

Abstract
Introduction:
Prader-Willi syndrome (PWS), is a complex genetic disease affecting 1/15,000 individuals, characterized by lack of expression of genes on the paternal chromosome 15q11-q13 region. Clinical features include central hypotonia, poor suck, learning and behavior problems, growth hormone deficiency with short stature, hyperphagia and morbid obesity. Despite significant advances in genetic testing, the mean age for diagnosis in PWS continues to lag behind.
Objective:
Our goal was to perform a pilot feasibility study to confirm the diagnosis utilizing different genetic technologies in a cohort of 34 individuals with genetically confirmed PWS and 16 healthy controls from blood samples spotted and stored on newborn screening (NBS) filter paper cards.
Methods:
DNA was isolated from NBS cards, and PWS testing performed using DNA methylation-specific PCR (mPCR) and the MS-MLPA chromosome 15 probe kit followed by DNA fragment analysis for methylation and copy number status.
Results:
DNA extraction was successful in 30 of 34 PWS patients and 16 controls. PWS methylation testing was able to correctly identify all PWS patients and MS-MLPA was able to differentiate between 15q11-q13 deletion and non-deletion status and correctly identify deletion subtype (i.e., larger Type I or smaller Type II).
Conclusion:
mPCR can be used to diagnose PWS and MS-MLPA testing to determine both methylation status as well as the type of deletion or non-deletion status from DNA extracted from NBS filter paper. We propose that PWS testing in newborns is possible and could be included in the Recommended Uniform Screening Panel after establishing a validated cost-effective method.
Keywords: Prader-Willi syndrome, newborn screening, DNA methylation, MLPA (Multiplex Ligand PCR Analysis)
INTRODUCTION
Prader-Willi syndrome (PWS) affects approximately one in 15,000 live births. PWS patients present with neonatal hypotonia with poor suckling, feeding difficulties, failure to thrive and mild dysmorphism. However in childhood hypogonadism/ hypogenitalism, intellectual disability, growth hormone deficiency with short stature, behavioral problems, hyperphagia and subsequent obesity with related complications including diabetes mellitus, hypertension and cardiovascular problems in late childhood and adulthood (Butler 1990; Butler et al., 2006; Cassidy et al., 2012; Driscoll et al., 2016).
The cause of PWS is the lack of expression of genes on the paternal chromosome 15q11.2 – 15q13 region. The majority of patients (60%) have the deletion, 36% have maternal uniparental disomy 15 (UPD) and 4% with an imprinting defect (ID) (Butler, 1990). Although not necessary to establish diagnosis of PWS, it is important to identify the genetic subtype to estimate the recurrence risk for future siblings and prognosis. The recurrence risk for PWS is low; about 1% for deletion and UPD however ID is associated with a 50% recurrence risk (Buiting et al., 1998; Buiting et al., 2003) if a microdeletion within the imprinting center is present. Knowing the phenotype also allows one to better prepare for complications associated with specific PWS genetic subtypes.
Comparisons of 15q11-q13 deletion versus UPD have led to genotype-phenotype correlations. UPD is associated with a higher verbal IQ (Roof et al., 2000) and more psychosis and autism. Individuals with a 15q11-q13 deletion have two main types based on the location of the proximal 15q11.2 breakpoint (e.g., BP1 or BP2). Forty percent of the patients with the larger 15q11-q13 Type I deletion with BP1 and distal 15q11-q13 breakpoint (BP3) and sixty percent of patients have the smaller Type II deletion involving breakpoints BP2 and BP3. Those with the 15q11-q13 Type I deletion is associated with more severe problems such as obsessive-compulsive disorder, skin picking and other aberrant behavior than the smaller type II deletion (Chai et al., 2003).
Currently, the clinical diagnosis is largely based on clinical presentation and findings but then confirmed with genetic testing which often leads to significant delays. The age at diagnosis of individuals with PWS was noted between 7 and 9 years, approximately three decades ago (Butler, 1990). Recently due to increase in the availability of molecular diagnostic testing, PWS is diagnosed at earlier ages ranging from 18 days to 4.9 years (Bar et al., 2017); Dobrescu et al., 2016; Gold et al., 2018. It is critical to detect individuals with PWS in the newborn period in order to begin intervention early. Early treatment includes growth hormone (GH) which not only improves stature and body composition (lowers body fat and increases muscle), but decreases morbidity and mortality associated with obesity-related complications. Newborn screening (NBS) program tests done vary from state to state with California testing for more than 80 medical conditions with a goal to improve prognosis by early diagnosis and treatment. We propose that PWS fulfills the criteria for newborn screening of rare disorders and have undertaken a pilot, feasibility study in a cohort of PWS individuals and controls using fresh and stored blood placed on NBS filter paper cards.
