

Abstract
Sphingosine-1-phosphate lyase (SPL) is an intracellular enzyme that controls the final step in the sphingolipid degradative pathway, the only biochemical pathway for removal of sphingolipids. Specifically, SPL catalyzes the cleavage of sphingosine 1-phosphate (S1P) at the C2–3 carbon bond, resulting in its irreversible degradation to phosphoethanolamine (PE) and hexadecenal. The substrate of the reaction, S1P, is a bioactive sphingolipid metabolite that signals through a family of five G protein-coupled S1P receptors (S1PRs) to mediate biological activities including cell migration, cell survival/death/proliferation and cell extrusion, thereby contributing to development, physiological functions and — when improperly regulated — the pathophysiology of disease. In 2017, several groups including ours reported a novel childhood syndrome that featured a wide range of presentations including fetal hydrops, steroid-resistant nephrotic syndrome (SRNS), primary adrenal insufficiency (PAI), rapid or insidious neurological deterioration, immunodeficiency, acanthosis and endocrine abnormalities. In all cases, the disease was attributed to recessive mutations in the human SGPL1 gene. We now refer to this condition as SPL Insufficiency Syndrome, or SPLIS. Some features of this new sphingolipidosis were predicted by the reported phenotypes of Sgpl1 homozygous null mice that serve as vertebrate SPLIS disease models. However, other SPLIS features reveal previously unrecognized roles for SPL in human physiology. In this review, we briefly summarize the biochemistry, functions and regulation of SPL, the main clinical and biochemical features of SPLIS and what is known about the pathophysiology of this condition from murine and cell models. Lastly, we consider potential therapeutic strategies for the treatment of SPLIS patients.
Keywords: SGPL1, sphingosine phosphate lyase, sphingolipid, nephrotic syndrome, focal segmental glomerulosclerosis, NPHS14, sphingosine-1-phosphate, neuropathy, lymphopenia, Charcot Marie Tooth disease, immunodeficiency
Introduction
Sphingolipids represent a highly conserved family of membrane lipids that share a sphingoid (also known as a long chain) base backbone as their common structural feature (Figure 1). Complex sphingolipids including sphingomyelin and the glycosphingolipids contribute to cell identity and to membrane organization and function (Hannun and Obeid, 2008; Merrill et al., 1997). Sphingolipids are degraded to bioactive intermediates that can be recycled back into complex sphingolipids or can participate in signal transduction pathways that regulate cell survival, migration, programmed cell death and intracellular functions (Fyrst and Saba, 2010). A highly conserved set of enzymes facilitates the degradation and recycling of sphingolipids, deficiencies of which result in storage disorders called “sphingolipidoses” marked by the accumulation of different classes of sphingolipids (Arenz, 2017; Grassi et al., 2018).
Figure 1. Basic structures of sphingolipids.
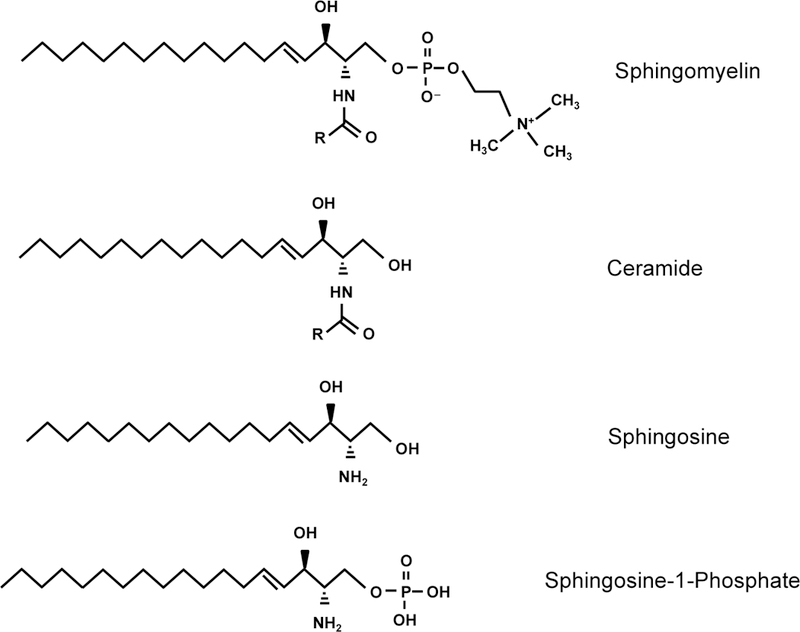
Sphingolipids contain a long chain amino base, also known as a sphingoid base. Sphingosine is the most common sphingoid base in mammals. Ceramides contain sphingosine in amide linkage with a fatty acid of variable chain length and saturation. Sphingosine can be phosphorylated, giving rise to the bioactive signaling molecule, sphingosine-1-phosphate (S1P). Sphingomyelin is formed when a phosphocholine head group is added to the hydroxyl group at the C1 position of ceramide.
Sphingosine-1-phosphate lyase (SPL) is an intracellular enzyme that controls the ultimate step in the sphingolipid degradative pathway, the only biochemical pathway for removal of sphingolipids. Specifically, SPL catalyzes the cleavage of sphingosine 1-phosphate (S1P) at the C2–3 carbon bond, resulting in its irreversible degradation to phosphoethanolamine (PE) and hexadecenal. It also catalyzes the degradation of other sphingoid base phosphates (i.e., phytosphingosine-1-phosphate, dihydrosphingosine-1-phosphate, etc.) to PE and the corresponding long chain aldehyde. The main substrate of the reaction in mammals, S1P, is a bioactive sphingolipid metabolite that signals through a family of G protein-coupled receptors, the S1PRs, to mediate diverse biological activities. These include cell migration, cell survival, cell death, cell proliferation, cell extrusion, neuronal and cardiac development, angiogenesis, inflammation, immune cell trafficking, gene regulation and tumorigenesis (Fyrst and Saba, 2010; Garris et al., 2014; Gudipaty and Rosenblatt, 2017; Kono et al., 2008; Takuwa et al., 2010). S1P levels are high in blood and lymph but are maintained at low concentration in tissues by SPL. The resulting S1P chemotactic gradient facilitates lymphocyte egress from thymus and lymphoid organs and likely contributes to other biological effects mediated by S1P. In the blood, S1P is mostly bound to protein carriers including albumin and ApoM-containing high density lipoprotein (HDL), creating distinct signaling pools of S1P (Blaho et al., 2015).
The first SPL-encoding gene, “bestower of sphingosine tolerance” (BST1)/dihydros phingosine phosphate lyase (DPL1), was cloned from Saccharomyces cerevisiae in 1997 (Saba et al., 1997). Soon after, orthologous genes from invertebrate genetic models, plants and mammals were identified and functionally confirmed by complementation in yeast including Sgpl1/SGPL1, the mouse and human SPL-encoding genes of the same name (Herr et al., 2003; Mendel et al., 2003; Van Veldhoven et al., 2000; Zhou and Saba, 1998). Based on the lack of additional homologous DPL1-like genes within each species and the significant sphingolipid accumulation and severe phenotypes observed in corresponding mutant organisms, it was concluded that each species harbors a single and essential SPL-encoding gene. In 2007, a mouse model was generated in which Sgpl1 was disrupted by gene trapping (Schmahl et al., 2007). Murine SPL loss of function affected hematopoietic cells, kidneys, bone, cartilage and uniformly resulted in severe runting and premature death around the time of weaning. Subsequently, Sgpl1 gene disruption in inducible and conditional mouse models have revealed important roles for SPL in kidney, platelet, gut, immune and skin functions (Degagné et al., 2014; Schumann et al., 2015; Vogel et al., 2009; Zamora-Pineda et al., 2016).
In 2017–2018, a series of reports identified a novel childhood syndrome caused by recessive mutations in the human SGPL1 gene that encodes SPL (Atkinson et al., 2017; Bamborschke et al., 2018; Janecke et al., 2017; Linhares et al., 2017; Lovric et al., 2016; Prasad et al., 2017). We now refer to this condition as SPL Insufficiency Syndrome, or SPLIS. One of the signal features of SPLIS is steroid-resistant nephrotic syndrome (SRNS). In SRNS, the kidney glomerulus fails to perform the critical function of filtering the blood, allowing small molecular weight proteins to be excreted into the urine, while retaining important blood proteins such as albumin, clotting factors and immunoglobulins (Nourbakhsh and Mak, 2017). The histological feature most closely associated with SRNS is called focal segmental glomerulosclerosis (FSGS), and the two terms are often used interchangeably. Glomerular cells called podocytes are required for the kidney’s filtration function, and their morphological alteration, damage and loss has been implicated as central to the pathophysiology of SRNS/FSGS (Fogo, 2015; Peev et al., 2017). This disease frequently progresses to kidney failure, requiring dialysis or kidney transplantation. Another feature of SPLIS is neuropathy, including central and peripheral neurological systems. Patients have exhibited microcephaly, seizures, cranial nerve defects, developmental anomalies of the brain, spasticity and a variety of sensory/motor neuropathies that are collectively referred to as “Charcot Marie Tooth Disease” (CMT) (Pareyson et al., 2017). Some patients also exhibited primary adrenal insufficiency (PAI), a life-threatening condition that can present with failure to thrive, hypersensitivity to illness, hypoglycemia, hypotension, vomiting and hyponatremia due to the inability of the adrenal cortex to produce cortisol and aldosterone. Other features of SPLIS included acanthosis/ichthyosis (thickening and discoloration of the skin), lymphopenia and other immune defects, other endocrine and gonadal defects, and repeated gastrointestinal illnesses and infections.
In this review, we briefly summarize SPL’s function and expression pattern, then describe the main clinical and biochemical features of SPLIS, disease models of SPLIS, and new insights regarding the role of sphingolipids in human biology that are revealed by the SPLIS phenotypes. Lastly, we consider potential therapeutic strategies for the treatment of SPLIS patients.
