

Abstract
Neuropathic pain caused by nerve injury is debilitating and difficult to treat. Current systemic pharmacological therapeutics for neuropathic pain produce limited pain relief and have undesirable side effects, while current local anesthetics tend to nonspecifically block both sensory and motor functions. Calcitonin gene related peptide (CGRP), a neuropeptide released from sensory nerve endings, appears to play a significant role in chronic neuropathic pain. In this study, an analgesic microneedle (AMN) patch was developed using dissolvable microneedles to transdermally deliver selective CGRP antagonist peptide in a painless manner for the treatment of localized neuropathic pain. Local analgesic effects were evaluated in rats by testing behavioral pain sensitivity in response to thermal and mechanical stimuli using neuropathic pain models such as spared-nerve injury and diabetic neuropathy pain, as well as neurogenic inflammatory pain model induced by ultraviolet B (UVB) radiation. Unlike several conventional therapies, the AMN patches produced effective analgesia on neuropathic pain without disturbing the normal nociception and motor function of the rat, resulting from the high specificity of the delivered peptide against CGRP receptors. The AMN patches did not cause skin irritation or systemic side effects. These results demonstrate that dissolvable microneedle patches delivering CGRP antagonist peptide provide an effective, safe, and simple approach to mitigate neuropathic pain with significant advantages over current treatments.
Keywords: microneedle, CGRP antagonist peptide, neuropathic pain, analgesia, drug delivery
Pain is one of the most frequent complaints for patients. Certain types of peripheral nerve injury produce localized persistent neuropathic pain, which can result from trauma, infections, inflammation, tumors, metabolic disease, or endocrine diseases (e.g., diabetes mellitus).1,2 Neuropathic pain is often associated with a stimulating or burning sensation in a specific area,3 which is one of the most difficult chronic pain symptoms to treat successfully with pharmacotherapy or surgery.2,4–6 Neuropathic pain forms a large subset of the chronic pain-related patient population; this condition affects approximately 10 million people in the US and 26 million people worldwide.7 Current pharmacotherapy for neuropathic pain is principally derived from other medications such as antiepileptics (e.g., gabapentin, carbamazepine and lamotrigine),8 antidepressants (amitriptyline and duloxetine),6,9 nonsteroidal anti-inflammatory drugs (NSAIDs, e.g., tramadol),10 and narcotic analgesics (e.g., oxycontin).11,12 However, these treatment options provide pain relief only in around 50% of patients and have significant systemic side effects.8–11,13
Transdermal injection of a local anesthetic such as lidocaine or bupivacaine has long been used to relieve localized acute nociceptive pain,14,15 such as post-operative pain. However, this has less efficacy for treating chronic neuropathic pain. Moreover, due to its nonspecific blockage of voltage-gated sodium channels in both sensory and motor nerve fibers, local anesthetics tend to numb skeletal muscles or peripheral tissues leading to unwanted side effects. Thus, transdermal injection of a local anesthetic is not suitable for long-term treatment of persistent neuropathic pain (Supporting Information S1).16,17
There is a need for alternative treatments that can provide more selective and safer analgesia for neuropathic pain without affecting normal nociception. There is also a need for improved delivery techniques, particularly for large molecules, such as peptides and proteins to minimize systemic side effects and toxicity. Calcitonin gene-related peptide (CGRP), a neuro-peptide synthesized and released by nociceptive sensory neurons, appears to be critical in the development and maintenance of neuropathic pain states (Supporting Information S2).18,19 Nerve or tissue injury caused by trauma or inflammation, for example, triggers the release of CGRP from nociceptive sensory nerve endings and enhances nociceptive neuronal activity by augmenting voltage-gated sodium channels and transient receptor potential cation channel subfamily V member 1, which leads to thermal hyperalgesia and mechanical allodynia.19,20 Selective inhibition of CGRP signaling by systemic blocking of CGRP receptors has demonstrated a promising therapy to treat migraines.21–23 Although orally available small molecule CGRP antagonists mitigate migraines, they also produced unwanted systemic side effects leading to suspension of clinical trials.24–26
CGRP receptor antagonist peptides (or anti-CGRP peptides), such as CGRP8–37, have been long studied in basic research as a CGRP biological probe (Supporting Information S3). Peptides used as therapeutics are generally safer compared to small molecular drugs due to their higher selectivity and effective metabolism after action. However, there are many challenges to peptide delivery including limited absorption in the gastrointestinal tract and a short half-life in blood circulation, leading to low potency and a short duration of action.30–32 Therefore, special delivery routes are often required. For example, intrathecal administration of CGRP8–37 has been shown to produce an antinociceptive effect, and CGRP receptors in the spinal cord may be involved.25,27,28 One report indicates that local injection using a metal needle produces analgesic effects in a rodent model of chronic central neuropathic pain,29 but this conventional delivery method itself can cause pain. Challenges remain in developing an alternative therapy that meets the following criteria: (i) selective and effective analgesia for persistent pain without interfering with normal nociception or motor function, (ii) local or special delivery avoiding systemic side effects, and (iii) convenience of application.