MATERIALS AND METHODS
Subjects and Materials
Blood samples from 34 patients with PWS and a median age of 18 years (age range 5–56 years) and 16 healthy controls were obtained (Table 1). Of the blood samples collected from patients with PWS, eight were freshly collected and 26 were stored at −20 C for periods as long as 13 years. Approximately 5–10 cc of blood was collected in a purple top (EDTA) tube for use in our study and placed on NBS filter paper cards. All PWS patients had a genetically confirmed diagnosis of PWS by methylation studies, FISH, chromosomal microarray and/or genotyping testing analysis for UPD identification. Human subjects IRB committee approval was obtained from the University of California-Irvine and informed consent or assent was obtained from patients, if minors and consent from their parents.
Table 1:
Demographics of patients with Prader-Willi syndrome
| Patient number | Molecular class | Age (y) | Sex | Storage duration (y) |
|---|---|---|---|---|
| 1 | 15q11-q13 Deletion | 18 | F | Fresh |
| 2 | 15q11-q13 Deletion | 23 | M | Fresh |
| 3 | 15q11-q13 Deletion | 21 | F | Fresh |
| 4 | 15q11-q13 Deletion | 18 | M | 5 |
| 5 | 15q11-q13 Deletion | 10.5 | M | 5 |
| 6 | 15q11-q13 Deletion | 23 | M | 8 |
| 7 | 15q11-q13 Deletion | 12 | M | 8 |
| 8 | 15q11-q13 Deletion | 30 | F | 5 |
| 9 | 15q11-q13 Deletion | 12 | M | 3 |
| 10 | 15q11-q13 Deletion | 21 | F | 8 |
| 11 | 15q11-q13 Type I deletion | 38 | M | 8 * |
| 12 | 15q11-q13 Type I deletion | 19 | F | 10 * |
| 13 | 15q11-q13 Type I deletion | 26 | F | 12 |
| 14 | 15q11-q13 Type I deletion | 13 | M | 10 * |
| 15 | 15q11-q13 Type II deletion | 11 | M | 3 |
| 16 | 15q11-q13 Type II deletion | 21 | F | 8 |
| 17 | 15q11-q13 Type II deletion | 14 | F | 10 |
| 18 | 15q11-q13 Type II deletion | 10 | F | 7 |
| 19 | 15q11-q13 Type II deletion | 8 | M | 11 |
| 20 | 15q11-q13 Type II deletion | 18 | M | 10 * |
| 21 | 15q11-q13 Type II deletion | 18 | M | Fresh |
| 22 | 15q11-q13 Type II deletion | 5 | F | Fresh |
| 23 | 15q11-q13 Type II deletion | 40 | M | 4 |
| 24 | UPD15 | 11 | F | 3 |
| 25 | UPD15 | 17 | F | 5 |
| 26 | UPD15 | 11 1 | M | 6 |
| 27 | UPD15 | 7 | M | Fresh |
| 28 | UPD15 | 24 | F | Fresh |
| 29 | UPD15 | 13 | M | Fresh |
| 30 | UPD15 | 8 | F | 4 |
| 31 | UPD15 | 14 | M | 5 |
| 32 | UPD15 | 47 | M | 10 * |
| 33 | UPD15 | 14 | F | 8 |
| 34 | UPD15 | 56 | F | 13 |
Samples with very low DNA concentrations and that failed to generate results
Fresh blood samples were directly placed on NBS filter papers circle (0.5cm x 0.5 cm) (Whatman GE Healthcare Life Sciences, Pittsburgh, PA, USA) shortly after collection in eight individuals with PWS. Frozen blood samples were thawed and similarly placed on NBS filter papers in the remaining individuals. DNA was then extracted from the dried blood spots on NBS filter paper for both controls and Prader-Willi syndrome participants. The following protocols and DNA isolation kits were used: GenSolve kit (GenTegra, Pleasanton, CA, USA) for Phase I to remove blood elements from the filter paper and Qiagen DNA micro kit (Germantown, MD, USA) for Phase II to isolate DNA. Briefly, the GenSolve procedure consists of incubating 3 punches of 6 mm filter paper discs at 65º C with a protease combined with a proprietary (high pH) solution and 1% lithium dodecyl sulfate. The incubation releases DNA, proteins and cellular debris from the matrix in a highly efficient manner. After centrifugation the eluate was purified using Qiagen DNA micro kit for DNA isolation and concentration.