Biochemistry of SPL
Sphingolipid metabolic pathway:
The de novo biosynthesis of sphingolipids is initiated by a condensation reaction between serine and palmitoyl-CoA resulting in the formation of 3-keto-dihydrosphingosine, a rate-limiting step catalyzed by the endoplasmic reticulum (ER) resident enzyme serine palmitoyltransferase (SPT) (Figure 2) (Bourquin et al., 2011). Mutations in one of the SPT subunits are responsible for the peripheral neuropathy called hereditary sensory neuropathy type 1. A reduction step converts the keto intermediate into dihydrosphingosine which is then acylated at the free amino group of the sphingoid base to form dihydroceramide. A desaturase then converts dihydroceramide to ceramide, a central structure of sphingolipid metabolism (Kitatani et al., 2008). Ceramide is a bioactive molecule recognized for its roles in promoting programmed cell death and contributing to insulin resistance (Stancevic and Kolesnick, 2010). Ceramide can alternatively be formed by the hydrolysis of sphingomyelin by three pH-dependent sphingomyelinases. Acid sphingomyelinase deficiency is the cause of Niemann-Pick disease types A and B. Ceramide is the anchor upon which higher order sphingolipids are formed via addition of complex carbohydrates (to form glycosphingolipids) or phosphocholine (to form sphingomyelin) at the C1 position. Ceramide can also be phosphorylated by ceramide kinase to produce the signaling molecule ceramide-1-phosphate (C1P), an inflammatory mediator that activates phospholipase A2α to generate arachidonate (Lamour and Chalfant, 2008). De-acylation of ceramide by pH-dependent ceramidases produces the sphingoid base sphingosine, an inhibitor of protein kinase C. Although all ceramidases catalyze the same biochemical step, each has a distinct expression pattern and function, with diverse effects on dietary sphingolipid metabolism, inflammation, carcinogenesis, cell proliferation and responses to DNA damage and other stress conditions (Coant et al., 2017). In the penultimate step of sphingolipid metabolism, S1P is produced when sphingosine is phosphorylated by the enzyme sphingosine kinase (Pyne et al., 2009). S1P can then undergo one of two fates: 1) reversible dephosphorylation by S1P phosphatases or non-specific lipid phosphatases (LPPs); 2) irreversible degradation by SPL into a hydrophobic aldehyde (hexadecenal) and the hydrophilic compound PE (Kumar and Saba, 2009). Both products of the SPL reaction can feed into phospholipid biosynthesis and have their own biological activities. For example, PE deficiency causes a block in autophagic flux in the neurons of Sgp11 null pups, as described further below (Mitroi et al., 2017). Hexadecenal is a highly reactive aldehyde that can bind to DNA or form protein adducts (Upadhyaya et al., 2012; Schumacher et al., 2017). It has been shown to bind directly to BAX, promoting its oligomerization, and sensitizing cells to mitochondrial membrane permeability and apoptosis (Chipuk et al., 2012). It has also been shown to modulate histone H3/H4 acetylation, thereby impacting inflammatory gene expression (Ebenezer et al., 2017). Further, the neurocutaneous disorder Sjogren-Larsson syndrome — which is associated with congenital ichthyosis, mental retardation, and spasticity — is caused by a genetic defect that results in failure to catabolize SPL-derived hexadecenal (Nakahara et al., 2012). Importantly, the degradation of sphingolipids from all sources ultimately requires passage through the metabolic step catalyzed by SPL, which is the only exit point of the sphingolipid metabolic pathway. When SPL is not present or operational, not only S1P but other upstream sphingolipid intermediates can accumulate and lead to cellular and organ dysfunction.
Figure 2. Sphingolipid metabolic pathway.
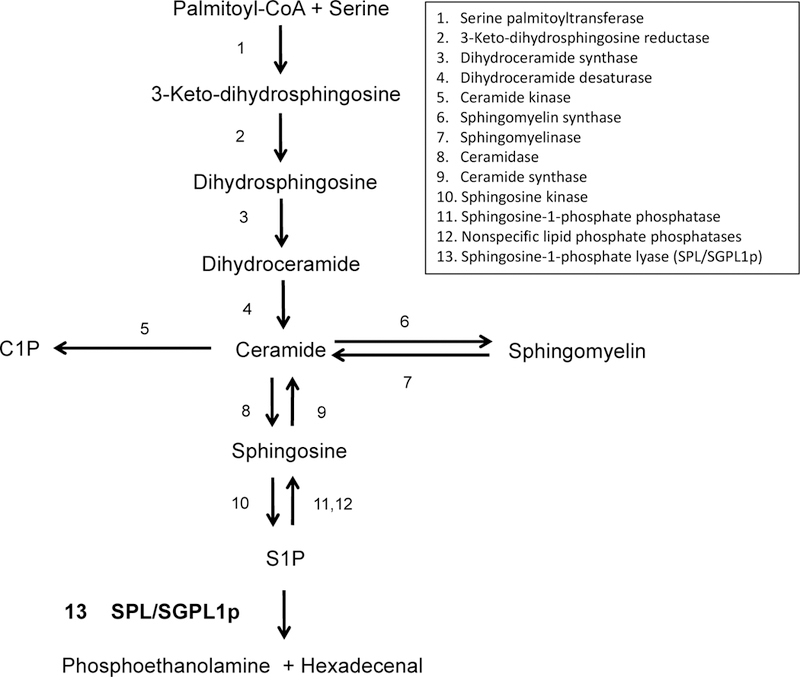
Enzymes catalyzing the conversion steps are listed numerically. Of note, serine palmitoyltransferase (SPT) catalyzes the condensation reaction between serine and palmitoyl-CoA, the rate limiting induction step in sphingolipid biosynthesis. Sphingosine phosphate lyase (SPL) catalyzes the irreversible cleavage of sphingosine-1-phosphate (S1P), the final and essential step in sphingolipid catabolism.
S1PR-dependent and S1PR-independent signaling:
The effects that S1P exerts on cell recruitment, cell fate and cell-cell interactions contribute to a wide variety of developmental and physiological processes including but not limited to brain, inner ear and heart development, angiogenesis, vascular maturation and permeability, blood pressure regulation, allergic responses, mucosal barrier integrity, bone homeostasis, neuronal functions, immunity and lymphocyte trafficking. S1P signaling is mediated through interactions with a family of five heterotrimeric guanine nucleotide-binding protein (G protein)-coupled receptors, designated S1P1–5. Through these cell surface receptors, S1P can act in autocrine or paracrine fashion to stimulate key intracellular signaling pathways that control actin cytoskeletal organization, gene expression, and proliferation. A recent study demonstrated an additional, unique mechanism of exosome-based S1PR release from cancer cells that mediates S1P’s effects on the tumor niche (Pyne et al., 2018). The regulation and biology of S1P receptors is beyond the scope of this review. Interested readers are referred to several excellent recent reviews on this subject (Allende and Proia, 2002; Bryan and Del Poeta, 2018; Garris et al., 2014; Rosen et al., 2013). There is evidence that S1P also acts independently of S1P receptors. Tumor-necrosis factor (TNF) receptor-associated factor 2 (TRAF2) is a key component in NFκB signaling triggered by TNF-α. S1P specifically binds to the TRAF2 E3 ubiquitin ligase and induces RIP1 polyubiquitination, phosphorylation of IκB kinase, leading to activation of transcription factor NFκB (Alvarez et al., 2010). S1P also specifically binds to histone deacetylases and inhibits their activities, linking nuclear S1P to epigenetic regulation of gene expression (Hait et al., 2009).
SPL, a pyridoxal 5′- phosphate (PLP) dependent enzyme:
SPL is located in the ER membrane and is an integral membrane protein with one transmembrane domain at the N-terminus. The N-terminus faces into the ER lumen, whereas the large hydrophilic domain containing active sites is located in the cytosol (Ikeda et al., 2004). SPL requires PLP (vitamin B6) as a cofactor and is sensitive to heavy metal ions and detergents (Van Veldhoven and Mannaerts, 1993). A lysine residue (Lys 353 in human SPL) located in the enzyme’s active site forms an internal Schiff base with the aldehyde group of PLP (Figure 3). Yeast and bacterial SPL proteins have been crystallized (Bourquin et al., 2010). Two subunits were shown to form a tightly interlocking dimer in which both chains contribute to the formation of the catalytic cavity defined by the covalently bound PLP. For more details on the structure/function relationships of SPL and similarity to other PLP-dependent class of enzymes, please refer to an excellent recent review on the subject (Bourquin et al., 2011).
Figure 3. Sites of all identified SGPL1 mutations in exon and protein structures.
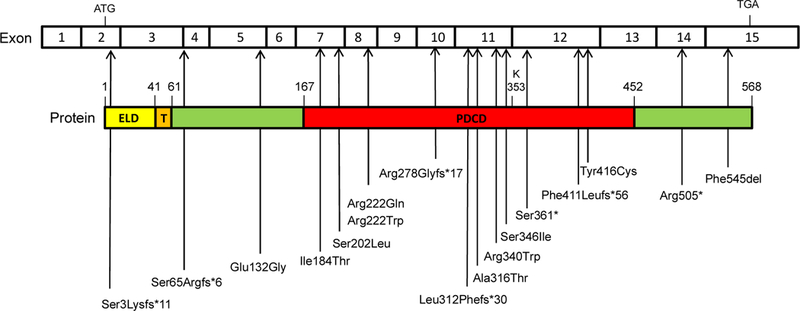
SGPL1 cDNA contains 15 exons, and the coding sequence is 568 amino acids long. Pyridoxal-dependent decarboxylase conserved domain (PDCD) contains active site residue K353 whichbinds to pyridoxal 5′-phosphate (PLP). ELD, Endoplasmic lumenal domain; T, Transmembrane domain; Cytoplasmic domain in green.
SPL tissue expression
The expression pattern of SPL was examined in adult and embryonic murine tissues. Analysis of β-galactosidase activity and immunohistochemistry of adult murine tissues in an SPL LacZ reporter mouse showed that SPL expression is highest in small intestine and thymus, intermediate in stomach and low in liver, colon, brain, lung, kidney, and heart (Borowsky et al., 2012). High levels of SPL expression in intestinal and colonic epithelia are consistent with its established role in the metabolism of dietary sphingolipids (Duan and Nilsson, 2000). However, more recent studies implicate SPL in the regulation of host-pathogen interactions and gut mucosal biology, extending its likely functions in the gut well beyond the handling of dietary lipids (Degagné et al., 2014; Liang et al., 2013).Further, SPL may regulate the role S1P plays in cell extrusion, an important process by which apoptotic cells are removed without causing a breach of the epithelial barrier (Gudipaty and Rosenblatt, 2017). Although thymus and jejunum showed by far the highest levels of SPL expression, β-galactosidase staining was observed in many SPL reporter mouse organs, including strong signal in various compartments of the central nervous system (CNS). Discrete expression was observed in arachnoid lining cells, spinal cord, choroid plexus, trigeminal nerve ganglion, and specific neurons of the olfactory bulb, cerebral cortex, midbrain, hindbrain and cerebellum. SPL was also found to be expressed in pituitary gland, brown fat, adrenal gland, kidney, bladder, ovaries, stomach, eyes and skin, revealing possible roles for SPL in the function of each of these organs.
