

Abstract
Contemporary chemoenzymatic approaches can provide highly complex multi-antennary N-linked glycans. These procedures are, however, very demanding and typically involve as many as 100 chemical steps to prepare advanced intermediates that can be diversified by glycosyltransferases in a branch selective manner to give asymmetrical structures commonly found in Nature. Only highly specialized laboratories can perform such syntheses, which greatly hampers progress in glycoscience. Here we describe a biomimetic approach in which a readily available bi-antennary glycopeptide can be converted in 10 or fewer chemical and enzymatic steps into multi-antennary N-glycans that at each arm can be uniquely extended by glycosyltransferases to give access to highly complex asymmetrically branched N-glycans. A key feature of our approach is the installation of additional branching points using recombinant MGAT4 and MGAT5 in combination with unnatural sugar donors. At an appropriate point in the enzymatic synthesis, the unnatural monosaccharides can be converted into their natural counterpart allowing each arm to be elaborated into a unique appendage.
Graphical Abstract
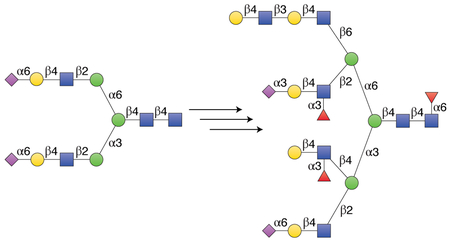
N-glycosylation of proteins is one of the most complex and diverse post-translational modifications that can influence a multitude of biological processes such as signal transduction, embryogenesis, neuronal development, fertilization, hormone activity, immune regulation and the proliferation of cells and their organization into specific tissues1. It has been implicated in the etiology of many human diseases such as pathogen recognition, inflammation, immune responses, the development of autoimmune diseases, and cancer2,3. Although it is widely accepted that N-glycans contain high information content, the limited accessibility of well-defined structures makes it difficult to uncover the molecular basis by which they regulate biological and disease processes4–6. Consequently, diverse collections of well-defined N-glycans are needed as standards for glycan structure determination of heterogeneous biological samples, as ligands to study interactions with glycan-binding proteins, as probes to examine the molecular basis of glycoconjugate biosynthesis and as starting materials for glycoprotein synthesis7–10.
To address this demand, chemoenzymatic methodologies are emerging that can provide multi-antennary N-glycans having highly complex branched architectures11–14. However, these approaches require demanding organic synthetic procedures to provide advanced intermediates suitable for enzymatic diversification. For instance, an advanced intermediate that can give entry into tetra-antennary structures having unique appendages at each arm required the preparation of twelve properly protected monosaccharide building blocks and a further thirty steps for assembly and deprotection, totaling well over 100 chemical manipulations15. Only highly specialized laboratories can perform such syntheses, and even in hands of specialists, it is very challenging to obtain sufficient quantities of precursors required for the preparation of large numbers of targets.
Herein, we present a synthetic strategy inspired by the biosynthesis of N-glycans that can provide highly complex, asymmetrically branched bi-, tri- and tetra-antennary N-glycans from a readily available bi-antennary glycopeptide. Only 10 facile chemical and enzymatic steps are needed to convert the starting material into a tetra-antennary intermediate that at each branching point can be uniquely extended by a panel of easy to express glycosyltransferases to give access to highly complex structures. Even fewer steps are needed to prepare complex bi- and tri-antennary glycans for enzymatic diversification, thereby opening the possibility to prepare diverse libraries of N-glycans without the need to chemically synthesize highly complex synthetic intermediates.
The biosynthesis of glycans is a non-template mediated process that occurs in the secretory pathway where glycosyltransferases catalyze the transfer of monosaccharides from activated sugar-nucleotides to specific hydroxyls of a growing oligosaccharide chain16. The complexity of N-glycans arises from the modification of a common core pentasaccharide by a family of N-acetylglucosaminyltransferases (termed mannosyl-glycoprotein N-acetylglucosaminyltransferases or MGATs)17 resulting in oligosaccharides having various numbers and patterns of branching N-acetylglucosamine (GlcNAc) moieties (Fig. 1a). Galactosyltransferases (GalT) can convert these GlcNAc residues into N-acetyllactosamine (LacNAc, Gal β(1,4)GlcNAc), which can then be elaborated by an array of glycosyltransferases into appendages with enormous structural diversity (Fig. 1b)18. Mature N-glycans usually have architectures in which each branching point is extended by a unique epitope11 and may also contain a core-fucose modification where an α1,6-fucoside is present on the reducing chitobiose residue1.
Fig. 1.

Structure of N-glycans and a bio-inspired strategy for their preparation. a, MGAT enzymes responsible for installing GlcNAc at different branching points. b, Enzyme classes involved in the biosynthesis of complex N-glycans. c, Structure of unnatural UDP-GlcNTFA (4). d, Bio-inspired strategy for the synthesis of asymmetric N-glycans. Symmetrical bi-antennary glycan 1, which can easily be obtained from a glycopeptide isolated from egg yolk, can be further branched by recombinant MGAT4 and MGAT5. The use of unnatural UDP-GlcNTFA makes it possible to prepare 2 bearing GlcNAc, GlcN3 and GlcNH2 branching moieties. Compound 2 is the key intermediate for preparing complex targets such as 3. e, Transformation of GlcNTFA, installed by MGAT4 and MGAT5, into GlcNH2 or GlcN3 “stops” further enzymatic extension of these moieties until they are converted into natural GlcNAc (go) that can then be elaborated by glycosyltransferases into complex appendages.