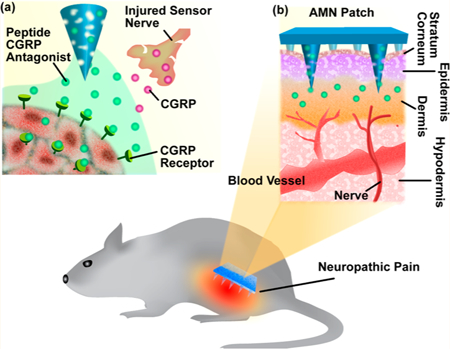
The present study demonstrates that analgesic microneedle (AMN) patches utilizing dissolvable microneedles (MNs) can transdermally deliver an anti-CGRP peptide locally, producing effective and safe analgesic effects for localized neuropathic pain (Figure 1). The anti-CGRP peptide was employed to produce selective antihypersensitivity through antagonism of peripheral CGRP receptors. Dissolvable MNs delivered the anti-CGRP peptide directly to a painful area in a painless and convenient way, avoiding systemic exposure and its resultant side effects. The short MNs used were designed to penetrate the stratum corneum (the outermost layer of the epidermis), making minimal contact with blood capillaries or nerve endings in the dermis layer, enabling painless application (Supporting Information S4).33–36 While MN patches have been previously demonstrated to be successful, particularly in vaccination, insulin delivery, and tumor therapies,33,37–41 using MNs to achieve local analgesia with neuropathic pain treatment has rarely been explored.35,42,43
Figure 1.
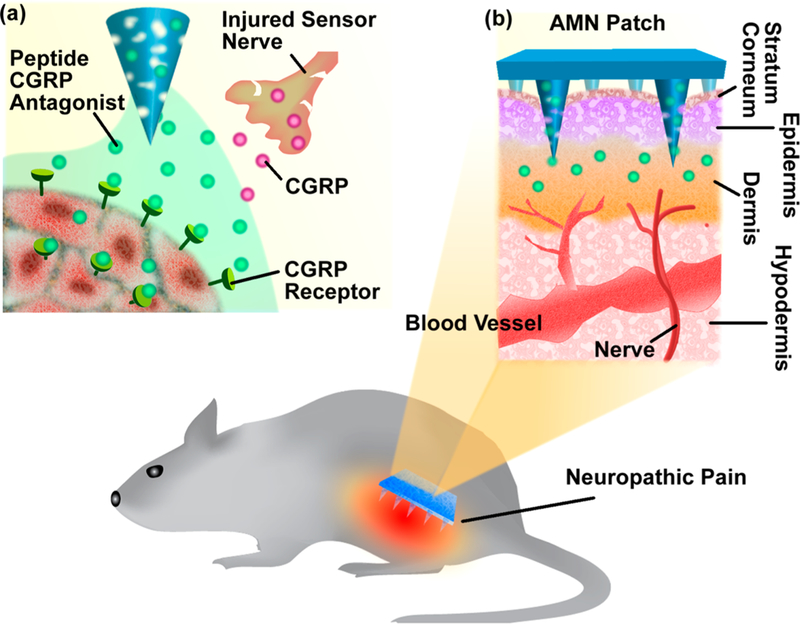
Schematic of dissolvable MNs mediating local delivery of peptide CGRP antagonist to produce analgesia for the treatment of neuropathic pain. (a) Peptide CGRP antagonist mitigates neuropathic pain by selectively blocking CGRP receptors and inhibiting CGRP signaling. (b) Dissolvable MNs transdermally delivered CGRP antagonist peptide directly to the local neuropathic pain area in a painless manner.
Biodegradable AMN patches were fabricated with a centrifugation casting method using an inverted cone-shaped template.37,44 Local analgesic effects of the released anti-CGRP peptide were evaluated by testing the thermal and mechanical behavioral responsiveness of rats. Our technique produced effective analgesia on multiple persistent pain models including rat spared-nerve injury and diabetic neuropathy, as well as neurogenic inflammatory pain induced by UVB radiation. Normal nociception and motor functions were spared by the anti-CGRP peptide. Negligible skin inflammation and neuro-behavioral toxicity were induced by the MN-mediated delivery. These results demonstrate that dissolvable MN patches delivering CGRP antagonist peptide can provide a safe, effective, and simple approach to relieve localized neuropathic pain and have significant potential advantages compared to currently available clinical treatments.