Methylation-specific PCR (mPCR)
DNA extracted from NBS filter paper was then treated with sodium bisulfite by using (EZ DNA Methylation-Lightning kit) from (Zymo research, CA, USA). DNA treated with sodium bisulfite to convert cytosine to uracil except when cytosine is methylated as found on the maternal chromosome as 5-methyl cytosine is resistant to bisulfite treatment and remains unchanged. PCR amplification was done using SNRPN primers generated specifically for the maternal and paternal allele methylation sites (Muralidhar & Butler, 1998). DNA oligonucleotides used for methylation PCR (mPCR) analysis are commercially synthesized and represent the maternally methylated sequences at genomic position +111 and +284 of the SNRPN gene and the paternally unmethylated sequences at genomic position +140 and +239 from sequence data referenced in Genbank (Bethesda, MD) and reported previously (Kubota et al., 1996; Muralidhar and Butler, 1996). The two primers are specifically designed to amplify the unmodified maternally methylated sequence and a second modified paternally unmethylated sequence after treatment with sodium bisulfite. PCR products were subjected to electrophoresis using 2% agarose gel in 0.5 × trisboric acid-ethylene diamine tetra acetate (TBE) buffer, stained in ethidium bromide visualized under ultra-violet illumination, and photographed. The PWS/AS critical region was thereafter examined by methylation-specific PCR (mPCR). The test was considered positive if the maternal band was visualized at 174 bp and the paternal fragment was not visualized at 100 bp.
Methylation specific-multiplex ligation dependent probe amplification (MS-MLPA)
MS-MLPA was performed on 10 samples using the ME028 B2 and X3 kits specifically designed to determine the methylation status and copy number of probes from chromosome 15. PCR products were then sent to Genewiz laboratory in Rockville, Maryland, for fragment analysis as a fee for service. The process involves DNA denaturation, probe hybridization, ligation and ligation digestion with final PCR reaction to generate 45 different size DNA fragments at low cost and hence feasible for not only determination of chromosome 15 methylation status but copy number for the 15q11-q13 region utilized for diagnosing PWS and Angelman syndrome. Results of fragment analysis were analyzed using Peak Scanner Software v1.0. Patients with PWS were identified by an abnormal methylation status. The SALSA MS-MLPA ME028 kit (MRC-Holland, Amsterdam, Netherlandshttp://www.falco-genetics.com/salsa/pdf/dl/me028.pdf) will detect copy number changes as well as analyze CpG island methylation of the 15q11-q13 region. At least five of these probes are specific for the imprinting center sequence and contains a recognition site for the methylation sensitive Hha1 enzyme.
Methylation-specific-multiplex ligation-dependent probe amplification (MS-MLPA) is used to determine the copy number of up to 50 DNA sequences in a single multiplex PCR-based reaction. Each MS-MLPA reaction results in a set of unique PCR amplicons between 64–500 nt in length, which are separated by capillary electrophoresis. In MS-MLPA, not sample DNA is amplified during PCR, but MS-MLPA probes hybridize to the sample DNA. In contrast to a standard multiplex PCR, MS-MLPA uses a single PCR primer pair to amplify all probes, making the method very robust. The hybridizing sequence of each probe is ~55–80 nt long. Each MS-MLPA probe consists of two oligonucleotides that must bind to adjacent target DNA sequences to be ligated. Upon ligation, a single molecule is formed that can be amplified during PCR. Because hybridization and ligation are essential for the probe to be amplified and give a signal, unbound oligonucleotides do not give a signal. Following PCR, the amplicons are separated and quantified by capillary electrophoresis. Each probe in an MS-MLPA probe set has a unique length and can therefore be easily identified.