Our biomimetic approach employs symmetric bi-antennary glycosyl asparagine derivative 1 as the key intermediate (Fig. 1d), which can be prepared from a glycopeptide isolated from egg yolk powder19,20 in six robust chemical and enzymatic steps. Next, we take advantage of recombinant MGAT4, MGAT5, and unnatural uridine 5′-diphospho-N-trifluoroacetyl glucosamine (UDP-GlcNTFA (4), Fig 1c) to transform 1 into tetra-antennary glycosyl asparagine 2 in five steps. The latter compound can be selectively extended at each antenna by a panel of mammalian glycosyltransferases to provide glycosyl asparagines, such as 3, endowed with unique epitopes at each antenna. A key strategic principle of our approach is the conversion of the newly installed GlcNTFA moieties at the MGAT4 and MGAT5 arms into glucosamine (GlcNH2) and 2-deoxy-2-azido-glucose (GlcN3), respectively, which are inert to modifications by our panel of mammalian glycosyltransferases (Fig. 1e). Subsequent elaboration of the GlcNAc residues at the MGAT1 or MGAT2 arm is then possible by exploiting inherent branch selectivities of glycosyltransferases and hydrolases. At the next stage of synthesis, the GlcNH2 and GlcN3 can be sequentially “unmasked” to give natural GlcNAc termini for further enzymatic elaboration into complex appendages.
Results
Universal starting material and differentiating the MGAT1 and MGAT2 arms.
Sialoglycopeptide 5 (SGP, Fig. 2), which was previously used for the preparation of simple symmetrical N-glycans21–25, can be routinely isolated from egg yolk powder in multi-gram quantities19,20. This compound was converted into bi-antennary glycosyl asparagine 1 by subsequent pronase treatment to remove the peptide, protection of the α-amine of the remaining asparagine (Asn) with a benzyloxycarbamate (Cbz) (→6), hydrolysis of the sialosides and galactosides by treatment with neuraminidase from C. perfringens and galactosidase from A. niger, and finally core fucosylation using recombinant α-fucosyltransferases 8 (FUT8) and guanosine 5′-diphospho-β-L-fucose (GDP-fucose).
Fig. 2.

Two strategies for desymmetrizing N-glycans using the branch selectivity of the sialyltransferase ST6Gal1 and the galactosidase from E. coli, and subsequent preparation of asymmetric branched bi-antennary glycans such as 13. The α2,6-sialoside of 8 blocks further modification of the MGAT1 antenna allowing selective elaboration of the MGAT2 arm. The MGAT1 and MGAT2 arms of asymmetrically branched glycan 9 can selectively be extended by exploiting that many glycosyltransferases modify LacNAc but not terminal GlcNAc moieties making it was possible to first elaborate the MGAT2 arm without affecting the MGAT1 arm. The peptide sequence of SGP is NH2-Lys-Val-Ala-Asn-Lys-Thr-COOH with the glycan connected to Asn. Each intermediate was purified by HPLC on an XBridge HILIC column. The transformation of 10 into 13 was also performed without intermediate compound purification and by only subjecting 13 to P2 size exclusion column chromatograph, an improved yield of 79% over three steps was accomplished.
Removal of the sialosides and galactosides of 5 was essential because FUT8 acts early in the biosynthetic pathway and requires a terminal GlcNAc at the MGAT1 arm26. Furthermore, we opted to maintained the asparagine aglycone because it locks the reducing anomeric center in the β-configuration, which is another requirement for FUT8 activity27. In addition, it simplifies HPLC purification because the synthetic intermediates exist as only one anomer. At the final stage of synthesis, the CBz group on the asparagine can be removed by hydrogenation to reveal a free amine that can be exploited for the installation of a tag of interest such as biotin or a fluorophore, or can be used for immobilization on carboxy-activated glass slides for creating glycan microarrays.
Next, we explored two strategies for the selective modification of the MGAT1 and MGAT2 arm by exploiting inherent branch selectivities of glycosyltransferases and glycosidases to prepare asymmetrically branched bi-antennary glycosyl asparagines (Fig. 2). The terminal GlcNAc residues of 1 were galactosylated with β−1,4-galactosyltransferase-1 (B4GALT1) and uridine 5′-diphosphogalactose (UDP-Gal) to install two LacNAc moieties yielding symmetrically branched derivative 7. First, we exploited the ST6 β-galactoside α−2,6-sialyltransferase 1 (ST6GAL1), which prefers the MGAT1 arm28 to desymmetrize 7 into mono-sialoside 8. It is known that an α2,6-sialoside blocks further modification of the resulting antenna by mammalian glycosyltransferases, thus it was possible to selectively elaborate the MGAT2 arm into a complex structure bearing an extended sialyl Lewisx-Lewisx (SLex-Lex) epitope (see Supplementary Fig. 1).
A second orthogonal strategy for branch specific modification involved the selective removal of the galactoside at the MGAT1 arm of 7. This was achieved using a galactosidase from E. coli29 to give 9, which has a terminal GlcNAc and LacNAc moiety at the MGAT1 and MGAT2 antennae, respectively. Many glycosyltransferases modify LacNAc but not terminal GlcNAc, thus it was possible to first elaborate the MGAT2 arm without affecting the MGAT1 arm. For instance, compound 9 was fucosylated with FUT5 to give 10 bearing a Lex functionality, which is not recognized by most mammalian glycosyltransferases. Inactivation of the MGAT2 branch allowed for selective extension of the terminal GlcNAc at the MGAT1 arm of 10 into a di-LacNAc moiety by subsequent addition of galactose (Gal), GlcNAc, and Gal units using β−1,4-galactosyltransferase 1 (B4GALT1) (→11), β−1,3-N-acetylglucosaminyltransferase 2 (B3GNT2) (→12), and B4GALT1 to ultimately provide complex glycosyl asparagine 13. Compounds 11, 12, and 13 are biologically relevant and have been observed on ovarian cancer cell lines30.
