

Abstract
Staphylococcus aureus secretes a family of potent, non-covalent inhibitory proteins that selectively target the neutrophil serine proteases neutrophil elastase, cathepsin-G, and proteinase-3. A majority of our understanding of these so-called EAP domain proteins has come from structure/function studies on EapH1 and its effects on human neutrophil elastase (hNE). Inspection of the EapH1/hNE co-crystal structure suggested that EapH1 residues R89, E94, and K95 are positioned near the EapH1/hNE interface and might contribute to the potent inhibition of hNE by EapH1. In this study, we used site directed mutagenesis, kinetic evaluation, and surface plasmon resonance to probe the individual contributions of R89, E94, and K95 to EapH1 function. We found that the wild-type EapH1/hNE complex is characterized by a fast association rate (2.0×106 M−1s−1) and a very slow dissociation rate (4.3×10−5 s−1), yielding an apparent inhibition constant of 21 pM. The slow dissociation rate of EapH1 from hNE resulted in a time-dependent inhibition pattern. Although conservative mutants E94Q and K95M, as well as the E94Q/K95M double mutant, had on- and off-rates comparable to wild-type EapH1, mutation of R89 to methionine resulted in a 15,000-fold decrease in inhibition (321 nM) and loss of the time-dependent inhibition characteristic. The double mutants R89M/E94Q and R89M/K95M, as well as the triple mutant R89M/E94Q/K95M were similarly perturbed. Mutation of R89 to lysine restored a portion of the inhibition of hNE (27 nM). Given these observations, we conclude that R89 is a primary contributor to EapH1 function vis-à-vis time-dependent inhibition of hNE.
Keywords: EapH1, surface plasmon resonance, time-dependent inhibition, neutrophil, elastase
Graphical Abstarct
Introduction
Neutrophils are the sentries of the immune system, and comprise ~ 60 percent of the leukocytes in normal human blood.2 A primary role of neutrophils lies in the initial innate immune response against invading bacteria.3, 4 Neutrophils are characterized by a preponderance of granules within their cytosol. Perhaps the best studied among these are the azurophilic granules, which contain high levels of myeloperoxidase5 and other bactericidal enzymes including the neutrophil serine proteases neutrophil elastase6, Cathepsin-G7, and proteinase-3.8 Neutrophils respond to biochemical gradients of various chemotactic signals to arrive at the site of potential bacterial infection. Thereafter, they ultimately engulf opsonized bacteria through the process of phagocytosis. The newly created phagosome fuses rapidly with azurophilic granules, whereby the antimicrobial components of these granules are solubilized and affect the eventual destruction of the bacterial cell.9
Staphylococcus aureus is a prominent human pathogen that causes a wide variety of diseases, ranging from superficial infections of the skin and minor wounds to potentially life-threatening infections such as sepsis, endocarditis, and necrotizing pneumonia. It has been estimated that ~30 percent of the human population is commensally colonized by this bacterium.10, 11 The relative contribution of various innate immune components to the overall response against S. aureus remains a topic or investigation and debate. However, recent discoveries reporting numerous staphylococcal innate immune evasion proteins that inhibit prominent constituents of the neutrophil’s anti-bacterial arsenal strongly suggest that neutrophils play an essential role in our defense against S. aureus.1, 12, 13
Stapels et al. reported the identification of a family of three secreted proteins from S. aureus that inhibit activity of the neutrophil serine proteases.1 These so-called “EAP domain proteins” consist of the founding member, the 50–70 kDa Extracellular Adherence Protein (i.e. Eap), as well as its two homologs EapH1 (~12 kDa) and EapH2 (~13 kDa).14 They are highly selective for the proteases found in azurophilic granules (i.e. neutrophil elastase, cathepsin-G, and proteinase-3), and appear to have no inhibitory activity against related chymotrypsin-like molecules found in the bloodstream.1 EAP domain proteins are defined by an eponymous motif of approximately ~110 residues that adopts a β-grasp fold.14 Their overall structure consists of a prominent α-helix superimposed on a five-stranded mixed β-sheet, and has been extensively characterized by both X-ray crystallography14 and solution NMR spectroscopy.15, 16 Since the amino and carboxy-termini lay on opposite sides of the fold14, the structure of the EAP domain allows for tandem repeats of this motif to occur within the same molecule. Such an arrangement is seen in the multi-domain Eap protein17, 18, and appears to impart additional innate immune evasion properties to this molecule aside from inhibition of neutrophil serine proteases.19–21
Although Eap, EapH1 (UniProtKB: A0A0H3K0M1) and EapH2 are all potent inhibitors of the canonical neutrophil serine proteases1, 22, a majority of the structure/function investigations thus far have come from studies on EapH1 and its effects on human neutrophil elastase (hereafter hNE, UniProtKB: P08246). A 1.85 Å resolution co-crystal structure of EapH1 bound to hNE has been solved (Fig. 1A).1 This structure shows that EapH1 binds non-covalently across the hNE active site channel, thereby preventing substrate access.1 EapH1 residues I56 – R61 contribute 628 Å2 of the 830 Å2 interface area buried in this complex, with the backbone carbonyl of L59 coming within 3.5 Å distance of the hNE catalytic serine.22
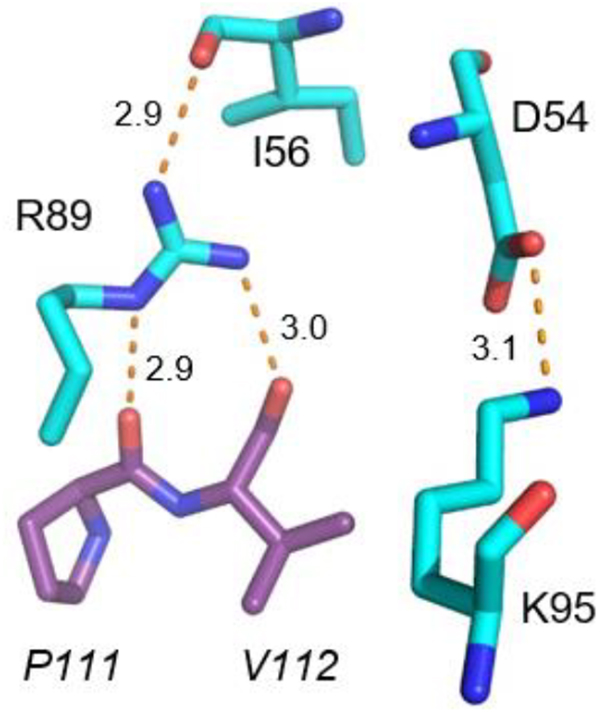
Figure 1. R89 is the Key Residue Involved in EapH1 Time-Dependent Inhibition of Human Neutrophil Elastase.