DNA fragment analysis
Fragment analysis was performed by Genewiz (Rockville, MD, USA). Data analysis was done by exporting the peak heights and areas into the Excel program (Microsoft, Seattle, WA). For copy number quantification, the height and area of each peak from chromosome 15 was divided by the closest smaller and larger peaks from the printout and outside of chromosome 15. The copy number was obtained by comparing this ratio with the same peak ratio obtained from a control individual. By taking the mean normalized ratio and rounding to 0.5, 1, 1.5 or 2 and then multiplying by 2 (normal or biallelic copy number), the copy number can be determined. Thus, our subjects with 15q11-q13 deletions having normalized ratios of approximately 0.5 with a copy number of 1 indicating they have a single allele (Bittel et al., 2008; Henkhaus et al., 2012).
RESULTS
DNA extraction
We were able to successfully extract DNA from samples of 29 of 34 patients with PWS (88%) and all 16 controls (100%) with a concentration range of 16–820 ng/microL, mean of 181 ng/microL. Five PWS samples showed very low DNA concentration and did not meet DNA concentration (< 10 ng/microl) required for MS-MLPA analysis. The length of storage of failed samples was 8–10 years. We suggest that the poor DNA quality and quantity may be related to prolonged storage time, inappropriate preservation conditions and/or bacterial contamination.
DNA methylation PCR
Out of 45 samples studied with adequate DNA concentration (29 PWS patients and 16 controls), DNA methylation confirmed the PWS status in all PWS individuals by exhibiting only the 174-bp PCR product from the maternal allele. All 16 control samples showed both the 174-bp PCR product from the methylated maternal chromosome and the 100-bp PCR product from the unmethylated paternal chromosome (Figure 1).
Figure 1.
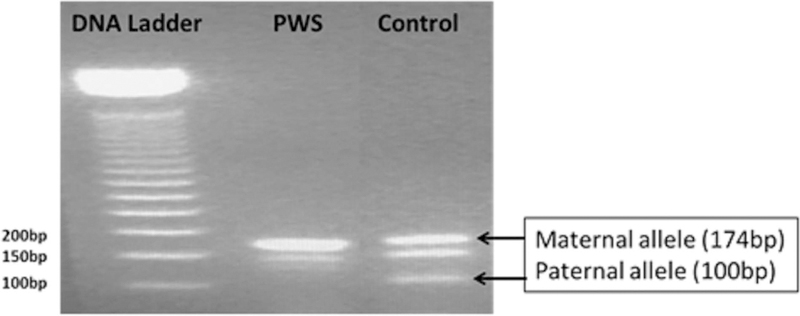
Photograph of gel electrophoresis of methylation SNRPN gene analysis by methylation PCR showing a positive methylation result for a PWS subject with absence of paternal allele (100 bp) and a normal control subject with presence of both methylated maternal (174 bp) and unmethylated paternal (100 bp) DNA signals.
Methylation specific-multiplex ligation dependent probe amplification (MS-MLPA)
Of the PWS specimens tested by MS-MLPA, Samples with methylation status at approximately 50% (range between 40–60%) for PWS specific probes were identified as normal controls and PWS if methylation status at approximately 100% (greater than 80%) (Figure 2).The genetic subtype was correctly identified in two PWS patients with UPD and four with the 15q11-q13 deletion (two with Type II and one with Type I deletion and one was unidentified). MS-MLPA was negative in four tested controls indicating a normal methylation status and a normal copy number. Therefore, using the copy number data, we were able to correctly identify the genetic subtype in PWS study participants. Using MS-MLPA we were also able to correctly identify the type of deletion in PWS deletion patients. Deletion patients who had only one copy number for BP1-BP2 region probe had Type 1 deletion and patients who had two copy numbers for this region had Type II deletion (Figure 3).
Figure 2.

Gene methylation status using MS-MLPA of a control subject (A) showing normal methylation status (about 50%) for five methylated probes (in bold) and a PWS subject (B) showing abnormal methylation status (about 100%) in the five methylated probes (in bold).
Figure 3.

MS-MLPA copy number status determination for PWS subjects. (A) PWS subject with maternal uniparental disomy 15 and a copy number for PWS probes at approximately 2 (non-deletion status). (B) PWS subject with 15q11-q13 Type I deletion and a copy number for PWS probes at approximately 1 (deletion status) and reference probes at approximately 2 (non-deletion status). (C) PWS subject with 15q11-q13 Type II deletion and the chromosome 15q11.2 BP1-BP2 region involving TUBGCP5 and NIPA1 probes not deleted while other PWS 15q11-q13 probes are deleted.