The formation of compounds 8 and 9 was accompanied by a small amount of starting material or by-product that could readily be removed by semi-preparative HPLC using an XBridge hydrophilic interaction liquid chromatography (HILIC) column31 employing ESI mass spectrometry for compound detection (see Supplementary Figs. 7 and 8). While the other enzymatic reactions proceeded to completion, each compound was purified by HILIC-HPLC, and it was found that this type of column chromatography provided base-line separation for all targets. Mass spectrometry and multi-dimensional NMR confirmed the homogeneity of the compounds. It is well known that purification of glycans by HILIC column chromatography results in loss of material32 providing a rationale for isolated yields ranging from 60–70%. Substantial higher overall yields were achieved by performing several enzymatic transformations without intermediate compound purification. For example, the conversion of 10 into 13 proceeded with an overall yield of 79% when only the final product was purified by size exclusion column chromatography over Bio-Gel® P2.
The Cbz protecting group of the asparagine moiety of compounds such as 13 could readily be removed by hydrogenation over Pd(OH)2/C to give derivatives having a free amine (Supplementary Section 6b). To demonstrate the resulting compounds can be used for glycan microarray development, a number of symmetrical and asymmetrical glycosyl asparagine amines were printed on N-hydroxysuccinimide (NHS) activated glass slides, and then examined for binding to the plant lectins Aleuria aurantia lectin (AAL) and Sambucus nigra agglutinin (SNA), the immune regulatory proteins Galectin-3 and −9 (Gal-3 and Gal-9), and several H7N2 flu viruses (Supplementary Fig. 13). The plant lectins gave an expected binding pattern demonstrating the compounds had been properly printed. Interestingly, although Gal-9 preferentially binds extended LacNAc epitopes, it did not show affinity to an asymmetrically branched compound having such an epitope on the MGAT1 arm. Strong binding was only observed for symmetrically branched glycans endowed with extended LacNAc units on both arms. Furthermore, it was observed that Gal-3 prefers α2,3-linked sialosides whereas Gal-9 showed strong responsiveness to α2,6-linked sialosides.
Synthesis of asymmetrical branched tri-antennary glycosyl asparagines.
Next, the focus was on the preparation of complex tri-antennary compounds (e.g. 22, Fig. 3) starting from 1 and employing an MGAT enzyme to install an additional branching point. These enzymes require terminal GlcNAc moieties at the MGAT1 or MGAT2 arms1, thus the third branching GlcNAc must be installed before these positions are diversified. For the successful installation of a GlcNAc branch that can be uniquely extended, our strategy required a modified UDP-GlcNAc donor that is readily accepted as a substrate by MGAT4 and MGAT5, but gives a product in which the newly introduced antenna is not recognized by glycosyltransferases. The latter feature would make it possible to selectively extend GlcNAc moieties at the MGAT1 and MGAT2 arms by exploiting inherent branch specificities of glycosyltransferases or glycosidases (as for bi-antennary 7). Next, it should be possible to convert the modified GlcNAc at the MGAT4 or MGAT5 arms into natural GlcNAc, which can then be extended into a complex appendage.
Fig. 3.

Synthesis of asymmetric branched tri-antennary glycosyl asparagines using MGAT5 and UDP-GlcNTFA. MGAT5 readily accepts UDP-GlcNTFA to give a tri-antennary glycan that upon base treatment provides a compound having a GlcNH2 at β6-arm (15). The latter residue is not a substrate for the galactosyl transferase B4GalT1 and therefore it is possible to selectively elaborate the MGAT1 and MGAT2 arm by exploiting inherent branch selectivities of glycosidases and glycosyl transferases. Once the MGAT1 and MGAT2 arms were capped with Neu5Ac preventing these positions from further elongation, the GlcNH2 could be acetylated to give natural GlcNAc capable of being extended by a series of glycosyl transferase. Compounds 16 - 19 were purified by HPLC using a HILIC column. Compound 19 was subjected to three enzymatic transformations to yield 22, which was purified by P2 size-exclusion chromatography.
To demonstrate the feasibility of this strategy, we employed MGAT5 and the unnatural sugar nucleotide UDP-GlcNTFA (4)33. This donor was readily transferred by MGAT5 to give tri-antennary glycosyl asparagine 14 (Fig. 3). Unfortunately, the terminal GlcNTFA was recognized as an acceptor for B4GALT1, and treatment with this enzyme resulted in all three antennae being galactosylated. Gratifyingly, galactosylation of the MGAT5 branch could be blocked by a GlcNH2 residue (15), which was obtained by clean removal of the TFA group in 14 with NaOH (pH = 10). Therefore, it was possible to selectively galactosylate the GlcNAc residues at the MGAT1 and MGAT2 arms of 15 to provide 16. The LacNAc moiety at the MGAT1 branch of 16 could selectively be sialylated with ST6GAL1 providing 17 (Supplementary Fig. 9), and the remaining LacNAc on the MGAT2 arm was capped with an α2,3 sialyloside (→18) using ST3 β-galactoside α−2,3-sialyltransferase 4 (ST3GAL4). Next, the MGAT5 arm was “unmasked” by acetylation of the amine of GlcNH2 with N-acetylsuccinimide (AcOSu) to give 19 having a natural GlcNAc moiety. This arm was then galactosylated by B4GALT1 and the resulting LacNAc moiety was extended by subsequent glycosylation with B3GNT2 followed by B4GALT1 to give asymmetrically branched tri-antennary structure 22. It was also possible to selectively remove the galactoside at the MGAT1 antenna of 16 providing an asymmetrically branched precursor that could be transformed into another series of compounds (Supplementary Fig. 2).
