; g,f).
5-(2-thiophene)-1,10-phenanthroline (phen-1T).
The starting material 2-(tributylstannyl)thiophene (IT-SnBu3) was prepared according to a published method with some exceptions (41). In summary, a 100 mL dry, argon purged, 3-hole round-bottom flask with stir bar was charged with anhydrous tetrahydrofuran (30 mL). Thiophene (320 μL, 4 mmol) was added via a dry 1 mL gas-tight syringe and the flask was immersed in a dry ice/acetone bath (approximately −80 °C) while slowly adding n-BuLi (2.8 mL, 1.6 M, 4.4 mmol) dropwise via a dry 5 mL gas-tight syringe. This was stirred for 30 min, then the reaction was quenched with a dropwise addition of tributyltinchloride (1.3 mL, 4.8 mmol). The flask was stirred for 45 min more, then brought slowly to room temp and stirred for 3 days. TLC revealed no spot for the thiophene starting material (using various solvent conditions with silica or alumina plates) but the reaction showed a new spot under short wavelength 254 nm. The mixture was transferred to a 200 mL beaker and saturated NH4Cl was added (20 mL) while stirring, producing an aqueous NH4Cl cloudy white bottom layer and a clear, pale yellow top layer. The mixture was transferred to a separatory funnel and an ether extraction (30–50 mL) was done, where the top ether layer was dried over anhydrous Na2SO4, then filtered using a medium glass-sintered frit. The solvent was removed by rotary evaporation and dried overnight to give a clear, pale yellow oil (1.4395 g, 95%). Rf = 0.87 (alumina, 100% hexanes).
To make the phen-1T ligand, a 50 mL dry Schlenk flask was charged with 5-bromo-1,10-phenanthroline (518 mg, 2 mmol), 1T-SnBu3 (756 mg, 2 mmol), tetrakis(triphenylphosphine)palladium(0) (69 mg, 0.06 mmol, 3 mol%), and dry DMF (20 mL). The flask was argon purged and the reaction refluxed at 100 °C for 3 days. The pale brown product was cooled then poured into a separatory flask holding saturated NH4Cl (50 mL), followed by two DCM extractions and one water wash. The DCM layer was dried over anhydrous Na2SO4, filtered, then concentrated under reduced pressure to obtain a pale orange oil. Purification was performed using alumina column chromatography, starting with 100% DCM and increasing polarity to 5% MeOH/DCM. This resulted in a clear brown/red oil (185 mg, 35%). Rf = 0.31 (alumina, 5% MeOH/DCM). 1H NMR (300 MHz, (CD3)2SO): δ 9.08 (s, 1H; g), 8.49 (dd, J = 48.3, 8.7 Hz, 2H; a,d), 7.93 – 7.66 (m, 3H; c,f,j), 7.64 – 7.46 (m, 4H; b,e,i,h).
5-(2,2′-bithiophene)-1,10-phenanthroline (phen-2T).
5-(tributylstannyl)-2,2′-bithiophene (2T-SnBu3) was prepared according to a published method (41). Briefly, a 100 mL dry, argon purged, 3-neck round-bottom flask with stir bar was charged with anhydrous THF (30 mL), then 2,2′-bithiophene (582 mg, 3.5 mmol) was added, and the flask was immersed in a dry ice/acetone bath and cooled to −80 °C followed by dropwise addition of n-BuLi (2.4 mL, 1.6 M, 3.9 mmol) dropwise via a dry 5 mL gas-tight syringe. This was stirred for 30 min, then the reaction was quenched with a dropwise addition of tributyltinchloride (1.14 mL, 4.2 mmol). The flask was stirred for 40 min more, then brought slowly to room temp and stirred overnight. The colour changed from clear light brown to clear light orange/brown. The mixture was transferred to a 200 mL beaker and saturated NH4Cl (20 mL) was added while stirring, resulting in a bilayer of aqueous NH4Cl cloudy white bottom layer and a clear amber/green top layer. The mixture was transferred to a separatory funnel, an ether extraction (30–50 mL) was done, and the top clear green ether layer was dried over anhydrous Na2SO4, then filtered using a medium glass-sintered frit. The solvent was removed by rotary evaporation and dried overnight to give a clear amber-green oil (1.14 g, 71%). Rf = 0.30 (Silica, 100% hexanes). The product was used without further purification.
The ligand phen-2T was then synthesized by charging a dry 50 mL Schlenk flask with 5-bromo-1,10-phenanthroline (518 mg, 2 mmol), 2T-SnBu3 (756 mg, 2 mmol), tetrakis(triphenylphosphine)palladium(0) (69 mg, 0.06 mmol, 3 mol%), and dry DMF (20 mL). The flask was argon purged and the reaction refluxed at 100 °C for 3 days. The product changed from a clear beige/brown to dark brown colour. It was cooled then poured into a separatory flask containing saturated NH4Cl (75 mL) followed by two DCM extractions and one water wash. The DCM layer was dried over anhydrous Na2SO4, filtered, and then concentrated down resulting in a brown/orange oil (5.631 g). Purification was performed using alumina column chromatography, starting with 100% DCM and increasing polarity from 2% to 5% MeOH/DCM. This resulted in a clear brown oil (263 mg, 58%). Rf = 0.5 (alumina, 5% MeOH/DCM). 1H NMR (300 MHz, (CD3)2SO): δ 9.10 (ddd, J = 7.6, 4.3, 2.0 Hz, 2H; a,d), 8.67 (ddd, J = 14.2, 8.4, 1.7 Hz, 1H; l), 8.52 (ddd, J = 17.2, 8.0, 1.8 Hz, 3H; c,f,g), 7.79 (ddd, J = 10.0, 8.1, 4.3 Hz, 2H; b,e), 7.57 (m, 1H; h), 7.48 (d, J = 3.7 Hz, 1H; i), 7.43 (dd, J = 3.7, 1.1 Hz, 1H; j), 7.14 (dd, J = 5.1, 3.6 Hz, 1H; k).
5-(2,2′:5′,2′′-terthiophene)-1,10-phenanthroline (phen-3T).
The starting material 5-(tributylstannyl)-5′-(2,2′:5′,2′′-terthiophene) was prepared according to a published method with some exceptions (41). A 100 mL dry, argon purged, 3-hole round-bottom flask with stir bar was charged with anhydrous THF (30 mL), then 2,2′:5′2:-terthiophene (496 mg, 2 mmol) was added and the flask was immersed in a dry ice/acetone bath and cooled to −80 °C while slowly adding n-BuLi (1.4 mL, 1.6 M, 2.2 mmol) dropwise via a dry 5 mL gas-tight syringe. This was stirred for 30 min, then the reaction was quenched with a dropwise addition of tributyltinchloride (651 μL, 2.4 mmol). The flask was stirred for 45 min more, then brought slowly to room temp and stirred for 3 days. The mixture was transferred to a 200 mL beaker and saturated NH4Cl (30 mL) was added while stirring, resulting in a bilayer of aqueous NH4Cl cloudy white bottom layer and a clear pale green top layer. The mixture was transferred to a separatory funnel, an ether extraction (30–50 mL) was done, and the top clear green ether layer was dried over anhydrous Na2SO4, then filtered using a medium glass-sintered frit. The solvent was removed by rotary evaporation and dried overnight to give a clear emerald green oil (977 mg, 91%). Rf = 0.35 (silica, 100% hexanes) that was used without further purification.
The ligand phen-3T was then synthesized by charging a dry 50 mL Schlenk flask containing 10 mL dry DMF with 5-bromophenanthroline (324 mg, 1.25 mmol) and 3T-SnBu3 (672 mg, 1.25 mmol). After argon purging, tetrakis(triphenylphosphine)palladium(0) (43 mg, 0.04 mmol, 3 mol%). The flask was argon purged again and the reaction refluxed at 100 °C for 3 days. The product changed from clear pale brown to a dark brown/red color. It was cooled and then poured into a separatory funnel containing saturated NH4Cl (75 mL) followed by two DCM extractions and one water wash. The DCM layer was dried over anhydrous Na2SO4, filtered, and then concentrated to give a dark brown/orange oil (2.42 g). Purification was performed using alumina column chromatrography, starting with 100% DCM and increasing polarity from 2% to 5% MeOH/DCM. This resulted in a clear orange oil (405 mg, 76%). Rf = 0.84 (alumina, 1% MeOH/DCM). 1H NMR (300 MHz, DMSO-d6) δ 9.15 (ddd, J = 10.8, 4.4, 1.7 Hz, 2H; a,d), 8.66 (dd, J = 8.4, 1.6 Hz, 1H; c), 8.53 – 8.47 (m, 2H; g,f), 7.93 (dd, J = 8.4, 4.3 Hz, 1H; b), 7.81 (dd, J = 8.1, 4.3 Hz, 1H; e), 7.66 – 7.50 (m, 7H; h,i,j,k,l,m,n).