Data Summary Analysis
We successfully extracted DNA from all samples freshly collected from PWS patients and 22 of 26 archived samples of up to 13 years. DNA extraction from control samples were successful in all subjects. It is possible that DNA degradation of the archived samples resulted in difficulty in extracting DNA for the methylation studies and impacted quality and quantity of DNA isolated. Several of these samples were in storage for periods ranging from 3 to 13 years. Once DNA was successfully extracted with quality and quantity required, we were able to correctly assign PWS status in all affected individuals.
DISCUSSION
There has been significant progress and acceptance of expanded newborn screening nationwide including the addition of several new metabolic and genetic disorders, e.g., severe combined immune deficiency (SCID), Pompe disease, Krabbe disease, adrenoleukodystrophy and fragile X syndrome among others in several states (Inaba et al., 2014). PWS is a significant health problem that results in significant medical, behavioral and mental disability and costs, particularly if treatment is delayed. The reported incidence of PWS is 1/10,000–15,000 live births, making it more common than several disorders included in the newborn screening (Burd, Vesely, Martsolf, & Kerbeshian, 1990); e.g., SCID with a frequency of 1/53,000 (Francisco, 2017) and Pompe disease with a frequency of 1/ 40,000 (Lawrence, 2017). Both have recently been added to the Recommended Universal Screening Panel (RUSP) and newborn screening list in several states. If all infants with PWS were diagnosed at birth with a proposed simple and cost-effective newborn test, they would benefit from early growth hormone and behavioral treatments as well as decrease costly medical evaluations and hospitalization. Individuals with PWS experience a wide variety of medical challenges throughout their life span. Infants with PWS classically have central hypotonia, a poor suck leading to feeding difficulties that may warrant placement of a gastric tube and mild dysmorphic features requiring extensive and costly evaluations with additional perinatal care if the diagnosis is not entertained immediately after delivery. In severe cases, neonatal intensive care may last for weeks to months (Cassidy et al., 2012). Bachere et al. (2008) noted that early diagnosis and multidisciplinary care in PWS resulted in decreasing hospital stay and the duration of tube feeding, optimizing screening for endocrine dysfunction, preventing growth delay and identifying early onset of obesity. In a survey of medical records and health costs of 2,030 patients with PWS from 2009 to 2014, the mean annual in-patient costs were greater than $10,000 (Shoffstall et al., 2016). In-patient care costs for PWS across all ages ranged between 10.7 to 4.8 times depending on insurance coverage when compared with individuals without PWS. During the first year of life for in-patients with PWS, the medical costs were doubled compared to later years (e.g., $51,000 per year from 0–1 yr. compared with $23,000 per year from 2 to 4 years) which reflects the in-patient costs for NICU care, delayed diagnosis with increasing hospitalization time, diagnostic testing and consultation costs.
In this study, we were able to extract DNA from 45 samples from both frozen and fresh blood specimens on NBS cards from patients with PWS and controls. DNA methylation testing confirmed PWS status in all patients from whom DNA was useable and furthermore MS-MLPA testing correctly identified the genetic subtypes in all specimens.
Prader-Willi syndrome diagnosis is currently based on clinical suspicion and traditionally confirmed post-natally by DNA methylation, FISH for 15q11-q13 deletions, SNP chromosomal microarray, MS-MLPA and/or genotyping testing, the latter requiring parental samples. Formal cost effective studies would need to be undertaken using a validated testing model before the financial and clinical impact of newborn screening for this disorder could be properly evaluated We believe that the costs would decrease with large scale, automated testing when newborn screening is implemented for PWS.