Synthesis of asymmetrical branched tetra-antennary glycosyl asparagines.
The final challenge was to adapt the synthetic approach to tetra-antennary glycosyl asparagines having unique glyco-epitopes on each appendage, and our focus was on the preparation of 3 which is a putative glycan structure observed on human cytolytic T lymphocytes34. Building on our bi- and tri-antennary findings, a strategy was envisaged in which MGAT4 and MGAT5 would install modified glucosamine moieties that could be sequentially “unmasked” to natural GlcNAc for subsequent selective enzymatic extension (Fig. 4). For this purpose, we selected 2-azido-glucose (GlcN3) and GlcNH2 that can both be derived from GlcNTFA, which in turn can be installed by MGAT4 and MGAT5 using UDP-GlcNTFA. Thus, compound 15, which was obtained by glycosylation of 1 using UDP-GlcNTFA with MGAT5 could be converted into GlcN3 by a two-step procedure. Compound 15 was first reacted with NaOH to remove the TFA protecting group, and then imidazole-1-sulfonyl azide35 was used to convert the free amine into the desired azide 23. Gratifyingly, the latter modification was tolerated by MGAT4 and after transfer of UDP-GlcNTFA, and treatment with NaOH, the key intermediate tetra-antennary glycosyl asparagine 2 was obtained. As expected, only the natural terminal GlcNAc moieties at the MGAT1 and MGAT 2 antennae were galactosylated with B4GALT1 to give 24, which was subsequently α2,6-sialylated at the MGAT1 arm with ST6GAL1 and α2,3-sialylated at the MGAT2 arm with ST3GAL4 to provide 25. Next, the GlcNH2 at the MGAT4 arm was acetylated, providing a GlcNAc residue that was converted to LacNAc by B4GALT1. Subsequent fucosylation with α-fucosyltransferases 5 (FUT5) resulted in the simultaneous installation of Lex and sLex epitopes at the MGAT4 and MGAT2 arm, respectively. Finally, the MGAT5 branch was activated for enzymatic extension by reduction of the azide with 1,3-dithiolpropane36, followed by acetylation of the resulting amine with AcOSu furnishing 28. This antenna was then extended to a di-LacNAc motif by subsequent treatment with B4GALT1, B3GNT2 then B4GALT1 to provide tetra-antennary 20-mer 3 bearing unique glyco-epitopes at each antenna.
Fig. 4.

Synthesis of asymmetric branched tetra-antennary N-glycans using MGAT4 and MGAT5 in combination with UDP-GlcNTFA and subsequent conversion of the transferred GlcNTFA into GlcN3 or GlcNH2. The latter moieties are temporary disabled from modification by glycosyl transferases making it possible to selectively elaborate the MGAT1 and MGAT 2 arms. At an appropriate point in the synthesis, the unnatural GlcN3 or GlcNH2 moieties can be converted into natural GlcNAc allowing each arm to be uniquely extended. Compounds 26, 28, and 3 were purified by HPLC using a HILIC column and derivatives 23, 2, and 24 were purified by P2 size-exclusion chromatography.
Initially, we attempted to install GlcNH2 and GlcN3 via a one-step procedure using UDP-GlcNH2 and UDP-GlcN3 as sugar nucleotide donors, respectively. It was found that these modifications were not tolerated by MGAT4 and MGAT5, and it is likely that the N-acetyl functionality of UDP-GlcNAc makes key interactions with the MGAT enzymes limiting tolerable modifications. A trifluoro-N-acetamido is, however, sufficiently isosteric, and thus UDP-GlcNTFA is readily employed as a substrate by these enzymes. Next, the TFA can be selectively removed to give an amine, which can be modified by an azido transfer reaction. Acceptors bearing an amine or azide are substantially altered so that they are not recognized by B4GALT1. In this respect, kinetic analysis of mutants of the latter enzyme has demonstrated that Tyr284, Tyr309, and Trp310 make essential binding interactions with the GlcNAc acceptor37. Furthermore, Tyr309 also appears to be involved in UDP-Gal binding suggesting a strictly organized microenvironment necessary for glycosylation. Thus, the two-step procedure makes it possible to deal with the limited tolerability of MGAT enzymes to modify the UDP-GlcNAc donor yet provide acceptors that can be altered in such a way that they are not recognized by the subsequent galactosyl transferase catalyzed reaction.
Discussion.
Although N-glycans have often different appendages at their antennae11, efforts to prepare this class of compounds have almost exclusively focused on simpler derivatives having symmetrically branched architectures38–44. This stems from the difficulties of controlling diversification at the various sites of branching, especially when several different complex terminal structures need to be appended.
Asymmetrically branched N-glycans have been prepared by chemo-enzymatic methods in which an advanced precursor is chemically synthesized in such a way that it can uniquely be extended at each branching point by glycosyl transferase catalyzed tranformations11,12,14,15. The synthesis of such precursors is very cumbersome requiring as many as 100 chemical steps hindering the preparation of large collections of compounds.
