

Abstract
Background
BRCA1-mutated cancers exhibit deficient homologous recombination (HR) DNA repair, resulting in extensive copy number alterations and genome instability. HR deficiency can also arise in tumors without a BRCA1 mutation. Compared with other breast tumors, HR-deficient, BRCA1-like tumors exhibit worse prognosis but selective chemotherapeutic sensitivity. Presently, patients with triple negative breast cancer (TNBC) who do not respond to hormone endocrine-targeting therapy are given cytotoxic chemotherapy. However, more recent evidence showed a similar genomic profile between BRCA1-deficient TNBCs and hormone-receptor-positive tumors. Characterization of the somatic alterations of BRCA1-like hormone-receptor-positive breast tumors as a group, which is currently lacking, can potentially help develop biomarkers for identifying additional patients who might respond to chemotherapy.
Methods
We retrained and validated a copy-number-based support vector machine (SVM) classifier to identify HR-deficient, BRCA1-like breast tumors. We applied this classifier to The Cancer Genome Atlas (TCGA) and Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) breast tumors. We assessed mutational profiles and proliferative capacity by covariate-adjusted linear models and identified differentially methylated regions using DMRcate in BRCA1-like hormone-receptor-positive tumors.
Results
Of the breast tumors in TCGA and METABRIC, 22% (651/2925) were BRCA1-like. Stratifying on hormone-receptor status, 13% (302/2405) receptor-positive and 69% (288/417) triple-negative tumors were BRCA1-like. Among the hormone-receptor-positive subgroup, BRCA1-like tumors showed significantly increased mutational burden and proliferative capacity (both P < 0.05). Genome-scale DNA methylation analysis of BRCA1-like tumors identified 202 differentially methylated gene regions, including hypermethylated BRCA1. Individually significant CpGs were enriched for enhancer regions (P < 0.05). The hypermethylated gene sets were enriched for DNA and chromatin conformation (all Bonferroni P < 0.05).
Conclusions
To provide insights into alternative classification and potential therapeutic targeting strategies of BRCA1-like hormone-receptor-positive tumors we developed and applied a novel copy number classifier to identify BRCA1-like hormone-receptor-positive tumors and their characteristic somatic alteration profiles.
Electronic supplementary material
The online version of this article (10.1186/s13058-018-1090-z) contains supplementary material, which is available to authorized users.
Keywords: BRCA1, BRCAness, Breast cancer, DNA methylation, Homologous recombination, Precision medicine
Background
Germline mutation in the BRCA1 gene is associated with an increased lifetime risk of breast cancer alongside earlier disease onset and predisposition to the more aggressive triple-negative disease subtype [1–4]. The enhanced risk and high penetrance of breast cancer due to a BRCA1 germline mutation are attributable to the tumor-suppressor role of the BRCA1 protein, which modulates homologous recombination (HR)-dependent DNA repair [4–6]. BRCA1-related HR deficiency is associated with large-scale chromosomal breaks, extensive copy number alterations, and genome instability [7, 8]. However, HR deficiency is not limited to cancers carrying a BRCA1 mutation. Epigenetic inactivation of BRCA1, as well as germline or somatic alteration of other HR-family genes, can serve as alternative mechanisms driving HR deficiency, resulting in a BRCA1-like phenotype also known as BRCAness [3, 4, 9–11]. Similar to BRCA1-mutated cancers, BRCA1-like cancers are aggressive and typically exhibit poor prognosis. However, BRCA1-like cancers are more sensitive to chemotherapy, evidenced in both experimental work and patient studies [4, 10–12]. Lacking HR DNA repair can selectively sensitize BRCA1-deficient cancer cells to DNA cross-linking, alkylating, and double stranded break-inducing agents as well as poly-ADP ribose polymerase (PARP) inhibitors [5, 6], and improved survival outcomes are observed after high-dose platinum-based chemotherapeutic and PARP inhibitor treatment in patients with BRCA1-like breast tumors [10, 11, 13]. The TALORx trial evaluating the potential benefit of chemo-endocrine versus endocrine therapy alone in patients with hormone-receptor-positive human epidermal growth factor 2 receptor (HER2)-negative cancer and intermediate OncotypeDX, recurrence scores showed mostly equivocal results [14]. However, some benefit of chemotherapy was observed in younger women with intermediate scores, potentially attributable to responses in patients with hormone-receptor-positive BRCA1-like tumors, which are diagnosed at a significantly younger age than non-BRCA1-like tumors.
BRCA1-like breast tumors harbor extensive, characteristic genomic alterations. Genomic analyses show the distinct molecular patterning of BRCA1-mutated, HR-deficient cancers compared to BRCA2-mutated amd HR-proficient cancers [15–17]. Another pronounced feature of BRCA1-like, HR-deficient cancers is the extensive copy number alterations. This molecular hallmark motivated the classification of HR-deficient tumors based on their copy number profiles. Initially, array comparative genomic hybridization (aCGH) copy number was used to characterize the BRCA1-like phenotype and led to the development of a tool to predict breast cancer in patients with a BRCA1 mutation or promoter hypermethylation [10, 11, 18]. The aCGH copy-number features that distinguish BRCA1-like tumors led to the development of the BRCA1ness-MLPA assay, an experimental gold standard currently being tested in the clinical setting [19, 20]. More recently, the classification of HR deficiency has been adapted to measurement of copy number using higher-resolution approaches [11, 21].
A few studies have begun to characterize the molecular differences associated with BRCA1-related HR deficiency. HR-deficient cancers tend to exhibit more severe mutational burden and distinct mutational signatures [3, 15, 22]. Transcriptome-wide alterations have also been reported and used for defining HR-deficient gene signatures [12, 23, 24]. Further, HR deficiency is associated with global epigenetic changes and aberrant methylation of several HR family genes in cultured cells [25, 26]. However, these initial assessments of BRCA1-like molecular or cellular profiles often had limited sample sizes and varying results. Moreover, a description of biological differences between BRCA1-like and non-BRCA1-like tumors in large-scale cancer cohorts is currently lacking. Further, while prior work has shown the highly dysregulated epigenetic landscape in breast tumors compared to the normal breast, especially at early stages of cancer [2, 27], little is known about the epigenetic patterning of HR-deficient, BRCA1-like breast tumors relative to their non-BRCA1-like counterpart.
Here, we retrained and evaluated a classifier to identify BRCA1-like tumors using genome-wide copy number profiles, which can be measured by multiple platforms including genotyping array, methylation array, and next-generation sequencing [21]. We then applied this classifier to identify tumors exhibiting the HR-deficient, BRCA1-like phenotype in two large-scale breast cancer cohorts: The Cancer Genome Atlas (TCGA) [2] and the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) cohorts [28, 29]. In TCGA, for example, we detected nearly one third of breast tumors ofh the BRCA1-like phenotype, while only approximately 3% tumors had a BRCA1 somatic alteration. Subsequently, we compared molecular, clinical, and epigenetic characteristics associated with the BRCA1-like phenotype, restricting our analyses to hormone-receptor-positive breast cancer (i.e. breast tumors expressing estrogen receptor (ER), progesterone receptor (PR), and/or human epidermal growth factor receptor 2 (HER2). We focused on these tumors because we anticipate their distinct molecular profile, which could render them responsive to the cytotoxic chemotherapy typically given to patients with triple negative breast cancer (TNBC) [30–32].