Ru(bpy)2(phen-1T)PF6 (1).
Ru(bpy)2Cl2 (364 mg, 0.71 mmol) and phen-1T (185 mg, 0.71 mmol) were added to a microwave vessel containing 99% ethanol (1.5 mL) and ethylene glycol (1.5 mL). The mixture was subjected to microwave irradiation at 180 °C for 15 min. TLC (silica, 7.5% H2O/MeCN + 0.5% KNO3) indicated that the starting materials were exhausted, therefore the mixture was cooled and transferred (using dI water) to a 500 mL separatory funnel and slightly diluted with water. DCM extractions removed excess Ru(bpy)2Cl2, then the aqueous layer containing product was introduced to saturated KPF6 (approximately 2–3 mL). Further DCM extractions containing the desired product were concentrated down to dry crude product (457.2 mg). The crude product was purified by silica flash chromatography, starting with 100% MeCN and the polarity was gradually increased (up to 7.5% H2O/MeCN + 0.5% - 1% KNO3). This had to be repeated three times to yield the purest possible product. Yield: 30.9 mg, 6.4%. Rf = 0.30 (10% H2O in MeCN + 0.5% KNO3). 1H NMR (500 MHz, CD3CN): δ 8.84 (dd, J = 8.5, 1.2 Hz, 1H; c), 8.62 (dd, J = 8.3, 1.2 Hz, 1H; f), 8.56 (d, J = 7.8 Hz, 2H; 3’), 8.53 (d, J = 8.1 Hz, 2H; 3), 8.36 (s, 1H; g), 8.16 – 8.11 (m, 4H; 4’,a,d), 8.03 (td, J = 7.9, 1.4 Hz, 2H; 4), 7.88 (dd, J = 5.4, 1.3 Hz, 2H; 6’), 7.79 – 7.74 (m, 3H; b,e,j), 7.61 (dd, J = 9.7, 5.6 Hz, 2H; 6), 7.51 (dd, J = 3.6, 1.2 Hz, 1H; h), 7.48 (ddd, J = 7.7, 5.6, 1.4 Hz, 2H; 5′), 7.37 (dd, J = 5.1, 3.6 Hz, 1H; i), 7.30 – 7.25 (m, 2H; 5). MS (ESI+) m/z: 821.07 [M-(PF6)]+, 338.33 [M-2(PF6)]2+. HRMS (ESI+) m/z for C36H26N6RuS: calc’d 338.0486, found: 338.0479. HPLC retention time: 16.15 min (97%).
Ru(bpy)2(phen-2T)PF6 (2).
Ru(bpy)2Cl2 (151 mg, 0.29 mmol) and phen-2T (99.7 mg, 0.29 mmol) were added to a microwave vessel containing 99% ethanol (1.5 mL) and ethylene glycol (1.5 mL). The mixture was subjected to microwave irradiation at 180 °C for 15 min. TLC (silica, 7.5% H2O/MeCN + 0.5% KNO3) indicated that the starting materials were exhausted, therefore the mixture was cooled and transferred (using dI water) to a 500 mL separatory funnel containing 100 mL dI water. DCM extractions removed excess Ru(bpy)2Cl2, then the aqueous layer containing product was introduced to saturated KPF6 (approximately 2–3 mL). Further DCM extractions removed the desired compound which was concentrated down to dry crude product (148.9 mg). The crude product was purified by silica flash chromatography, starting with 100% MeCN and the polarity was gradually increased (up to 7.5% H2O/MeCN + 0.5% to 1% KNO3). This was repeated twice to afford the purest possible product. Yield: 18.1 mg, 6.0%. Rf = 0.34 (7.5% H2O in MeCN + 0.5% KNO3). 1H NMR (500 MHz, CD3CN): δ 8.94 (dd, J = 8.5, 1.2 Hz, 1H; c), 8.63 (dd, J = 8.2, 1.2 Hz, 1H; f), 8.57 (dd, J = 8.3, 1.0 Hz, 2H; 3’), 8.53 (dd, J = 8.2, 1.2 Hz, 2H; 3), 8.39 (s, 1H; g), 8.17 – 8.11 (m, 4H; 4’,a,d), 8.04 (td, J = 8.0, 1.5 Hz, 2H; 4), 7.88 (d, J = 5.2 Hz, 2H; 6’), 7.78 (ddd, J = 8.2, 5.2, 1.6 Hz, 2H; b,e), 7.61 (ddt, J = 9.4, 5.7, 1.2 Hz, 2H; 6), 7.51 – 7.47 (m, 4H; 5′,h,i), 7.45 (d, J = 3.8 Hz, 1H; l), 7.43 (dd, J = 3.6, 1.1 Hz, 1H; j), 7.28 (tdd, J = 5.9, 3.3, 1.6 Hz, 2H; 5), 7.17 (dd, J = 5.1, 3.6 Hz, 1H; k). MS (ESI+) m/z: 903.00 [M-PF6]+, 379.33 [M-2(PF6)]2+. HRMS (ESI+) m/z for C40H28N6RuS: calc’d 379.0425, found 379.0412. HPLC retention time: 23.36 min (96%).
Ru(bpy)2(phen-3T)PF6 (3).
Ru(bpy)2Cl2 (210 mg, 0.4 mmol) and phen-3T (167 mg, 0.4 mmol) were added to a microwave vessel containing 99% ethanol (1.5 mL) and ethylene glycol (1.5 mL). The mixture was subjected to microwave irradiation at 180 °C for 15 min. TLC (silica, 7.5% H2O/MeCN + 0.5% KNO3) indicated that the starting materials were exhausted, therefore the mixture was cooled and transferred (using dI water) to a 500 mL separatory funnel containing 100 mL dI water. DCM extractions removed excess Ru(bpy)2Cl2, then the aqueous layer containing product was introduced to saturated KPF6 (approximately 2–3 mL). Further DCM extractions removed the desired compound which was concentrated down to dry crude product (328 mg). The crude product was purified by silica flash chromatography, starting with 100% MeCN and the polarity was gradually increased (up to 7.5% H2O/MeCN + 0.5% to 1% KNO3). This was repeated twice to yield the purest possible product. Yield: 20.4 mg, 4.5%. Rf = 0.33 (7.5% H2O in MeCN + 0.5% KNO3). 1H NMR (500 MHz, CD3CN): δ 8.93 (dd, J = 8.6, 1.2 Hz, 1H; c), 8.63 (dd, J = 8.3, 1.2 Hz, 1H; f), 8.56 (d, J = 8.3 Hz, 2H; 3’), 8.52 (d, J = 8.3 Hz, 2H; 3), 8.39 (s, 1H; g), 8.16 – 8.10 (m, 4H; 4’,a,d), 8.03 (td, J = 7.9, 1.5 Hz, 2H; 4), 7.88 (d, J = 5.9 Hz, 2H; 6’), 7.77 (ddd, J = 8.3, 5.2, 2.4 Hz, 2H; b,e), 7.63 – 7.58 (m, 2H; 6), 7.50 – 7.45 (m, 4H; 5′,h,k), 7.43 (dd, J = 5.1, 1.1 Hz, 1H; n), 7.36 (d, J = 3.8 Hz, 1H; i), 7.35 (dd, J = 3.6, 1.1 Hz, 1H; l), 7.30 – 7.25 (m, 3H; 5,j), 7.13 (dd, J = 5.1, 3.6 Hz, 1H; m). MS (ESI+) m/z: 985.00 [M-PF6]+, 420.33 [M-2(PF6)]2+. HRMS (ESI+) m/z for C44H30N6RuS: calc’d 420.0363, found 420.0366. HPLC retention time: 25.51 min (100% purity).
Photophysical methods.
Photophysical measurements were performed on dilute solutions (5‒20 μM) of the metal complexes as their PF6− salts in spectroscopic grade MeCN. Molar extinction coefficients (ε) were determined from the slopes of absorption versus concentration plots for five concentrations, and measured in duplicate. Quantum yields for emission (Φem) and singlet oxygen (ΦΔ) were measured relative to [Ru(bpy)3](PF6)2 as the standard (s) according to eq 1, where I, A, and η are the integrated emission intensity, the absorbance at the excitation wavelength, and the refractive index of the solvent, respectively. Reference quantum yields used for [Ru(bpy)3](PF6)2 were: Φem = 0.012 at 298 K in aerated MeCN (42), Φem = 0.095 at 298 K in deaerated MeCN (43), Φem = 0.38 at 77 K in frozen 4:1 v/v EtOH:MeOH (18), and ΦΔ = 0.56 in aerated MeCN (44).
| (1) |
Solutions were degassed by argon sparging in a long-stemmed cuvette (Luzchem QSC10S) for steady-state emission experiments. Samples for 77 K measurements were prepared in 4:1 EtOH:MeOH solution in a 5 mm i.d. NMR tube, and then frozen in liquid nitrogen in a quartz-tipped cold finger Dewar (Wilmad Labglass). UV-vis absorption spectra were recorded on a Jasco V-730 spectrometer, and steady-state emission spectra were measured on a PTI Quantamaster equipped with a K170B PMT for measuring visible emission from the metal complex and a Hamamatsu R5509–42 NIR PMT for measuring NIR emission (<1400 nm) from 1O2 (centered around 1276 nm). Emission and excitation spectra were corrected for the wavelength dependence of lamp output and detector response.