RESULTS
AMN Patch Fabrication, Application, And Characterization.
Dissolvable MN patches were fabricated using sodium carboxylmethyl cellulose (SCMC, molecular weight ~90,000), a safe and highly biocompatible material.45 The MNs were prepared by centrifugation casting SCMC solution onto an inverted cone-shaped polydimethylsiloxane (PDMS) mold and then allowed to dry. 33,46 Briefly, CGRP8–37, a model anti-CGRP peptide, was dissolved in 8% (w/w) SCMC solution, which was then spun onto the mold at 4000 rpm for 5 min to form the needle tips. A second layer of gel containing 8% (w/ w) SCMC alone was applied onto the mold to create a mechanically robust substrate (Figure 2a). The as-fabricated MN patches were air-dried overnight and separated from the mold (Figure 2b). The density of MNs in the patch (0.785 cm2) was 56 needles/cm2, with MN length of 800 μm. When gently inserted to the skin of the rat, the MNs were readily dissolved and gradually released anti-CGRP peptide. The length was found to be reduced by ~3/4 at the time of removal 20 min after the application (Figure 2c). To visualize the distribution of drug molecules within the MN patch, Rhodamine B-labeled (red fluorescence) dextran, a polysaccharide with a similar molecular weight (MW ~ 3000) to CGRP8–37 (MW ~ 3128), was used as a surrogate marker of CGRP8–37 and loaded in the MNs. Confocal fluorescence microscopy revealed that most of the fluorescent molecules were distributed in the MN tips (Figure 2d). The amount of CGRP8–37 in the AMN patch was determined with enzyme immunoassay (EIA, Supporting Information S5) and found to be 1.42 ± 0.65 μg in the MN tips and 0.064 ± 0.037 μg in the patch substrate base (Figure 2e), confirming that most of the peptide was loaded in the MN tips. To evaluate the stability of the peptide CGRP8–37 retained in the MNs, the effective amounts of intact CGRP8–37 after MN fabrication and storage were evaluated using EIA (Figure 2f).47 In this experiment, when 10.4 ± 2.3 μg CGRP8–37 was loaded in an MN patch, 91.5 ± 15.3% of the peptide remained stable after MN fabrication and 81.1 ± 23.2% remained stable following storage of MN patch at 4 °C for 1 week (Supporting Information S5). These are in contrast to the negative control samples where the peptide was destabilized to 3.1 ± 0.2% when the MN patch was shortly stored at high temperature (at 90 °C for 30 min). This result confirms the stability of the anti-CGRP peptide within MN patch under general storage conditions.
Figure 2.
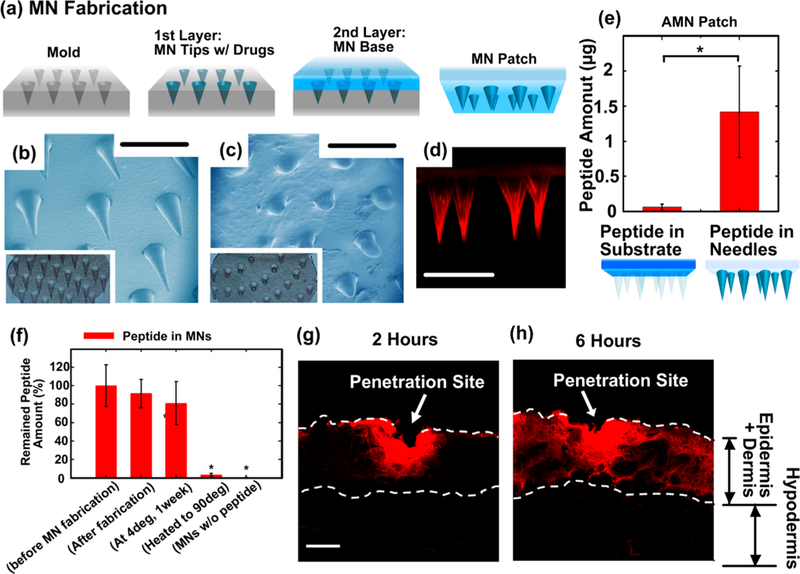
(a) Schematic of MN fabrication process. (b and c) Optical images and SEM images of (b) as-fabricated MNs and dissolved MNs (c) after inserting into skin for 20min. (d) Confocal fluorescence microscopy image showing MNs-loaded with dextran-Rhodamine B (red). The fluorescent molecules were localized compared to the group “Peptide in Substrate” at the level of p < 0.05 using a Student’s t-test; n= 4/group. (f) The stability of CGRP8–37 peptides retained in the MNs, determined with EIA. The * indicates statistically significant compared to the group “in water, before MN fabrication” at the level of p < 0.05 using ANOVA followed by a post hoc test; n = 3/group. (g and h) Confocal fluorescence microscopy images shwing the release of fluorescent molecules from the MNs into rat skin and their spatial distribution. The skin near the penetration sites were dissected (g) 2 h or (h) 6 h after MNs insertion and then prepared for imaging. Scale bar: 1000 μm in (b –d) and 100 μm in (g and h).