(A) Crystal Structure of the wild-type EapH1 (cyan)/Elastase (purple) complex. PDB: 4nzl.1 (B) Stick representation of R89, K95 and likely interacting residues. EapH1 residues (normal font), Elastase residue (italicized). Potential interactions depicted as red dashes.
Remarkably, the effect on EapH1 affinity for hNE was minimal when these six residues were collectively mutated to alanine.22 By contrast, EapH1 residues R89, E94, and K95 are some distance from the hNE active site in the co-crystal structure1, but appear to contribute potentially important interactions (Fig. 1B). When R89, E94 and K95 of EapH1 were collectively mutated to alanine, the binding affinity for hNE decreased by ~74,000-fold22, while the IC50 of this mutant could not be determined due to its low activity.
In this study, we explored the steady-state kinetics of elastase inhibition by wild-type EapH1 and determined the individual contributions of R89, E94, and K95. Each residue was interrogated by conservative mutation (R89M, E94Q, K95M) in an effort maintain the overall residue volume. Double and triple mutants were also generated to examine whether any functional coupling exists between the residues (e.g. synergy, antagonism, etc.). We then conducted kinetic evaluation by activity assays and surface plasmon resonance to ascertain residue-specific quantitative contributions to binding and inhibition of hNE. Collectively, these experiments have identified R89 as a major contributor to EapH1 function as a time-dependent, picomolar-affinity inhibitor of hNE.
Materials and Methods
Materials
Mutagenic oligonucleotides were purchased from Integrated DNA Technologies. Phusion High-Fidelity DNA Polymerase, dNTPs and Dpn I were purchased from New England Biolabs. Kanamycin Monosulfate, isopropyl-β-D-thiogalactopyranoside (IPTG), tris(hydroxymethyl)aminomethane (TRIS) and Nickel-IDA agarose were purchased from Gold Biotechnology. Human neutrophil elastase was purchased from Elastin Products Company. Methoxysuccinyl-Ala-Ala-Pro-Val-P-nitroanilide (MSA2PV-pNA), Triton X-100, dimethylsulfoxide (DMSO), succinic acid and Luria Broth powder were purchased from Sigma-Aldrich. Imidazole was purchased from Alfa Aesar. Nickel (II) Chloride-hexahydrate was purchased from Acros Organics. 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) was purchased from MP Biomedical, LLC. Plasmid DNA was purified using the Wizard Plus SV Miniprep DNA Purification System (Promega).
Mutagenesis, protein expression and purification
A full-circle site-directed PCR strategy was utilized to generate the desired mutations. The integrity of the coding sequence and the presence of the mutation was verified by DNA sequencing (GENEWIZ). The double mutants were made using the single mutant (R89M) as a template in the PCR. The triple mutant was made using the R89M/K95M plasmid as the template.
The wild-type and mutant EapH1 proteins were expressed in E. coli BL21 (λDE3). Briefly, 2 L LB was inoculated with 10 mL/L of an overnight culture. The cultures were incubated, shaking at 37°C to an optical density of ~ 0.7, then induced with 1 mM IPTG and incubated, shaking overnight at 18°C. The bacterial cells were harvested by centrifugation and the pellet was re-suspended in 80 mL of buffer A (20 mM TRIS-Cl, 500 mM NaCl, 5 mM Imidazole (pH:8.0 (RT))). The cells were lysed by passing the suspension through a microfluidizer (Microfluidics) at a 17 kpsi chamber pressure. The lysate was clarified by centrifugation (~27,000×g). The cell-free extract was loaded onto 4 mL Nickel-IDA agarose. The column was washed with ~200 mL buffer A, 100 mL of buffer B (20 mM TRIS, 1 M NaCl, 10 mM Imidazole (pH: 8.0(RT))), followed by 100 mL buffer A. Protein was eluted with buffer A supplemented with 150 mM Imidazole. The eluted protein was applied to a HiPrep 26/10 Desalting column (Akta pure Chromatography System, GE Healthcare Life Sciences) to exchange the protein into buffer A. The N-terminal hexa-Histidine tag was cleaved using the Tobacco Etch Virus (TEV) protease; β-mercaptoethanol (β–ME) was added to a final concentration of 1 mM prior to the cleavage reaction. The cleavage reaction was allowed to proceed overnight at RT. The cleaved protein was applied to the Nickel-IDA column and the flow-through collected. The concentrations of wild-type/mutant EapH1 proteins were determined spectrophotometrically (DS-11 Spectrophotometer, DeNovix) using an extinction coefficient calculated from the deduced protein composition (EapH1: ε280 = 7,450 M−1 cm−1).
Preparation of human neutrophil elastase
Lyophilized human neutrophil elastase (2 mg) was dissolved in 200 μL 20 mM Sodium Acetate/50 % (v/v) glycerol (pH: 5.0, RT). The concentration of the dissolved elastase was determined using the deduced protein composition (ε280 = 20,230 M−1 cm−1). The enzyme was separated into 10 μL aliquots and stored at −20°C until use.
Steady-state kinetic analysis of human neutrophil elastase
The substrate, Methoxysuccinyl-Ala-Ala-Pro-Val-p-nitroanalide (MSA2PV-pNA), was quantified by end-point assays (ε400 =12.3 mM−1 cm−1). Initial velocities of the elastase (4nM) reaction were measured spectrophotometrically at 400nm (Cary 50Bio UV-Vis, Varian) at substrate concentrations ranging from 0.1 × Km to 5 × Km. These data (n = 3) were fit to a Michaelis-Menten equation to determine the kcat and Km (SigmaPlot 10.0/Enzyme Kinetics Module 1.3, Systat Software, Inc.). The steady-state kinetic constants were also determined by a plate-based assay using a Versamax tunable microplate reader (Molecular Devices). These data (n = 9) were fit to a Michaelis-Menten equation. The Km determined from the plate assay was used in the script for progress curve analysis. The reaction buffer contained 50 mM HEPES/140 mM NaCl/0.05 % (v/v) Triton X-100 (pH: 7.4, RT).