Emission lifetimes (λex=457 nm, λem=λem max) were collected on degassed MeCN solutions at room temperature or on 4:1 v/v EtOH:MeOH glass at 77 K using a PTI LaserStrobe system, equipped with an R928 stroboscopic detector integrated with a GL-3300 nitrogen/GL-301 dye laser (2−3 nm fwhm). The nanosecond transient difference absorption (TA) spectra and decays were measured in degassed MeCN solutions on an Edinburgh LP920 laser flash photolysis spectrometer. The third harmonic output (355 nm) of a Nd:YAG laser (Quantel Brilliant, pulse width = 4.1 ns, repetition rate = 1 Hz) was used as the excitation source. Each sample was purged with argon for 45 min prior to measurement.
Metal compound solutions for cellular assays.
Stock solutions of the chloride salts of the Ru(II) complexes were prepared at 5 mM in 10% DMSO in water and kept at −20 °C prior to use. Working dilutions were made by diluting the aqueous stock with pH 7.4 Dulbecco’s phosphate buffered saline (DPBS). DPBS is a balanced salt solution of 1.47 mM potassium phosphate monobasic, 8.10 mM sodium phosphate dibasic, 2.68 mM potassium chloride, and 0.137 M sodium chloride (no Ca2+ or Mg2+). DMSO in the assay wells was under 0.1% at the highest complex concentration.
Cell culture reagents.
For cellular assays, characterized FBS (VWR), RPMI 1640 media (Corning Cellgro), EMEM (Corning Cellgro), and Dulbecco’s phosphate buffered saline (DPBS 1×, Mediatech, 21–031-CV), were purchased from VWR. Trypsin-EDTA solution (0.25% w/v Trypsin/0.53 mM EDTA, 30–2101), human promyelocytic leukemia cells (HL-60) and human malignant melanoma cells (SK-MEL-28) were procured from ATCC.
Cell culture of HL-60 cells.
HL-60 human promyelocytic leukemia cells (ATCC CCL-240) were cultured at 37 °C under 5% CO2 in RPMI 1640 (Mediatech Media MT-10–040-CV) supplemented with 20% FBS (PAA Laboratories, A15–701) and were passaged 3−4 times per week according to standard aseptic procedures. Cultures were started at 200,000 cells mL−1 in 25 cm2 tissue culture flasks and were subcultured when growth reached 800,000 cells mL−1 to avoid senescence associated with prolonged high cell density. Complete growth medium was prepared in 200 mL portions as needed by combining RPMI 1640 (160 mL) and FBS (40 mL, prealiquoted and heat inactivated) in a 250 mL Millipore vacuum stericup (0.22 μm), and filtering.
Cell culture of SK-MEL-28.
Adherent SK-MEL-28 malignant melanoma cells (ATCC HTB-72) were cultured in Eagle’s Minimum Essential Medium (EMEM, Mediatech Media MT-10–009-CV) supplemented with 10% FBS. They were incubated at 37 °C under 5% CO2, and were passaged 2−3 times per week according to standard aseptic procedures. SK-MEL-28 cells were started at 200,000 cells mL−1 in 75 cm2 tissue culture flasks and were subcultured when growth reached 550,000 cells mL−1 by removing old culture medium and rinsing the cell layer once with DPBS, followed by dissociation of the cell monolayer with trypsin-EDTA solution. Complete growth medium was added to the cell suspension to allow appropriate aliquots of cells to be transferred to new cell vessels. Complete growth medium was prepared in 250 mL portions as needed by combining EMEM (225 mL) and FBS (25 mL, prealiquoted and heat inactivated) in a 250 mL Millipore vacuum stericup (0.22 μm) and filtering.
Cell culture of CCD-1064Sk cells.
Adherent CCD-1064Sk normal skin fibroblast cells (ATCC CRL-2076) were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM, ATCC 30–2005) supplemented with 10% FBS. They were incubated at 37 °C under 5% CO2, and were passaged 2−3 times per week according to standard aseptic procedures. CCD-1064Sk cells were started at approximately 200,000 cells mL−1 in 75 cm2 tissue culture flasks and were subcultured when growth reached confluency, approximately 500,000 cells mL−1 by removing old culture medium and rinsing the cell layer once with DPBS, followed by dissociation of the cell monolayer with trypsin-EDTA solution. Complete growth medium was added to the cell suspension to allow appropriate aliquots of cells to be transferred to new cell vessels. Complete growth medium was prepared in 250 mL portions as needed by combining IMDM (225 mL) and FBS (25 mL, prealiquoted and heat inactivated) in a 250 mL Millipore vacuum stericup (0.22 μm) and filtering.
Cytotoxicity and photocytotoxicity assays.
Cell viability experiments were performed in triplicate in 96-well ultra-low attachment flat bottom microtiter plates (Corning Costar, Acton, MA), where outer wells along the periphery contained 200 μL of DPBS to minimize evaporation from sample wells. Cells growing in log phase (HL-60 cells: ~800,000 cells mL−1. SK-MEL-28 cells: ~550,000 cells mL−1. CCD-1064Sk cells: ~500,000 cells mL−1) with at least 93% viability were transferred in 50 μL aliquots to inner wells containing warm culture medium (25 μL) and placed in a 37 °C, 5% CO2 water-jacketed incubator (Thermo Electron Corp., FormaSeries II, Model 3110, HEPA Class 100) for 3 h to equilibrate (and allow for efficient cell attachment in the case of both the SK-MEL-28 and CCD-1064Sk adherent cells). Metals compounds were serially diluted with DPBS and prewarmed at 37 °C before 25 μL aliquots of the appropriate dilutions were added to cells. PS-treated microplates were incubated at 37 °C under 5% CO2 for 16 h drug-to-light intervals. Control microplates not receiving a light treatment were kept in the dark in an incubator, and light-treated microplates were irradiated under one of the following conditions: visible light (400−700 nm, 44 mW∙cm−2) using a 190 W BenQ MS 510 overhead projector or red light (625 nm, 52.5 mW∙cm−2) from an LED array (PhotoDynamic Inc., Halifax, NS). Irradiation times using these two light sources were approximately 38 and 32 min, respectively, to yield total light doses of 100 J cm−2. Both untreated and light-treated microplates were incubated for another 48 h before 10 μL aliquots of prewarmed, sterile filtered 0.6 mM resazurin reagent (Sigma Aldrich Canada), according to a standard protocol (45), were added to all sample wells and subsequently incubated for another 3 h. Cell viability was determined on the basis of the ability of the resazurin redox indicator to be metabolically converted to a fluorescent dye by only live cells. Fluorescence was quantified with a Cytofluor 4000 fluorescence microplate reader with the excitation filter set at 530 ± 25 nm and emission filter set at 620 ± 40 nm. EC50 values for cytotoxicity (dark) and photocytotoxicity (light) were calculated from sigmoidal fits of the dose-response curves using Graph Pad Prism 6.0 according to eq 2, where yi and yf are the initial and final fluorescence signal intensities. For cells growing in log phase and of the same passage number, EC50 values are generally reproducible to within ±25% in the submicromolar regime, ±10% below 10 μM, and ±5% above 10 μM. Phototherapeutic indices (PIs), a measure of the therapeutic window, were calculated from the ratio of dark to light EC50 values obtained from the dose-response curves.
| (2) |
DNA photocleavage assays.
DNA photocleavage experiments were performed according to general plasmid DNA gel mobility shift assay with 20 μL total sample volumes in 0.5 mL microcentrifuge tubes. Transformed pUC19 plasmid (1.3 μL, > 95% form I) was added to 10 μL of 10 mM Tris-HCl buffer (pH 7.5). Dilutions of the Ru(II) compound(s) were prepared in ddH2O and added in 5 μL aliquots to the appropriate tubes to yield final Ru(II) concentrations ranging from 5 to 100 μM. Then, ddH2O (3.7 μL) was added to bring the final assay volumes to 20 μL. Control samples with no metal complex received 8.7 μL of water. Sample tubes were kept at 37 ˚C in the dark or irradiated with broadband visible light (14 J cm−2 delivered from a Luzchem LZC-4X photo reactor over the course of 30 min). After treatment, all samples (dark and light) were quenched by the addition of 4 μL of gel loading buffer (0.025% bromophenol blue, 40% glycerol). Samples volumes of 11.8 μL were loaded onto 1% agarose gels cast with 1x TAE (40 mM Tris-acetate, 1 mM EDTA, pH 8.2) containing ethidium bromide (0.75 μg mL−1) and electrophoresed for 30 min at 8 V cm−1 in 1x TAE. The bands were visualized using the Gel Doc-It Imaging system (UVP) with Vision Works software.