To observe drug molecule distribution of drug released from the MNs following skin penetration, MNs containing dextranRhodamine B were used. Confocal fluorescence microscopy was conducted to visualize the release of fluorescent molecules from the MNs into the rat skin and their spatial distribution. MNs were applied to the rat’s dorsal surface for 20 min and then removed. Two or 6 h after MNs insertion, the skin near the penetration sites was dissected and prepared for imaging. As shown in Figure 2g, the MNs disrupted the epidermis, and the fluorescent molecules were found to deposit in the dermis layer (at depths of approximately 100 μm or greater) and diffused locally near the needle penetration site 2 h after MNs application. After 6 h, the fluorescent molecules were observed to continue to diffuse and spread over a larger area within the dermis layer (Figure 2h), in the vicinity of the needle penetration site (Supporting Information S6). These results indicate the successful utilization of MNs for stably carrying anti-CGRP peptide and locally delivering drug molecules into the skin.
Spared Nerve Injury (SNI) Model.
We employed a relatively localized neuropathic pain model, the SNI model in rodents,48,49 to assess the analgesic effects produced by the AMN patch which mediated CGRP8–37 delivery through MNs (i.e., MN/CGRP8–37). The SNI in the rat involves a lesion of two of the three terminal branches of the sciatic nerve (tibial and common peroneal nerves), leaving the sural nerve intact. After the peripheral nerve lesion, the remaining primary afferent develops spontaneous abnormal excitability and heightened sensitivity to thermal and mechanical stimuli. The hindpaw is one of the most commonly tested areas when dealing with rodent models of inflammatory pain or chronic neuropathic pain.49 Nociception on the hindpaws of the SNI rats in response to thermal and mechanical stimuli was tested using the Hargreaves thermal stimulator and von Frey monofilaments,50,51 which measured the ability of the rats to withstand thermal or mechanical stimulation, respectively. The MN/CGRP8–37 patch was applied to the hindpaws of the rats for 20 min to allow the microneedles to dissolve and subsequent peptide release, and then the patch base was removed. The nociceptive response after MN treatment was reassessed to evaluate pain responses compared to pretreatment (Figure 3a). Each rat’s thermal and mechanical measurements were expressed as a percentage of the rat’s individual normal nociception measured before SNI surgery (baseline).
Figure 3.
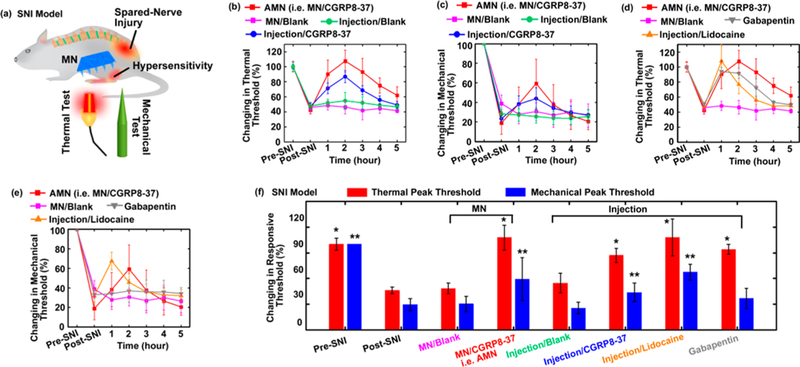
SNI model in the rat and different treatments. (a) Illustration of the experiment on testing MN-mediated CGRP8–37 delivery for the treatment of neuropathic pain on SNI model. Rats were subjected to SNI operation and allowed to develop thermal and mechanical hypersensitivity over 1–8 weeks. After treatment on the hindpaws, the rats’ nociception to thermal and mechanical stimuli was tested. (b) The thermal pain threshold and (c) mechanical pain threshold were tested every hour for up to 5 h after the treatment with AMN (i.e., MN/ CGRP8–37), compared to MN/blank (control), injection/CGRP8–37, and injection/blank (control)/ (d) The thermal pain threshold and (e) mechanical pain threshold were tested every hour for up to 5 h after the treatments with MN/CGRP8–37, compared to MN/ blank (control), injection/lidocaine, and IP administration of gababpentin. In (b-e), each rat’s thermal and mechanical measurements were expressed as a percentage of the rat’s individual normal nociception before SNI surgery (baseline); n = 8/ group. (f) The measured peak thresholds of each 0.05 using ANOVA followed by a post hoc test. The ** indicates statistically compared to the group “Post-SNI (Mechanical)” at the level of p < 0.05 using ANOVA followed by a post hoc test; n = 8/ group.