During murine development, SPL was expressed by E15 in the developing brain and neural tube and in other developmental structures including Rathke’s pouch (which gives rise to the anterior pituitary gland), first and third pharyngial arches (which give rise to bones of the lower face and jaw as well as a muscle of the pharynx and larynx), optic stalk (which gives rise to the optic nerves), midgut loops, and lung buds (Newbigging et al., 2013). SPL’s embryonic expression pattern augured a possible role in the development of the mammalian CNS and other organs. SPL expression was observed in cells of the innate and adaptive immune system including splenocytes, circulating T and B lymphocytes, granulocytes, and monocytes, whereas thymocytes had notably low SPL expression (Borowsky et al., 2012). In addition to the high expression in thymic stroma, SPL was expressed in other lymphoid tissues including Peyer’s Patches of the jejunum, lymph nodes and colonic lymphoid aggregates. These findings are consistent with the important role of SPL in controlling neutrophil migration and chemotactic gradients involved in lymphocyte egress from thymus and peripheral lymphoid organs (Allende et al., 2011; Schwab et al., 2005; Zamora-Pineda et al., 2016).
SPL Insufficiency Syndrome (SPLIS)
In 2017, six different research groups reported patients and kindred harboring autosomal recessive mutations in SGPL1 and exhibiting a variety of presentations including congenital NS or steroid-resistant nephrotic syndrome (SRNS), PAI and central and peripheral neurological defects (Atkinson et al., 2017; Bamborschke et al., 2018; Janecke et al., 2017; Linhares et al., 2017; Lovric et al., 2016; Prasad et al., 2017). Additional features included ichthyosis/acanthosis, immunodeficiency, gonadal dysgenesis and other endocrine defects. The patients were all found to harbor inactivating mutations in SGPL1. In each case, SGPL1 disease-associated mutations were present on both alleles of the index case and all affected individuals within the kindred, but were not found to affect both alleles in unaffected family members or the general population. Further, patients with the disease did not harbor any other mutations known to cause the main disease feature. Therefore, it was concluded that the SGPL1 mutations were responsible for the disease. SGPL1 mutations determined by whole exome sequencing (WES) and Sanger sequencing are summarized in Table 1 and Figure 3.
Table 1:
SPLIS cases reported to date *
| Case/Family | Mutation | Phenotype | Reference |
|---|---|---|---|
| 1 | c.7dup; p.Ser3Lysfs*11 | ichthiosis, steroid resistant nephrotic syndrome (SRNS) | Lovric et al., 2017. |
| 2 | c.7dup; p.Ser3Lysfs*11 | cranial nerve, gait problems, SRNS, adrenal insufficiency (AI), immune deficiency, bony defects |
Lovric et al., 2017 |
| 3 | c.7dup; p.Ser3Lysfs*11 | SRNS, ichthyosis, AI, macrosomia, bony abnormalities, scoliosis | Lovric et al., 2017 |
| 4 | c.7dup; p.Ser3Lysfs*11 | SRNS, AI | Prasad et al., 2017 |
| 5 | c.261+1G>A; p.Ser65Argfs*6 | SRNS, AI, neurological symptoms, immune deficiency, gonadal defects, endocrine defects |
Prasad et al., 2017 |
| 6 | c.261+1G>A; p.Ser65Argfs*6 | developmental delay, SRNS, AI, cataracts | Prasad et al., 2017 |
| 7 | c.395A>G; p.Glu132Gly c.832delA;p.Arg278Glyfs*17 |
SRNS, ichthyosis, AI | Lovric et al., 2017 |
| 8 | c.395A>G; p.Glu132Gly c.832delA;p.Arg278Glyfs*17 |
SRNS, ichthyosis, AI | Lovric et al., 2017 |
| 9 | c.551T>C; p.Ile184Thr c.1082C>G; p.Ser361* |
peripheral neuropathy | Atkinson et al., 2017 |
| 10 | c.551T>C; p.Ile184Thr c.1082C>G; p.Ser361* |
peripheral neuropathy | Atkinson et al., 2017 |
| 11 | c.605C>T; p.Ser202Leu c.946G>A; p.Ala316Thr (Wash U) |
peripheral neuropathy, SRNS, immune deficiency, endocrine defects, amblyopia, strabismus |
Lovric et al., 2017 |
| 12 | c.664C>T; p.Arg222Trp | developmental defects of the CNS, AI, immune deficiency | Lovric et al., 2017 |
| 13 | c.664C>T; p.Arg222Trp | CNS, fetal hydrops (FH), died in infancy | Lovric et al., 2017 |
| 14 | c.664C>T; p.Arg222Trp | (Fetal loss) | Lovric et al., 2017 |
| 15 | c.664C>T; p.Arg222Trp | (Fetal loss) | Lovric et al., 2017 |
| 16 | c.665G>A; p.Arg222Gln | SRNS, AI | Lovric et al., 2017 |
| 17 | c.665G>A; p.Arg222Gln | cranial nerve and peripheral neurological defects, SRNS, AI, short stature |
Lovric et al., 2017 |
| 18 | c.665G>A; p.Arg222Gln | SRNS, AI | Lovric et al., 2017 |
| 19 | c.665G>A; p.Arg222Gln | SRNS, AI | Prasad et al., 2017 |
| 20 | c.665G>A; p.Arg222Gln | SRNS, AI | Prasad et al., 2017 |
| 21 | c.665G>A; p.Arg222Gln | SRNS, AI | Prasad et al., 2017 |
| 22 | c.665G>A; p.Arg222Gln | SRNS, AI, immune deficiency | Prasad et al., 2017 |
| 23 | c.934delC; p.Leu312Phefs*30 |
CNS, AI, gonadal defects | Janecke et al., 2017 |
| 24 | c.1018C>T; p.Arg340Trp |
AI, nephrotic syndrome (NS), dilated heart | Linhares et al., 2017 |
| 25 | c.1018C>T; p.Arg340Trp |
AI, hyperpigmentation, nephrotic syndrome, anemia |
Pezzuti et al., 2014 (this case diagnosed retrospectively after diagnosis of a sibling) |
| 26 | c.1037G>T; p.Ser346Ile | neurodevelopmental delay, cranial nerve/CNS, ichthyosis, adrenal calcifications, endocrine defects, malrotation, dysmorphic features, hypocalcemia, immune deficiency |
Lovric et al., 2017 |
| 27 | c.1037G>T; p.Ser346Ile | CNS, ichthyosis, immune deficiency, neurodevelopmental delay, deafness |
Lovric et al., 2017 |
| 28 | c.1037G>T; p.Ser346Ile | (Fetal loss with HF) | Lovric et al., 2017 |
| 29 | c.1037G>T; p.Ser346Ile | (Fetal loss with HF) | Lovric et al., 2017 |
| 30 | c.1037G>T; p.Ser346Ile | CNS defects, seizures, adrenal calcifications, endocrine defects, hypocalcemia, immune deficiency |
Lovric et al., 2017 |
| 31 | c.1233delC; p.Phe411Leufs*56 |
severe CNS developmental birth defects, adrenal seizures, adrenal calcificationscalcification, gonadal defects, immune deficiency, endocrine defects |
Bamborschke et al.,2018 |
| 32 | c.1247A>G; p.Tyr416Cys | CNS, AI, immune deficiency, endocrine defects, dilated seizures, adrenal calcificationscardiomyopathy |
Lovric et al., 2017 |
| 33 | c.1513C>T; p.Arg505* | CNS, adrenal calcification, gonadal defects | Janecke et al., 2017 |
| 34 | c.1513C>T; p.Arg505* | CNS, adrenal calcification, gonadal defects | Janecke et al., 2017 |
| 35 | c.1633_1635delTTC; p.F545del |
Delayed development, SRNS, AI, CNS, sensorineural seizures, adrenal calcificationsdeafness, immune deficiency |
Prasad et al., 2017 |
Note: we are aware of four additional cases of SPLIS. They will be described in a separate report.
The largest cohort was reported by Lovric and colleagues, in which 9 different mutations in SGPL1 were identified by WES among a large, worldwide set of patients with SRNS (Lovric et al., 2017) On renal biopsies, FSGS and diffuse mesangial sclerosis were observed. The index patients exhibited a syndromic form of SRNS that presented with additional clinical features. Extrarenal manifestations included ichthyosis, PAI, other endocrine or gonadal defects and neurological deficits including microcephaly, cranial nerve defects and peripheral neuropathy Approximately half of the patients with the condition were noted to have severe immunodeficiency manifesting as lymphopenia and multiple bacterial infections (often gastrointestinal maladies). One patient underwent a thorough immunological evaluation and was found to have markedly reduced CD4 and CD8 T lymphocytes and B lymphocytes. The patient’s lymphocytes exhibited deficient functional responses to stimulation. Some patients exhibited failure to thrive. Many families reported a history of prior fetal loss.
Patient-derived skin fibroblasts exhibited reduced or absent SPL activity and expression and migrated more slowly than control fibroblasts. Overexpression of SPLIS-associated mutant SGPL1 cDNAs in HEK293 cells resulted in essentially no increase in SPL activity above endogenous levels. Some of the mutations were found in the PLP-binding domain of SGPL1, and all were located in highly conserved regions. SGPL1 cDNAs harboring patient-associated mutations failed to complement the phenotypes of yeast and Drosophila SPL mutant organisms, whereas wild type human SPL abrogated these phenotypes, establishing the functional insufficiency of the mutant SPL proteins.