Reversible behavior of EapH1 inhibition of human neutrophil elastase
To probe the reversible nature of EapH1 inhibition of hNE, 200 nM hNE was pre-incubated with 400 nM EapH1 then diluted to a final concentration of 14.3 nM hNE, 28.5 nM EapH1 with 50 mM HEPES/140 mM NaCl/0.05 % (v/v) Triton X-100 (pH: 7.4, RT) containing 557 μM MSA2PV-pNA. The reaction was followed at 400 nm for 140 minutes. As a control, the reaction was separately followed without the pre-incubation of enzyme and inhibitor. In this scenario, the concentrations of enzyme and inhibitor were the same as the pre-incubation concentrations post-dilution.
Steady-state inhibition kinetics of human neutrophil elastase by wild-type/mutant EapH1 proteins
Progress curves were collected for 30 minutes using a VersaMax tunable microplate reader (Molecular Devices). A total of 57 progress curves were collected for each inhibitor (three hNE concentrations (~4, 8, 12 nM), three substrate concentrations ((~Km, 2 × Km, 3 × Km) per hNE concentration, five inhibitor concentrations per substrate concentration). To obtain a more accurate estimate of the substrate on-rate, two hNE concentrations with 6 substrate concentrations per hNE concentration were included in the global fitting. The data were fit to a rapid-equilibrium competitive inhibition mechanism (Scheme 1, Fig. S2) using Dynafit (Biokin Ltd).23
Scheme 1.

To decrease the search space of fitting the progress curves, the substrate off-rate (k2) was linked to the on-rate by the estimated Michaelis constant (Km) as determined by steady-state kinetic analysis using the plate-based assay (Fig. S2). The inhibition constant was calculated from the on- and off-rate estimates determined from the fits. The values reported are the average of three different sets of data, while the errors are the standard deviation.
The inhibition constants for R89M-containing EapH1 mutants were determined using a classical 5×5 assay. The initial velocity of hNE (~4 nM) was determined at five substrate concentrations (~0.5 × Km − 4 × Km) using five inhibitor concentrations (0 – ~5 × Ki) per substrate concentration. The data (n = 3) were globally fit to a competitive inhibition model using SigmaPlot 10.0/Enzyme Kinetics Module 1.3 (Systat Software, Inc). Errors reported are standard error of the global fits.
Determination of on- and off-rate estimates of the R89M EapH1 mutants
The association/dissociation rates for the R89-containing mutants could not be determined by progress curve analysis. As a consequence, Surface Plasmon Resonance (SPR) was utilized as an alternative approach to obtain these values. hNE, previously modified to contain an N-linked glycan ethylenediamine moiety22, was immobilized on a CMD-200M sensor chip (Xantec Bioanalytics; Dusseldorf, Germany) via EDC/NHS coupling chemistry to ~1000 RU. A reference flow cell was created by flowing the EDC/NHS solution over the surface followed by quenching the activated carboxymethyldextran with 1 M ethanolamine (pH: 9.0, RT).
All injections were carried out using a running buffer of 20 mM HEPES/140 mM NaCl/0.05 % (v/v) Tween-20 (pH: 7.4, RT). The parameters for binding of wild-type EapH1 to hNE were estimated by both single-cycle kinetics ([EapH1] (nM): 0.04, 0.2, 1, 5, 25) and conventional dose-response kinetics ([EapH1] (nM): 1.5625, 3.125, 12.5, 25, 100, 1000). The analyte contact time for the single-cycle approach was 120 s with a final dissociation phase of 3600 s at a flow rate of 60 μL/min; similarly, the analyte contact time for the conventional dose-response runs was 120 s followed by a dissociation phase of 3600 s at a flow rate of 60 μL/min for each analyte concentration. The microscopic rate constants for the R89-containing mutants were estimated by conventional dose-response kinetics ([EapH1] (nM): 62.5, 125, 250, 500, 1000, 2000, 4000), with the exception of R89K ([EapH1] (nM): 7.8, 15.6, 31.25, 62.5, 125, 250, 500, 1000). The analyte contact time was 120 s with a dissociation time of 240 s at a flow rate of 30 μL/min and 60 μL/min. The chip surface was regenerated after each injection with 10 mM glycine/1.5 M NaCl (pH: 2.2, RT) for 30 s at a flow rate of 20 μL/min. All reference-subtracted sensograms were fit to a 1:1 Langmuir binding model using the BiaCore T-200 Evaluation Software (GE Life Sciences). The microscopic rate estimates reported are the average of either four individual sensogram fits (WT) or six individual semsogram fits (mutants). The errors on the microscopic rate constants are the standard deviation. The dissociation constant was estimated from the averaged microscopic rate constants. The Kd errors were propagated from the on- and off-rate standard deviations.
Structural modeling of EapH1 mutant hNE complexes
Homology models for the R89Q and R89K mutants of EapH1 bound to hNE were constructed using the SWISS-MODEL server.24 All structural representations were rendered from the corresponding PDB files using PyMol (Schrödinger, LLC).
Results
EapH1 is a reversible, time-dependent inhibitor of human neutrophil elastase
Whereas prior functional studies of EAP domain proteins made use of a fluorometric assay for hNE activity1, we initially set out to adopt a more economical and higher throughput chromogenic method to support our current work. We first determined steady-state kinetic constants for hNE as a precursor to further experimental design (Fig. S1). hNE turnover of the substrate MSA2PV-pNA was estimated to be 8.7 ± 0.3 s−1 and 7.8 ± 0.3 s−1 in the cuvette and plate-based assay, respectively. The Michaelis constant was estimated to be 47 ± 5 μM and 57 ± 6 μM in the cuvette and plate-based assay, respectively. We note that these values are in agreement with the manufacturer’s specifications (7.4 − 9.3 s−1, Elastin Products Co.). The Km estimates are slightly lower than those previously reported (180 μM25, 140 μM26).