At 2 weeks post-surgery, the SNI rats showed reduced thermal pain thresholds to 46.1 ± 4.2% of pre-SNI baseline, confirming the presence of hyperalgesia. Four groups received either treatment of MNs containing CGRP8–37 (~1.4 μg/ patch), MNs without drugs as blank control, subcutaneous (SC) injection of CGRP8–37 (~1.6 μg in 50 μL), or SC injection of blank vesicles on the left hindpaws of the rats. After 20 min of MN treatment or conventional SC injections using metal needles, the SNI rats were tested every hour for up to 5 h (Figure 3b). Of the four treatment groups, the rats treated with MN/CGRP8–37 exhibited a reversal to normal thermal nociception as early as the first time-point 1 h after the MNs application. The observed antihyperalgesic effect reached a maximal effect at 2 h post-MN application and lasted for up to 5 h. Similarly, the rats treated with SC injection/CGRP8–37 showed substantial and significant alleviation of hyperalgesia, peaking at 1 h post-injection, which confirms the analgesic effects of CGRP8–37 are independent of delivery methods. In contrast, the MN/blank and SC injection/blank controls had no observable analgesic effects.
In addition to examining thermal responsivity, mechanical nociceptive thresholds were measured (Figure 3c). At 2 weeks post-surgery, the rats exhibited significant mechanical allodynia with a mechanical threshold reduced to 29.7 ± 8.2% of pre-SNI baseline. The rats that received CGRP8–37 exhibited a peak analgesia recovering to 59.2 ± 24.9% with MNs, and 43.9 ± 11.0% by SC injection, lasting up to 3 h. After 3 h post-treatment, the antiallodynia effect began to gradually subside. The treatments with blank controls did not show any analgesic effects. Thus, application of CGRP8–37 either by MNs or SC injection can mitigate both thermal hyperalgesia and mechanical allodynia resulting from peripheral nerve injury.
To compare with current treatments, SC injection of lidocaine (1.5% w/w, a typical concentration used in clinic, 50 μL)52 and intraperitoneal (IP) administration of gabapentin53 (100 g/kg) were used as a benchmark for comparison (Supporting Information S7 and S8). Both treatments with lidocaine and gabapentin achieved full analgesic effects on thermal hyperalgesia that peaked at 1 h post-administration and gradually declined over a 3 h period (Figure 3d). Lidocaine produced a peak elevation of allodynia, as indicated by an increase in the mechanical pain threshold from 31.9 ± 6.3% to 67.5 ± 9.1% of presurgical thresholds on mechanical pain; while gabapentin produced only a small analgesic effect on mechanical allodynia 36.9 ± 11.5% compared to the 33.1 ± 4.0% pretreatment (Figure 3e). The measured peak effects of different groups in Figure 3b-e are summarized n Figure 3f. These results suggest that the CGRP8–37 delivered by MNs was able to achieve analgesic effects against neuropathic pain as effective as CGRP8–37 or lidocaine delivered by SC injection, and CGRP8–37 delivered by MNs was superior to systematic gabapentin treatment.
Diabetic Neuropathy Model.
Effects of the treatments were also evaluated in animals with peripheral neuropathy induced by streptozotocin (STZ)-induced damage to the pancreas in the rat resulting from high blood glucose levels (Figure 4a).54 Prior to STZ administration, each rat’s normal nociceptive responsiveness of their left hindpaws was assessed as a baseline. Two weeks post-STZ administration (50–55 mg/ kg, IP), the rats developed significant peripheral neuropathy, manifested as thermal hyperalgesia (thermal pain threshold dropped to 52.7 ± 5.4% of baseline) and mechanical allodynia (mechanical pain threshold dropped to 15.9 ± 10.3% of baseline). In addition, each rats’ blood glucose levels were measured using a basic glucose level monitor to verify hyperglycemia status (pre-STZ 115 ± 17 mg/100 mL vs post-STZ 405 ± 25 mg/100 mL).
Figure 4.