N-glycans can be prepared by fewer steps starting from a N-glycan precursor obtained from natural sources such as a glycopeptide from egg yolk21–25, an invertase glycoprotein from wild-type S. cerevisiae45, a lipid-linked oligosaccharide from glyco-engineered E. coli45, or bovine fetuin46. After deglycosylation of these natural products, glycan cores are obtained that can be modified by a series of glycosidase and glycosyl transferase catalyzed transformations to give more complex structures. Unfortunately, these methods provide precursors that have several terminal GlcNAc or Gal moieties and therefore cannot be elaborated in a branch selective manner to yield compounds having unique appendages at each antenna. Ingenuous protecting group manipulations have been employed to convert a naturally derived oligosaccharide into glycosyl acceptors that can then be diversified by chemical glycosylation23,47. However, this approach involves several low yielding steps and has limited capability to prepare compounds with complex branching patterns. We recently described an enzymatic strategy for accessing complex human milk oligosaccharides by strategically installing a branching GlcNAc moiety on a linear precursor to give a compound with an asymmetrical architecture that at each arm could be uniquely extended by glycosyl transferases48. Such a strategy cannot be adapted to the preparation of N-glycans because MGAT4 and MGAT5 require terminal GlcNAc moieties at the MGAT1 and MGAT2 arms1. Thus, the modification of an appropriate bi-antennary glycan (e.g. compound 1) by these enzymes provides multi-antennary glycans having several terminal GlcNAc moieties that cannot be selectively extended.
The chemoenzymatic methodology presented here makes it possible to prepare, using a relatively small number of easy to implement steps, N-glycans precursors that at each arm can be uniquely extended to provide complex bi-, tri-, and tetra-antennary glycans. The cornerstone of the approach is the use of unnatural sugar-nucleotide donors that can be utilized by MGAT4 and MGAT5 and provide products that temporarily are “stopped” from further enzymatic extension. At an appropriate point in the synthesis, the unnatural sugars can be converted into their natural counterparts “go” for subsequent elaboration into complex appendages. Further control of selective antennae modification can be achieved by exploiting inherent selectivities of glycosidases and glycosyltransferases. The so-called “stop and go” strategy makes it possible to create considerable structural diversity starting from a common precursor. It can provide positional isomers (e.g. 2,4,2,- and 2,2,6-branched tri-antennary glycans by using either MGAT4 or MGAT5) and offers substantial flexibility to selectively modify the various arms with different forms of sialylation and fucosylation.
We envisage that the “stop and go” strategy can be extended to other glycosyl transferases and sugar nucleotide donors providing an additional level of regioselective control. For example, this strategy may find use for the selective modification of oligo-LacNAc residues. Termini of such compounds can readily be transformed into important glyco-epitopes via selective fucosylation and sialylation, however, difficulties remain in the selective modification of internal LacNAc residues (e.g. Lex moieties).
In this study, glycosyl asparagine 1 was employed as the starting material for the preparation of bi-, tri- and tetra-antennary structures. After completion of a specific target, the Cbz protecting group on the asparagine moiety can be removed by hydrogenation to reveal an amine that can, for example, be used for glycan array development. The asparagine aglycone locks the reducing GlcNAc moiety in the β-configuration, which is a prerequisite of the enzyme FUT8 activity27 (e.g. conversion of 6 into 1, Fig. 2), and simplifies HILIC-HPLC purification. The use of well-defined glycans as analytical standards may require compounds lacking the anomeric asparagine moiety. The methodology described can also furnish free-reducing glycans by performing all transformations on glycopeptide 5 (Fig. 2). Once a target has been synthesized, the peptide can be released by N-glycosidase F (PNGase F) treatment. We have already established that a series of enzymatic transformations can be performed on SGP and that acetylation of the lysine residues, which will occur during activation of a GlcNH2 residue (e.g. conversion of 18 into 19, Fig. 3), does not interfere with the activity of PGNase F (see Supplementary Figs. 11 and 12).
In addition to well-defined glycans, there is a need for glycan-defined glycoproteins for detailed structure-function relationship studies or for the development of therapeutics. Recent advances in host glyco-engineering and in vitro enzymatic remodeling makes it possible to prepare specific glycoforms of glycoproteins8. The methodologies described here cannot directly be employed for the remodeling of glycans covalently attached to proteins because the acetylation or azido transfer step would result in derivatization of lysine residues. However, the strategy can be combined with a powerful approach for glycoprotein synthesis in which a preassembled oligosaccharide is enzymatically transferred en bloc to a protein having a GlcNAc moiety8. The combined approaches will make it possible to prepare glycoproteins having complex branched glycan architectures.