Mutant SPL proteins were not detectable by fluorescence microscopy in patient-derived fibroblasts. When overexpressed in HEK293 cells, they were detectable but appeared punctate, suggesting the possibility of protein aggregation. This phenotype raises the possibility that the mutant SPL proteins may undergo misfolding, a common mechanism of disease in many inborn errors of metabolism (see below). Patient-derived fibroblast conditioned medium showed accumulation of sphingolipids, as expected with a block in the sphingolipid degradative pathway. SPL was determined by immunofluorescence microscopy to be expressed in murine glomerular podocytes and mesangial cells, with little detection in endothelial cells. Sgpl1 null mice were shown to exhibit kidney podocyte foot process effacement and an elevated urine albumin/creatinine ratio (ACR), consistent with the presence of nephrotic syndrome (NS). Although no effect was detected in SPL-silenced podocytes, a migration defect was observed in SPL-silenced mesangial cells, and this phenotype was reversed by pretreatment with an S1P1/3 antagonist which implicates aberrant S1PR signaling as a potential mechanism of SRNS in SPLIS patients. Future studies will be required to further delineate the pathophysiology of renal disease in SPLIS. Altogether, Lovric et al established a causal role for inactivating SGPL1 mutations as the molecular etiology of a syndromic form of SRNS/FSGS. This study provided the first description of SPLIS and added SGPL1 mutation (originally named NPHS14) to a list of over 40 gene mutations now recognized to cause monogenic SRNS/FSGS (Fogo, 2015).
In a back-to-back report in the same journal, Prasad et al. identified 4 different mutations of SGPL1 in five families in which patients were diagnosed with PAI, SRNS, primary hypothyroidism, neurological symptoms and/or cryptorchidism (Prasad et al., 2017). In this report, the focus was primarily on the PAI. Most affected patients presented with glucocorticoid deficiency requiring hydrocortisone replacement. Others also had mineralocorticoid deficiency and adrenal androgen deficiency. Adrenal calcifications and bilateral enlarged adrenal glands were observed. Investigation of the adrenal glands of Sgpl1 null mice revealed imperfect adrenal gland zonation with a reduction in expression of steroidogenic enzymes and loss of vacuolization in the zona fasciculata (ZF). Fewer lipid droplets were contained in cells in ZF of Sgpl1 null mice than controls. In normal adult human adrenal gland, SPL was found to be expressed throughout the cortex with the highest expression in zona reticularis, the region responsible for adrenal androgen secretion.
Janecke et al. identified two homozygous truncating SGPL1 mutations present in two separate kindred with congenital NS associated with adrenal calcification, hypogonadism and vascular alterations (Janecke et al., 2017). Congenital NS features massive proteinuria, hypoalbuminemia, and generalized edema developing within the first 3 months of life. Renal autopsies showed either FSGS or global sclerosis, hypertrophic thickened vessel walls and perivascular sclerosis with foci of calcification within both interstitial and glomerular compartments. The patients also exhibited small penis and small or no palpable testes and low testosterone levels. Some of the patients showed elevations of serum cholesterol and triglycerides. Bilateral adrenal calcifications were observed on radiological exam. S1P and sphingosine levels were increased in patient’s blood and fibroblasts.
Linhares et al also identified one variant in SGPL1 in a 3-year-old girl diagnosed with NS and PAI (Linhares et al., 2017). Taken together, these reports confirm an important and previously unrecognized role of SPL in the adrenal gland associated with adrenal calcification and vascular abnormalities. The combination of the two findings suggests the possibility of calcification due to prior hemorrhage. However, since S1P signaling influences calcium homeostasis, it is also possible that a more discrete reason for calcification due to aberrant S1P signaling is responsible.
Atkinson et al. focused on the neurological features of SPLIS, further establishing SGPL1 mutations as a cause of the hereditary peripheral neuropathy syndrome known as CMT (Atkinson et al., 2017). CMT is caused by degeneration of peripheral motor and sensory neurons, resulting in distal muscle weakness and wasting, motor impairment, sensory loss, paresthesias and secondary skeletal deformities. Autosomal recessive forms account for <10% of the European CMT population. Some SPLIS patients with the CMT phenotype presented with macute/subacute onset and mononeuropathy and have remained alive in their third and fourth decades.
Bamborschke et al. identified a single patient with SGPL1 mutations associated with a congenital brain malformation and fetal hydrops (Bamborschke et al., 2018). The affected patient exhibited microcephaly, generalized cortical atrophy with simplified gyral pattern, hypoplastic temporal lobes, and cerebellar hypoplasia. Additional features included dysmorphic features, hypothyroidism, adrenal calcifications, congenital NS and immunodeficiency with lymphopenia and hypogammaglobulinemia. The patient died at 6 weeks of age. The patient was found to be homozygous for a frameshift mutation in SGPL1.
In the reported SPLIS patients, inactivation of SPL has resulted from recessive mutations that exert a range of molecular effects on the encoded mRNA or protein. They include mutations resulting in protein truncation, frameshifts, nonsense mediated mRNA decay, and splicing defects as well as missense mutations that would be predicted to influence protein localization, stability and/or catalytic activity. Interestingly, thus far no evidence for SPLIS mutations inhibiting protein dimerization has been shown. Not surprisingly, the variety of mutations and their consequences for SPL expression and activity have resulted in a wide range of severity in SPLIS presentations that could ultimately factor into decisions about treatment (see below).
Murine models of SPLIS
Subsequent to the identification of the yeast Dpl1 gene (Saba et al., 1997), the SPL genes of C. elegans (Mendel et al., 2003), mouse (Zhou and Saba, 1998), plant (Nishikawa et al., 2008), human (Van Veldhoven et al., 2000) and some pathogen species (Custodio et al., 2016; Degtyar et al., 2009; Zhang et al., 2007) have been identified. The characterization of SPL mutants and identification of their discrete phenotypes has shed light on SPL’s diverse functions. Although the majority of these phenotypes were identified prior to the discovery of SPLIS in humans, we now revisit vertebrate SPL mutants as useful models for dissecting the pathophysiology of SPLIS and exploring potential treatments of the condition.
Homozygous Sgpl1 null mice with Sgpl1 gene disruption fail to thrive after birth and have an average lifespan of 29 days (Schmahl et al., 2007). Runting is not evident at birth but becomes noticeable toward the end of the first week of life, consistent with the finding that sphingolipid accumulation in Sgpl1 null pups begins after birth (Billich et al., 2013). This is presumably due to a maternal effect that maintains normal sphingolipid degradation in utero. If this translates to humans with SPLIS, the post-natal onset would provide an opportunity for early medical intervention in order to prevent end organ damage, provided prenatal diagnosis and therapeutic options become available. However, some Sgpl1 null mice exhibit developmental abnormalities. For example, some homozygous null mice exhibited renal defects with swollen and hemorrhagic glomeruli (Schmahl et al., 2007). Reduction in the number of smooth muscle actin-positive mesangial cells in the glomeruli suggests their failure to migrate into the glomerular space (Schmahl et al., 2007). This finding is consistent with a migration defect observed in Sgpl1 null mouse embryonic fibroblasts and SPLIS patient-derived fibroblasts (Lovric et al., 2017; Schmahl et al., 2007). Vascular abnormalities leading to fetal hemorrhage and anemia, skeletal defects, improper palatal fusion, and thoracic malformations of the sternum, ribs and vertebrae were also observed at low frequency (Schmahl et al., 2007). Other rare phenotypes of Sgpl1 null mice included pulmonary, cardiac, urinary tract, and hepatic abnormalities (Schmahl et al., 2007; Vogel et al., 2009). In humans, some severely affected SPLIS patients have shown evidence of developmental defects including brain malformations, microcephaly, hypoplasia of corpus callosum, facial dysmorphism, pulmonary hypoplasia and fetal hydrops. These findings are consistent with the distinct expression pattern of SGPL1 in mammalian embryonic structures during early organ development.
Sgpl1 null mice uniformly exhibit blood and immunological defects such as platelet activation, lymphopenia, and reduced T cell egress (Schmahl et al., 2008; Vogel et al., 2009). Proia’s group showed the liver of Sgpl1 null mice to have high levels of cytokines, and neutrophil entry from blood into tissues was disrupted via a mechanism involving an IL23/IL17/granulocyte colony stimulating factor cytokine-controlled loop (Allende et al., 2011). Weber et al., characterized the thymii of Sgpl1 null mice as atrophic, with enhanced thymocyte apoptosis in association with high levels of thymic ceramides (Weber et al., 2009). As described above, various immunodeficiencies including T and B cell lymphopenias have been described in some SPLIS patients. However, hematological and immune profiles have not been systematically reported. Considering the major role of S1P signaling in immune cell trafficking and the sensitivity of lymphocyte trafficking to minor changes in S1P gradients (see below), a rigorous and prospective study of the natural history of SPLIS will be helpful in characterizing this novel primary immunodeficiency syndrome.
The Proia group also showed the Sgpl1 null mice had altered lipid metabolism (Bektas et al., 2010). Disruption of Sgpl1 increased sphingoid base phosphates and other sphingolipids including sphingosine, ceramide and sphingomyelin in serum and /or liver due to the inability to exit the sphingolipid metabolic pathway. SPL deficiency also increased the levels of serum and liver lipids including phospholipids, triacylglycerol, diacylglycerol, and cholesterol. This important feature of Sgpl1 null mice has not been studied extensively in SPLIS patients. Considering the physical and biochemical interactions between sphingolipids and other lipids such as cholesterol, phospholipids, eicosanoids and phosphoinositides that have come to light in recent years, it will be important to compare each of these lipid classes in the plasma, fibroblasts and other biological samples of SPLIS patients and controls (Banerji et al., 2010; Blind, 2014; Chalfant and Spiegel, 2005; Garcia-Arribas et al., 2016; Lemos et al., 2014; Pettus et al., 2005; Senkal et al., 2017).
A human SGPL1 knock-in mouse line was generated in the Sgpl1 null background by Lexicon (Vogel et al., 2009). Human SGPL1 expression (one or two human SGPL1 alleles) resulted in less than 10 and 20 percent of normal SPL activity, respectively, and conferred full protection from the rare defects in lung, heart, urinary tract and bone that were observed in Sgpl1 null mice. However, lymphodepletion is not prevented in human SGPL1 knock-in mice, indicating that lymphocyte trafficking is exquisitely sensitive to changes in the S1P chemotactic gradient. This study is important, because it demonstrates that restoring even as little as 10% of normal SPL may prevent severe consequences of SPL insufficiency. On the other hand, inhibiting SPL pharmacologically to 10% of normal levels can exert immunomodulatory therapeutic effects in patients with autoimmune disease without potentially harmful sequelae.