Despite significant diagnostic advances, the mean age for diagnosis of PWS continues to lag behind as syndromic specific testing ordered by the physician requires awareness and knowledge about the appropriate genetic test to order. Inappropriate choice of the initial genetic test from a selection of multiple tests has led to delayed diagnosis. Vogels and Fryns (2004) reported the mean age of diagnosis as 6.5 months with a range from 1 week to 8 years based on genetic testing. Other studies reported significant delayed age of diagnosis in the UPD group in comparison with the deletion group (GunayAygun et al., 1997; Dykens, 2002; Hartley et al., 2005; Cassidy et al., 2012), suggesting that subtle diagnostic features in early childhood in the UPD group lead to later diagnosis (Gunay-Aygun, Heeger, Schwartz, & Cassidy, 1997; Dykens, 2002; Hartley, Maclean, Butler, Zarcone, & Thompson, 2005; Cassidy, Schwartz, Miller, & Driscoll, 2012). Currently, DNA methylation is the gold standard test for confirming the diagnosis of PWS. DNA methylation can identify 99% of PWS patients and is less costly and effective, although, it cannot differentiate among the different PWS genetic subtypes. A second line test is needed for further clarification once the genetic diagnosis is made. The methylation positive PWS cases can then be examined by FISH, SNP array, MS-MLPA or UPD testing. We believe it is critical to detect those with PWS in the newborn period to begin care and treatment specific for PWS as early as possible. Our results provide evidence that DNA methylation can be performed on dried blood spots routinely as a proof of concept obtained for newborn screening for PWS and conversely for Angelman syndrome, a second genetic syndrome occurring at about the same prevalence but clinically distinct. Yet, Angelman syndrome can be detected with the same methylation DNA assay by showing loss of the maternal DNA methylation band pattern instead of the paternal band. Wide adoption of this principle would permit early and undisputed diagnosis even in premature babies with vague clinical features and those with atypical presentation, rather than relying solely on subjective clinical assessment for both PWS and AS. Early diagnosis is optimal for clinical care and treatment making both conditions candidate for newborn screening.
Early diagnosis and GH treatment affords the opportunity of proactive strategies for prevention of obesity and many of the associated co-morbidities such as diabetes, hypertension, and respiratory compromise; common in PWS without early recognition and treatment. The benefits of growth hormone therapy in infants, children and adults with PWS have been demonstrated in multiple well-designed and well-controlled studies (Lindgren & Lindberg, 2008; Mogul et al., 2008; Butler et al., 2013). When treatment begins in infancy, facial appearance and body habitus normalizes in conjunction with good dietary management generating improvement in quality of life and psychosocial status in those with PWS. GH replacement therapy improves linear growth velocity and height, body composition, muscle function and level of activity (Butler et al., 2008; 2016). GH treatment is now commonly prescribed in infancy and childhood once the diagnosis of PWS is confirmed genetically to increase stature, muscle tone and decrease fat, thereby lowering risk factors for diabetes and cardiopulmonary problems.
In summary, the ultimate goal is to change the face of PWS by early identification and awareness which will permit proactive monitoring of the diet, and exercise and treatment with growth hormone at an early age thus preventing co-morbidities associated with PWS and reducing the current high mortality rate (Butler et al., 2016; Manzardo et al., 2017). The methylation testing used for diagnosis of PWS would also have the added benefit of detecting Angelman syndrome (AS). AS is caused by chromosome 15q11-q13 deletion however unlike PWS the deletion is of maternal origin and is associated with microcephaly, intellectual disability, epilepsy, limited language development, dysmorphic features, ataxia and autistic behaviors. Early diagnosis also permits detection of the imprinting center type of PWS or AS which may be associated with a 50% recurrence risk in future offspring thus permitting early and accurate genetic counseling.
Our study is limited by the small number of samples tested and hence is a proof of concept. We failed to extract DNA from dried blood in 14% particularly in those with stored blood specimens for an extended length of time (e.g.,13 years). We hypothesize that failed DNA isolation could result from degradation from suboptimal storage condition, bacterial contamination, and effect of DNases compromising our specimen quality and quantity. None the less, it is possible to extract sufficient quantity and quality of DNA from NBS filter paper cards and feasible to correctly identify the PWS in 99% of patients and in 85% for AS with current technology (Butler et al., 2006) using inexpensive DNA methylation assays that can be generated at high volume required for newborn screening programs. However, this assay cannot differentiate between deletion vs. non-deletion (e.g., UPD) status for both PWS and AS and will require more sophisticated (and expensive) testing such as MS-MLPA or high resolution SNP microarrays to identify UPD subclasses as described and illustrated by Butler et al. (2018). Future studies are needed to focus on using high throughput cost-effective methods and analysis for PWS testing in the newborn period before it can be adopted on a national basis.