RESULTS AND DISCUSSION
Synthesis and characterization
An overview of the synthetic steps for preparing the Ru(II) dyads is shown in Scheme 1. The previously unreported thienyl-appended 1,10-phenanthroline ligands phen-1T, phen-2T, and phen-3T were synthesized via a Pd(0)-catalyzed Stille cross-coupling between the tributylstannane of thiophene (1T-SnBu3) or oligothiophene (2T-SnBu3 3T-SnBu3) and 5-bromo-1,10-phenanthroline in 35–60% yield after purification on alumina. The nT-SnBu3 starting materials were obtained in 70–95% yield by lithiation of the corresponding nT in THF followed by quenching with SnBu3Cl and were used without further purification (41). 5-Bromo-1,10-phenanthroline was prepared in 47% yield from freshly dried 1,10-phenanthroline and bromine in oleum (39).
Scheme 1.

Synthetic Scheme for Ru(II) Dyads 1–3 and Their Ligands.
The Ru(II) dyads were assembled from the corresponding phen-nT ligands and [Ru(bpy)2Cl2]∙2H20 in 1:1 ethanol:ethylene glycol using microwave irradiation and isolated as their PF6– salts. Purification was carried out on silica gel and was repeated 2–3 times to obtain very modest yields (5–6%) of the final complexes in >96% purity by HPLC. Characterization by 1D 1H and 2D 1H-1H NMR (Figures S1–S6) as well as ESI-MS (Figures S8–S13) confirmed the structures of the desired products. The hydrogen assignments in the 1H NMR spectra are compared for the three complexes in the Supporting Information (Figure S7). Photophysical characterization was done on the PF6– salts in MeCN for comparison with published photophysical data for other Ru(II) compounds and to be able to measure 1O2 quantum yields by emission at 1270 nm, which would otherwise be quenched in aqueous solution. For biological experiments, the compounds were converted to their water-soluble Cl– salts in quantitative yield using anion-exchange chromatography.
Photophysical properties
Absorption.
The electronic spectra of complexes 1‒3 at room temperature are shown in Figure 1a and tabulated in Table 1. Four key features appeared. The intense sharp peak near 284 nm for all three Ru(II) dyads, also a feature observed in Ru(bpy)32+ (18), can be attributed to the 1IL state (i.e., analogous to the ππ* transition) centered on the bpy ligands. Similarly, the peak near 450 nm was assigned to MLCT transitions between the Ru(II) center and the bpy ligands. The analogous transition between Ru(II) and the more π-extended ligands occurred around 420 nm and did not vary with the number of appended thiophene rings, suggesting that this MLCT transition was primarily associated with the phen portion of the ligand. A broad peak of increasing intensity occurred around 325 nm in 1, 380 nm in 2, and 400 nm in 3, and the decrease in energy with the increasing n points to ππ* transitions localized on the appended thienyl ring(s) of the phen ligands.
Figure 1.

Steady-state UV-vis absorption (a) and emission (b) spectra of compounds 1-3 at room temperature in MeCN. Emission spectra for compounds 1-3 at 77 K in 4:1 EtOH:MeOH glass (c).
Table 1.
Photophysical Data for 1–3.
| Cmpd | λabs [nm] (log ε) | λem [nm]a,b | Φa,b | τem [ns]a,b | τTA [ns] (λprobe. nm)a,b | ΦΔa,c | |||
|---|---|---|---|---|---|---|---|---|---|
| 298 K | 77 K | 298 K | 77 K | 298 K | 77 K | 298 K | 298 K | ||
| 1 | 450 (4.16), 426 (4.10), 332 (4.04), 285 (4.79), 254 (4.57), 231 (4.53) | 620 | 579, 631 | 0.12 | 0.66 | 1,050 | 6,160 | 950 (744) | 0.58 |
| 2 | 448 (3.88), 424 (3.86), 379 (3.84), 271 (4.29), 260 (4.20), 234 (4.17) | 617 | 580, 629 | 0.0092 | 0.14 | 1,030 | 6,600 | 43,200 (543) | 0.78 |
| 3 | 453 (4.33), 419 (4.43), 383 (4.41), 286 (4.76), 271 (4.64), 255 (4.53), 233 (4.58) | 618 | 578, 629 | 0.0016 | 0.0087 | 1,290 | 7,760 | 26,900 (590) | 0.74 |
λex= longest wavelength absorption maximum.
Measurements at 298 K were performed on argon-purged samples in MeCN; 77 K measurements were performed in air-saturated EtOH:MeOH (4:1) glass.
Air-saturated MeCN at 298 K assuming 21% O2.
Emission.
Complexes 1‒3 produced moderate (Φem ≈ 10−1–10−3, Table 1), broad and featureless emission near 618 nm at room temperature in Ar-sparged MeCN (Figure 1b). At 77 K (in 4:1 MeOH:EtOH glass), this emission shifted to shorter wavelengths (~580 nm), increased in quantum yield, and developed typical 3MLCT vibronic structure (Figure 1c). The vibronic intervals were around 1350 cm−1, which is characteristic of diimine involvement in the emissive state (40) and did not change significantly between the three complexes. The ΔEs values of around 1060 cm−1 compare well with ΔEs=1127 cm−1 for [Ru(bpy)3]2+ under the same conditions (15,23), and the ≈1 μs lifetime (τem) of all three approximates the 0.9 μs lifetime of [Ru(bpy)3]2+ (18), further supporting this assignment of the 3MLCT state. The fact that the emission maximum did not change with n (Figure 1b–c) indicates that the π* acceptor orbital of the 3MLCT state is the same in the three complexes: the 3MLCT phosphorescence is thus ascribed to the Ru(dπ) → bpy(π*) configuration.
Despite the similarities in the electronic configuration of the emissive states in the series, compound 1 exhibited much stronger 3MLCT phosphorescence (Table 1) at both room temperature and at 77K, at least an order of magnitude greater than 2 or 3, with no significant effect on the 3MLCT lifetime. This suggests that 2 and 3 may differ substantially from 1 in terms of their competing nonradiative 3MLCT relaxation pathways.
Transient absorption (TA).
The triplet excited states of complexes 1–3 were also probed using nanosecond transient absorption (TA) spectroscopy (Figure 2a and S14), and their spectroscopic signatures were analyzed in the context of the well-established TA profiles of similar Ru(II) complexes. The TA spectra are differential measurements, consisting of new excited-state absorption (ESA) bands (positive signals) that are superimposed on ground state transitions that are no longer available (negative signals). For typical Ru(II) polypyridyl complexes, these differential spectra usually comprise a prominent bleach in the mid-visible, due to the loss of the 1A1 → 1MLCT ground-state absorption, plus a new sharp absorption in the UV and a broader one in the red-NIR, both of which arise from the diimine ligand radical anion of the 3MLCT excited state (46).
Figure 2.

(a) Time-resolved nanosecond TA spectra collected for compounds 1-3 in MeCN (O.D.=0.4 in a 1-cm cuvette) with λex = 355 nm. (b) Nanosecond TA decay of the signal shown in (a) for Ru(II) dyad 3.
The TA signal of 1 (Figure 2a, S14a) exhibited these features, and the excited state lifetime (τTA) measured at different wavelengths across the TA spectrum was similar to that observed in the emission experiment, demonstrating that (i) the excited state of 1 probed by TA is the emitting state, and (ii) no additional states appear to contribute to the excited state relaxation pathway for 1 on this timescale. Compounds 2‒3 had very different profiles, characterized by a broad, and much longer-lived ESA in the red-NIR that partly overlapped the MLCT bleach and affected its apparent position. This signature has been observed previously for other oligothiophene-containing Ru(II) polypyridyl complexes and was attributed to 3IL states involving ππ* transitions localized on the thienyl groups (33). The lifetime of this new signal was 43 μs for 2 (Figure S14b) and 27 μs for 3 (Figure 2b), pointing to a long-lived 3IL state that can be populated only when n≥2 and that cannot (re)populate the 3MLCT state since its emissive lifetime did not vary between the complexes. In other words, all of the Ru(II) dyads exhibited 3MLCT emission (τem=≈1 μs), but only 2 and 3 displayed 3IL ESA with the prolonged lifetime.
Photophysical model.