Materials and methods
Training, testing, and experimental validation of the BRCA1-like classifier
Data sets and samples
The training data set for developing a support vector machine (SVM) BRCA1-like classifier consists of a relatively balanced number of BRCA1-like and non-BRCA1-like breast tumors from the Netherlands Cancer Institute (Joosse data set, GSE9021 and GSE9114, n = 74 total) [16, 17]. The BRCAx data set, which consists of BRCA1/2-like and non-BRCA1/2-like breast tumors measured on the same aCGH copy number array platform of the Netherland Cancer Institute (GSE18626, n = 106) [33], was used as the independent validation set (Table 1). Breast cancer cell lines with genome-wide copy number data (available from the Cancer Cell Line Encyclopedia (CCLE) [34]) and BRCA1ness-MLPA assay profiles (seven collected in-house, three recently published [35]) were used as an additional validation set (Additional file 1: Table S1).
Table 1.
Data sets used in this study. Number of samples (n) for a given data set indicates those with support vector machine (SVM)-predicted BRCA1-like status based on copy number data
| Data set (n) | Percentage ER positivitya | Percentage Predicted BRCA1-like | Purpose | Data types analyzed | Accession (if applicable) | Refs. |
|---|---|---|---|---|---|---|
| Joosse et al. (74) | 42.9 (27/63) | 47.3 (35/74) | BRCA1-like classifier training | Copy number | GSE9021, GSE9114 | [16, 17] |
| BRCAx (106) | 70.7 (41/58) | 19.8 (21/106) | BRCA1-like classifier validation | Copy number | GSE18626 | [33] |
| CCLE breast cancer cell lines (10) | 10.0 (1/10) | 60.0 (6/10) | BRCA1-like classifier experimental validation | Copy number, MLPA | Additional file 1: Table S1; portals.broadinstitute.org/ccle | [34, 35] |
| TCGA breast cancer (957) | 77.1 (704/913) | 32.2 (308/957) | BRCA1-like differential analyses | Copy number, mutation, gene expression, DNA methylation, clinical |
synapse.org
;
gdc.cancer.gov; gdac.broadinstitute.org ; |
[2, 39–41] |
| METABRIC (1968) | 76.3 (1501/1968) | 17.4 (343/1968) | BRCA1-like differential analyses | Copy number, gene expression, clinical | cbioportal.org | [28, 29] |
ER estrogen receptor, TCGA The Cancer Genome Atlas, CCLE Cancer Cell Line Encyclopedia, METABRIC Molecular Taxonomy of Breast Cancer International Consortium
aER status is not reported, is unknown, or is equivocal in a subset of Joosse et al., BRCAx and TCGA breast tumors. Such tumors were excluded from percentage ER positivity calculation
Classifier training and testing
Figure 1 summarizes the workflow for development and application of the BRCA1-like classifier in this study. Briefly, the training set hg18 copy number features previously known to distinguish BRCA1-like and non-BRCA1-like breast tumors [10, 16] were mapped onto the hg19 reference assembly. Genomic annotation files used for lift-over were downloaded from the UCSC Genome Browser (hgdownload.cse.ucsc.edu). We then used an in-house algorithm to map and normalize segmented copy numbers as shown in Additional file 2: Figure S1.
Fig. 1.

Workflow for developing a support vector machine (SVM) BRCA1-like classifier and application to publicly available datasets for biological discovery. In step 1, a new SVM-based BRCA1-like classifier is trained on re-processed and normalized array copy number data. In step 2, the receiver-operation characteristic (ROC) curves were used for evaluating our BRCA1-like classifier in a training and test set (AUC training = 1.00, AUC test = 0.75). In step 3, we applied the SVM classifier to tumors in the The Cancer Genome Atlas (TCGA) and Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) data sets. Finally, in step 4, we performed bioinformatics and statistical analyses attempting to understand the biological characteristics of hormone-receptor-positive breast tumors predicted to be BRCA1-like by our SVM classifier
Next, normalized gene copy number profiles were used to retrain a support vector machine (SVM) BRCA1-like classifier in a similar manner as in published reports [11, 21]. Briefly, our BRCA1-like classifier solves the following optimization problem:
where θi, θ0 are model weights and regression intercept for the i-th tumor, respectively. C (= 8.00) and ζi are SVM hyperparameters. We applied sigmoidal kernel smoothing and ten-fold cross validation. The probability of the i-th sample belonging to the BRCA1-like class was calculated as:
where xi is a vector of normalized copy number features and yi is the binary BRCA1-like class (yi = 1 indicates BRCA1-like). Above the probabilistic threshold of 0.50 (i.e. Pr(yi = 1) > Pr(yi = 0)), a tumor is defined as being BRCA1-like. The SVM classifier training and performance evaluation were implemented in the e1071 and pROC R package, respectively [36–38].
Experimental validation of the retrained BRCA1-like classifier
To further validate the SVM classifier, the BRCA1ness-MLPA assay (MRC-Holland catalog number P376), a BRCA1-like experimental gold standard, was used [19]. To this end, the SVM BRCA1-like classifier was first applied to published breast cancer cell line copy number data measured by Affymetrix SNP6.0 array (available from portals.broadinstitute.org/ccle) [34]. Next, the BRCA1ness-MLPA assay was performed on breast cancer cell lines BT549, CAL51, CAL120, HCC1806, HDQ-P1, MDA-MB-231 and MDA-MB-468 obtained from American Type Culture Collection (ATCC), thawed and cultured < 4 months prior to assay. Authenticated by ATCC, the cell lines used are assumed to be mycoplasma-free. No further mycoplasma testing or authentication was performed after thawing: 200 ng DNA was precipitated using 0.5 μL of 20 mg/mL glycogen, 15 μL of 3 M sodium acetate, and 495 μL pure ethanol at − 20 °C for 3 h, followed by precipitation at 14,000*g for 30 min at 4 °C and a 70% ethanol rinse. Precipitated DNA was re-suspended at a final concentration of 25 ng/μL and solubilized by incubating at 37 °C for 1 h. DNA from three fresh-frozen normal breast DNA (source: National Disease Research Interchange) and RPE1 cell line were used as copy number-neutral controls. The subsequent steps were based on the BRCA1ness-MLPA kit instructions (MRC-Holland, the Netherlands): 100 ng DNA was mixed with S4 Stabilizer and denatured at 98 °C for 5 min. Probe-DNA hybridization was performed at 60 °C for ≥ 16 h, followed by a 15-min ligation at 54 °C and 5-min ligase inactivation at 98 °C. Then, 35 cycles of PCR were performed, each with 30-s denaturation at 95 °C, 30-s annealing at 60 °C, and 60-s extension at 72 °C, followed by a 20-min final extension at 72 °C. Fragment analysis was performed by capillary electrophoresis in technical triplicate on the ABI3730 DNA Analyzer (Thermo Fisher Scientific, Waltham, MA, USA). The Coffalyser.Net software with default peak detection setting was used. BRCA1-like classification was executed by the nearest shrunken centroid method implemented in the pamr R package (v.1.55), and a probabilistic threshold of 0.50 recommended by the manufacturer was used to call BRCA1-like tumors. MLPA profiles for three additional cell lines (HCC70, MCF7, and MDA-MB-436) were recently published by Roig et al. [35]. SVM BRCA1-like status and MLPA profiles of the breast cancer cell lines used are listed in Additional file 1: Table S1.
Correlation of SVM BRCA1-like probability scores with published HR deficiency metrics
The Homologous Recombination Deficiency-Loss of Heterozygosity (HRD-LOH) [8, 13] and Large Scale Transition (LST) [7] scores for 717 TCGA breast tumors were downloaded from Marquard et al. [39]. The correlation between SVM BRCA1-like probability scores and HRD-LOH scores or LST scores was assessed by linear regression.
BRCA1-like analyses
Data sets and samples
For the remainder of the study, we compared BRCA1-like and non-BRCA1-like receptor-positive breast tumors in two large-scale breast cancer cohorts: TCGA and METABRIC. SVM BRCA1-like status was estimated in the same manner as mentioned.