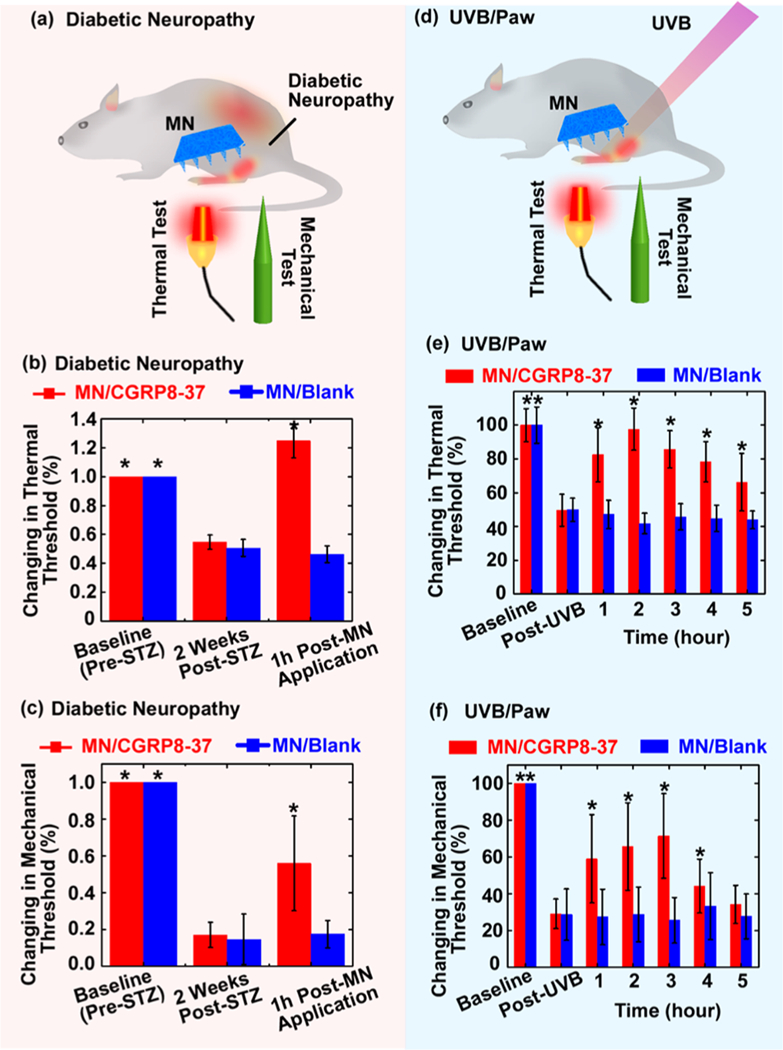
(a-c) Diabetic peripheral neuropathy model in rats and treatments. (a) Illustration of the experiments on testing MN-mediated CGRP8–37 delivery for the treatment of STZ-induced diabetic neuropathy. After treatments on the hindpaws, the rats’ nociception to thermal and mechanical stimuli was tested. (b) The thermal pain threshold and (c) mechanical pain threshold were tested 1 h after the treatments with MN/CGRP8–37 (i.e., AMN), compared to MN blank control. Each rat’s thermal and mechanical measurements were expressed as a percentage of the rat’s individual normal nociception before STZ treatments (baseline). The * indicates statistically significant compared to the group “2 Weeks Post-STZ (MN/blank)” at the level of p < 0.05 using ANOVA followed by a post hoc test; n = 6/group. (d-f) UVB/Paw model and treatments. (d) Illustration of the experiments on testing MN-mediated CGRP8–37 delivery for the treatment of UVB-induced inflammatory pain on hindpaws. The rats’ hindpaws were exposed to UVB and allowed to develop hypersensitivity over a 24 h time course. After the hindpaws were treated, the rats’ nociception to thermal and mechanical stimuli was tested. (e) The thermal pain thresholds and (c) mechanical pain thresholds were tested every hour for up to 5 h after the treatments with MN/ CGRP8–37, compared to MN blank control. Each rat’s thermal and mechanical measurements were expressed as a percentage of the rat’s individual normal nociception before UVB treatments (baseline). The * indicates statistically significance compared to the group “Post UVB (MN/blank)” at the level of p < 0.05 using ANOVA followed by a post hoc test; n = 6/group.
As described above, MN patches were applied on the rats’ left hindpaws for 20 min and then removed, and the rats were tested 1 h after removal of the patches. Similar to results with the SNI model, rats receiving MN/CGRP8–37 showed a full return to their baseline thermal pain thresholds in responding to thermal stimulation (Figure 4b), and the mechanical stimulation test indicated a significant analgesic effect in which the rats displayed a partial recovery to 55.9 ± 25.7% of baseline mechanical pain threshold (Figure 4c). In contrast, for the rats that received blank MN control patches, the hyperalgesia persisted, with the thermal thresholds remaining at 46.3 ± 5.9% and the mechanical thresholds at 17.5 ± 7.3%.
UVB Model.