The 3MLCT emission from all three compounds occurred at around 618 nm, or 2.1 eV. The triplet state energies ET(0‒0) of thiophene, bithiophene, and terthiophene have been reported as 3.50, 2.23, and 1.93 eV, respectively (32). Using these values as estimates for the 3IL states for the complexes, the major differences between the three complexes are the energies of their 3IL-based states. The 3IL state for 1 was inaccessible with λex=355 nm (3.49 eV), which presumably populates nonthermalized 1IL and/or 1MLCT states that rapidly undergo internal conversion (IC) to singlet states that can populate the 3MLCT state in all three complexes and the 3IL state for only 2 and 3, as illustrated on the Jabłonski diagram in Scheme 2. The current hypothesis is that the 3IL state for 1 is energetically uphill and inaccessible from the thermalized 1MLCT and/or 1IL states.
Scheme 2.

Jabłonski Diagram Depicting the Estimated 3IL Energies and Relevant Photophysical Relaxation Pathways for Complexes 1–3.
The fact that the emissive lifetimes of the 3MLCT states for all three complexes were similar and monoexponential (≈1 μs, typical for unsubstituted Ru(bpy)32+-type complexes) (4) suggests that the 3IL states did not act as “triplet reservoirs”, unlike some other π-expansive Ru(II) dyads based on pyrene (33,47) The observation of 3MLCT lifetimes for 2 and 3 that were not shorted or lengthened relative to the typical 1-μs lifetime for Ru(II) polylpyridyls supported the notion that the 3IL states are not populated directly from 3MLCT states (which would shorten τem, assuming the forward rate constants for this process were faster than the rate constants for emission) and that the 3IL states are not in thermal equilibrium with 3MLCT states (which would lengthen τem). The absence of a rise time (matching the decay of the 3MLCT state) for the formation of the 3IL state for 2 and 3 also suggests that the 3IL states were not populated directly from the 3MLCT manifold in these two systems. Nevertheless, the photophysical scheme is tentative without further testing with different excitation wavelengths and on shorter timescales.
Singlet oxygen sensitization.
The quantum yields for singlet oxygen generation (ΦΔ) were calculated for compounds 1-3 in air-saturated MeCN at room temperature using the integrated emission centered at 1268 nm (Table 1). Ru(bpy)32+, which is an efficient 1O2 sensitizer, was used as a standard (ΦΔ=0.56) (44). Compound 1 had a 1O2 quantum yield of 58%, implying good conversion efficiency from the 3MLCT state, comparable to the standard. Compounds 2 and 3 were more efficient still, 78% and 74%, respectively. Since the spectroscopic data suggests that the thiophene-centered 3IL state dominates in these two complexes and that it is not in equilibrium with the 3MLCT, one can conclude that the longer-lived 3IL state is more efficient at sensitizing 1O2 and that the 1O2 sensitization competes favorably with the nonradiative decay pathway from this state. This is not surprising, since terthiophene itself is an excellent 1O2 sensitizer, with ΦΔ=0.86 (31). The relatively large ΦΔ values in this series, especially for 2 and 3, suggest that these Ru(II) dyads may be photobiologically active and could be exploited for photodynamic applications such as PDT.
In vitro photobiological activity
The cytotoxicities and photocytotoxicities of Ru(II) dyads 1-3 were evaluated in two different cancer cell lines: SKMEL28 human melanoma cells as a representative cell line that grows as adherent 2D monolayers, and HL60 human leukemia cells as one that grows as a suspension. In addition, CCD-1064Sk human skin fibroblast cells were used to determine whether the compounds showed any cytotoxicity toward noncancerous cells. Briefly, cells growing in log phase were dosed with metal complex (1 nM-300 μM) in 96-well plates and incubated at for 16 h at 37 °C in an atmosphere containing 5% CO2 prior to a light or sham (dark) treatment. The light treatments were 100 J cm−2 delivered from visible (400–700 nm, 34 mw cm−2) or red (625 nm, 29 mW cm−2) LEDs (Figure S15); the compounds showed no observable photobleaching at these light doses. The samples were incubated for an additional 48 h (37 °C, 5% CO2) followed by the addition of Alamar Blue and its fluorescence quantification 16 h later. The dark and light cytotoxicities were calculated as the effective concentrations required to reduce cell viability to 50% (EC50) under a given condition. Photocytotoxicity indices (PIs) were determined as the ratio of dark to light EC50 values. Selectivity factors (SF) were defined as the ratio of the dark EC50 for CCD-1064Sk cells to the dark EC50 for a given cancerous cell line. PIs and SFs greater than one signify light-amplified cytotoxic effects and selectivity for cancer cells over normal cells, respectively. As long as the light EC50 value is significantly less than the dark EC50 value for the noncancerous cell line, it is not necessary to have selectivity for cancer cells over healthy cells because the selectivity can be achieved by spatiotemporal control of the light delivery. The SFs were determined to assess whether any of the compounds might be of interest as traditional chemotherapeutics.
All three Ru(II) dyads were considered nontoxic to the CCD-1064Sk normal skin fibroblast cells (EC50>100 μM) in the absence of a light trigger, and compound 1 was nontoxic toward the two cancer cell lines as well (Table 2). Compounds 2 and 3 were slightly cytotoxic toward SKMEL28 cells (EC50=32 and 50 μM, respectively), but nontoxic to HL60 cells (EC50>100 μM). At this point, it is uncertain whether the four-fold selectivity of 3 and eight-fold selectivity of 2 toward melanoma cells (but no selectivity toward HL60 cells) stems from an inherent difference in the identities of these two cancerous cell lines and their uptake of the compounds or whether it is simply due the difference in the way these two cell lines grow (dendritic adherent monolayers versus spherical suspension cells). It is also not clear why there is some selectivity for SKMEL28 cells over noncancerous cells with regard to the two oligothienyl dyads and no preference when n=1. Regardless, the cytotoxicities of these compounds are marginal, and thus they are of little interest as traditional chemotherapeutics. However, this property makes them potentially attractive as light-responsive prodrugs for photobiological applications such as PDT.
Table 2.
In vitro cytotoxicity and photocytotoxicity data for compounds 1–3 in three cell lines: CCD1064SK, SKMEL28, and HL60. PDT conditions: 16 hr drug-to-light interval, followed by 100 J cm−2 light irradiation; “vis” was incandescent white, “red” was LED at 625 nm. The phototherapeutic index (PI) is the ratio of dark to light EC50. The selectivity factor (SF) is the ratio of the dark EC50 of non-cancerous human fibroblast cells (CCD-1064SK) to the dark EC50 of the cancer line of interest.
| CCD | SKMEL28 | HL60 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| dark | dark | visible light | red light | dark | visible light | red light | |||||||
| EC50 | EC50 | SF | EC50 | PI | EC50 | PI | EC50 | SF | EC50 | PI | EC50 | PI | |
| 1 | > 300 | > 300 | 1 | 1.39 ± 0.02 | > 216 | > 300 | 1 | > 300 | 1 | 6.82 ± 0.19 | > 44 | > 300 | 1 |
| 2 | 253 ± 11 | 32.0 ± 1 | 8 | 0.214 ± 0.002 | 150 | 14.3 ± 0.3 | 2 | 259 ± 9 | 1 | 0.310 ± 0.010 | 835 | 13.5 ± 0.5 | 19 |
| 3 | 190 ± 4 | 50.0 ± 3 | 4 | 0.125 ± 0.003 | 400 | 2.03 ± 0.05 | 25 | 168 ± 3 | 1 | 0.210 ± 0.010 | 800 | 2.20 ± 0.08 | 76 |
The cytotoxicities of all the compounds toward both cancer cell lines were enhanced upon visible light activation (Table 2, Figure 3), yielding EC50 values as low as 125 nM (PI=400) for 3 toward SKMEL28 cells and PIs >800 (EC50=310 nM) for 2 toward HL60. Compound 1, with one thienyl group, was the least potent with EC50 values in the low micromolar range (1.4 and 6.8 μM toward SKMEL28 and HL60, respectively). However, the low dark cytotoxicity of 1 still led to PIs >210 in SKMEL28 and >44 in HL60. Compounds 2 and 3 exhibited light EC50 values in the range of 125–310 nM with PIs between 150 and 835. The activity plot shown in Figure 4 clearly demonstrates that phototoxicities toward both cell lines increase as the number of thienyl groups increases: 1 < 2 < 3, with PIs generally being larger in the HL60 cell line (except for 1).
Figure 3.

In vitro dose–response curves for compounds 1 (a), 2 (b), and 3 (c) in SK-MEL-28 cells (left) and HL-60 cells (right). Treatment conditions were dark (black), broadband visible light (blue), or single-wavelength red light at 625 nm (red). Light treatments were 100 J cm−2 delivered at a rate of approximately 28 mW cm−2.