S1P signaling regulates bone metabolism, and a recent study established the role of SPL in this process (Weske et al., 2018) . Inducible genetic disruption of Sgpl1 and pharmacological inhibition of SPL in mice led to increased bone formation, bone mass, strength and reduced white adipose tissue. The study found that S1P stimulates osteoblastogenesis at the expense of adipogenesis by activating osterix and inhibiting PPAR-γ, whereas S1P signaling through S1P2 inhibits osteoclastogenesis. Thus far, no specific description of alterations in bone density have been noted in SPLIS patients. However, it is interesting that some SPLIS patients exhibited short stature, craniotabes, and rachitic rosary, signs that could suggest impaired bone formation or mineralization (Lovric et al., 2017). More careful analysis of bone structure in SPLIS patients may be warranted.
In addition to the above phenotypes affecting the hematopoietic and immune systems, lipid metabolism, kidneys and neurological functions, Sgpl1 null mice also rarely exhibited impaired testicular and ovarian steroidogenesis and infertility (Schmahl et al., 2007), which is consistent with the gonadal dysgenesis observed in some SPLIS patients.
A number of inducible and tissue-specific mouse models of Sgpl1 gene disruption have been generated, providing further insight into the pathophysiology of SPLIS. Inducible global Sgpl1 disruption in mice and pharmacological inhibition of SPL in rats cause podocyte-based kidney toxicity with glomerular proteinuria (Schumann et al., 2015). SPL deficient mice exhibited increased mesangial matrix, obliteration of some capillary lumina, podocyte effacement and NS as determined by ACR and podocyte foot process effacement. Partial deficiency of SPL led to decreased lethality compared to control mice and defects in kidney, the immune system, platelets and skin. Combined with the findings of Lovric et al confirming podocyte effacement in Sgpl1 null mice and demonstrating migration defects in glomerular mesangial cells (Lovric et al., 2017), this earlier report provides evidence that SPL plays a key role in maintaining glomerular homeostasis and the morphology of podocytes. It will be important to dissect whether SPL plays a direct or indirect role in maintaining podocyte health and whether aberrant podocyte S1P signaling and a resulting disruption of normal cytoskeletal architecture are at fault, or some other cellular consequence of SPL inhibition.
Inducible disruption of SPL in the epithelial cells of the lower gastrointestinal tract enhances the severity of colitis and colitis-associated colon cancer in mice (Degagné et al., 2014). The effects are mediated through a pathway involving STAT3-induced microRNAs and their downstream impact on AKT/PTEN and CYLD/NFκB signaling pathways. Our unpublished observations show that SPL deficiency has a profound impact on gut barrier functions and gut mucosal gluthathione status. Together with the reported role of S1P in regulating cell extrusion, these findings suggest that SPL plays a critical role in maintaining the gut epithelial barrier and host-pathogen interactions. Whether these roles explain the gastrointestinal maladies of SPLIS patients, or whether this feature of the disease is specifically related to their immunodeficient state, has not yet been established.
S1P, when present at high levels in the brain, has been shown to be neurotoxic, causing ER-stress and increased intracellular calcium (Hagen et al., 2011; Hagen et al., 2009). The consequences of Sgpl1 disruption on behavior, motor function and brain morphology were examined by Mitroi and colleagues in studies employing “brain-specific” Nestin CreTg+; Sgpl1 flox/flox mice in which Sgpl1 is specifically and constitutively disrupted in neuronal and glial cell precursors of the central and peripheral nervous system (CNS/PNS) (Mitroi et al., 2016). In addition, postnatal forebrain-restricted knockout (KO) mice were generated for comparison. Only the brain-specific KO mice accumulated S1P in brain tissues and exhibited cognitive and motor deficits. This phenotype was associated with a disruption of normal presynaptic architecture and activation of ubiquitin-mediated proteasomal machinery. A reduction in levels of the deubiquitinating enzyme USP14 and key presynaptic proteins was observed in the KO mouse brains, and inhibition of the proteasome restored presynaptic protein levels and synaptic function. In a second study, the authors found that Nestin CreTg+; Sgpl1 flox/flox mice brains exhibited a block in autophagic flux (Mitroi et al., 2017). This occurred concomitantly with a reduction in the SPL product, PE. PE serves as a critical factor in autophagic processes including lipidation of LC3 and is likely also involved in phagophore expansion (Rockenfeller et al., 2015). Blocked autophagy was associated with increased levels of the aggregate-prone proteins amyloid β [A4] precursor protein and SNCA/α-synuclein (Mitroi et al., 2017). When PE was added exogenously to cultured neurons of KO mice, autophagic flux was restored.
This study suggests that both S1P accumulation and PE depletion may contribute to SPL’s role in maintaining normal synaptic architecture, plasticity and neuronal autophagic flux. Further, since postnatal disruption of SPL in forebrain did not have the same impact, it suggests that SPL activity may be critical for certain processes during prenatal brain development, consistent with the importance of S1P signaling in neurogenesis (Mizugishi et al., 2005).
The egress of mature T cells from the thymus is a well-established S1P/SPL/S1P1-dependent process. In a study investigating the role of SPL in this process, thymic epithelial cell and hematopoietic cell-specific Sgpl1 KO mouse lines were generated and evaluated for an egress phenotype (Zamora-Pineda et al., 2016) . They found that dendritic cells, which make up only a fraction of a percent of thymic cells, serve as gatekeepers of T cell egress, by controlling the local S1P gradient near the site of egress at the thymic corticomedullary junction. T cell Sgpl1 KO mice also exhibited an egress phenotype, but this was found to be a minor contributor to egress, as double KO mice did not exhibit an additive effect, and the phenotype was rescued when dendritic cell Sgpl1 KO mice were administered wild type dendritic cells which homed to the thymus. These studies reveal how SPL insufficiency in mice and SPLIS patients contributes to their primary immunodeficiency state. Whether SPLIS patients exhibit additional defects of immune and hematopoietic cell development, trafficking or function will require more intensive analysis. Based on the rescue of dendritic cell KO mice, it would seem plausible that the immunodeficiency state and possibly other features of SPLIS could be attenuated by bone marrow transplantation (Kumar et al., 2017; Zamora-Pineda et al., 2016).
Sgpl1 null mice may serve as SPLIS disease models for specific indications including studying developmental phenotypes, further elucidation of SPL’s role in the pathophysiology of SPLIS, and for testing the efficacy of therapeutic strategies including substrate reduction, gene therapy and enzyme replacement (see below). However, the KO mice do not produce a mutant Sgpl1 mRNA or protein product. Therefore, they cannot be used to study the potential therapeutic impact of interventions aimed at improving mutant SPL folding, stability and activity or correcting a point mutation through gene editing. The latter will require the development of patient-specific transgenic mouse models of SPLIS.
Therapeutic strategies to treat patients with SPLIS
A variety of different approaches ranging from small molecules to gene therapies could be considered for the treatment of SPLIS patients. SPLIS presentations vary widely with regard to age of onset, disease manifestations and severity. Some patients die of fetal hydrops, others succumb in infancy, while still others have presented later in the first decade of life and are living into adulthood, albeit requiring kidney replacement therapy or transplantation. The diverse manifestations of this syndrome are likely due to mutational pleiotropy, i.e., the specific genetic determinants that influence SPL expression, conformation, subcellular localization, mRNA and protein stability, cofactor binding, enzyme activity. Clearly, inheriting two mutations that result in failure to produce a full-length polypeptide is associated with the worst outcome. Mutation position in the open reading frame will likely affect the polypeptide folding in different ways, and/or may influence substrate or cofactor binding. In addition, other genetic or environmental modifiers may influence the phenotypic expression of disease. Different treatment strategies and combination strategies might be appropriate for different SPLIS patients. A personalized approach may prove necessary to maximize therapeutic efficacy while minimizing risks.
Supportive therapy.
There is no standard medical treatment for SPLIS. Targeted therapeutic strategies using FDA-approved agents (see below) remain theoretical options or are in the earliest experimental stages. Therefore, the mainstays of therapy are focused on supportive care. These include standardized approaches aimed at compensating for end organ failure, such as dialysis or kidney transplantation for end-stage renal disease, hydrocortisone supplementation for adrenal insufficiency, and physical therapy and/or seizure control for neurological impairment. It is interesting to consider whether kidney transplantation could — by introducing an organ with wild type SPL expression and activity— compensate f or the SPL deficiency in other organs. S1P is carried in the bloodstream in association with albumin and Apo-M containing HDL. It is therefore conceivable that the donor kidney, while filtering the recipient’s blood, could degrade circulating S1P, reduce accumulated sphingolipids and provide a source of SPL products in SPLIS patients. Similarly, nonmyeloablative bone marrow transplantation could provide SPL- competent hematopoietic cells that could migrate throughout the body and degrade S1P. The utility of cell or organ transplantation to correct the underlying biochemical deficiency in SPLIS patients will depend on the expression level of SPL in the donor organ, the ability of donor cells to traffic to distant sites (lack of a trafficking or egress block) and the ability of donor cells to restore highly localized S1P gradients that are critical to sustaining S1P-mediated physiological functions. This information should be forthcoming as transplanted SPLIS patients are followed over time.
Chemical chaperones.