The target compounds described here are prepared in quantities sufficient for glycan microarray development or to serve as analytical standards (0.1 – 0.5 mg). The technology has the potential to provide larger quantities of material that for example will be required for glycoprotein synthesis. The starting glycosyl asparagine 1 can routinely be prepared in multi-gram quantities and sugar nucleotide donors can conveniently and inexpensively be enzymatically prepared in-situ49. Furthermore, the recent generation of a library of expression constructs encoding all human glycosylation enzymes for production in mammalian or insect cells provides easy access to recombinant glycosyltransferases50. The glycosyltransferases employed in the present study include GlcNAc transferases (MGAT4 and MGAT5, and B3GNT2), Gal transferases (B4GALT1), Neu5Ac transferases (ST6GAL1 and ST3GAL4) and Fuc transferases (FUT5 and FUT8) were readily generated as soluble secreted catalytic domain forms in purified quantities ranging from 11 – 140 mg/L at high specific activities (see Supplementary Table 1). Enzymes with other donor and acceptor specificities are also available for further elaboration of asymmetric glycan branches through expression from this library source. In addition, many microbial glycosyltransferases have been described that can be employed for the preparation of specific epitopes of N-glycans51,52. An impediment for the large-scale synthesis of the complex glycans is the loss of material during HILIC-HPLC purification32. Most enzymatic reactions proceeded to completion as determined by LC-MS, and Bio-Gel® P2 size-exclusion column chromatography provided an attractive alternative for compound purification that did not lead to substantial loss of material. This technique is still time consuming and future efforts should focus on the development of tagging methods that will facilitate rapid target purification.
In conclusion, the “stop and go” strategy described here makes it possible to prepare complex, multi-antennary glycosyl asparagine targets without a need for highly-specialized synthetic skills. It is expected that this strategy will permit greater access to this class of biologically important compounds, which are urgently needed in many areas of Glycoscience.
Methods
General procedure for the installation of β1,6-GlcNTFA using MGAT5.
Glycosyl asparagine acceptor 1 (17.6 mg, 10.2 μmol) and UDP-GlcNTFA (13.5 mg, 20.4 μmol) were dissolved at a final acceptor concentration of 10 mM in a sodium cacodylate buffered solution (100 mM, pH 6.5) containing MnCl2 (10 mM) and BSA (1% total volume, stock solution = 10 mg mL−1). Calf intestine alkaline phosphatase (CIAP, 1% total volume, stock solution = 1kU mL−1) and MGAT5 (40 μg/μmol acceptor) were added, and the reaction mixture was incubated overnight at 37 °C with gentle shaking. Reaction progress was monitored by MALDI-TOF MS and if starting material remained after 18 h another portion of MGAT5 was added until no starting material could be detected. The reaction mixture was centrifuged over a Nanosep® Omega ultrafiltration device (10 kDa MWCO) to remove reaction proteins and the filtrate was lyophilized. Purification by HPLC using a HILIC column (Supplementary Section 2f) provided desired product 14 as a white fluffy solid (18.6 mg, 92%).
General procedure for the installation of β1,4-GlcNTFA using MGAT4B.
Glycosyl asparagine- acceptor 2 (4.0 mg, 2.1 μmol) and UDP-GlcNTFA (2.75 mg, 4.2 μmol) were dissolved at a final acceptor concentration of 5 mM in a Tris buffered solution (100 mM, pH 7.5) containing MnCl2 (5 mM) and BSA (1% total volume). CIAP (1% total volume) and MGAT4B (400 μg/μmol acceptor) were added, and the reaction mixture was incubated overnight at 37 °C with gentle shaking. Reaction progress was monitored by ESI-TOF MS, and if starting material remained after 18 h another portion of MGAT4B was added until no starting material could be detected. The reaction mixture was centrifuged over a Nanosep® Omega ultrafiltration device (10 kDa MWCO) to remove reaction proteins, and the filtrate was lyophilized. Purification by HPLC using a HILIC column (Supplementary Section 2f) provided the desired product S6 as a white fluffy solid (3.8 mg, 85%).
General procedure for removal of TFA protecting group of an N-glycan.
The GlcNTFA moiety of S6 was converted to GlcNH2 by dissolving the substrate (3.8 mg, 1.8 μM) in H2O to a final concentration of 10 mM. The pH of the solution was adjusted to 10 using μL aliquots 1 M NaOH. The reaction mixture was incubated overnight at 37 °C with gentle shaking. Progress of the reaction was monitored by MALDI-TOF MS and once complete the solvent was removed by lyophilization. The reaction was neutralized by μL aliquots of 1 M acetic acid and purified by P2 size-exclusion chromatography eluting with 50 mM ammonium bicarbonate to yield the desired target 2 as a white fluffy solid (3.5 mg, 92%).
General procedure for the conversion of GlcNH2 to GlcN3.
Substrate 15 (9.3 mg, 5 μmol, 1 eq) was dissolved in water (1.6 mL) and to this solution was added imidazole-1-sulfonyl azide hydrogen sulfate (13.4 mg, 50 μmol), K2CO3 (6.8 mg, 50 μmol) and catalytic CuSO4.5H2O. The reaction mixture was incubated overnight at 37 °C with gentle shaking. Reaction progress was monitored by MALDI-TOF MS and if starting material remained, an additional 1/2 portion of the imidazole-1-sulfonyl azide hydrogen sulfate, K2CO3, and CuSO4 was added until no starting material could be observed. The reaction solvent was removed by lyophilization and the salts were removed by P2 size-exclusion chromatography eluting with 50 mM ammonium bicarbonate to yield 23 as a white fluffy solid (7.2 mg, 76%).
General procedure for reduction of GlcN3.
Intermediate 27 (2.3 mg, 0.66 μmol, 1 eq) was dissolved in a solution of 9:1 pyridine / triethylamine to give a final concentration of 5 mM. The mixture was vortexed until all solids dissolved and 10 eq. 1,3-dithiolpropane (0.7 mg, 6.6 μmol, 10 eq) were added in one portion. The reaction mixture was kept at 37 °C was until no azide could be detected by ESI-TOF-MS. Reaction was carried forward to acetylate the amine without further purification.