Acknowledgements
We wish to thank the participants for donation of the blood samples which were spotted on filter paper cards for genetic analysis. We acknowledge support from National Institute of Child Health and Human Development (NICHD) grant number HD02528 and the UC Irvine Institute for Clinical and Translational Science. Funding for this research project was also provided by the Neonatology Division, Department of Pediatrics, University of California-Irvine
Footnotes
The authors claim no conflict of interest.
REFERENCES
- Angulo MA, Castro-Magana M, Lamerson M, Arguello R, Accacha S, & Khan A (2007). Final adult height in children with Prader-Willi syndrome with and without human growth hormone treatment. Am J Med Genet A, 143A(13), 1456–1461. [DOI] [PubMed] [Google Scholar]
- Bar C, Diene G, Molinas C, Bieth E, Casper C, & Tauber M (2017). Early diagnosis and care is achieved but should be improved in infants with Prader-Willi syndrome. Orphanet J Rare Dis, 12(1), 118. doi: 10.1186/s13023-017-0673-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachere N, Diene G, Delagnes V, Molinas C, Moulin P, & Tauber M (2008). Early diagnosis and multidisciplinary care reduce the hospitalization time and duration of tube feeding and prevent early obesity in PWS infants. Horm Res, 69(1), 45–52. [DOI] [PubMed] [Google Scholar]
- Buiting K, Dittrich B, Gross S, Lich C, Farber C, Buchholz T, Horsthemke B (1998). Sporadic imprinting defects in Prader-Willi syndrome and Angelman syndrome: implications for imprint-switch models, genetic counseling, and prenatal diagnosis. Am J Hum Genet, 63(1), 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buiting K, Gross S, Lich C, Gillessen-Kaesbach G, el-Maarri O, & Horsthemke B (2003). Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet, 72(3), 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd L, Vesely B, Martsolf J, & Kerbeshian J (1990). Prevalence study of Prader-Willi syndrome in North Dakota. Am J Med Genet, 37(1), 97–99. [DOI] [PubMed] [Google Scholar]
- Butler MG (1990). Prader-Willi syndrome: current understanding of cause and diagnosis. Am J Med Genet, 35(3), 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Smith BK, Lee J, Gibson C, Schmoll C, Moore WV, & Donnelly JE (2013). Effects of growth hormone treatment in adults with Prader-Willi syndrome. Growth Horm IGF Res, 23(3), 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrel AL, Myers SE, Whitman BY, & Allen DB (2001). Sustained benefits of growth hormone on body composition, fat utilization, physical strength and agility, and growth in Prader-Willi syndrome are dose-dependent. J Pediatr Endocrinol Metab, 14(8), 1097–1105. [DOI] [PubMed] [Google Scholar]
- Cassidy SB, Schwartz S, Miller JL, & Driscoll DJ (2012). Prader-Willi syndrome. Genet Med, 14(1), 10–26. [DOI] [PubMed] [Google Scholar]
- Chai JH, Locke DP, Greally JM, Knoll JH, Ohta T, Dunai J, … Nicholls RD (2003). Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet, 73(4), 898–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrescu AI, Chirita-Emandi A, Andreescu N, Farcas S, & Puiu M (2016). Does the Genetic Cause of Prader-Willi Syndrome Explain the Highly Variable Phenotype? Maedica (Buchar), 11(3), 191–197. [PMC free article] [PubMed] [Google Scholar]
- Driscoll DJ, Miller JL, Schwartz S et al. (1998 October 6, 2016. February 4). Prader-Willi syndrome Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1330/ [DOI] [PubMed]
- Dykens EM (2002). Are jigsaw puzzle skills ‘spared’ in persons with Prader-Willi syndrome? J Child Psychol Psychiatry, 43(3), 343–352. [DOI] [PubMed] [Google Scholar]
- Gunay-Aygun M, Heeger S, Schwartz S, & Cassidy SB (1997). Delayed diagnosis in patients with Prader-Willi syndrome due to maternal uniparental disomy 15. Am J Med Genet, 71(1), 106–110. [PubMed] [Google Scholar]