Figure 4.

Cytotoxic/photocytotoxic activity plot for compounds 1-3 against SK-MEL-28 melanoma cells (filled triangles), HL60 leukemia cells (filled diamonds), and CCD-1064Sk (unfilled triangles). Treatment conditions: dark (black), visible light (blue), or red light (red). Only the dark condition is shown for CCD-1064Sk.
The visible light used for the photocytotoxicity experiments included higher-energy blue wavelengths, which overlap well with the 1MLCT absorption band for all the Ru(II) dyads (Figure 1a) and give rise to photocytotoxic effects. However, there is more focus on using red and near-infrared wavelengths for in vivo photobiological applications, especially when there is a need to penetrate tissue and minimize scatter. Therefore, we also determined EC50 values and PIs using single-wavelength red LEDs emitting at 625 nm. Despite the fact that absorption of light for the series at this wavelength is minimal (ε625 nm <<100 M−1 cm−1), we have previously shown that red PDT effects are possible even when molar extinction coefficients are <100 M−1 cm−1 for a variety of Ru(II) dyads derived from oligothienyls (7,36) and other π-expansive systems (48). The criterion appears to be a critical degree of π-conjugation on one of the ligands that lowers the energy of the 3IL state below that of the 3MLCT state (<2.1 eV), making the former accessible.
Based on our previous studies, it was expected that compound 1 (with a single thienyl group) would not give rise to photocytotoxic effects with red light in either cell line. However, compounds 2 and 3 yielded EC50 values of approximately 14 and 2 μM, respectively, in both cancer cell lines. The PIs measured in SKMEL28 cells were less than those in HL60 cells due to the higher dark cytotoxicity toward this cell line. Notably, the PIs for compound 3 were 25 and and 76 with red light. Even when any dark cytotoxic contribution to the observed photocytotoxicity is considered, the red photobiological activity increased with π-expansion in this series. The theoretical basis for this phenomenon, from computational methods on related Ru(II) dyads containing thienyl groups, appears to be direct population of a low-lying 3IL state for n≥2, but the increased organic character of these states lead to small SOC values (on the order of a few tens of cm−1) and thus very weak S-T absorption (36). The fact that any red photocytotoxicity is observed at all suggests that these 3IL states must be highly photosensitizing, particularly in the case of 3. We hypothesize that the prolonged intrinsic triplet excited state lifetimes, characteristic of Ru(II) dyads (13) leads to efficient bimolecular quenching to form 1O2 (or other reactive intermediates) that are important for photobiological effects. This assertion is partially supported by the larger 1O2 quantum yields for 2 and 3.
DNA photocleavage
The in vitro light-mediated cytotoxic effects described above could arise in part from photogenerated reactive intermediates, such as 1O2, that damage biological macromolecules and cause cell death. Supercoiled plasmid DNA (pUC19) was used as a general probe for assessing the abilities of Ru(II) dyads 1-3 to photodamage biomolecules (regardless of whether DNA is the actual cellular target) in an agarose gel electrophoretic mobility-shift assay (49–51). Plasmid DNA migration rates are acutely sensitive to size and topology, which can be affected by exogenous agents, and typically increase in the order: aggregated/condensed DNA (Form IV) < nicked circular (Form II, single-strand breaks) < linear (Form III, frank double-strand breaks or single-strand breaks in close proximity on opposite strands) < supercoiled (Form I, native structure). It is also possible to discern other DNA interactions. Noncovalent DNA intercalation or covalent cross-linking agents unwind the DNA helix in a concentration-dependent manner, resulting in a gradual change in DNA mobility rather than an abrupt change between forms as occurs with strand breaks or aggregation (52). With regard to the present study, the goal was to compare the photodamage by 1-3 in the form of 1O2-induced single-strand breaks at characteristic metal complex (MC) to nucleotide phosphate (MC:NP) ratios by monitoring the conversion of undamaged DNA (Form I) to nicked DNA (Form II) triggered by the photoactivated metal complex. This is a very sensitive assay since it takes only one single strand break to produce Form II DNA.
Briefly, pUC19 plasmid DNA (20 μM NPs) was exposed to increasing concentrations of 1-3 (5–100 μM), and then kept in the dark or treated with visible light (14 J cm−2, 7.8 mW cm−2). These samples (Figure 5, lanes 3–8) were added to agarose gels cast with ethidium bromide for DNA visualization, and electrophoresed alongside control wells containing untreated DNA (lane 1), DNA treated with light but no compound (lane 2), and DNA treated with the highest concentration of compound but no light (lane 9). The range for comparison (MC:NP=0.25–5) was chosen so that complete photocleavage by 1, which required MC:NP=5, could be captured. The results show that the Ru(II) dyads of this study are capable of photodamaging biological macromolecules. Using DNA as the model system, 2 and 3 were better photocleavage agents than 1 by comparing the amount of Form II DNA relative to Form I DNA in lane 3 for all of the compounds. At MC:NP=1, compound 2 caused complete conversion to Form II, while a five-fold higher concentration of 1 was required to effect a similar change. Compound 3 appeared to be slightly more potent than 2 according to the relative band densities of Forms I and II in lane 3, but a loss of the ethidium bromide fluorescence at ≥20 μM for 3 precluded a more quantitative comparison. Treatment of DNA with compounds 2 and 3 in the dark also caused the DNA bands to disappear (lane 9), and was attributed to interference with the ethidium bromide fluorescence by high concentrations of the metal complexes (a known phenomenon). Nevertheless, a small signal corresponding to aggregated DNA could be discerned for compounds 1 (lane 7) and 2 (lane 9). None of the compounds caused frank double-strand breaks, underscored by the absence of Form III DNA on the gel.
Figure 5.

DNA photocleavage of pUC19 DNA (20 μM bases) dosed with metal complex (MC) 1 (a), 2 (b), or 3 (c) and visible light (14 J·cm−2). Gel mobility shift assays employed 1% agarose gels (0.75 μg·mL−1 ethidium bromide) electrophoresed in 1× TAE at 9 V·cm−1 for 30 min. Lane 1, DNA only (-hν); lane 2, DNA only (+hν); lane 3, 5 μM MC (+hν); lane 4, 20 μM MC (+hν); lane 5, 40 μM MC (+hν); lane 6, 60 μM MC (+hν); lane 7, 80 μM MC (+hν); lane 8, 100 μM MC (+hν); lane 9, 100 μM MC (-hν). Light treatments were broadband visible light (14 J cm−2, 7.8 mW cm−2).
The estimated photoreactivities of 1-3 from the DNA gel mobility experiment paralleled the in vitro photobiological activities of the compounds with both visible and red light treatments in two different cell lines, with 3 being the most potent and 1 being the least. The results also corroborated the 1O2 quantum yields, where compounds 2 and 3 were 74 and 78%, respectively, and 1 was slight lower at 58%. While the positive correlations between cell-free DNA photocleavage assays and 1O2 measurements do not definitively establish 1O2 as the most important mediator of the in vitro photobiological activity or DNA as the intracellular target, they do strongly suggest that 1O2 is involved and that the process becomes more efficient as the number of thiophene rings appended to 1,10-phenanthroline increases from n=1 to 3.
CONCLUSION
A series of three new bis(bpy)-heteroleptic Ru(II) dyads, differing in the number of thienyl groups (n=1, 2, or 3) directly appended to their 1,10-phenanthroline coligands, were synthesized and characterized to evaluate the effect of n on their photophysical and photobiological properties. The complexes were characterized by HPLC, 1D 1H and 2D 1H-1H NMR, and ESI-MS. Together, a careful assignment of all of the hydrogens by 1D and 2D NMR alongside detection of the predicted mass by ESI-MS confirmed the desired structures.
The room temperature absorption and emission profiles of compounds 1-3 were similar to those of the parent Ru(II) polypyridyl compounds [Ru(bpy)3]2+ or [Ru(phen)3]2+, with the lowest energy MLCT absorption and emission transitions assigned to the Ru(dπ) → bpy(π*) configuration. The steady-state 77 K and time-resolved room temperature emission spectra also supported the assignment of the emissive state as 3MLCT for all three of the compounds. While n had little impact on the nature of the emissive state for this series, the ESA signature of 1 differed markedly from that of 2 and 3. The TA signal for 1 was typical of 3MLCT states at all wavelengths, while that for 2 and 3 showed an additional nonemissive component with features consistent with 3IL states: a broad and intense ESA across the visible region (extending into the NIR) that decayed with a lifetime on the order of tens of microseconds. The 3IL state energies were estimated from triplet state energies of the free 1T, 2T, and 3T groups, which decrease on going from n=1 to 3. It appears that the energy of the 3IL state must be in proximity to that of the 3MLCT state (near 2.1 eV) in order for this state to play an important role in the excited state dynamics of the compound. Thus, 2 and 3 have the requisite 3IL energies for extending the intrinsic lifetimes of their excited states and thus producing higher 1O2 quantum yields.