The TCGA breast cancer molecular and clinical data were downloaded from FireBrowse (gdac.broadinstitute.org), cBioPortal (cbioportal.org), SynapseTCGAlive (synapse.org), and the Genomic Data Commons (gdc.cancer.gov). We excluded 100 tumors for which TNBC status could not be ascertained and 18 additional BRCA1/2-mutated or BRCA1-hypermethylated tumors predicted to be non-BRCA1-like, leaving a total of 837 tumors (Additional file 1: Table S2A). DNA methylation and gene expression, measured by Illumina 450 K and RNA-seqV2/miRseq, respectively, were available for a subset of TCGA tumors.
METABRIC breast cancer clinical data, copy number, gene expression, and BRCA1/2 somatic mutation profiles were downloaded from cBioPortal (cbioportal.org). DNA methylation and mutational burden/signature were not available for this data set. Among breast tumors with SVM BRCA1-like status, a large proportion (519/1985) with missing tumor stage and 12 classified as stage 0 (ductal carcinoma in situ) were excluded. Additionally, 37 non-BRCA1-like tumors with a BRCA1/2 mutation were considered misclassified and were excluded, leaving 1429 tumors (Additional file 1: Table S2B).
Relation of genomic burden with BRCA1-like status
Mutational rates per mega base-pairs (Mb) were published by Kandoth et al. [40] and are available for 662 TCGA tumors. Given the large number of outliers and substantial variability in mutation rates, the association between mutation rates per Mb and SVM BRCA1-like status was assessed by a linear model with robust variance adjusting for subject age, tumor stage, and ER, PR, and HER2 positivity, implemented in R packages MASS (v.7.3.49), sandwich (2.4.0), and lmtest (v.0.9.35) with type = HC0. Somatic Mutational Signature 3, which is related to HR deficiency, and Mutational Signature 1, which is related to global CpG methylation, were published by Rosenthal et al. [41] and are available for 745 TCGA breast tumors. A linear model adjusting for the same potential confounders was used for comparison.
Survival analysis
A Cox proportional hazards regression model was used to test the relationship between overall survival and BRCA1-like status. For this analysis, we combined TCGA and METABRIC data sets and restricted our analysis to ER-positive or PR-positive and HER2-negative patients as follows:
where λi and λ0 are hazard for i-th patient and baseline hazard assumed to be constant, respectively. t represents overall survival time in months, SVM_BRCA1 represents the BRCA1-like status, Age represents age at diagnosis in years, and Stage is a binary variable representing early stage (II or lower) or late stage (III or higher). Administrative censoring was imposed at 5 years (60 months) of follow up. Cox regression and data visualization were implemented in R packages survival (v.2.41.3) and survminer (v.0.4.2).
DNA methylation processing and analysis
In the TCGA breast cancer data set, level 1 methylation intensity data files from the Illumina 450 K array were preprocessed by the minfi R/Bioconductor package (v.1.20). Based on the overall methylated-to-unmethylated intensity ratio, four samples classified as poor-performing outliers were excluded. CpG probes with P > 0.05 for detection in more than 20% samples were excluded from downstream analysis. The quality control procedure left 464,028 high-quality CpG probes in the final data set. BRCA1 promoter hypermethylation was determined using four array CpGs (cg19531713, cg19088651, cg08993267, and cg04658354) as previously described; samples with mean beta values ≥ 0.20 were defined as BRCA1 promoter-hypermethylated [42, 43].
To identify differentially methylated CpGs and gene regions in BRCA1-like tumors relative to non-BRCA1-like tumors predicted by the SVM classifier (n = 322 with no missing covariates), we applied the DMRcate algorithm (v.1.14.10) to 464,028 CpGs adjusting for subject age, tumor stage, and ER, PR, and HER2 status [44]. DMRcate first identified individually significant CpGs at a false discovery rate (FDR) < 0.05. A differentially methylated region (DMR) was then called if a given gene region had ≥ 10 significant CpGs within a 1-kb bandwidth and a Benjamini-Hochberg FDR < 0.05. Autosomal and chromosome X DMRs were identified separately to avoid bias driven by imprinting. Individually significant CpGs were used for genomic context enrichment against the 464,028-CpG universe set using the two-tailed Fisher’s exact test. CpGs with |%∆beta| ≥ 12.5% were used for unsupervised hierarchical clustering with Euclidean distance and complete linkage. DMR-associated genes were used as input for Gene Ontology: Biological Processes against the whole genome via the WebGestalt tool (webgestalt.org) [45]. The minimum and maximum number of genes required per pathway were 5 and 2000, respectively (default). Raw P values were adjusted by the Bonferroni method.
DNA methylation age (“epigenetic clock”) was inferred by applying the Horvath algorithm [46] to 450 K methylation beta-values in 322 breast tumors tested for differential methylation. To compare methylation age or chronological age between BRCA1-like and non-BRCA1-like tumors, separate linear models were built adjusting for tumor stage, ER, PR, and HER2 positivity.
Relation of candidate gene expression with BRCA1-like status
Linear models were used to compare Ki-67 (MKI67), DNMT1/3A/3B, and miR124–2 gene expression between BRCA1-like and non-BRCA1-like receptor-positive tumors adjusting for age, tumor stage, and ER, PR, and HER2 positivity.
Results
Development of a copy-number-based BRCA1-like classifier
Prior studies demonstrated the utility of array comparative genomic hybridization (aCGH) copy number profiles for BRCA1-like classification in breast tumors [16, 17, 21, 33]. To enable identification of BRCA1-like tumors measured by a non-aCGH copy number platform or analyzed by a more up-to-date genome assembly, such as The Cancer Genome Atlas (TCGA) data set, we first mapped copy number features previously used for BRCA1-like classification to the human hg19 reference genome followed by data re-normalization using an in-house pipeline (Additional file 2: Figure S1A). We then retrained a BRCA1-like classifier using support vector machine (SVM), a robust supervised-learning method that seeks a class-separating hyperplane within higher dimensional data [36]. Our SVM BRCA1-like classifier achieved acceptable performance in the training and the independent test set (AUC training = 1.00, AUC test = 0.75, Additional file 2: Figure S1B). In the Cancer Cell Line Encyclopedia (CCLE) data set with publicly available copy number data, SVM BRCA1-like status in breast cancer cell lines is 80% (8/10) concordant with the BRCA1ness-MLPA assay profile measured in house (Additional file 1: Table S1). When applied to TCGA breast tumors, SVM BRCA1-like probability scores were highly correlated with existing HR deficiency metrics [39] (both P < 2.2E-16; Additional file 2: Figure S1C-D). Figure 1 summarizes the workflow for the present study.
Molecular and clinical characteristics related with the BRCA1-like phenotype
As expected, a large proportion (69%, 288/417) of all triple negative breast cancer (TNBC) was predicted to be BRCA1-like [47, 48] (Table 2, Additional file 1: Table S3 and Fig. 2). Among all BRCA1-like tumors, 36% (237/651) were positive for estrogen receptor (ER) and 14% (93/651) for human epidermal growth factor 2 (HER2). In addition, in the TCGA data set where race information is available, 21.3% (60/282) of African American subjects were classified as having BRCA1-like tumors compared to 10.4% (61/585) classified as having non-BRCA1-like tumors (Additional file 1: Table S3A). In other words, there is a 2.32-fold (95% CI = 1.54–3.49, P = 2.59E-5) increase in the proportion of African American subjects classified as having BRCA1-like compared to non-BRCA1-like tumors, and the increased prevalence of TNBC in African Americans is established [22, 49] (Additional file 1: Table S3A). We also noted the difference in BRCA1-like probability score distribution between TCGA and METABRIC. This difference could be explained by the differential subject characteristics including younger age at diagnosis and higher cancer stage in the TCGA than the METABRIC data set (linear regression P = 2.62-E7 and Fisher’s exact test P < 2.2E-16, respectively).
Table 2.