SPLIS mutations are found in many locations throughout the SGPL1 open reading frame. The direct impact of most missense mutations is likely on protein folding and protein stability rather than protein dimerization or enzyme kinetics (Lovric et al., 2017). This presumption is based on several findings, including: 1) mutant SPL proteins are found in low abundance in patient fibroblasts; 2) overexpression of mutant SPL proteins in HEK293 cells leads to a punctate appearance suggestive of protein aggregation; 3) co-immunoprecipitation of mutant SPL proteins did not show effects on protein dimerization (Lovric et al., 2017). The notion that mutations in SGPL1 lead to protein misfolding is consistent with the mechanisms implicated in the enzyme dysfunction of many inborn errors of metabolism (Bross et al., 1999). Based on this notion, chemical chaperones that increase the likelihood a nascent mutant SPL polypeptide will fold into its proper three- and four-dimensional conformation could be useful in SPLIS (Cortez and Sim, 2014). The most well-established chemical chaperone is 4-phenylbutyrate (Kolb et al., 2015). SPL’s cofactor PLP has also been shown to function as a chaperone that can facilitate the folding of PLP-dependent mutant enzymes including alanine-glyoxylate:aminotransferase, which is mutated in patients with primary hyperoxaluria (PH) (Cellini et al., 2014). In a small clinical trial, patients with one of the common PH mutations showed sustained responses to high dose PLP supplementation (Lorenz et al., 2014). PLP effects on mutant SPL could be tested in individual patient fibroblasts to determine whether a subset of SPLIS patients might be responsive to vitamin supplementation.
Inhibiting protein degradation pathways.
Another strategy that could increase SPL enzyme activity in SPLIS patients is to prolong the half-life of mutant SPL by blocking pathways involved in the clearance of misfolded proteins. In patients harboring mutations that lead to misfolding but do not ablate enzyme activity, stabilizing the mutant protein by blocking its rapid clearance could potentially boost enzyme activity to levels that could be clinically relevant. It is important to note that mice with only 10% of normal SPL activity are essentially normal and lack most of the manifestations of SPLIS with the exception of lymphopenia. Many proteasome inhibitors are in clinical use in cancer patients and could be repurposed for SPLIS.
S1P-targeted therapies.
Aberrant S1P signaling contributes to some of the disease manifestations of SPLIS. The lymphopenic status of SPLIS patients is a direct cause of the disruption of S1P chemotactic gradients that regulate lymphocyte egress from thymus and peripheral lymphoid organs. Aberrant S1P signaling through S1PRs may also contribute to the development of FSGS in SPLIS patients. This notion is supported by the observation that kidney mesangial cells in which SGPL1 is silenced exhibit an S1P-receptor dependent migration defect that is corrected by pretreatment with the nonselective S1P1/3 antagonist VPC23109 (Lovric et al., 2017). The finding of podocyte foot process effacement in mice and humans with SPL insufficiency also indicates a direct or indirect effect of SPL on podocyte health. To what extent S1P signaling contributes to the other manifestations of SPLIS remains to be established. Sphingosine kinase inhibitors, monoclonal S1P specific antibodies (O’Brien et al., 2009) and S1PR antagonists or blocking antibodies may eventually have a place in SPLIS treatment. Of these, only the nonselective S1PR functional antagonist Gilenya is FDA-approved — for clinical use in chronic relapsing multiple sclerosis (Kappos et al., 2006). However, it should be noted that such strategies will not address the consequences of ceramide accumulation and SPL product deficiency that may contribute to the pathology of SPLIS patients.
Product replacement.
Supplementation with SPL products could potentially have a role in SPLIS treatment. Mitroi and colleagues showed that the neurological abnormalities in mice lacking SPL specifically in brain tissues is attributed at least in part to a block in autophagic flux. Autophagic flux was also blocked in SPL-deficient cultured hippocampal neurons but was restored by the addition of PE. Whether a deficiency in long chain aldehydes contributes to SPLIS pathology is unknown, and most of the effects attributed to reactive aldehydes relates to their inappropriate accumulation and formation of adducts with DNA or protein. Hexadecenal and EP deficiencies are unlikely to cause all of the disease manifestations of SPLIS. As with S1P-targeted therapy, product replacement could become a part of the armamentarium of treatments for SPLIS patients but would not likely be curative.
Substrate depletion/reduction.
Substrate depletion and similar strategies aimed at reducing toxic sphingolipid intermediates have been used effectively in mouse models and in patients with other disorders of sphingolipid metabolism (Biswas et al., 2003; Garofalo et al., 2011; Shayman, 2018). In treating SPLIS, the goal is to balance a deficiency of the sphingolipid degradative pathway with a reduction in sphingolipid biosynthesis. In fruitflies, Sply mutants exhibit muscle patterning defects, and introduction of a hypomorphic mutation in lace, which encodes one subunit of the rate limiting enzyme of sphingolipid biosynthesis SPT, attenuates the Sply defects (Herr et al., 2003). This suggests that substrate depletion strategies could attenuate disease features in SPLIS. L-cycloserine is an FDA-approved drug that inhibits SPT and could be considered for substrate depletion in SPLIS. However, this drug also unfortunately inhibits SPL activity (Dominic Campopiano, personal communication). This could result in reduction of any residual enzyme activity provided by a mutant form of SPL. Thus, L-cycloserine would be problematic for all but the most severely affected individuals, namely those patients harboring homozygous or compound heterozygous nonsense SGPL1 mutations resulting in complete lack of SPL expression, or other mutations that result in a completely inactive enzyme.
Gene correction, gene replacement and enzyme replacement.
Although the pathogenesis of SPLIS is likely to be complex, its single enzyme basis makes it an ideal candidate for exploring gene therapy. Gene correction, gene replacement or enzyme replacement therapies are the only strategies with the potential to resolve all the biochemical defects associated with SPL insufficiency. SPL is an intracellular, integral membrane protein that localizes to the ER. Developing an effective enzyme replacement therapy that can deliver SPL protein to the ER membrane or provide a surrogate membrane system would be challenging. However, some bacterial organisms produce soluble SPL proteins, and truncation of the N-terminus of human SPL yields a soluble protein that could be deployed as a therapeutic. It has been shown that delivery of bacterial (soluble) SPL proteins to mice can reduce some disease-related endpoints and therefore was considered for potential therapeutic application in hyperproliferative conditions (Huwiler et al., 2011). Such approaches could also be applicable to SPLIS. Gene editing technology is developing rapidly and represents a potential therapeutic avenue for SPLIS, as well as all monogenic forms of disease. Problems with off-target effects and efficient delivery in an intact organism remain to be solved, but gene editing remains highly promising(Lux and Scharenberg, 2017). Gene therapy strategies represent another potential therapeutic with the potential to correct all of the biochemical consequences of SPL insufficiency. The most severe forms of SPLIS lead to high mortality in the first years of life. For such patients, the risk/benefit ratio for gene therapy is likely to be considered acceptable. One of the greatest challenges in treating severe SPLIS patients in hopes of preventing irreversible sequelae and death will be early detection, thereby creating a window of opportunity for medical intervention. Genetic counseling for parents who have had a child or family member affected by SPLIS, and early WES or whole genome sequencing of all newborns suspected of having a monogenic disorder would help to identify SPLIS patients early in their course. WES has recently been shown to outperform standard, disease-focused investigations and significantly improved the rate of early diagnosis (Stark et al., 2016).
Summary
SPL is an intracellular enzyme that guards the exit of the sphingolipid metabolic pathway by irreversibly converting S1P to PE and hexadecenal. S1P signaling is mediated by S1P receptor-dependent and receptor-independent actions. SPL expression is detected in many tissues in adult and embryonic murine tissues. Combined with the features of SPLIS and the reported phenotypes of global and conditional Sgpl1 KO mice, the ubiquitous expression of SPL indicates its importance in many physiological and developmental processes. The defects observed in SPL-deficient states in mice and humans resemble one another in many aspects, including NS, adrenal pathology, neurological defects, blood and immunological defects, acanthosis, and disordered lipid metabolism. Critical remaining questions include: 1) whether the discovery of SPLIS is a harbinger of a role for S1P metabolism and signaling in sporadic SRNS/FSGS, CMT, adrenal insufficiency and immunodeficiencies, 2) what are the specific mechanisms by which SPL dysfunction contributes to the pathophysiology of each feature of SPLIS, and 3) what are the ideal therapeutic strategies for the treatment of individual SPLIS patients, taking into consideration where their condition falls within the wide spectrum of SPLIS severity. Since the most severely affected (fetal death) and least severely affected SPLIS patients likely remain under-diagnosed, it may take time before the percentage of SPLIS patients whose disease course could potentially be fully or partially averted by early medical intervention can be determined.
Acknowledgments
This manuscript was supported by funds from the Swim Across America Foundation (JDS) and by NIH Public Health Services grant DK115669 and DK115669-S1 (JDS).