Previous investigations into EAP domain proteins’ effects on hNE activity were carried out by pre-incubating inhibitor with hNE prior to adding the fluorometric substrate.22 However, when we performed traditional steady-state kinetics by exposing the enzyme to the substrate and inhibitor simultaneously, we observed that the progress curve of the hNE reaction was non-linear in the presence of EapH1. When the inhibitor and enzyme were not pre-incubated, the velocity of the initial phase was approximately equal to the rate of the non-inhibited reaction but also decreased as a function of time to a steady-state inhibited rate. This pattern is consistent with time-dependent inhibition.27 Furthermore, when we pre-incubated hNE with EapH1 and then diluted the reaction with a saturating concentration of substrate, we observed a pronounced lag followed by a steady-state phase that corresponded to the rate of the reaction determined at the diluted hNE/EapH1 concentrations (Fig. 2). Together, these data suggest that EapH1 is a reversible, time-dependent inhibitor of hNE.
Figure 2. EapH1 is a Time-Dependent,Reversible Inhibitors of Human Neutrophil Elastase.

() 557 μM MSA2PV-pNA incubated with 28.5 nM EapH1, reaction started with 14.3 nM elastase; () 400 nM EapH1 pre-incubated with 200 nM elastase and diluted to 28.5 nM EapH1, 14.3 nM elastase with buffer containing 557 μM MSA2PV-pNA.
Steady-state inhibition kinetics of human neutrophil elastase wild-type/mutant EapH1 proteins
Initial velocity analysis could not be used to further investigate the impact of wild-type EapH1 on hNE activity, due to the time-dependent nature of the EapH1 inhibitory effect. Consequently, we instead analyzed globally the progress curves of the hNE reaction across a series of enzyme, substrate, and inhibitor concentrations to derive further insight into the EapH1 inhibitory mode. According to this formalism, the microscopic rate constants relating to the hNE mechanism should be consistent between wild-type and mutant EapH1 proteins, while those constants relating to the EapH1 inhibitor may vary between the wild-type and mutant proteins. In this regard, we found that the kcat values (Table 1, k3) for substrate turnover by hNE estimated during inhibition with wild-type and mutant EapH1 were in good agreement to those determined by steady-state kinetics and the manufacturer’s specifications (6.5 − 9.5 s−1, Table 1). The hNE substrate on-rates (Table 1, k1) were likewise consistent between the wild-type and mutant EapH1 proteins (3.2 × 105 − 7.2 × 105 M−1 s−1, Table 1).
Table 1.
Kinetic Rate Constant Estimates determined by Progress Curve Analysisa
| Inhibitor | k1 (M−1s−1) | SD | k3 (s−1) | SD | k4 (M−1s−1) | SD | k5 (s−1) | SD | Ki (nM) | SD |
|---|---|---|---|---|---|---|---|---|---|---|
| EapHl WT | 6.0×105 | 2.2×105 | 7.4 | 0.8 | 2.0×106 | 1.4×105 | 4.3×10−5 | 1.3×10−5 | 0.021 | 0.005 |
| R89Mc | - | 7.0 | 0.1 | - | - | 321 | 12 | |||
| R89K | - | 9.2 | 0.2 | - | - | 26.8 | 1.3 | |||
| R89Q | - | 9.2 | 0.2 | - | - | 944 | 46 | |||
| E94Q | 3.3 ×105 | 5.1×104 | 9.3 | 1.0 | 1.8×106 | 1.6×105 | 5.6×10−5 | 5.8×10−6 | 0.031 | 0.006 |
| K95M | 7.2×105 | 1.9×105 | 7.7 | 0.7 | 2.2×106 | 2.9×105 | 6.1×10−5 | 1.0×10−6 | 0.028 | 0.003 |
| R89M/E94Qc | - | 7.0 | 0.1 | - | - | 236 | 11 | |||
| R89M/K95Mc | - | 9.5 | 0.2 | - | - | 437 | 25 | |||
| E94Q/K95M | 5.7×105 | 4.×105 | 9.2 | 2.3 | 2.4×106 | 4.4×105 | 7.2×10−5 | 2.3×10−5 | 0.032 | 0.015 |
| R89M/E94Q/K95Mc | - | 6.5 | 0.1 | - | - | 406 | 20 | |||
| (-)b | 4.0×105 | 2.2×105 | 7.4 | 0.8 | - | - | - |
Microscopic rate constant estimates are the average of three individual sets of 57 progress curves (three [elastase], three [substrate] per [elastase], and five [inhibitor] per [substrate].
Microscopic rate constant estimates are the average of 10 individual sets of 12 progress curves (two [elastase], six [substrate] per [elastase].
Due to the loss of the time-dependent inhibition characteristic, R89M mutants were analyzed by a classical 5×5 inhibition assay. Ki estimated by globally fitting three assay sets. Error is the standard error of the global fit. k4 and k5 were estimated by following R89M binding to immobilized elastase using surface plasmon resonance (See Table 2).
We subsequently determined the estimated on-rate (k4) for wild-type EapH1 to be 2.0 × 106 ± 1.4 × 105 M−1s−1, while the estimated off-rate (k5) for the wild-type protein was 4.3 × 10−5 ± 1.3 × 10−5 s−1. Using these microscopic rate constants, we arrived at a value of 21 ± 5 pM for the macroscopic inhibition constant (Ki) for wild-type EapH1.