Prevalence of BRCA1-like phenotype in the large-scale breast cancer cohorts, TCGA and METABRIC
| TCGA | METABRIC | |||||
|---|---|---|---|---|---|---|
| BRCA1-like | non-BRCA1-like | P value § | BRCA1-like | non-BRCA1-like | P value § | |
| n | 308 | 649 | 343 | 1625 | ||
| Age, years (mean (sd)) | 56.57 (13.26) | 59.33 (13.06) | 0.002 | 56.74 (13.93) | 62.02 (12.56) | < 0.001 |
| Stage (%) | 0.22 | 0.35 | ||||
| Stage I-II | 238 (77.3) | 473 (72.9) | 220 (64.1) | 1106 (68.1) | ||
| Stage III-IV | 63 (20.5) | 165 (25.4) | 26 (7.6) | 102 (6.3) | ||
| (Missing) | 7 (2.3) | 11 (1.7) | 97 (28.3) | 417 (25.7) | ||
| ER (%) | < 0.001 | < 0.001 | ||||
| Positive | 129 (41.9) | 575 (88.6) | 108 (31.5) | 1393 (85.7) | ||
| Negative | 166 (53.9) | 43 (6.6) | 235 (68.5) | 232 (14.3) | ||
| (Missing) | 13 (4.2) | 31 (4.8) | 0 (0.0) | 0 (0.0) | ||
| PR (%) | < 0.001 | < 0.001 | ||||
| Positive | 97 (31.5) | 514 (79.2) | 57 (16.6) | 980 (60.3) | ||
| Negative | 195 (63.3) | 104 (16.0) | 286 (83.4) | 645 (39.7) | ||
| NA | 16 (5.2) | 31 (4.8) | 0 (0.0) | 0 (0.0) | ||
| HER2 (%) | 0.59 | 0.98 | ||||
| Positive | 50 (16.2) | 93 (14.3) | 43 (12.5) | 200 (12.3) | ||
| Negative | 161 (52.3) | 333 (51.3) | 300 (87.5) | 1425 (87.7) | ||
| (Missing) | 97 (31.5) | 223 (34.4) | 0 (0.0) | 0 (0.0) | ||
| Any ER, PR, or HER2 positivity (%) | < 0.001 | < 0.001 | ||||
| Yes | 159 (51.6) | 595 (91.7) | 143 (41.7) | 1508 (92.8) | ||
| No | 88 (28.6) | 12 (1.8) | 200 (58.3) | 117 (7.2) | ||
| Cannot be determined | 61 (19.8) | 42 (6.5) | 0 (0.0) | 0 (0.0) | ||
TCGA The Cancer Genome Atlas, METABRIC Molecular Taxonomy of Breast Cancer International Consortium, ER estrogen receptor, PR progesterone receptor, HER2 human epidermal growth factor receptor 2
§For any continuous measure (i.e. age), the two-tailed t-test was used. For any categorical measure, the two-tailed Fisher’s exact test was used
Fig. 2.
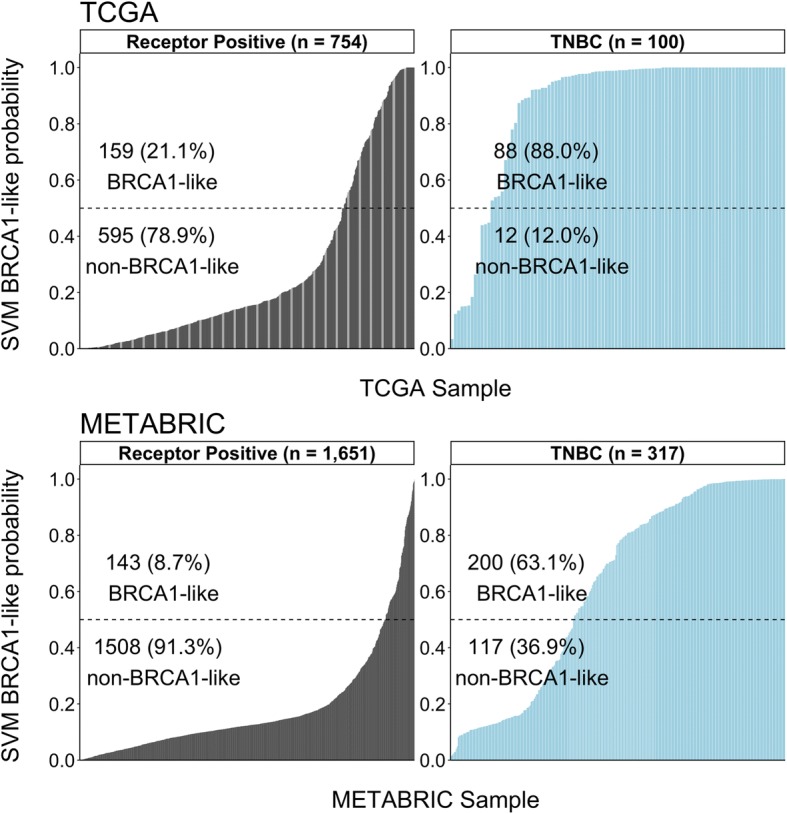
Distribution of BRCA1-like probability scores in The Cancer Genome Atlas (TCGA) (top panel) and Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) (bottom panel) data sets, stratified by hormone-receptor-based subtypes. Each vertical bar represents a patient. The height of the bar represents the probability score of being BRCA1-like assigned by our support vector machine (SVM) copy number classifier. TNBC, triple negative breast cancer
We detected 13% (302/2405) hormone-receptor-positive patients with breast cancer to have BRCA1-like tumors. Recent studies suggest that hormone-receptor-positive tumors could also exhibit HR deficiency and potentially benefit from chemotherapeutic treatments [30–32]. Hereafter, we restricted our analyses to hormone-receptor-positive breast tumors (Table 2 and Additional file 1: Table S3). In TCGA, BRCA1-like tumors exhibit greater mutational burden than their non-BRCA1-like counterpart (P < 0.01, Fig. 3a). Somatic Mutational Signature 3, inferred from exome sequencing and strongly related to HR deficiency [3, 15, 22, 50], was significantly elevated in BRCA1-like tumors (P < 0.001, Fig. 3b). These tumors also demonstrated enhanced proliferative capacity, indicated by increased Ki-67 gene expression (P < 0.001, Fig. 3c and Additional file 2: Figure S2). In addition, BRCA1-like status appeared to have a harmful association with 5-year overall survival based on Kaplan-Meier analysis in ER/PR-positive, HER2-negative tumors, and although the hazards ratio estimate indicated poorer prognosis in models adjusted for potential confounders, the results were not statistically significant (Additional file 2: Figure S3).
Fig. 3.

Molecular characteristics of support vector machine (SVM) BRCA1-like hormone-receptor-positive tumors in The Cancer Genome Atlas (TCGA). Comparison of whole exome sequencing-based mutation rates per mega base-pair (Mb) (a) and Somatic Mutational Signature 3 related with HR-deficiency (b), and Ki-67 (MKI67) gene expression as a surrogate marker for cellular proliferation (c). Variations in the number of tumors were due to data availability (see “Materials and methods”). *P < 0.05, **P < 0.01, ***P < 0.001
Epigenetic characteristics of hormone-receptor-positive BRCA1-like breast tumors
Promoter hypermethylation of BRCA1 or other HR-family genes (e.g. RAD51C) is a known mechanism driving HR deficiency and BRCAness [2–4, 15, 22, 31, 42]. Inactivation of BRCA1 by promoter hypermethylation also shows higher prevalence in the triple-negative subtype [2, 4, 48]. However, the relationships between the genome-scale DNA methylation pattern and the HR-deficient phenotype in hormone-receptor-positive breast cancer has not been previously reported. We applied the DMRcate algorithm to identify individual cytosine-phosphate-guanine (CpG) sites and genomic regions harboring differential methylation in BRCA1-like relative to non-BRCA1-like tumors identified by our SVM BRCA1-like classifier [44]. This approach identified 350 CpGs with a FDR < 0.05 and |% ∆beta| ≥ 12.5%, which we define as differentially methylated. Unsupervised hierarchical clustering of these differentially methylated loci separated tumors into two major clusters. Methylation of CpGs in the “BRCA1-like Cluster” exhibited greater heterogeneity compared to CpGs in the “non-BRCA1-like cluster” (Additional file 2: Figure S5; mean inter-sample variances in methylation beta-values for BRCA1-like cluster are 0.0527 and 0.0371, respectively).