General procedure for amine acetylation.
18 (1.3 mg, 0.5 μmol, 1 eq) was dissolved in water to a final concentration of 2 mM. The pH was adjusted to 8 using μL aliquots of 1M NaOH. To this solution was added solid AcOSu (0.7 mg, 5 μmol, 10 eq) in one portion. The reaction mixture was vortexed vigorously until all solids were dissolved. The reaction was kept at 37 °C until full acetylation was observed by ESI-TOF-MS. In the event starting amine was detected, additional AcOSu (5 eq) was added until complete conversion was observed. The reaction was lyophilized and purified by HPLC using a HILIC column (Supplementary Section 2f) to afford 19 as a white fluffy solid (0.9 mg, 67%).
Data availability
All data related with this study are included in this article and the Supplementary Information, and also available from the authors upon request.
Supplementary Material
Acknowledgments
This research was supported by the National Institute of General Medical Sciences (P01GM107012, P41GM103390, and U01GM120408 to G.-J.B. and KM) and the National Cancer Institute (F31CA180478 to A.R.P.) from the US National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The research benefitted from instrumentation provided by an NIH grant, S10 RR027097.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information (Materials and Methods, Supplementary Figures 1–13, Supplementary Table 1 and copies of NMR spectra) is available.
Correspondence and requests for materials should be addressed to G.J.B.
References
- 1.Moremen KW, Tiemeyer M & Nairn AV Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol 13, 448–462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohtsubo K & Marth JD Glycosylation in cellular mechanisms of health and disease. Cell 126, 855–867 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Lauc G, Pezer M, Rudan I & Campbell H Mechanisms of disease: The human N-glycome. Biochim. Biophys. Acta 1860, 1574–1582 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Hart GW & Copeland RJ Glycomics hits the big time. Cell 143, 672–676 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiessling LL & Splain RA Chemical approaches to glycobiology. Annu. Rev. Biochem 79, 619–653 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Cummings RD & Pierce JM The challenge and promise of glycomics. Chem. Biol 21, 1–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pilobello KT & Mahal LK Deciphering the glycocode: the complexity and analytical challenge of glycomics. Curr. Opin. Chem. Biol 11, 300–305 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Wang LX & Lomino JV Emerging technologies for making glycan-defined glycoproteins. ACS Chem. Biol. 7, 110–122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofmann J & Pagel K Glycan analysis by ion mobility-mass spectrometry. Angew. Chem. Int. Ed 56, 8342–8349 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Hyun JY, Pai J & Shin I The glycan microarray story from construction to applications. Acc. Chem. Res 50, 1069–1078 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Wang Z et al. A general strategy for the chemoenzymatic synthesis of asymmetrically branched N-glycans. Science 341, 379–383 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li L et al. Efficient chemoenzymatic synthesis of an N-glycan isomer library. Chem. Sci 6, 5652–5661 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li T et al. Divergent chemoenzymatic synthesis of asymmetrical-core-fucosylated and core-unmodified N-glycans. Chem. Eur. J 22, 18742–18746 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shivatare SS et al. Modular synthesis of N-glycans and arrays for the hetero-ligand binding analysis of HIV antibodies. Nat. Chem 8, 338–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gagarinov IA et al. Chemoenzymatic approach for the preparation of asymmetric bi-, tri-, and tetra-antennary N-glycans from a common precursor. J. Am. Chem. Soc 139, 1011–1018 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schachter H & Freeze HH Glycosylation diseases: Quo vadis? Biochim. Biophys. Acta 1792, 925–930 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kizuka Y & Taniguchi N Enzymes for N-glycan branching and their genetic and nongenetic regulation in cancer. Biomolecules 6, pii: E25 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cummings RD The repertoire of glycan determinants in the human glycome. Mol. BioSyst 5, 1087–1104 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Seko A et al. Occurence of a sialylglycopeptide and free sialylglycans in hen’s egg yolk. Biochim. Biophys. Acta 1335, 23–32 (1997). [DOI] [PubMed] [Google Scholar]
- 20.Liu L, Prudden AR, Bosman GP & Boons GJ Improved isolation and characterization procedure of sialylglycopeptide from egg yolk powder. Carbohydr. Res 452, 122–128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Umekawa M et al. Efficient transfer of sialo-oligosaccharide onto proteins by combined use of a glycosynthase-like mutant of Mucor hiemalis endoglycosidase and synthetic sialo-complex-type sugar oxazoline. Biochim. Biophys. Acta 1800, 1203–1209 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Huang W, Giddens J, Fan SQ, Toonstra C & Wang LX Chemoenzymatic glycoengineering of intact IgG antibodies for gain of functions. J. Am. Chem. Soc 134, 12308–12318 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maki Y, Okamoto R, Izumi M, Murase T & Kajihara Y Semisynthesis of intact complex-type triantennary oligosaccharides from a biantennary oligosaccharide isolated from a natural source by selective chemical and enzymatic glycosylation. J. Am. Chem. Soc 138, 3461–3468 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Alexander SR, Lim D, Amso Z, Brimble MA & Fairbanks AJ Protecting group free synthesis of glycosyl thiols from reducing sugars in water; application to the production of N-glycan glycoconjugates. Org. Biomol. Chem 15, 2152–2156 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Peng W et al. Recent H3N2 viruses have evolved specificity for extended, branched human-type receptors, conferring potential for increased avidity. Cell Host Microbe 21, 23–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paschinger K, Staudacher E, Stemmer U, Fabini G & Wilson IB Fucosyltransferase substrate specificity and the order of fucosylation in invertebrates. Glycobiology 15, 463–474 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Voynow JA, Kaiser RS, Scanlin TF & Glick MC Purification and characterization of GDP-L-fucose-N-acetyl beta-D-glucosaminide alpha 1-->6fucosyltransferase from cultured human skin fibroblasts. Requirement of a specific biantennary oligosaccharide as substrate. J. Biol. Chem 266, 21572–21577 (1991). [PubMed] [Google Scholar]