R89 is a major contributor to EapH1 binding and inhibition of human neutrophil elastase
Using the EapH1/hNE co-crystal structure1 and previous alanine-scanning mutagenesis data as a guide22, we next prepared a panel of site-directed single, double, and triple mutants of EapH1 affecting positions R89, E94, and K95. We then kinetically characterized these EapH1 variants. The E94Q and K95M mutants, as well as the E94Q/K95M double mutant, had on- and off-rates similar to the wild-type protein (Table 1). By contrast, mutation of R89 to methionine resulted in a substantial decrease in inhibition of hNE (Ki:321 nM). Interestingly, loss of the arginine-specific guanidinium moiety also abolished the time-dependent characteristic of hNE inhibition (Fig. 3). The double mutants R89M/E94Q and R89M/K95M, as well as the triple mutant R89M/E94Q/K95M, all had similar hNE inhibition profiles to the R89M mutant (Ki:236 nM, 437 nM, and 406 nM, respectively, Table 1, Figure 4). Together, these apparently similar inhibition constants suggest that no functional coupling exists between these EapH1 residues (e.g synergistic, antagonistic, etc.).
Figure 3. Representative Fitted Inhibition Progress Curves.

(A) WT EapH1 inhibition: assay contained 3.9 nM E lastase, 147 μM MSA2PV-pNA. (B) R89M EapH1 inhibition: 4.5 nM elastase, 156 μM MSA2PV-pNA. The respective inhibitor to enzyme concentration ratios are inset at the right hand side of each panel.
Figure 4. Competitive Inhibition of Elastase by EapH1 R89 Mutants.

(A) R89M, (B) R89M/E94Q, (C) R89M/K95M, (D) R89M/E94Q/K95M, (E) R89Q, (F) R89K. All assays were done with ~4 nM elastase in 50 mM HEPES/140 mM NaCl/0.05 % triton X-100. The respective inhibitor concentrations are inset within the legend box at the left hand side of each panel.
Since the inhibition constants were determined via a classical 5×5 assay, the individual microscopic rate constants responsible for the decrease in activity of the R89M mutants could not be directly obtained. To circumvent this limitation, we used surface plasmon resonance to investigate the interaction between various EapH1 proteins and immobilized hNE.22 Wild-type EapH1 was flowed over the hNE sensor surface using both single cycle and dose response kinetic methods to establish the reproducibility of previous results. The measured on- and off-rates (Table 2, 4.8×105 M−1s−1; 7.0×10−5 s−1, respectively) were comparable to values previously determined (3.7×105 M−1s−1, 6.4×10−5 s−1).22 However, in this study, a more extensive dose-response kinetic analysis (i.e. long duration dissociation phase (3600 s) following each concentration injection) of wild-type EapH1 binding to hNE estimated a Kd of 0.271 nM (Fig. 5b, Table 2).
Table 2.
Kinetic Rate Constant Estimates determined by Surface Plasmon Resonancea
| Protein | kon (M−1s−1) | SD | koff (s−1) | SD | Kd (nM) | errorb | Ki (nM)c | Fold differencee |
|---|---|---|---|---|---|---|---|---|
| EapH1 WT | 2.23×105 | 5.0×103 | 6.03×10−5 | 3.1×10−6 | 0.271 | 0.015 | 0.021 | 12.9 |
| EapH1 WTd | 4.85×105 | 2.06×105 | 7.10×10−5 | 5.6×10−6 | 0.146 | 0.063 | 0.021 | 7.0 |
| R89M | 2.85×104 | 5.13×103 | 0.0737 | 0.0115 | 2586 | 615 | 321 | 8.1 |
| R89K | 8.72×104 | 7.91×103 | 0.0165 | 0.0020 | 189 | 29 | 26.8 | 7.0 |
| R89Q | 2.32×104 | 2.32×103 | 0.1397 | 0.0271 | 6028 | 1316 | 944 | 6.4 |
| R89M/E94Q | 5.27×104 | 2.99×103 | 0.0876 | 0.0084 | 1662 | 185 | 236 | 7.0 |
| R89M/K95M | 2.49×104 | 4.61×103 | 0.0904 | 0.0166 | 3632 | 945 | 437 | 8.3 |
| R89M/E94Q/K95M | 2.08×104 | 1.14×103 | 0.0855 | 0.0124 | 4108 | 637 | 406 | 10.1 |
Microscopic rate constant estimates are the average of either four individual sensograms (WT) or six individual sensograms (mutants).
The dissociation constant error was propagated from the on- and off-rate standard deviations.
The inhibition constants determined by progress curve analysis (WT) or 5×5 assay (R89M mutants, Fig. 4).
Microscopic rate constant estimates determined by single-cycle binding kinetics.
The fold difference depicts the difference between the dissociation constant determined by SPR and the inhibition constant determined by kinetic analysis.
Figure 5. Surface Plasmon Resonance of Wild-Type and Mutant EapH1 Binding to Immobilized hNE.

(A) WT (single-cycle kinetics, [EapH1] ranging from ~ 0.2× − 130× the published Kd22), (B) WT (dose-response kinetics with elongated dissociation phase, [EapH1] ranging from ~ 0.5× − 34× the published Kd22), (C) R89M, (D) R89M/D94Q, (E) R89M/K95M, (F) R89M.D94Q/K95M, (G) R89Q (All R89M [EapH1] ranging from ~ 0.2× − 12× the R89M Ki), (H) R89K ([EapH1] ranging from ~ 0.3× − 18× the R89K Ki). All sensorgram series were evaluated with a Langmuir kinetic model, as described in the Materials and Methods section. Data analysis parameters are found in Table 2. Note the intermediate dissociation rate for the R89K mutant (H), when compared to WT EapH1 (A, B), and the other R89 mutants (C-G). Absolute values of each concentration series can be found in the methods section.
While the on-rates estimated for the R89M mutants were consistently slower (~10-fold, Table 2) than for wild-type EapH1, the R89M mutant off-rates were substantially faster than for the wild-type protein (~1600-fold, Table 2). Thus, while impaired on-rates and enhanced off-rates both seem to contribute to impairment of EapH1 R89 mutants, loss of R89 has a more than 100-fold greater impact on the dissociation rate constant.
Partial restoration of EapH1 function in an R89K mutant.