We next investigated the biological relevance of the differentially methylated CpG loci and gene sets. Hypermethylated CpGs associated with SVM BRCA1-like status determined from the SVM classifier were enriched for CpG islands but not for promoter regions. Stratified by direction of the change in methylation, 202 of 350 hypomethylated CpGs overrepresented gene promoters (OR = 1.68, 95% CI = 1.25–2.24) and underrepresented enhancers (OR = 0.46, 95% CI = 0.30–0.68). There were 48 out of 350 differentially hypermethylated CpGs that were enriched for DNase I hypersensitivity sites associated with active chromatin and gene transcription (OR = 2.95, 95% CI = 2.02–4.23). Intriguingly, the hypermethylated and hypomethylated CpGs overrepresented and underrepresented the “CpG Island” genomic context, respectively (OR Hyper = 2.75, 95% CI Hyper = 1.96–3.87; OR Hypo = 0.39, 95% CI Hypo = 0.26–0.57). Both sets significantly underrepresented the “Open Sea” genomic context that has low CpG density (OR Hyper = 0.45, 95% CI Hyper = 0.29–0.67; OR Hypo = 0.67, 95% CI Hypo = 0.48–0.92) (Fig. 4a).
Fig. 4.

Differential DNA methylation in groups with BRCA1-like tumors relative to groups with non-BRCA1-like tumors defined by the support vector machine (SVM) BRCA1-like classifier among hormone-receptor-positive tumors in The Cancer Genome Atlas (TCGA). a Genomic context enrichment analysis of hypermethylated and hypomethylated CpGs. Solid dots and horizontal segments indicate odds ratios and 95% confidence intervals. P values were from the two-tailed Fisher’s exact test. b Correlation of the Horvath methylation age (“epigenetic clock”) with patient chronological age, colored by BRCA1-like status. c Comparison of chronological age or the Horvath methylation age between BRCA1-like and non-BRCA1-like hormone-receptor-positive tumors. P value indicates statistical significance from the covariate-adjusted linear model. n.s., not significant
There were 108 and 94 gene regions that were hypermethylated and hypomethylated in BRCA1-like tumors, respectively (all with FDR < 0.05 and a minimum of 10 CpGs per kb; Additional file 1: Table S5A-B). The BRCA1 locus had increased methylation in BRCA1-like tumors. In line with this key observation, FANCF, another member of the BRCA1/Fanconi anemia pathway [51], was also hypermethylated (Additional file 1: Table S5A). Hypermethylation of developmentally related genes, including HOXB13, HOXD3/12, and FOXR1, suggested that developmental signaling might be dysregulated in BRCA1-like tumors (Additional file 1: Table S5A). Many histone genes were also hypermethylated, hinting at potentially elevated DNA damage and genome instability in BRCA1-like tumors. miR124–2, a microRNA that negatively regulates cellular proliferation in breast cancer [52], showed hypermethylation and reduced expression in BRCA1-like tumors (P < 0.05; Additional file 2: Figure S4). Among the hypomethylated genes, E3 ubiquitin ligases (HUWE1, UNKL, and VHL) that also play a role in cell-cycle regulation [53] showed reduced methylation in BRCA1-like receptor-positive tumors (Additional file 1: Table S5B). Applying Gene Ontology: Biological Processes to 158 genes associated with the hypermethylated DMRs, we identified DNA conformation and chromatin assembly-related gene sets to be most hypermethylated (all with FDR < 0.05; Table 3). Gene sets related to developmental signaling and the cell cycle had mild enrichment (Additional file 1: Table S6A).
Table 3.
Gene Ontology: Biological Processes for genes associated with hypermethylated gene regions
| Accession | Description | Number of genes | Number of input genes | Expected ratio | Observed ratio | P value | Bonferroni P value |
|---|---|---|---|---|---|---|---|
| GO:0071103 | DNA conformation change | 248 | 10 | 1.39 | 7.20 | 1.24E-06 | 0.011 |
| GO:0006323 | DNA packaging | 165 | 8 | 0.92 | 8.66 | 3.95E-06 | 0.017 |
| GO:0031497 | Chromatin assembly | 128 | 7 | 0.72 | 9.77 | 7.44E-06 | 0.021 |
| GO:0065004 | Protein-DNA complex assembly | 197 | 8 | 1.10 | 7.26 | 1.45E-05 | 0.031 |
| GO:0006333 | Chromatin assembly or disassembly | 153 | 7 | 0.86 | 8.17 | 2.38E-05 | 0.041 |
| GO:0006334 | Nucleosome assembly | 111 | 6 | 0.62 | 9.66 | 3.68E-05 | 0.045 |
| GO:0071824 | Protein-DNA complex subunit organization | 224 | 8 | 1.25 | 6.38 | 3.65E-05 | 0.045 |
The complete list of pathways can be found in Additional file 1: Table S6A
Given the hypermethylation of many developmentally related genes and detection of developmental gene sets, we compared the DNA methylation age (“epigenetic clock”), a metric related to aging, cell-culture passage, and differentiation potential [46], between BRCA1-like and non-BRCA1-like tumors. Adjusting for tumor stage and hormone-receptor positivity, DNA methylation age was significantly lower in patients with BRCA1-like tumors (median 60.7 years, compared to non-BRCA1-like median 70.5 years; P < 0.05; Fig. 4b-c).
To confirm a distinct global DNA methylation landscape in BRCA1-like tumors, we compared gene expression of the de novo methyltransferases DNMT3A/3B and the maintenance methyltransferase DNMT1 [54] between BRCA1-like and non-BRCA1-like tumors. All three methyltransferases were overexpressed in BRCA1-like tumors (all P < 0.001; Additional file 2: Figure S6). Likewise, Mutational Signature 1, contributed to by genome-wide cytosine-to-thymine deamination that acts on unmethylated NpCpG sequences [50], was significantly lower in BRCA1-like tumors (P < 0.01; Additional file 2: Figure S7).
Discussion
In this study, we retrained a BRCA1-like classifier using genome-wide copy number and validated the classifier by in silico and experimental approaches. We estimated that 22% of all TCGA and METABRIC breast tumors were BRCA1-like, consistent with existing literature [4, 15]. Notably, 13% hormone-receptor-positive tumors were BRCA1-like. Therapeutic strategies such as cytotoxic chemotherapy more commonly used in the triple-negative disease setting might be an effective alternative for treating these tumors.
Among hormone-receptor-positive breast tumors, the BRCA1-like phenotype is associated with increased mutational burden, as demonstrated by elevated mutation rates. Expression of Ki-67, a surrogate marker for cellular proliferation, was increased in BRCA1-like receptor-positive tumors. These molecular hallmarks serve as evidence supporting the more aggressive character of BRCA1-like tumors.
The genome-scale DNA methylation profile of BRCA1-like tumors, identified by our SVM classifier, appeared distinct. Furthermore, we detected hypermethylation of gene sets related to chromatin and nucleosome assembly. Of note, the BRCA1 locus showed increased DNA methylation in BRCA1-like tumors, supporting the existing concept that hypermethylation of HR-family genes could serve as a driver for HR deficiency. Detecting hypermethylation and reduced gene expression of miR124–2, a negative regulator of cell proliferation [52], is consistent with our Ki-67 gene expression analysis and further supports BRCA1-like tumors having a more aggressive phenotype.