Abbreviations:
- ACR
Albumin Creatinine Ratio
- CMT
Charcot Marie Tooth Disease
- CNS
Central Nervous System
- C1P
Ceramide-1-Phosphate
- ELD
Endoplasmic Lumenal Domain
- ER
Endoplasmic Reticulum
- FSGS
Focal Segmental Glomerulosclerosis
- HDL
High Density Lipoprotein
- ID
Immunodeficiency
- KO
knockout
- NS
Nephrotic Syndrome
- PAI
Primary Adrenal Insufficiency
- PE
Phosphoethanolamine
- PH
Primary Hyperoxaluria
- PDCD
Pyridoxal Dependent Decarboxylase Conserved Domain
- PLP
Pyridoxal 5′-Phosphate
- PNS
Peripheral Nervous System
- S1P
Sphingosine-1-Phosphate
- S1PR
S1P Receptor
- SPL
Sphingosine Phosphate Lyase
- SPLIS
SPL Insufficiency Syndrome
- SPT
Serine Palmitoyltransferase
- SRNS
Steroid Resistant Nephrotic Syndrome
- TRAF2
TSH Thyroid Stimulating Hormone Tumor-necrosis factor receptor-associated factor 2
- WES
Whole Exon Sequencing
- ZF
Zona Fasciculata
Footnotes
Competing interests: The authors have no competing interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allende M, Bektas M, Lee B, Bonifacino E, Kang J, Tuymetova G, Chen W, Saba J, and Proia R (2011). Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. J Biol Chem 286, 7348–7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allende ML, and Proia RL (2002). Sphingosine-1-phosphate receptors and the development of the vascular system. Biochim Biophys Acta 1582, 222–227. [DOI] [PubMed] [Google Scholar]
- Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T, et al. (2010). Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465, 1084–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenz C (2017). Recent advances and novel treatments for sphingolipidoses. Future Med Chem 9, 1687–1700. [DOI] [PubMed] [Google Scholar]
- Atkinson D, Nikodinovic Glumac J, Asselbergh B, Ermanoska B, Blocquel D, Steiner R, Estrada-Cuzcano A, Peeters K, Ooms T, De Vriendt E, et al. (2017). Sphingosine 1-phosphate lyase deficiency causes Charcot-Marie-Tooth neuropathy. Neurology 88, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamborschke D, Pergande M, Becker K, Koerber F, Dotsch J, Vierzig A, Weber LT, and Cirak S (2018). A novel mutation in sphingosine-1-phosphate lyase causing congenital brain malformation. Brain Dev 40, 480–483. [DOI] [PubMed] [Google Scholar]
- Banerji S, Ngo M, Lane CF, Robinson CA, Minogue S, and Ridgway ND (2010). Oxysterol binding protein-dependent activation of sphingomyelin synthesis in the golgi apparatus requires phosphatidylinositol 4-kinase IIalpha. Mol Biol Cell 21, 4141–4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bektas M, Allende ML, Lee BG, Chen W, Amar MJ, Remaley AT, Saba JD, and Proia RL (2010). Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J Biol Chem 285, 10880–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billich A, Baumruker T, Beerli C, Bigaud M, Bruns C, Calzascia T, Isken A, Kinzel B, Loetscher E, Metzler B, et al. (2013). Partial deficiency of sphingosine-1-phosphate lyase confers protection in experimental autoimmune encephalomyelitis. PLoS One 8, e59630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S, Biesiada H, Williams TD, and LeVine SM (2003). Substrate reduction intervention by L-cycloserine in twitcher mice (globoid cell leukodystrophy) on a B6;CAST/Ei background. Neurosci Lett 347, 33–36. [DOI] [PubMed] [Google Scholar]
- Blaho VA, Galvani S, Engelbrecht E, Liu C, Swendeman SL, Kono M, Proia RL, Steinman L, Han MH, and Hla T (2015). HDL-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature 523, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blind RD (2014). Disentangling biological signaling networks by dynamic coupling of signaling lipids to modifying enzymes. Advances in biological regulation 54, 25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowsky AD, Bandhuvula P, Kumar A, Yoshinaga Y, Nefedov M, Fong LG, Zhang M, Baridon B, Dillard L, de Jong P, et al. (2012). Sphingosine-1-phosphate lyase expression in embryonic and adult murine tissues. J Lipid Res 53, 1920–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourquin F, Capitani G, and Grutter MG (2011). PLP-dependent enzymes as entry and exit gates of sphingolipid metabolism. Protein Sci 20, 1492–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourquin F, Riezman H, Capitani G, and Grutter MG (2010). Structure and function of sphingosine-1-phosphate lyase, a key enzyme of sphingolipid metabolism. Structure 18, 1054–1065. [DOI] [PubMed] [Google Scholar]
- Bross P, Corydon TJ, Andresen BS, Jorgensen MM, Bolund L, and Gregersen N (1999). Protein misfolding and degradation in genetic diseases. Hum Mutat 14, 186–198. [DOI] [PubMed] [Google Scholar]
- Bryan AM, and Del Poeta M (2018). Sphingosine-1-phosphate receptors and innate immunity. Cell Microbiol 20, e12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellini B, Montioli R, Oppici E, Astegno A, and Voltattorni CB (2014). The chaperone role of the pyridoxal 5’-phosphate and its implications for rare diseases involving B6-dependent enzymes. Clin Biochem 47, 158–165. [DOI] [PubMed] [Google Scholar]
- Chalfant CE, and Spiegel S (2005). Sphingosine 1-phosphate and ceramide 1-phosphate: expanding roles in cell signaling. J Cell Sci 118, 4605–4612. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, and Green DR (2012). Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148, 988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coant N, Sakamoto W, Mao C, and Hannun YA (2017). Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv Biol Regul 63, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez L, and Sim V (2014). The therapeutic potential of chemical chaperones in protein folding diseases. Prion 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custodio R, McLean CJ, Scott AE, Lowther J, Kennedy A, Clarke DJ, Campopiano DJ, Sarkar-Tyson M, and Brown AR (2016). Characterization of secreted sphingosine-1-phosphate lyases required for virulence and intracellular survival of Burkholderia pseudomallei. Mol Microbiol 102, 1004–1019. [DOI] [PubMed] [Google Scholar]
- Degagné E, Pandurangan A, Bandhuvula A, Kumar A, Eltanawy A, Zhang M, Yoshinaga Y, Nefedov M, de Jong P, Fong L, et al. (2014). Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J Clin Invest 124, 5368–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degtyar E, Zusman T, Ehrlich M, and Segal G (2009). A Legionella effector acquired from protozoa is involved in sphingolipids metabolism and is targeted to the host cell mitochondria. Cell Microbiol 11, 1219–1235. [DOI] [PubMed] [Google Scholar]
- Duan RD, and Nilsson A (2000). Sphingolipid hydrolyzing enzymes in the gastrointestinal tract. Methods Enzymol 311, 276–286. [DOI] [PubMed] [Google Scholar]
- Ebenezer DL, Fu P, Suryadevara V, Zhao Y, and Natarajan V (2017). Epigenetic regulation of pro-inflammatory cytokine secretion by sphingosine 1-phosphate (S1P) in acute lung injury: Role of S1P Lyase. Adv Biol Regul 63, 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogo AB (2015). Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol 11, 76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyrst H, and Saba JD (2010). An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat Chem Biol 6, 489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Arribas AB, Alonso A, and Goni FM (2016). Cholesterol interactions with ceramide and sphingomyelin. Chem Phys Lipids 199, 26–34. [DOI] [PubMed] [Google Scholar]
- Garofalo K, Penno A, Schmidt BP, Lee HJ, Frosch MP, von Eckardstein A, Brown RH, Hornemann T, and Eichler FS (2011). Oral L-serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1. J Clin Invest 121, 4735–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris CS, Blaho VA, Hla T, and Han MH (2014). Sphingosine-1-phosphate receptor 1 signalling in T cells: trafficking and beyond. Immunology 142, 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi S, Chiricozzi E, Mauri L, Sonnino S, and Prinetti A (2018). Sphingolipids and neuronal degeneration in lysosomal storage disorders. J Neurochem [DOI] [PubMed] [Google Scholar]
- Gudipaty SA, and Rosenblatt J (2017). Epithelial cell extrusion: Pathways and pathologies. Semin Cell Dev Biol 67, 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen N, Hans M, Hartmann D, Swandulla D, and van Echten-Deckert G (2011). Sphingosine-1-phosphate links glycosphingolipid metabolism to neurodegeneration via a calpain-mediated mechanism. Cell Death Differ 18, 1356–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen N, Van Veldhoven PP, Proia RL, Park H, Merrill AH Jr., and Van Echten-Deckert G (2009). Subcellular origin of sphingosine-1-phosphate is essential for its toxic effect in lyase deficient neurons. J Biol Chem 284, 11346–11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, et al. (2009). Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun YA, and Obeid LM (2008). Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Bio 9, 139–150. [DOI] [PubMed] [Google Scholar]
- Herr DR, Fyrst H, Phan V, Heinecke K, Georges R, Harris GL, and Saba JD (2003). Sply regulation of sphingolipid signaling molecules is essential for Drosophila development. Development 130, 2443–2453. [DOI] [PubMed] [Google Scholar]
- Huwiler A, Bourquin F, Kotelevets N, Pastukhov O, Capitani G, Grutter MG, and Zangemeister-Wittke U (2011). A prokaryotic S1P lyase degrades extracellular S1P in vitro and in vivo: implication for treating hyperproliferative disorders. PLoS One 6, e22436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Kihara A, and Igarashi Y (2004). Sphingosine-1-phosphate lyase (SPL) is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5’-phosphate binding domain exposed to the cytosol. Biochem Biophys Res Commun 325, 338–343. [DOI] [PubMed] [Google Scholar]
- Janecke AR, Xu R, Steichen-Gersdorf E, Waldegger S, Entenmann A, Giner T, Krainer I, Huber LA, Hess MW, Frishberg Y, et al. (2017). Deficiency of the sphingosine-1-phosphate lyase SGPL1 is associated with congenital nephrotic syndrome and congenital adrenal calcifications. Hum Mutat 38, 365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappos L, Antel J, Comi G, Montalban X, O’Connor P, Polman C, Haas T, Korn A, Karlsson G, and Radue E (2006). Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med 355, 1124–1140. [DOI] [PubMed] [Google Scholar]
- Kitatani K, Idkowiak-Baldys J, and Hannun YA (2008). The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal 20, 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb PS, Ayaub EA, Zhou W, Yum V, Dickhout JG, and Ask K (2015). The therapeutic effects of 4-phenylbutyric acid in maintaining proteostasis. Int J Biochem Cell Biol 61, 45–52. [DOI] [PubMed] [Google Scholar]