- Hartley SL, Maclean WE Jr., Butler MG, Zarcone J, & Thompson T (2005). Maladaptive behaviors and risk factors among the genetic subtypes of Prader-Willi syndrome. Am J Med Genet A, 136(2), 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoybye C (2007). Five-years growth hormone (GH) treatment in adults with Prader-Willi syndrome. Acta Paediatr, 96(3), 410–413. doi: 10.1111/j.1651-2227.2006.00051.x [DOI] [PubMed] [Google Scholar]
- Inaba Y, Schwartz CE, Bui QM, Li X, Skinner C, Field M, … Godler DE (2014). Early detection of fragile X syndrome: applications of a novel approach for improved quantitative methylation analysis in venous blood and newborn blood spots. Clin Chem, 60(7), 963–973. [DOI] [PubMed] [Google Scholar]
- Gold JA, Mahmoud R, Cassidy SB, & Kimonis V (2018). Comparison of perinatal factors in deletion versus uniparental disomy in Prader-Willi syndrome. Am J Med Genet A, 176(5), 1161–1165. doi: 10.1002/ajmg.a.38679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T, Sutcliffe JS, Aradhya S, Gillessen-Kaesbach G, Christian SL, Horsthemke B, … Ledbetter DH (1996). Validation studies of SNRPN methylation as a diagnostic test for Prader-Willi syndrome. Am J Med Genet, 66(1), 77–80. [DOI] [PubMed] [Google Scholar]
- Lindgren AC, & Lindberg A (2008). Growth hormone treatment completely normalizes adult height and improves body composition in Prader-Willi syndrome: experience from KIGS (Pfizer International Growth Database). Horm Res, 70(3), 182–187. [DOI] [PubMed] [Google Scholar]
- Lionti T, Reid SM, White SM, & Rowell MM (2015). A population-based profile of 160 Australians with Prader-Willi syndrome: trends in diagnosis, birth prevalence and birth characteristics. Am J Med Genet A, 167A(2), 371–378. doi: 10.1002/ajmg.a.36845 [DOI] [PubMed] [Google Scholar]
- Mogul HR, Lee PD, Whitman BY, Zipf WB, Frey M, Myers S, … Southren AL (2008). Growth hormone treatment of adults with Prader-Willi syndrome and growth hormone deficiency improves lean body mass, fractional body fat, and serum triiodothyronine without glucose impairment: results from the United States multicenter trial. J Clin Endocrinol Metab, 93(4), 1238–1245. [DOI] [PubMed] [Google Scholar]
- Muralidhar B, Butler MG (1998). Methylation PCR analysis of Prader-Willi syndrome, Angelman syndrome, and control subjects. Am J Med Genet, 80(3):263–5 [PMC free article] [PubMed] [Google Scholar]
- Vogels A, & Fryns JP (2004). Age at diagnosis, body mass index and physical morbidity in children and adults with the Prader-Willi syndrome. Genet Couns, 15(4), 397–404. [PubMed] [Google Scholar]
- Francisco AB (2017) Severe combined immunodeficiency (SCID): An overview. In TePas E (ED), UpToDate. Retrieved from https://www.uptodate.com/contents/severe-combined-immunodeficiency-scid-an-overview
- Lawrence M (2017) Lysosomal acid alpha-glucosidase deficiency (Pompe disease, glycogen storage disease II, acid maltase deficiency). In TePas E. (ED), UpToDate. Retrieved from https://www.uptodate.com/contents/lysosomal-acid-alpha-glucosidase-deficiency-pompe-disease-glycogen-storage-disease-ii-acid-maltasedeficiency?search=pompe%20disease&source=search_result&selected Title=1~34&usage_type=default&display_rank=1
- Henkhaus RS, Kim S-J, Kimonis VE, et al. Methylation-specific multiplex ligation-dependent probe amplification and identification of deletion genetic subtypes in Prader-Willi syndrome. Genet Test Mol Biomarkers, 2012;16(3):178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roof E, Stone W, MacLean W, Feurer ID, Thompson T, Butler MG. (2000). Intellectual characteristics of Prader-Willi syndrome: comparison of genetic subtypes. J Intellect Disabil Res, 44 ( Pt 1):25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoffstall AJ, Gaebler JA, Kreher NC, Niecko T, Douglas D, Strong TV, … Butler MG (2016). The High Direct Medical Costs of Prader-Willi Syndrome. J Pediatr, 175, 137–143. doi: 10.1016/j.jpeds.2016.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]