Subsequently, differential methylation analysis comparing SVM-predicted BRCA1-like and non-BRCA1-like tumors identified 202 hypomethylated and 148 hypermethylated CpGs. Unsupervised hierarchical clustering of all 350 CpGs revealed a distinct “BRCA1-like cluster”, implying the potential utility of genome-scale DNA methylation as another biomarker to identify HR-deficient cancers, possibly in breast cancer biopsies shown to have similar methylation profiles to larger surgical blocks [55]. We also noticed the increased heterogeneity in this cluster relative to the “non-BRCA1-like cluster”, a hallmark of aggressive cancers [56]. In addition, this finding parallels the prior observation that when compared to normal-adjacent breast tissue, breast tumors exhibit increased heterogeneity [2]. Moreover, our precise identification of differentially methylated CpGs, genes and gene sets allows focused investigation in the future, thereby enabling the identification of effective pharmacologic and therapeutic strategies in the future.
We observed members of the HOX gene cluster to be hypermethylated. In line with this, many developmentally related pathways were found to be mildly enriched though not statistically significantly. These findings indicate that development and differentiation-related signaling pathways are characteristic of the HR-deficient, BRCA1-like phenotype. We followed up with this postulate by comparing DNA methylation age - a metric inferred from genome-scale DNA methylation profiles and related to cellular differentiation potential [46] – between BRCA1-like and non-BRCA1-like tumors. In line with our differential methylation and pathway analysis, DNA methylation age was significantly lower in BRCA1-like tumors indicating a more poorly differentiated tumor state. These observations were overall consistent with prior works demonstrating that tumors with BRCA1/2-related HR deficiency tend to be poorly differentiated or undifferentiated [57].
Recent studies have shown that BRCA1-deficient and BRCA2-deficient genomes, despite both having HR loss, may nevertheless differ [15–17]. Therefore, to better understand HR deficiency and chemotherapeutic sensitivity, development and characterization of molecular signatures that more broadly characterize the HR-deficient phenotype may be necessary.
One challenge was identifying HR-deficient, BRCA1-like tumors using a strict probabilistic threshold. Here, we used the cutoff of 0.50, which could be rather conservative. Despite having used a robust cross validation-based machine learning approach, there will be opportunities in the future to potentially improve the performance of our SVM classifier, with better balance among breast cancer subtypes in the training data. We acknowledge the limitation of cell lines in the experimental validation set, and note that future studies would benefit from inclusion of larger human sample sets for validation. Biologically, as seen in the TALORx trial where younger patients (age < 50 years) had improved chemotherapy response [14], we also suspected that confounders such as patient age strata could influence the performance of BRCA1-like classifiers and the molecular characteristics of these tumors. While our Kaplan-Meier analysis showed some evidence that ER/PR-positive, HER2-negative breast cancer with the BRCA1-like phenotype was associated with worse overall survival, our results were not statistically significant in a covariate-adjusted Cox regression model. A possible explanation is the heterogeneity of the treatment regimens among study participants. We therefore anticipate that the application of our SVM BRCA1-like classifier to cohorts with more consistent treatment will have greater clinical value.
Conclusions
In this work, we applied a copy-number-based classifier to identify breast tumors with the BRCA1-like phenotype. Among breast tumors expressing ER, PR, and/or HER2, we found evidence for previously unknown molecular alterations, including enhanced mutational burden and proliferative capacity, to be associated with the BRCA1-like phenotype. Importantly, we demonstrated that genome-wide DNA methylation profiles differ substantially in HR-deficient, BRCA1-like cancers. The BRCA1-like phenotype may ultimately contribute to increased heterogeneity of molecular alterations in this tumor subset [56], a common characteristic of aggressive but more treatable cancers.
Additional files
Table S1. SVM BRCA1-like status and BRCA1ness-MLPA profiles in 10 breast cancer cell lines. Table S2A. TCGA breast tumors with SVM BRCA1-like status. Table S2B. METABRIC breast tumors with SVM BRCA1-like status. Table S3. Complete subject characteristics of TCGA and METABRIC breast tumors with SVM BRCA1-like status. Table S4. Differentially methylated CpGs in BRCA1-like tumors identified by DMRcate. Table S5A. Hypermethylated DMRs (n = 108) from DMRcate analysis. Table 5SB. Hypomethylated DMRs (n = 94) from DMRcate analysis. Table S6A. GOBP terms associated with 158 unique genes from 108 hypermethylated DMRs. Table S6B. GOBP terms associated with 131 unique genes from 94 hypermethylated DMRs. (XLSX 923 kb)
Figure S1. Details of SVM BRCA1-like classifier. (A) Overview of copy number mapping algorithm for generating the input for training the SVM BRCA1-like classifier. (B) Receiver-operation characteristic curves (ROC) of the classifier applied to training and test set (AUC = 1.00 and 0.75, respectively). (C-D) Correlation of SVM BRCA1-like probability scores with published HR-deficiency metrics (HRD-LOH and LST scores). ***P < 0.001. Figure S2. Comparison of Ki-67 (MKI67) gene expression as a surrogate marker for cellular proliferation in METABRIC hormone-receptor-positive breast tumors. P value indicates statistical significance from a linear model adjusting for age, tumor stage, ER, PR and HER2 positivity. ***P < 0.001. Figure S3. Five-year overall survival comparison between BRCA1-like and non-BRCA1-like ER-positive/PR-positive, HER2-negative breast tumors in TCGA and METABRIC (combined). Table inset shows hazards ratio (95% CI) and P value from Cox proportional hazards regression adjusting for potential confounders. ***P < 0.001. Figure S4. miR124–2 with hypermethylation exhibit reduced gene expression in TCGA BRCA1-like receptor positive tumors. P value indicates statistical significance from a linear model adjusting for age, tumor stage, and ER, PR, and HER2 positivity. *P < 0.05. Figure S5. Comparison of heterogeneity between the “BRCA1-like methylation cluster” and “non-BRCA1-like methylation cluster” generated by hierarchical clustering of 350 most differential CpGs identified by DMRcate (all FDR <0.05 and |log2∆beta| ≥ 3.50). (A) Heat map showing unsupervised clustering (Euclidean distance, complete linkage) of the 350 DMRcate-identified CpGs. (B) Rank-ordered inter-sample variance in beta-values of the 350 differentially methylated CpGs. Horizontal dotted lines indicate mean inter-sample variance distribution for each group. Figure S6. Differential gene expression of DNA methyltransferases (DNMT1/3A/3B) in TCGA receptor-positive BRCA1-like breast tumors. A P value indicates statistical significance from linear model adjusting for age, tumor stage, and ER, PR, and HER2 positivity. ***P < 0.001. Figure S7. Comparison of Somatic Mutational Signature 1 contributed to by genome-wide cytosine-to-thymine (C > T) deamination events in TCGA hormone-receptor-positive breast tumors. A P value indicates statistical significance from linear model adjusting for age, tumor stage, and ER, PR, and HER2 positivity. **P < 0.01. (PDF 3058 kb)
Acknowledgements
We acknowledge the U.S. National Institute of Health for funding support (R01DE022772, R01CA216265, and P20GM104416/6369 to BCC, KL2TR001088 to CC, and P20GM113132 to ANK), the Norris Cotton Cancer Center Prouty Pilot Funding to ANK and TWM, and the Burroughs-Wellcome Big Data in the Life Sciences Fellowship to YC. We thank the Dartmouth Molecular Biology Shared Resources and Research Computing for assistance.