- Kono M, Allende ML, and Proia RL (2008). Sphingosine-1-phosphate regulation of mammalian development. Biochim Biophys Acta 1781, 435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, and Saba JD (2009). Lyase to live by: sphingosine phosphate lyase as a therapeutic target. Expert Opin Ther Targets 13, 1013–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Zamora-Pineda J, Degagné E, and Saba JD (2017). S1P lyase regulation of thymic egress and oncogenic inflammatory signaling. Mediators Inflamm 2017, 7685142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamour NF, and Chalfant CE (2008). Ceramide kinase and the ceramide-1-phosphate/cPLA2alpha interaction as a therapeutic target. Curr Drug Targets 9, 674–682. [DOI] [PubMed] [Google Scholar]
- Lemos T, Verdoorn KS, Nogaroli L, Britto-Borges T, Bonilha TA, Moreno PA, Silva OF, Tortelote GG, and Einicker-Lamas M (2014). Biphasic regulation of type II phosphatidylinositol-4 kinase by sphingosine: cross talk between glycero- and sphingolipids in the kidney. Biochim Biophys Acta 1838, 1003–1009. [DOI] [PubMed] [Google Scholar]
- Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, et al. (2013). Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 23, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhares N, Arantes R, Araujo S, and Pena S (2017). Nephrotic syndrome and adrenal insufficiency caused by a variant in SGPL1. Clinical Kidney Journal 1.-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz EC, Lieske JC, Seide BM, Meek AM, Olson JB, Bergstralh EJ, and Milliner DS (2014). Sustained pyridoxine response in primary hyperoxaluria type 1 recipients of kidney alone transplant. Am J Transplant 14, 1433–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovric S, Ashraf S, Tan W, and Hildebrandt F (2016). Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant 31, 1802–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovric S, Goncalves S, Gee HY, Oskouian B, Srinivas H, Choi WI, Shril S, Ashraf S, Tan W, Rao J, et al. (2017). Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J Clin Invest 127, 912–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux CT, and Scharenberg AM (2017). Therapeutic Gene Editing Safety and Specificity. Hematol Oncol Clin North Am 31, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel J, Heinecke K, Fyrst H, and Saba JD (2003). Sphingosine phosphate lyase expression is essential for normal development in Caenorhabditis elegans. J Biol Chem 278, 22341–22349. [DOI] [PubMed] [Google Scholar]
- Merrill AH Jr., Schmelz EM, Dillehay DL, Spiegel S, Shayman JA, Schroeder JJ, Riley RT, Voss KA, and Wang E (1997). Sphingolipids--the enigmatic lipid class: biochemistry, physiology, and pathophysiology. Toxicol Appl Pharmacol 142, 208–225. [DOI] [PubMed] [Google Scholar]
- Mitroi DN, Deutschmann AU, Raucamp M, Karunakaran I, Glebov K, Hans M, Walter J, Saba J, Graler M, Ehninger D, et al. (2016). Sphingosine 1-phosphate lyase ablation disrupts presynaptic architecture and function via an ubiquitin- proteasome mediated mechanism. Sci Rep 6, 37064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitroi DN, Karunakaran I, Graler M, Saba JD, Ehninger D, Ledesma MD, and van Echten-Deckert G (2017). SGPL1 (sphingosine phosphate lyase 1) modulates neuronal autophagy via phosphatidylethanolamine production. Autophagy 13, 885–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizugishi K, Yamashita T, Olivera A, Miller G, Spiegel S, and Proia R (2005). Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol 25, 11113–11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara K, Ohkuni A, Kitamura T, Abe K, Naganuma T, Ohno Y, Zoeller RA, and Kihara A (2012). The Sjogren-Larsson syndrome gene encodes a hexadecenal dehydrogenase of the sphingosine 1-phosphate degradation pathway. Molecular Cell 46, 461–471. [DOI] [PubMed] [Google Scholar]
- Newbigging S, Zhang M, and Saba JD (2013). Immunohistochemical analysis of sphingosine phosphate lyase expression during murine development. Gene Expr Patterns 13, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa M, Hosokawa K, Ishiguro M, Minamioka H, Tamura K, Hara-Nishimura I, Takahashi Y, Shimazaki K, and Imai H (2008). Degradation of sphingoid long-chain base 1-phosphates (LCB-1Ps): functional characterization and expression of AtDPL1 encoding LCB-1P lyase involved in the dehydration stress response in Arabidopsis. Plant Cell Physiol 49, 1758–1763. [DOI] [PubMed] [Google Scholar]
- Nourbakhsh N, and Mak RH (2017). Steroid-resistant nephrotic syndrome: past and current perspectives. Pediatric Health Med Ther 8, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien N, Jones ST, Williams DG, Cunningham HB, Moreno K, Visentin B, Gentile A, Vekich J, Shestowsky W, Hiraiwa M, et al. (2009). Production and characterization of monoclonal anti-sphingosine-1-phosphate antibodies. J Lipid Res 50, 2245–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareyson D, Saveri P, and Pisciotta C (2017). New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr Opin Neurol 30, 471–480. [DOI] [PubMed] [Google Scholar]
- Peev V, Hahm E, and Reiser J (2017). Unwinding focal segmental glomerulosclerosis. F1000Res 6, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettus BJ, Kitatani K, Chalfant CE, Taha TA, Kawamori T, Bielawski J, Obeid LM, and Hannun YA (2005). The coordination of prostaglandin E2 production by sphingosine-1-phosphate and ceramide-1-phosphate. Mol Pharmacol 68, 330–335. [DOI] [PubMed] [Google Scholar]
- Pezzuti IL, Silva IN, Albuquerque CTM, Duarte MG, and Silva JMP (2014). Adrenal insufficiency in association with congenital nephrotic syndrome: A case report. J Pediatr Endocr Met 27(5–6): 565–567. [DOI] [PubMed] [Google Scholar]
- Prasad R, Hadjidemetriou I, Maharaj A, Meimaridou E, Buonocore F, Saleem M, Hurcombe J, Bierzynska A, Barbagelata E, Bergada I, et al. (2017). Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. J Clin Invest 127, 942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne NJ, El Buri A, Adams DR, and Pyne S (2018). Sphingosine 1-phosphate and cancer. Adv Biol Regul 68, 97–106. [DOI] [PubMed] [Google Scholar]
- Pyne S, Lee SC, Long J, and Pyne NJ (2009). Role of sphingosine kinases and lipid phosphate phosphatases in regulating spatial sphingosine 1-phosphate signalling in health and disease. Cell Signal 21, 14–21. [DOI] [PubMed] [Google Scholar]
- Rockenfeller P, Koska M, Pietrocola F, Minois N, Knittelfelder O, Sica V, Franz J, Carmona-Gutierrez D, Kroemer G, and Madeo F (2015). Phosphatidylethanolamine positively regulates autophagy and longevity. Cell Death Differ 22, 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen H, Stevens RC, Hanson M, Roberts E, and Oldstone MB (2013). Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Annu Rev Biochem 82, 637–662. [DOI] [PubMed] [Google Scholar]
- Saba JD, Nara F, Bielawska A, Garrett S, and Hannun YA (1997). The BST1 gene of Saccharomyces cerevisiae is the sphingosine-1-phosphate lyase. J Biol Chem 272, 26087–26090. [DOI] [PubMed] [Google Scholar]
- Schmahl J, Raymond CS, and Soriano P (2007). PDGF signaling specificity is mediated through multiple immediate early genes. Nat Genet 39, 52–60. [DOI] [PubMed] [Google Scholar]
- Schmahl J, Rizzolo K, and Soriano P (2008). The PDGF signaling pathway controls multiple steroid-producing lineages. Genes Dev 22, 3255–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher F, Neuber C, Finke H, Nieschalke K, Baesler J, Gulbins E, and Kleuser B (2017). The sphingosine 1-phosphate breakdown product, (2E)-hexadecenal, forms protein adducts and glutathione conjugates in vitro. J Lipid Res 58, 1648–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann J, Grevot A, Ledieu D, Wolf A, Schubart A, Piaia A, Sutter E, Cote S, Beerli C, Pognan F, et al. (2015). Reduced activity of sphingosine-1-phosphate lyase induces podocyte-related glomerular proteinuria, skin irritation, and platelet activation. Toxicol Pathol 43, 694–703. [DOI] [PubMed] [Google Scholar]
- Schwab S, Pereira J, Matloubian M, Xu Y, Huang Y, and Cyster J (2005). Lymphocyte sequestration through S1P lyase inhibition an disruption of S1P gradients. Science 309, 1735–1739. [DOI] [PubMed] [Google Scholar]
- Senkal CE, Salama MF, Snider AJ, Allopenna JJ, Rana NA, Koller A, Hannun YA, and Obeid LM (2017). Ceramide Is Metabolized to Acylceramide and Stored in Lipid Droplets. Cell Metab 25, 686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayman JA (2018). Targeting Glucosylceramide Synthesis in the Treatment of Rare and Common Renal Disease. Semin Nephrol 38, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancevic B, and Kolesnick R (2010). Ceramide-rich platforms in transmembrane signaling. FEBS Lett 584, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, Yeung A, Peters H, Mordaunt D, Cowie S, et al. (2016). A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med 18, 1090–1096. [DOI] [PubMed] [Google Scholar]
- Takuwa Y, Du W, Qi X, Okamoto Y, Takuwa N, and Yoshioka K (2010). Roles of sphingosine-1-phosphate signaling in angiogenesis. World J Biol Chem 1, 298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya P, Kumar A, Byun HS, Bittman R, Saba JD, and Hecht SS (2012). The sphingolipid degradation product trans-2-hexadecenal forms adducts with DNA. Biochem Biophys Res Commun 424, 18–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Veldhoven PP, Gijsbers S, Mannaerts GP, Vermeesch JR, and Brys V (2000). Human sphingosine-1-phosphate lyase: cDNA cloning, functional expression studies and mapping to chromosome 10q22. Biochim Biophys Acta 1487, 128–134. [DOI] [PubMed] [Google Scholar]
- Van Veldhoven PP, and Mannaerts GP (1993). Sphingosine-phosphate lyase. Adv Lipid Res 26, 69–98. [PubMed] [Google Scholar]
- Vogel P, Donoviel MS, Read R, Hansen GM, Hazlewood J, Anderson SJ, Sun W, Swaffield J, and Oravecz T (2009). Incomplete inhibition of sphingosine 1-phosphate lyase modulates immune system function yet prevents early lethality and non-lymphoid lesions. PLoS One 4, e4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C, Krueger A, Munk A, Bode C, Van Veldhoven PP, and Graler MH (2009). Discontinued postnatal thymocyte development in sphingosine 1-phosphate-lyase-deficient mice. J Immunol 183, 4292–4301. [DOI] [PubMed] [Google Scholar]
- Weske S, Vaidya M, Reese A, von Wnuck Lipinski K, Keul P, Bayer JK, Fischer JW, Flogel U, Nelsen J, Epple M, et al. (2018). Targeting sphingosine-1-phosphate lyase as an anabolic therapy for bone loss. Nat Med 24, 667–678. [DOI] [PubMed] [Google Scholar]
- Zamora-Pineda J, Kumar A, Suh J, Zhang M, and Saba J (2016). Dendritic cell sphingosine-1-phosphate lyase regulates thymic egress. J Exp Med 213, 2773–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Pompey J, Hsu F, Turk J, Bandhuvula P, Saba J, and Beverley S (2007). Redirection of sphingolipid metabolism towards de novo synthesis of ethanolamine in Leishmania EMBO J 26, 1094–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, and Saba JD (1998). Identification of the first mammalian sphingosine phosphate lyase gene and its functional expression in yeast. Biochem Biophys Res Commun 242, 502–507. [DOI] [PubMed] [Google Scholar]