The photocytotoxicity and dark cytotoxicity for 1-3 were assessed in HL60 human leukemia and SKMEL28 human melanoma cells, and dark cytotoxicity was also measured in CCD-1064Sk normal skin fibroblasts. Ru(II) dyads 2 and 3 were more powerful phototoxins (compared to 1) in both cell lines, and generally had larger PI values. Their in vitro photobiological activities correlated positively with their cell-free 1O2 quantum yields and DNA photodamaging capacities, suggesting that 1O2 may be the mediator of photocytotoxicity via direct damage to biological macromolecules. Compounds 2 and 3 also had the unusual property of being activatable by red light, at wavelengths where their molar extinction coefficients were vanishingly small. Thus, these Ru(II) dyads derived from α-oligothiophenes represent another example of blue-green absorbing photosensitizers that can be activated with red light to yield potent photobiological effects, presumably due to accessible 3IL states centered on the π-extended ligands.
Efforts are currently underway to understand these red photobiological effects from a theoretical standpoint and to elucidate the in vitro mechanisms of action for this series and related compounds. While this study points toward the possible utility of 2 and 3 as PDT agents, their low-yielding syntheses would need to be optimized for the cost-effective production of the larger quantities of material required for additional biological testing.
Supplementary Material
ACKNOWLEDGEMENTS
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number R01CA222227. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also acknowledge financial support from the University of North Carolina at Greensboro, the Natural Sciences and Engineering Council of Canada, the Canadian Institutes of Health Research, the Canadian Foundation for Innovation, the Nova Scotia Research and Innovation Trust, and Acadia University.
Footnotes
This article is part of a Special Issue celebrating Photochemistry and Photobiology’s 55th Anniversary.
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article:
REFERENCES
- (1).Juris A; Balzani V; Barigelletti F; Campagna S; Belser P; von Zelewsky A Ru(II) Polypyridine Complexes: Photophysics, Photochemistry, Eletrochemistry, and Chemiluminescence. Coord. Chem. Rev 1988, 84, 85–277. [Google Scholar]
- (2).Juris A; Campagna S; Balzani V; Gremaud G; Von Zelewsky A Absorption Spectra, Luminescence Properties, and Electrochemical Behavior of Tris-Heteroleptic Ruthenium(II) Polypyridine Complexes. Inorg. Chem 1988, 27, 3652–3655. [Google Scholar]
- (3).Balzani V; Juris A Photochemistry and Photophysics of Ru(II)polypyridine Complexes in the Bologna Group. From Early Studies to Recent Developments. Coord. Chem. Rev 2001, 211, 97–115. [Google Scholar]
- (4).Campagna S; Puntoriero F; Nastasi F; Bergamini G; Balzani V Photochemistry and Photophysics of Coordination Compounds: Ruthenium In Photochemistry and Photophysics of Coordination Compounds I; Balzani V, Campagna S, Eds.; Topics in Current Chemistry; Springer Berlin Heidelberg: Heidelberg, 2007; pp 117–214. [Google Scholar]
- (5).Glazer EC Light-Activated Metal Complexes That Covalently Modify DNA. Isr. J. Chem 2013, 53, 391–400. [Google Scholar]
- (6).White JK; Schmehl RH; Turro C An Overview of Photosubstitution Reactions of Ru(II) Imine Complexes and Their Application in Photobiology and Photodynamic Therapy. Inorg. Chim. Acta 2017, 454, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Shi G; Monro S; Hennigar R; Colpitts J; Fong J; Kasimova K; Yin H; DeCoste R; Spencer C; Chamberlain L; Mandel A; Lilge L; McFarland SA Ru(II) Dyads Derived from Alpha-Oligothiophenes: A New Class of Potent and Versatile Photosensitizers for PDT. Coord. Chem. Rev 2015, 282–283, 127–138. [Google Scholar]
- (8).Knoll JD; Turro C Control and Utilization of Ruthenium and Rhodium Metal Complex Excited States for Photoactivated Cancer Therapy. Coord. Chem. Rev 2015, 282–283, 110–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mari C; Pierroz V; Leonidova A; Ferrari S; Gasser G Towards Selective Light-Activated Ru II -Based Prodrug Candidates: Towards Selective Light-Activated RuII-Based Prodrug Candidates. Eur. J. Inorg. Chem 2015, 2015, 3879–3891. [Google Scholar]
- (10).Heinemann F; Karges J; Gasser G Critical Overview of the Use of Ru(II) Polypyridyl Complexes as Photosensitizers in One-Photon and Two-Photon Photodynamic Therapy. Acc. Chem. Res 2017, 50, 2727–2736. [DOI] [PubMed] [Google Scholar]
- (11).Mari C; Pierroz V; Ferrari S; Gasser G Combination of Ru(II) Complexes and Light: New Frontiers in Cancer Therapy. Chem. Sci 2015, 6, 2660–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Poynton FE; Bright SA; Blasco S; Williams DC; Kelly JM; Gunnlaugsson T The Development of Ruthenium(II) Polypyridyl Complexes and Conjugates for in Vitro Cellular and in Vivo Applications. Chem. Soc. Rev 2017, 46, 7706–7756. [DOI] [PubMed] [Google Scholar]
- (13).McClenaghan ND; Leydet Y; Maubert B; Indelli MT; Campagna S Excited-State Equilibration: A Process Leading to Long-Lived Metal-to-Ligand Charge Transfer Luminescence in Supramolecular Systems. Coord. Chem. Rev 2005, 249, 1336–1350. [Google Scholar]
- (14).Monro S; Scott J; Chouai A; Lincoln R; Zong R; Thummel RP; McFarland SA Photobiological Activity of Ru(II) Dyads Based on (Pyren-1-Yl)Ethynyl Derivatives of 1,10-Phenanthroline. Inorg. Chem 2010, 49, 2889–2900. [DOI] [PubMed] [Google Scholar]
- (15).Lincoln R; Kohler L; Monro S; Yin H; Stephenson M; Zong R; Chouai A; Dorsey C; Hennigar R; Thummel RP; McFarland SA Exploitation of Long-Lived 3IL Excited States for Metal–Organic Photodynamic Therapy: Verification in a Metastatic Melanoma Model. J. Am. Chem. Soc 2013, 135, 17161–17175. [DOI] [PubMed] [Google Scholar]
- (16).Reichardt C; Pinto M; Wächtler M; Stephenson M; Kupfer S; Sainuddin T; Guthmuller J; McFarland SA; Dietzek B Photophysics of Ru(II) Dyads Derived from Pyrenyl-Substitued Imidazo[4,5-f][1,10]Phenanthroline Ligands. J. Phys. Chem. A 2015, 119, 3986–3994. [DOI] [PubMed] [Google Scholar]
- (17).Reichardt C; Sainuddin T; Wächtler M; Monro S; Kupfer S; Guthmuller J; Gräfe S; McFarland S; Dietzek B Influence of Protonation State on the Excited State Dynamics of a Photobiologically Active Ru(II) Dyad. J. Phys. Chem. A 2016, 120, 6379–6388. [DOI] [PubMed] [Google Scholar]
- (18).Reichardt C; Schneider KRA; Sainuddin T; Wächtler M; McFarland SA; Dietzek B Excited State Dynamics of a Photobiologically Active Ru(II) Dyad Are Altered in Biologically Relevant Environments. J. Phys. Chem. A 2017, 121, 5635–5644. [DOI] [PubMed] [Google Scholar]
- (19).Stephenson M; Reichardt C; Pinto M; Wächtler M; Sainuddin T; Shi G; Yin H; Monro S; Sampson E; Dietzek B; McFarland SA Ru(II) Dyads Derived from 2-(1-Pyrenyl)-1H-Imidazo[4,5-f][1,10]Phenanthroline: Versatile Photosensitizers for Photodynamic Applications. J Phys Chem A 2014, 118, 10507–10521. [DOI] [PubMed] [Google Scholar]