Funding
NIH grants R01DE022772, R01CA216265, and P20GM104416/6369 to BCC, NIH/NCATS grant KL2TR001088 to CC, NIH/NIGMS grant P20GM113132 to ANK, NIH/NCI grants R01CA200994 and R01CA211869 to TWM, the Norris Cotton Cancer Center Prouty Pilot Funding to ANK/TWM/BCC, and the Burroughs-Wellcome Big Data in the Life Sciences Fellowship to YC.
Availability of data and materials
Breast tumor data sets used to develop the BRCA1-like classifier are publicly available in Gene Expression Omnibus [GEO:GSE9021, GEO:GSE9114, and GEO:GSE18626]. The Cancer Genome Atlas (TCGA) breast tumor molecular and clinical data are publicly available in the Genomic Data Commons (gdc.cancer.gov), FireBrowse (gdac.broadinstitute.org) and SynapseTCGAlive (synapse.org). The METABRIC breast tumor molecular and clinical data are publicly available in the cBioPortal database (cbioportal.org). Breast cancer cell line copy number profiles are available in the Cancer Cell Line Encyclopedia (CCLE) database (portals.broadinstitute.org/ccle).
Abbreviations
- 450 K
Illumina HumanMethylation450 BeadChip
- aCGH
Array comparative genomic hybridization
- CCLE
Cancer Cell Line Encyclopedia
- CpG
Cytosine-phosphate-guanine
- DMR
Differentially methylated region
- ER
Estrogen receptor
- HER2
Human epidermal growth factor receptor 2
- HR
Homologous recombination
- METABRIC
Molecular Taxonomy of Breast Cancer International Consortium
- PR
Progesterone receptor
- SVM
Support vector machine
- TCGA
The Cancer Genome Atlas
- TNBC
Triple negative breast cancer
Authors’ contributions
YC, YW, CC, and BCC conceived the study design and approach. YW, KM, and CC identified publicly available data sets, retrained, evaluated, and applied the copy number SVM BRCA1-like classifier. YC, JDM, TWM, and ANK contributed to design, collection, and interpretation of experiments. YC, YW, LS, CC, and BCC contributed to bioinformatics and statistical analysis. ANK, CC, and BCC supervised the project. YC, YW, CC, and BCC wrote the manuscript, and all authors reviewed the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Youdinghuan Chen, Email: Youdinghuan.Chen.GR@dartmouth.edu.
Yue Wang, Email: Yue.Wang@dartmouth.edu.
Lucas A. Salas, Email: Lucas.A.Salas.Diaz@dartmouth.edu
Todd W. Miller, Email: Todd.W.Miller@dartmouth.edu
Kenneth Mark, Email: Kenneth.M.Mark.GR@dartmouth.edu.
Jonathan D. Marotti, Email: Jonathan.D.Marotti@hitchcock.org
Arminja N. Kettenbach, Email: Arminja.N.Kettenbach@dartmouth.edu
Chao Cheng, Phone: +1 (713) 798-3332, Email: Chao.Cheng@bcm.edu.
Brock C. Christensen, Phone: +1 (603) 650-1828, Email: Brock.C.Christensen@dartmouth.edu
References
- 1.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nik-Zainal S, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chalasani P, Livingston R. Differential chemotherapeutic sensitivity for breast tumors with ‘BRCAness’: a review. Oncologist. 2013;18:909–916. doi: 10.1634/theoncologist.2013-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 6.Fong PC, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 7.Popova T, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 8.Abkevich V, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107:1776–1782. doi: 10.1038/bjc.2012.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 10.Vollebergh MA, et al. An aCGH classifier derived from BRCA1-mutated breast cancer and benefit of high-dose platinum-based chemotherapy in HER2-negative breast cancer patients. Ann Oncol. 2011;22:1561–1570. doi: 10.1093/annonc/mdq624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vollebergh MA, et al. Genomic patterns resembling BRCA1- and BRCA2-mutated breast cancers predict benefit of intensified carboplatin-based chemotherapy. Breast Cancer Res. 2014;16:R47. doi: 10.1186/bcr3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Severson TM, et al. The BRCA1ness signature is associated significantly with response to PARP inhibitor treatment versus control in the I-SPY 2 randomized neoadjuvant setting. Breast Cancer Res. 2017;19:99. doi: 10.1186/s13058-017-0861-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Telli ML, et al. Phase II study of gemcitabine, carboplatin, and iniparib as neoadjuvant therapy for triple-negative and BRCA1/2 mutation-associated breast cancer with assessment of a tumor-based measure of genomic instability: PrECOG 0105. J Clin Oncol. 2015;33:1895–1901. doi: 10.1200/JCO.2014.57.0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sparano, J. A. et al. Adjuvant chemotherapy guided by a 21-gene expression assay in breast cancer. N. Engl. J. Med. NEJMoa1804710 (2018). 10.1056/NEJMoa1804710 [DOI] [PMC free article] [PubMed]
- 15.Davies H, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23:517–525. doi: 10.1038/nm.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joosse SA, et al. Prediction of BRCA1-association in hereditary non-BRCA1/2 breast carcinomas with array-CGH. Breast Cancer Res Treat. 2009;116:479–489. doi: 10.1007/s10549-008-0117-z. [DOI] [PubMed] [Google Scholar]
- 17.Joosse SA, et al. Prediction of BRCA2-association in hereditary breast carcinomas using array-CGH. Breast Cancer Res Treat. 2012;132:379–389. doi: 10.1007/s10549-010-1016-7. [DOI] [PubMed] [Google Scholar]
- 18.Lips EH, et al. Indicators of homologous recombination deficiency in breast cancer and association with response to neoadjuvant chemotherapy. Ann Oncol. 2011. 10.1093/annonc/mdq468. [DOI] [PubMed]
- 19.Lips EH, et al. Quantitative copy number analysis by multiplex ligation-dependent probe amplification (MLPA) of BRCA1-associated breast cancer regions identifies BRCAness. Breast Cancer Res. 2011;13:R107. doi: 10.1186/bcr3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The Netherlands Cancer Institute. Neo adjuvant chemotherapy in triple negative breast cancer (neo-TN). ClinicalTrials.gov NCT01057069 (2017). Available at: https://clinicaltrials.gov/ct2/show/NCT01057069.