In a third model, the antihyperalgesic or antiallodynic effects of MN/CGRP8–37 were studied in a rat neuroinflammation model adapted from a simple pain model in healthy subjects55 (Figure 4d). The rat’s hindpaws were exposed to an inflammatory dose of UVB radiation (1200mJ/cm2 for 25 s). UVB exposure caused significant thermal hyperalgesia and mechanical allodynia, as indicated by decreases in the rats’ paw withdrawal latencies in response to noxious heat and von Frey hair stimulation, respectively. One day post-UVB exposure, the thermal pain thresholds decreased to 49.7 ± 8.1%, and the mechanical threshold decreased to 29.0 ± 11.0%, compared to responsiveness before UVB radiation. MN/CGRP8–37 or blank MNs were applied to the UVB-radiated hindpaws, and the thermal and mechanical pain thresholds were measured once every hour for 5 h. The MN/ CGRP8–37 produced both antihyperalgesic and antiallodynic effects in the rats. The rats that received MN/CGRP8–37 exhibited a complete return on thermal pain thresholds (Figure 4e) and a maximal recovery to 71.4 ± 23.0% of baseline mechanical thresholds (Figure 4f) at 2–3 h post-treatment, with significant analgesic effects that lasted for 5 h. In contrast, the rats treated with blank MNs exhibited no pain attenuation. These results suggest that locally applied CGRP8–37 is an effective analgesic in neuroinflammatory pain.
The MN patches could be flexibly applied on different surfaces of the rat body, owing to the superior mechanical strength and malleability of the patches. Given these features, we also tested the MN/CGRP8–37 patch on the rat cheek, a larger and flatter surface than the hindpaw. The latency of each rat to withdraw its cheek from a beam of noxious heat which was applied directly to the rat’s cheeks was measured and treated as a baseline (100%; Figure 5a). UVB (1200 mJ/cm2 for 25 s) was applied to rat’s depilated cheeks (both left and right) to produce significant neuroinflammation. One day post-UVB radiation, the rat’s cheeks exhibited hypersensitivity to thermal stimuli (Figure 5bl), as indicated by the cheek withdrawal latencies decreased from 10.4 ± 0.2 s (blue curve, prior to UVB) to 6.8 ± 0.2 s (pink curve, post-UVB), and the degree of hypersensitivity was consistent among all separate experiments.
Figure 5.
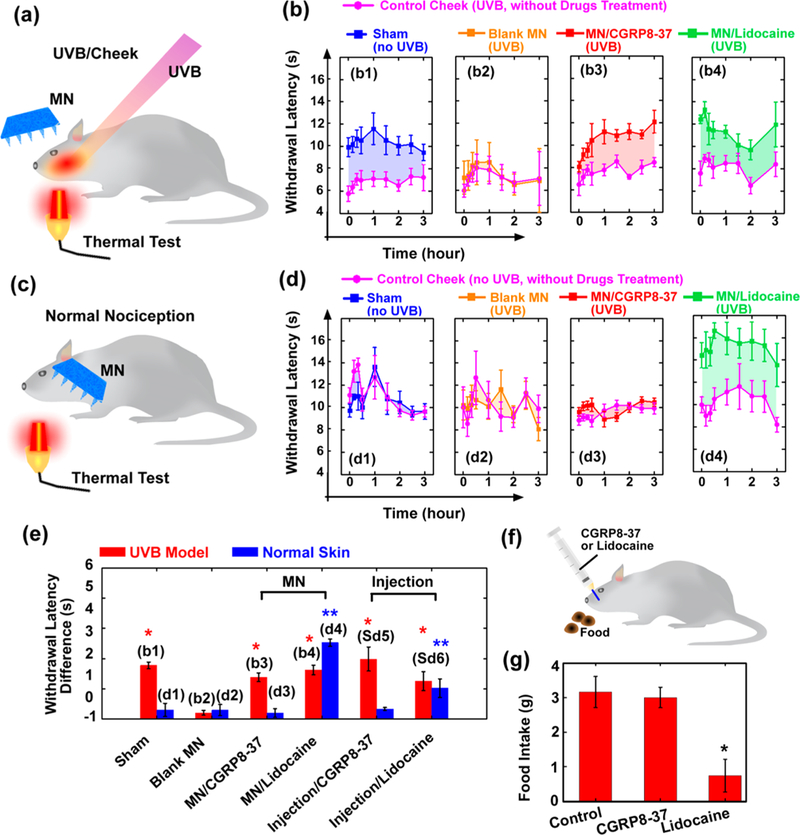
(a and b) UVB/cheek model and treatments. (a) Illustration of the experiments on testing MN-mediated CGRP8–37 delivery for the treatment of UVB-induced inflammatory pain on cheeks. The rat’s left and right cheeks were exposed to UVB and allowed to develop hypersensitivity over a 24 h time course. After treatment, the rats’ nociception to thermal stimuli was tested. (b) The ability of the rats to withstand thermal stimulation (withdraw latency) was tested for up to 3 h after the treatments with MN/CGRP8–37 (i.e., AMN), compared to a sham control, MN blank control, and MN/lidocaine. In each group, treatment was applied on the left cheek, while the right cheek was untreated as control (pink curves). (c and d) Normal nociception model. (c) Illustration of the experiments on testing the interference on normal sensation after MN-mediated CGRP8–37 delivery. Both of the rat’s cheeks were not UVB irradiated. After treatments, the rats’ nociception to thermal stimuli was tested. (d) The ability of the rats to withstand thermal stimulation (withdraw latency) was tested for up to 3 h after the treatments with MN/CGRP8–37, compared to a sham control, MN blank control, and MN/lidocaine. In each group, treatment was applied on the left cheek, while the right cheek was untreated as control (pink curves). (e) The results in (b and d) are summarized. In each group, the withdrawal latencies after treatments were compared to those without treatment (pink curves), and the averaged differences, which indicate analgesic effects due to treatments, are shown. The * indicates statistically