- (20).Arenas Y; Monro S; Shi G; Mandel A; McFarland S; Lilge L Photodynamic Inactivation of Staphylococcus Aureus and Methicillin-Resistant Staphylococcus Aureus with Ru(II)-Based Type I/Type II Photosensitizers. Photodiag. Photodyn. Ther 2013, 10, 615–625. [DOI] [PubMed] [Google Scholar]
- (21).Ford WE; Rodgers MAJ Reversible Triplet-Triplet Energy Transfer within a Covalently Linked Bichromophoric Molecule. J Phys Chem 1992, 96, 2917–2920. [Google Scholar]
- (22).Harriman A; Hissler M; Khatyr A; Ziessel R A Ruthenium(II) Tris(2,2′-Bipyridine) Derivative Possessing a Triplet Lifetime of 42 μs. Chem. Commun 1999, 735–736. [Google Scholar]
- (23).Goze C; Kozlov DV; Tyson DS; Ziessel R; Castellano FN Synthesis and Photophysics of Ruthenium(II) Complexes with Multiple Pyrenylethynylene Subunits. New J. Chem 2003, 27, 1679–1683. [Google Scholar]
- (24).Ji S; Wu W; Wu W; Song P; Han K; Wang Z; Liu S; Guo H; Zhao J Tuning the Luminescence Lifetimes of Ruthenium(II) Polypyridine Complexes and Its Application in Luminescent Oxygen Sensing. J. Mater. Chem 2010, 20, 1953–1963. [Google Scholar]
- (25).Johansson T; Mammo W; Svensson M; Andersson MR; Inganäs O Electrochemical Bandgaps of Substituted Polythiophenes. J. Mater. Chem 2003, 13, 1316–1323. [Google Scholar]
- (26).Mauer R; Howard I; Laquai F Energy and Charge Transfer. In Semiconducting Polymer Composites: Principles, Morphologies, Properties and Applications; Wiley-VCH Verlag. [Google Scholar]
- (27).Köhler A; Beljonne D The Singlet–Triplet Exchange Energy in Conjugated Polymers. Adv. Funct. Mater 2004, 14, 11–18. [Google Scholar]
- (28).Scaiano JC; Redmond RW; Mehta B; Arnason JT Efficiency of the Photoprocesses Leading to Singlet Oxygen Generation by Alpha-Terthienyl: Optical Absorption, Optoacoustic Calorimetry and Infrared Luminescence Studies. Photochem. Photobiol 1990, 52, 655–659. [DOI] [PubMed] [Google Scholar]
- (29).Wasserberg D; Marsal P; Meskers SCJ; Janssen RAJ; Beljonne D Phosphorescence and Triplet State Energies of Oligothiophenes. J. Phys. Chem. B 2005, 109, 4410–4415. [DOI] [PubMed] [Google Scholar]
- (30).Becker RS; Seixas de Melo J; Maçanita AL; Elisei F Comprehensive Evaluation of the Absorption, Photophysical, Energy Transfer, Structural, and Theoretical Properties of α-Oligothiophenes with One to Seven Rings. J. Phys. Chem 1996, 100, 18683–18695. [Google Scholar]
- (31).Scaiano JC; MacEachern A; Arnason JT; Morand P; Weir D Singlet Oxygen Generating Efficiency of α-Terthienyl and Some of Its Synthetic Analogues. Photochem. Photobiol 1987, 46, 193–199. [Google Scholar]
- (32).de Melo JS; Silva LM; Arnaut LG; Becker RS Singlet and Triplet Energies of α-Oligothiophenes: A Spectroscopic, Theoretical, and Photoacoustic Study: Extrapolation to Polythiophene. J. Chem. Phys 1999, 111, 5427–5433. [Google Scholar]
- (33).Majewski MB; de Tacconi NR; MacDonnell FM; Wolf MO Ligand-Triplet-Fueled Long-Lived Charge Separation in Ruthenium(II) Complexes with Bithienyl-Functionalized Ligands. Inorg. Chem 2011, 50, 9939–9941. [DOI] [PubMed] [Google Scholar]
- (34).Majewski MB; de Tacconi NR; MacDonnell FM; Wolf MO Long-Lived, Directional Photoinduced Charge Separation in RuII Complexes Bearing Laminate Polypyridyl Ligands. Chem. Eur. J 2013, 19, 8331–8341. [DOI] [PubMed] [Google Scholar]
- (35).Moorlag C; Sarkar B; Sanrame CN; Bäuerle P; Kaim W; Wolf MO Conjugation Length Dependent Ground and Excited State Electronic Behavior in Oligothienyl Ru Complexes. Inorg. Chem 2006, 45, 7044–7046. [DOI] [PubMed] [Google Scholar]
- (36).Alberto ME; Pirillo J; Russo N; Adamo C Theoretical Exploration of Type I/Type II Dual Photoreactivity of Promising Ru(II) Dyads for PDT Approach. Inorg. Chem 2016, 55, 11185–11192. [DOI] [PubMed] [Google Scholar]
- (37).Tyson DS; Henbest KB; Bialecki J; Castellano FN Excited State Processes in Ruthenium(II)/Pyrenyl Complexes Displaying Extended Lifetimes. J. Phys. Chem. A 2001, 105, 8154–8161. [Google Scholar]
- (38).Sullivan BP; Salmon DJ; Meyer TJ Mixed Phosphine 2,2’-Bipyridine Complexes of Ruthenium. Inorg. Chem 1978, 17, 3334–3341. [Google Scholar]
- (39).Treadway CR Spectroscopic Characterization of DNA-Mediated Charge Transfer Ph.D. dissertation, California Institute of Technology, 2003. [Google Scholar]
- (40).Hissler M; Connick WB; Geiger DK; McGarrah JE; Lipa D; Lachicotte RJ; Eisenberg R Platinum Diimine Bis(Acetylide) Complexes: Synthesis, Characterization, and Luminescence Properties. Inorg. Chem 2000, 39, 447–457. [DOI] [PubMed] [Google Scholar]
- (41).Kitagawa T; Matsubara H; Okazaki T; Komatsu K Electrochemistry of the Self-Assembled Monolayers of Dyads Consisting of Tripod-Shaped Trithiol and Bithiophene on Gold. Mol. Basel Switz 2014, 19, 15298–15313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Foxon SP; Metcalfe C; Adams H; Webb M; Thomas JA Electrochemical and Photophysical Properties of DNA Metallo-Intercalators Containing the Ruthenium(II) Tris(1-Pyrazolyl)Methane Unit. Inorg. Chem 2007, 46, 409–416. [DOI] [PubMed] [Google Scholar]
- (43).Suzuki K; Kobayashi A; Kaneko S; Takehira K; Yoshihara T; Ishida H; Shiina Y; Oishi S; Tobita S Reevaluation of Absolute Luminescence Quantum Yields of Standard Solutions Using a Spectrometer with an Integrating Sphere and a Back-Thinned CCD Detector. Phys. Chem. Chem. Phys 2009, 11 (42), 9850–9860. [DOI] [PubMed] [Google Scholar]
- (44).DeRosa MC; Crutchley RJ Photosensitized Singlet Oxygen and Its Applications. Coord. Chem. Rev 2002, 233/234, 351–371. [Google Scholar]
- (45).Hanson BJ Composition and Assays for Determining Cell Viability. U.S. Patent 2013/0210063 A1, August 15, 2013.
- (46).McCusker CE; McCusker JK Synthesis and Spectroscopic Characterization of CN-Substituted Bipyridyl Complexes of Ru(II). Inorg. Chem 2011, 50, 1656–1669. [DOI] [PubMed] [Google Scholar]
- (47).Barbieri A; Ventura B; Flamigni L; Barigelletti F; Fuhrmann G; Bäuerle P; Goeb S; Ziessel R Binuclear Wirelike Dimers Based on Ruthenium(II)−Bipyridine Units Linked by Ethynylene−Oligothiophene−Ethynylene Bridges. Inorg. Chem 2005, 44, 8033–8043. [DOI] [PubMed] [Google Scholar]
- (48).Yin H; Stephenson M; Gibson J; Sampson E; Shi G; Sainuddin T; Monro S; McFarland SA In Vitro Multiwavelength PDT with 3IL States: Teaching Old Molecules New Tricks. Inorg. Chem 2014, 53, 4548–4559. [DOI] [PubMed] [Google Scholar]
- (49).Hintermann G; Fischer H-M; Crameri R; Hütter R Simple Procedure for Distinguishing CCC, OC, and L Forms of Plasmid DNA by Agarose Gel Electrophoresis. Plasmid 1981, 5, 371–373. [DOI] [PubMed] [Google Scholar]
- (50).Schmidt T; Friehs K; Schleef M; Voss C; Flaschel E Quantitative Analysis of Plasmid Forms by Agarose and Capillary Gel Electrophoresis. Anal. Biochem 1999, 274, 235–240. [DOI] [PubMed] [Google Scholar]
- (51).Shafirovich V; Singh C; Geacintov NE Photoinduced Oxidative DNA Damage Revealed by an Agarose Gel Nicking Assay: A Biophysical Chemistry Laboratory Experiment. J. Chem. Educ 2003, 80, 1297. [Google Scholar]
- (52).Palchaudhuri R; Hergenrother PJ DNA as a Target for Anticancer Compounds: Methods to Determine the Mode of Binding and the Mechanism of Action. Curr. Opin. Biotechnol 2007, 18, 497–503. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.