According to the EapH1/hNE co-crystal structure, the sidechain of EapH1 R89 contributes three potentially significant interactions toward hNE binding (Fig. 1B). The first interaction is an intramolecular hydrogen bond to the carbonyl oxygen of I56. I56 is noteworthy because it is positioned toward the middle of the loop that forms a bulk of the interface with hNE. The second and third interactions are intermolecular hydrogen bonds between the carbonyl oxygens of P111 and V112 of hNE, respectively (Fig. 1B). Mutation of R89 to methionine not only dramatically decreased inhibition of hNE by EapH1 (321 nM, Table 1), it also ablated the time-dependent characteristic of this inhibition (Fig. 3B). Although the unique chemical properties of the arginine sidechain cannot be perfectly mimicked by other amino acids, we nevertheless wondered whether mutation of R89 to either glutamine or lysine might restore some functionality to the EapH1 protein due to either the hydrogen-bonding ability or charge properties of these residues.
We tested this hypothesis by expressing and characterizing the EapH1 single mutants R89Q and R89K. Interestingly, we found that the R89Q mutant was approximately three-fold diminished when compared to the initial R89M mutant in both kinetic and surface plasmon resonance assays (Ki: 944 nM, Table 1, Kd: ~6 μM, Table 2, Fig. 4E, and Fig. 5G). Despite the fact that glutamine, like arginine, maintains the ability to form two hydrogen bonds, its sidechain is apparently not long enough to reconstitute either of the hydrogen bonds formed by arginine in the wild-type inhibitor (Fig. S3). On the other hand, the R89K mutant exhibited approximately 10-fold greater inhibitory capacity toward hNE than R89M in both assays (Ki: 26.8 nM, Table 1, Kd:189 nM, Table 2, Fig. 4F, Fig. 5H). Although the lysine sidechain can only participate in a single hydrogen bond or salt bridge, it appears to be capable of coming into interaction distance with the carbonyl oxygen of EapH1 156 (Fig. S3). The fact that EapH1 R89K displays non-time dependent inhibiton at a potency intermediate between that of wild-type EapH1 and other mutants characterized here suggests that the intermolecular interactions formed by R89 with P110 and V111 of hNE are required for the time-dependent inhibitory effect of wild-type EapH1.
Discussion
In a previous study, Stapels and co-workers mutated EapH1 residues R89, E94, and K95 simultaneously to alanine and showed that EapH1 binding and inhibition of hNE was dramatically reduced.22 Herein, we expanded upon this prior investigation by individually and conservatively mutating these EapH1 residues to determine their precise contribution(s) to EapH1 function. Since the inhibition of hNE by S. aureus EapH1 had yet to be fully evaluated by rigorous kinetic methods, we began by analyzing wild-type EapH1 using both cuvette- and plate-based chromogenic assays performed without pre-incubating the enzyme and inhibitor. As shown in Figure 2, we discovered that EapH1 inhibits hNE in a time-dependent manner. Time-dependent inhibition is characterized by a change in reaction velocity over time, whereby a final steady-state rate is ultimately achieved.27 There are several examples of low molecular weight peptides/proteins that inhibit various proteases in a time-dependent manner. Among these are inhibition of hNE by human proteinase inhibitor 9 (PI9)28, inhibition of pepsin by a low molecular weight peptide secreted by Streptomyces sp. MBR0429, and Thermomonospora sp. xylanase inhioition by an aspartic protease inhibitor secreted by an extremophilic Bacillus sp.30
Since none of the molecules mentioned above share any sequence relationship with S. aureus EapH1 or other EAP domains, it seems that there is no obviously unifying structural feature that gives rise to time-dependent inhibitory phenomena. Furthermore, while time-dependent inhibition is generally characterized by a slow inhibitor association rate27, this does not seem to be the case for wild-type EapH1. In fact, the progress curve analysis described here indicates that the on-rate for EapH1 is faster (2.0 × 106 M−1s−1, Table 1) than that of the chromogenic hNE substrate, MSA2PV-pNA (6.0 × 105 M−1s−1). Another characteristic of time-dependent inhibition is an extremely slow dissociation rate of the enzyme-inhibitor complex.27 In this regard, the off-rate of EapH1 (4.3 × 10−5 s−1, Table 1) appears to be the dominant effect that contributes to potent inhibition of hNE by wild-type EapH1 (21 pM, Table 1).
We found that mutation of E94 to glutamine had no significant effect on EapH1 function (Table 1), even though it is conserved between the NSP-inhibitors EapH1 and EapH222 and lies in proximity to the EapH1/hNE interface.1 Nearby, the sidechain of residue K95 forms an intra-molecular salt bridge with the sidechain of D54 (Fig. 1B). Since D54 is at the N-terminus of the loop that interacts with the hNE active site1, the K95-to-D54 interaction has the potential of organizing this interfacial loop for more productive binding to hNE. Despite this, we found that the K95M mutant retained on- and off-rates that were essentially wild-type (Table 1). The double mutant (E94Q/K95M) was also not perturbed (Table 1).
Of the three residues chosen for study here (i.e. R89, E94, and K95), our results demonstrated that only R89 makes significant contributions to EapH1 function (Table 1 and Table 2). Indeed, loss of R89 specifically eliminated both the picomolar affinity of EapH1 for hNE and its time-dependent inhibitory characteristic (Table 1, Table 2, and Fig. 3). Because measuring initial velocities does not provide any information on the microscopic rate constants, we used surface plasmon resonance to arrive at estimates of the on- and off-rates of the R89M mutants. During the early stages of this work, we noticed that the wild-type on-rate determined by SPR (2.23 × 105 M−1s−1) was reproducibly slower than that derived from progress curve analysis (2.0 × 106 M−1s−1). Although we initially attributed this difference to the mass transport limit for transition from solution- to solid-state in SPR (previously reported to be near 105 M−1s−1)31, subsequent characterization of the R89 mutants showed that their dissociation constants (Kd) estimated by SPR were consistently higher than the inhibition constants obtained by kinetic analysis by approximately a factor of 10 (Table 2).
Considering that the on-rates of the R89 mutants are well below the mass transport limit, we conclude that immobilized hNE is somewhat hindered in forming an inhibitor-enzyme complex when compared to a purely solution-state system. One potential explanation for this observation lies in the hNE immobilization approach. Since the matured hNE sequence is devoid of lysine residues, we introduced free amines onto hNE by oxidizing its N-linked glycans, forming a Schiff-base adduct with the ethylenediamine, and reducing this adduct with sodium cyanoborohydride.22 While this modification provided a facile way to couple hNE to the sensor chip surface through amine-based chemistries, it could very well be that the N-linked glycans facilitate EapH1 binding to its hNE target. If so, modification of these glycans might be expected to influence the rate of complex formation, as measured by SPR.