- 28.Meng L et al. Enzymatic basis for N-glycan sialylation: structure of rat alpha2,6-sialyltransferase (ST6GAL1) reveals conserved and unique features for glycan sialylation. J. Biol. Chem. 288, 34680–34698 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van den Eijnden DH, Blanken WM & van Vliet A Branch specificity of beta-D-galactosidase from Eschericha coli. Carbohydr. Res 151, 329–335 (1986). [DOI] [PubMed] [Google Scholar]
- 30.Choo M et al. Characterization of H type 1 and type 1 N-acetyllactosamine glycan epitopes on ovarian cancer specifically recognized by the anti-glycan monoclonal antibody mAb-A4. J. Biol. Chem 292, 6163–6176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zauner G, Deelder AM & Wuhrer M Recent advances in hydrophilic interaction liquid chromatography (HILIC) for structural glycomics. Electrophoresis 32, 3456–3466 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Lauber MA, Koza SM & Fountain KJ Optimization of GlycoWorks HILIC SPE for the quantitative and robust recovery of N-linked glycans from mAb-type samples. Waters Application Note, 720004717EN (2013). [Google Scholar]
- 33.Xu Y et al. Chemoenzymatic synthesis of homogeneous ultralow molecular weight heparins. Science 334, 498–501 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antonopoulos A et al. Loss of effector function of human cytolytic T lymphocytes is accompanied by major alterations in N- and O-glycosylation. J. Biol. Chem 287, 11240–11251 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goddard-Borger ED & Stick RV An efficient, inexpensive, and shelf-stable diazotransfer reagent: imidazole-1-sulfonyl azide hydrochloride. Org. Lett 9, 3797–3800 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Bayley H, Standring DN & Knowles JR Propane-1,3-dithiol - selective reagent for efficient reduction of alkyl and aryl azides to amines. Tetrahedron Lett., 3633–3634 (1978). [Google Scholar]
- 37.Aoki D, Appert HE, Johnson D, Wong SS & Fukuda MN Analysis of the substrate binding sites of human galactosyltransferase by protein engineering. EMBO J. 9, 3171–3178 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Unverzagt C Chemoenzymatic synthesis of a sialylated undecasaccharide-asparagine conjugate. Angew. Chem. Int. Ed. 35, 2350–2353 (1996). [Google Scholar]
- 39.Hanashima S, Manabe S & Ito Y Divergent synthesis of sialylated glycan chains: combined use of polymer support, resin capture-release, and chemoenzymatic strategies. Angew. Chem. Int. Ed 44, 4218–4224 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Jonke S, Liu KG & Schmidt RR Solid-phase oligosaccharide synthesis of a small library of N-glycans. Chem. Eur. J 12, 1274–1290 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Sun B, Srinivasan B & Huang XF Pre-activation-based one-pot synthesis of an alpha-(2,3)-sialylated core-fucosylated complex type bi-antennary N-glycan dodecasaccharide. Chem. Eur. J 14, 7072–7081 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Unverzagt C et al. Synthesis of multiantennary complex type N-glycans by use of modular building blocks. Chem. Eur. J 15, 12292–12302 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Serna S, Etxebarria J, Ruiz N, Martin-Lomas M & Reichardt NC Construction of N-glycan microarrays by using modular synthesis and on-chip nanoscale enzymatic glycosylation. Chem. Eur. J 16, 13163–13175 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Walczak MA & Danishefsky SJ Solving the convergence problem in the synthesis of triantennary N-glycan relevant to prostate-specific membrane antigen (PSMA). J. Am. Chem. Soc 134, 16430–16433 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamilton BS et al. A library of chemically defined human N-glycans synthesized from microbial oligosaccharide precursors. Sci. Rep 7, 15907 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calderon AD et al. An enzymatic strategy to asymmetrically branched N-glycans. Org. Biomol. Chem 15, 7258–7262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maki Y, Mima T, Okamoto R, Izumi M & Kajihara Y Semisynthesis of complex-type biantennary oligosaccharides containing lactosamine repeating units from a biantennary oligosaccharide isolated from a natural source. J. Org. Chem 83, 443–451 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Prudden AR et al. Synthesis of asymmetrical multiantennary human milk oligosaccharides. Proc. Natl. Acad. Sci. U. S. A 114, 6954–6959 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai L Recent progress in enzymatic synthesis of sugar nucleotides. J. Carbohydr. Chem 31, 535–552 (2012). [Google Scholar]
- 50.Moremen KW et al. Expression system for structural and functional studies of human glycosylation enzymes. Nat. Chem. Biol 14, 156–162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muthana S, Cao H & Chen X Recent progress in chemical and chemoenzymatic synthesis of carbohydrates. Curr. Opin. Chem. Biol 13, 573–581 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmaltz RM, Hanson SR & Wong CH Enzymes in the synthesis of glycoconjugates. Chem. Rev 111, 4259–4307 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data related with this study are included in this article and the Supplementary Information, and also available from the authors upon request.