- 21.Schouten PC, et al. Robust BRCA1-like classification of copy number profiles of samples repeated across different datasets and platforms. Mol Oncol. 2015;9:1274–1286. doi: 10.1016/j.molonc.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polak P, et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet. 2017;49:1476–1486. doi: 10.1038/ng.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larsen MJ, et al. Classifications within molecular subtypes enables identification of BRCA1/BRCA2 mutation carriers by RNA tumor profiling. PLoS One. 2013;8. [DOI] [PMC free article] [PubMed]
- 24.Wang Y, Ung MH, Cantor S, Cheng C. Computational investigation of homologous recombination DNA repair deficiency in sporadic breast cancer. Sci Rep. 2017;7:15742. doi: 10.1038/s41598-017-16138-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe Y, et al. Aberrant DNA methylation status of DNA repair genes in breast cancer treated with neoadjuvant chemotherapy. Genes Cells. 2013;18:1120–1130. doi: 10.1111/gtc.12100. [DOI] [PubMed] [Google Scholar]
- 26.Shukla V, et al. BRCA1 affects global DNA methylation through regulation of DNMT1. Cell Res. 2010;20:1201–1215. doi: 10.1038/cr.2010.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson KC, et al. DNA methylation in ductal carcinoma in situ related with future development of invasive breast cancer. Clin Epigenetics. 2015;7. [DOI] [PMC free article] [PubMed]
- 28.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pereira B, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7. [DOI] [PMC free article] [PubMed]
- 30.Timms KM, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014;16. [DOI] [PMC free article] [PubMed]
- 31.Manié E, et al. Genomic hallmarks of homologous recombination deficiency in invasive breast carcinomas. Int J Cancer. 2016;138:891–900. doi: 10.1002/ijc.29829. [DOI] [PubMed] [Google Scholar]
- 32.Lips EH, et al. BRCA1-mutated estrogen receptor-positive breast cancer shows BRCAness, suggesting sensitivity to drugs targeting homologous recombination deficiency. Clin Cancer Res. 2017;23:1236–1241. doi: 10.1158/1078-0432.CCR-16-0198. [DOI] [PubMed] [Google Scholar]
- 33.Didraga MA, et al. A non-BRCA1/2 hereditary breast cancer sub-group defined by aCGH profiling of genetically related patients. Breast Cancer Res Treat. 2011;130:425–436. doi: 10.1007/s10549-011-1357-x. [DOI] [PubMed] [Google Scholar]
- 34.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roig B, et al. Metabolomics reveals novel blood plasma biomarkers associated to the BRCA1-mutated phenotype of human breast cancer. Sci Rep. 2017;7. [DOI] [PMC free article] [PubMed]
- 36.Cortes C, Vapnik V. Support-vector networks. Mach Learn. 1995;20:273–297. [Google Scholar]
- 37.Chang C, Lin C. LIBSVM: a library for support vector machines. ACM Trans Intell Syst Technol. 2013;2:1–39. doi: 10.1145/1961189.1961199. [DOI] [Google Scholar]
- 38.Robin X, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marquard AM, et al. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark Res. 2015;3:9. doi: 10.1186/s40364-015-0033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kandoth C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenthal, R., McGranahan, N., Herrero, J., Taylor, B. S. & Swanton, C. deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, (2016). [DOI] [PMC free article] [PubMed]
- 42.Cancer Genome Atlas Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma P, et al. The prognostic value of BRCA1 promoter methylation in early stage triple negative breast cancer. J Cancer Ther Res. 2014;3:2. doi: 10.7243/2049-7962-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peters TJ, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6. doi: 10.1186/1756-8935-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang, J., Vasaikar, S., Shi, Z., Greer, M. & Zhang, B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 1–8 (2017). 10.1093/nar/gkx356 [DOI] [PMC free article] [PubMed]
- 46.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holstege H, et al. BRCA1-mutated and basal-like breast cancers have similar aCGH profiles and a high incidence of protein truncating TP53 mutations. BMC Cancer. 2010;10:654. doi: 10.1186/1471-2407-10-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lips EH, et al. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br J Cancer. 2013;108:2172–2177. doi: 10.1038/bjc.2013.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carey LA, et al. Race, breast cancer subtypes, and survival in the Carolina breast cancer study. JAMA. 2006. 10.1001/jama.295.21.2492. [DOI] [PubMed]
- 50.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.D’Andrea AD. BRCA1: a missing link in the Fanconi anemia/BRCA pathway. Cancer Discov. 2013;3:376–378. doi: 10.1158/2159-8290.CD-13-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng T, et al. miR-124 inhibits cell proliferation in breast cancer through downregulation of CDK4. Tumor Biol. 2015;36:5987–5997. doi: 10.1007/s13277-015-3275-8. [DOI] [PubMed] [Google Scholar]
- 53.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 54.Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009;10:805–811. doi: 10.1038/nrg2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Y, et al. Concordance of DNA methylation profiles between breast core biopsy and surgical excision specimens containing ductal carcinoma in situ (DCIS). Exp Mol Pathol. 2017. 10.1016/j.yexmp.2017.07.001. [DOI] [PMC free article] [PubMed]
- 56.Brocks D, et al. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep. 2014;8:798–806. doi: 10.1016/j.celrep.2014.06.053. [DOI] [PubMed] [Google Scholar]
- 57.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discovery. 2015;5:1137–1154. doi: 10.1158/2159-8290.CD-15-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. SVM BRCA1-like status and BRCA1ness-MLPA profiles in 10 breast cancer cell lines. Table S2A. TCGA breast tumors with SVM BRCA1-like status. Table S2B. METABRIC breast tumors with SVM BRCA1-like status. Table S3. Complete subject characteristics of TCGA and METABRIC breast tumors with SVM BRCA1-like status. Table S4. Differentially methylated CpGs in BRCA1-like tumors identified by DMRcate. Table S5A. Hypermethylated DMRs (n = 108) from DMRcate analysis. Table 5SB. Hypomethylated DMRs (n = 94) from DMRcate analysis. Table S6A. GOBP terms associated with 158 unique genes from 108 hypermethylated DMRs. Table S6B. GOBP terms associated with 131 unique genes from 94 hypermethylated DMRs. (XLSX 923 kb)
Figure S1. Details of SVM BRCA1-like classifier. (A) Overview of copy number mapping algorithm for generating the input for training the SVM BRCA1-like classifier. (B) Receiver-operation characteristic curves (ROC) of the classifier applied to training and test set (AUC = 1.00 and 0.75, respectively). (C-D) Correlation of SVM BRCA1-like probability scores with published HR-deficiency metrics (HRD-LOH and LST scores). ***P < 0.001. Figure S2. Comparison of Ki-67 (MKI67) gene expression as a surrogate marker for cellular proliferation in METABRIC hormone-receptor-positive breast tumors. P value indicates statistical significance from a linear model adjusting for age, tumor stage, ER, PR and HER2 positivity. ***P < 0.001. Figure S3. Five-year overall survival comparison between BRCA1-like and non-BRCA1-like ER-positive/PR-positive, HER2-negative breast tumors in TCGA and METABRIC (combined). Table inset shows hazards ratio (95% CI) and P value from Cox proportional hazards regression adjusting for potential confounders. ***P < 0.001. Figure S4. miR124–2 with hypermethylation exhibit reduced gene expression in TCGA BRCA1-like receptor positive tumors. P value indicates statistical significance from a linear model adjusting for age, tumor stage, and ER, PR, and HER2 positivity. *P < 0.05. Figure S5. Comparison of heterogeneity between the “BRCA1-like methylation cluster” and “non-BRCA1-like methylation cluster” generated by hierarchical clustering of 350 most differential CpGs identified by DMRcate (all FDR <0.05 and |log2∆beta| ≥ 3.50). (A) Heat map showing unsupervised clustering (Euclidean distance, complete linkage) of the 350 DMRcate-identified CpGs. (B) Rank-ordered inter-sample variance in beta-values of the 350 differentially methylated CpGs. Horizontal dotted lines indicate mean inter-sample variance distribution for each group. Figure S6. Differential gene expression of DNA methyltransferases (DNMT1/3A/3B) in TCGA receptor-positive BRCA1-like breast tumors. A P value indicates statistical significance from linear model adjusting for age, tumor stage, and ER, PR, and HER2 positivity. ***P < 0.001. Figure S7. Comparison of Somatic Mutational Signature 1 contributed to by genome-wide cytosine-to-thymine (C > T) deamination events in TCGA hormone-receptor-positive breast tumors. A P value indicates statistical significance from linear model adjusting for age, tumor stage, and ER, PR, and HER2 positivity. **P < 0.01. (PDF 3058 kb)
Data Availability Statement
Breast tumor data sets used to develop the BRCA1-like classifier are publicly available in Gene Expression Omnibus [GEO:GSE9021, GEO:GSE9114, and GEO:GSE18626]. The Cancer Genome Atlas (TCGA) breast tumor molecular and clinical data are publicly available in the Genomic Data Commons (gdc.cancer.gov), FireBrowse (gdac.broadinstitute.org) and SynapseTCGAlive (synapse.org). The METABRIC breast tumor molecular and clinical data are publicly available in the cBioPortal database (cbioportal.org). Breast cancer cell line copy number profiles are available in the Cancer Cell Line Encyclopedia (CCLE) database (portals.broadinstitute.org/ccle).