Recognizing the difference in the absolute value of these rate constants, we nevertheless observed that the trend in binding rates was consistent with the estimated inhibition constants (i.e. mutants with a low Ki displayed a low Kd and vice-versa). This allowed us to determine that R89, rather than E94 or K95, plays a key role in EapH1 function, and also that the main effect of mutating the positively charged arginine is a dramatic increase in the rate of EapH1/hNE dissociation. The off-rate of the R89 mutants are increased by ~1600-fold with the exception of the R89K mutant (273-fold). These data are consistent with the hypothesis that the intra- and inter-molecular hydrogen bonds formed between R89 and 156 (EapH1) and P111 and V112 (hNE) are critical for the incredibly potent inhibition of hNE. Mutation of R89 to lysine recovers some of the binding affinity for and inhibition of hNE (Table 1, 2), but does not restore the time-dependent characteristic. Although a crystal structure of the R89K/hNE complex was not determined, a homology model of this mutant based on the wild-type EapH1/hNE co-crystal structure strongly suggests that lysine maintains the intra-molecular hydrogen bond and restores the ordering of the interfacial loop (Figure S3). This also suggests that the inter-molecular hydrogen bonds between R89 and P111 and V112 are responsible for the time-dependent characteristic, and underlie the dominant contributions of R89 to the potent hNE inhibition by EapH1.
It should be noted that we have observed progressively tighter apparent Kd values for the EapH1/hNE interaction by using ever more sensitive assay techniques. Initially, the EapH1/hNE dissociation constant was estimated at 25 nM by isothermal titration calorimetry1, SPR studies provided values of 2.95 nM (dose-response) and 0.191 nM (single-cycle).22 The difference in these sets of SPR-derived values was due to a more accurate determination of the slow dissociation rate constant through an increased observation time. However, the label-free solution-state kinetic method of progress curve analysis yielded a Ki of 0.021 nM (Table 1,2). Since this latter approach investigates the biologically-relevant reaction without the restrictions imposed by mass-transport, we believe this value is probably the most accurate representation of the true affinity of EapH1 for hNE
Conclusion
In this manuscript, we evaluated the contribution of three EapH1 (R89, E94, K95) residues to the potent inhibition of human Neutrophil Elastase (hNE). Initial kinetic evaluation of hNE inhibition by wild-type EapH1 revealed a time-dependent characteristic. Based on the EapH1/hNE co-crystal structure, the guanidinium moiety of R89 in EapH1 forms one intramolecular and two intermolecular interactions. The EapH1 R89M mutation dramatically reduced the affinity for, as determined by SPR, and inhibition of hNE, as determined by steady-state kinetics. This mutation also abolished the time-dependent characteristic of inhibition. Mutation of R89 to lysine recovers a portion of the inhibition of hNE, but not the time-dependent characteristic. The homology model of R89K EapH1/hNE suggest that the intramolecular interaction is maintained. These data strongly suggest the intermolecular interactions are responsible for the majority of the inhibition potency and the time-dependent characteristic.
Supplementary Material
Acknowledgements:
We would like to thank Dr. D.J. Black, Principal Scientist, BioLogic USA, and Dr. Brandon Garcia, Assistant Professor, East Carolina University, for helpful discussions during this work.
Funding Source
Funding was provided by a grant from the National Institute of General Medical Sciences (R01-GM121511) to B.V.G.
Footnotes
UniProt Accession ID for Proteins
EapH1, UniprotKB: A0A0H3K0M1
Human Neutrophil Elastase, UniProtKB: P08246
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
Supporting information: Steady-state kinetics of hNE, representative Dynafit script for progress curve analysis, homology models of selected R89 mutants in EapH1 bound to hNE.
References
- [1].Stapels DA, Ramyar KX, Bischoff M, von Kockritz-Blickwede M, Milder FJ, Ruyken M, Eisenbeis J, McWhorter WJ, Herrmann M, van Kessel KP, Geisbrecht BV, and Rooijakkers SH (2014) Staphylococcus aureus secretes a unique class of neutrophil serine protease inhibitors, Proc Natl Acad Sci U S A 111, 13187–13192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Spaan AN, Surewaard BG, Nijland R, and van Strijp JA (2013) Neutrophils versus Staphylococcus aureus: a biological tug of war, Annu Rev Microbiol 67, 629–650. [DOI] [PubMed] [Google Scholar]
- [3].Amulic B, Cazalet C, Hayes GL, Metzler KD, and Zychlinsky A (2012) Neutrophil function: from mechanisms to disease, Annu Rev Immunol 30, 459–489. [DOI] [PubMed] [Google Scholar]
- [4].Nauseef WM (2007) How human neutrophils kill and degrade microbes: an integrated view, Immunol Rev 219, 88–102. [DOI] [PubMed] [Google Scholar]
- [5].Borregaard N, and Cowland JB (1997) Granules of the human neutrophilic polymorphonuclear leukocyte, Blood 89, 3503–3521. [PubMed] [Google Scholar]
- [6].Ohlsson K, and Olsson I (1974) The neutral proteases of human granulocytes. Isolation and partial characterization of granulocyte elastases, Eur J Biochem 42, 519–527. [DOI] [PubMed] [Google Scholar]
- [7].Odeberg H, and Olsson I (1975) Antibacterial activity of cationic proteins from human granulocytes, J Clin Invest 56, 1118–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kao RC, Wehner NG, Skubitz KM, Gray BH, and Hoidal JR(1988) Proteinase 3. A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamsters, J Clin Invest 82, 1963–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Segal AW (2005) How neutrophils kill microbes, Annu Rev Immunol 23, 197–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gorwitz RJ, Kruszon-Moran D, McAllister SK, McQuillan G, McDougal LK, Fosheim GE, Jensen BJ, Killgore G, Tenover FC, and Kuehnert MJ (2008) Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States, 2001–2004, J Infect Dis 197, 1226–1234. [DOI] [PubMed] [Google Scholar]
- [11].Krismer B, Weidenmaier C, Zipperer A, and Peschel A (2017) The commensal lifestyle of Staphylococcus aureus and its interactions with the nasal microbiota, Nat Rev Microbiol 15, 675–687. [DOI] [PubMed] [Google Scholar]
- [12].de Jong NWM, Ramyar KX, Guerra FE, Nijland R, Fevre C, Voyich JM, McCarthy AJ, Garcia BL, van Kessel KPM, van Strijp JAG, Geisbrecht BV, and Haas PA (2017) Immune evasion by a staphylococcal inhibitor of myeloperoxidase, Proc Natl Acad Sci U S A 114, 9439–9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Stapels DA, Geisbrecht BV, and Rooijakkers SH (2015) Neutrophil serine proteases in antibacterial defense, Curr Opin Microbiol 23, 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Geisbrecht BV, Hamaoka BY, Perman B, Zemla A, and Leahy DJ (2005) The crystal structures of EAP domains from Staphylococcus aureus reveal an unexpected homology to bacterial superantigens, J Biol Chem 280, 17243–17250. [DOI] [PubMed] [Google Scholar]
- [15].Herrera AI, Ploscariu NT, Geisbrecht BV, and Prakash O (2018) (1)H, (15)N, and (13)C resonance assignments of the third domain from the S. aureus innate immune evasion protein Eap, Biomol NMR Assign 12, 175–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Woehl JL, Takahashi D, Herrera AI, Geisbrecht BV, and Prakash O (2016) (1)H, (15)N, and (13)C resonance assignments of Staphylococcus aureus extracellular adherence protein domain 4, Biomol NMR Assign 10, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hammel M, Nemecek D, Keightley JA, Thomas GJ Jr., and Geisbrecht BV (2007) The Staphylococcus aureus extracellular adherence protein (Eap) adopts an elongated but structured conformation in solution, Protein Sci 16, 2605–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jonsson K, McDevitt D, McGavin MH, Patti JM, and Hook M (1995) Staphylococcus aureus expresses a major histocompatibility complex class II analog, J Biol Chem 270, 21457–21460. [DOI] [PubMed] [Google Scholar]
- [19].Chavakis T, Wiechmann K, Preissner KT, and Herrmann M (2005) Staphylococcus aureus interactions with the endothelium: the role of bacterial “secretable expanded repertoire adhesive molecules” (SERAM) in disturbing host defense systems, Thromb Haemost 94, 278–285. [DOI] [PubMed] [Google Scholar]
- [20].Woehl JL, Ramyar KX, Katz BB, Walker JK, and Geisbrecht BV (2017) The structural basis for inhibition of the classical and lectin complement pathways by S. aureus extracellular adherence protein, Protein Sci 26, 1595–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Woehl JL, Stapels DAC, Garcia BL, Ramyar KX, Keightley A, Ruyken M, Syriga M, Sfyroera G, Weber AB, Zolkiewski M, Ricklin D, Lambris JD, Rooijakkers SHM, and Geisbrecht BV (2014) The extracellular adherence protein from Staphylococcus aureus inhibits the classical and lectin pathways of complement by blocking formation of the C3 proconvertase, J Immunol 193, 6161–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Stapels DAC, Woehl JL, Milder FJ, Tromp AT, van Batenburg AA, de Graaf WC, Broll SC, White NM, Rooijakkers SHM, and Geisbrecht BV (2018) Evidence for multiple modes of neutrophil serine protease recognition by the EAP family of Staphylococcal innate immune evasion proteins, Protein Sci 27, 509–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kuzmic P (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase, Anal Biochem 237, 260–273. [DOI] [PubMed] [Google Scholar]
- [24].Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, and Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes, Nucleic Acids Res 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Virca GD, Metz G, and Schnebli HP (1984) Similarities between human and rat leukocyte elastase and cathepsin G, Eur J Biochem 144, 1–9. [DOI] [PubMed] [Google Scholar]
- [26].Nakajima K, Powers JC, Ashe BM, and Zimmerman M (1979) Mapping the extended substrate binding site of cathepsin G and human leukocyte elastase. Studies with peptide substrates related to the alpha 1-protease inhibitor reactive site, J Biol Chem 254, 4027–4032. [PubMed] [Google Scholar]
- [27].Strelow J, Dewe W, Iversen PW, Brooks HB, Radding JA, McGee J, and Weidner J (2004) Mechanism of Action Assays for Enzymes, In Assay Guidance Manual (Sittampalam GS, Coussens NP, Brimacombe K, Grossman A, Arkin M, Auld D, Austin C, Baell J, Bejcek B, Caaveiro JMM, Chung TDY, Dahlin JL, Devanaryan V, Foley TL, Glicksman M, Hall MD, Haas JV, Inglese J, Iversen PW, Kahl SD, Kales SC, Lal-Nag M, Li Z, McGee J, McManus O, Riss T, Trask OJ Jr., Weidner JR, Wildey MJ, Xia M, and Xu X, Eds.), Bethesda (MD). [Google Scholar]
- [28].Dahlen JR, Foster DC, and Kisiel W (1999) Inhibition of neutrophil elastase by recombinant human proteinase inhibitor 9, Biochim Biophys Acta 1451, 233–241. [DOI] [PubMed] [Google Scholar]
- [29].Menon V, and Rao M (2012) Slow-tight binding inhibition of pepsin by an aspartic protease inhibitor from Streptomyces sp. MBR04, Int J Biol Macromol 51, 165–174. [DOI] [PubMed] [Google Scholar]
- [30].Dash C, Vathipadiekal V, George SP, and Rao M (2002) Slow-tight binding inhibition of xylanase by an aspartic protease inhibitor: kinetic parameters and conformational changes that determine the affinity and selectivity of the bifunctional nature of the inhibitor, J Biol Chem 277, 17978–17986. [DOI] [PubMed] [Google Scholar]
- [31].Schuck P, and Zhao H (2010) The role of mass transport limitation and surface heterogeneity in the biophysical characterization of macromolecular binding processes by SPR biosensing, Methods Mol Biol 627, 15–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.