significant compared to the group “Blank MN (UVB model)” at the level of p < 0.05 using ANOVA followed by a post hoc test. The ** indicates statistically significant compared to the group “Blank MN (Normal Nociception)” at the level of p < 0.05 using ANOVA followed by a post hoc test; n = 6/group. (f and g) The assessment of local interference with active physical functions after oral mucosa injection of CGRP8–37 or lidocaine. The ability for food-deprived rats to eat and chew after injection was tested. (g) The food uptake was measured. The * indicates statistically significant compared to the group “Control” at the level of p < 0.05 using ANOVA followed by a post hoc test; n = 6/group.
MN/CGRP8–37 patches were applied on the rat’s left cheek for 20 min. After removing the patch, cheek withdrawal latencies for avoiding noxious heat were assessed at different time points up to 3 h. The rat’s right cheek was assessed as negative control (pink curve), which was UVB-exposed as well but without MN treatment. As shown in Figure 5b3, MN/ CGRP8–37 produced a potent antihyperalgesic effect (red curve), compared to the right cheek (pink curve). In contrast, the cheek treated with blank MN control patches did not produce any analgesic effects (orange curve in Figure 5b2). MNs containing 450 ± 87 μg lidocaine (comparable dose with SC injection of 50 μL 1–2% lidocaine solution; Supporting Information S8) produced significant thermal analgesia (Figure 5b4) and served as a positive control.
Ideal therapeutics of antihypersensitivity or analgesics should not interfere with normal physiological pain sensation.14,56 To investigate whether MN-mediated CGRP8–37 delivery alters normal nociceptive sensation, naϊve rats without induced neuropathic or inflammatory pain were subjected to the treatment procedure and the thermal pain test (Figure 5c). The rat’s left cheek was treated with MNs for 20 min, while the right cheek, without any treatments, was used as a negative control. After removing the MN patches, noxious heat was alternatively applied to both cheeks at different time points, and the cheek withdrawal latencies were measured. As shown in Figure 5d, the withdrawal latencies of the cheeks treated with MN/CGRP8–37 (~1.4 μg/patch, Figure 5d3, red curve) or blank MNs (Figure 5d2, orange curve) showed insignificant differences compared to the right cheeks without any treatments (pink curves), indicating that MN/CGRP8–37 did not produce discernible analgesic effects on normal nociception. In contrast, MNs containing lidocaine (450 ± 87 μg) produced local anesthetic effects on normal nociceptive pain as expected (Figure 5d4, green curve). In addition, SC injections of CGRP8–37 (1.6 μg in 50 μL) or lidocaine (750 μg in 50 μL, ~ 1.5%) in the rats’ left cheeks were conducted to verify the differential effects between CGRP8–37 and lidocaine (Supporting Information S9). To make a more clear comparison among all groups tested, the left cheek withdrawal latencies with treatments were subtracted from the individual right cheek without treatment, and the mean difference indicates analgesic effects resulting from the treatment (Figure 5e). Taken together, these results demonstrate that CGRP8–37 can preferentially produce antihyperalgesia without affecting normal pain sensation.
In addition to differential modulation of hypersensitivity and normal nociception, local application of CGRP8–37 was investigated to determine whether and to what extent this treatment might interfere with motor function (as do local anesthetics). In this experiment, rats were fasted for 1 day and then received an injection into the oral mucosa of either a high dose of CGRP8–37 (~5 μg in 50 μL) or a standard clinical dose of lidocaine (~750 μg in 50 μL, ~ 1.5%) for comparison. After injection, the rats were given 20 min to to freely eat one piece of ordinary solid rodent chow placed on the homecage floor (Figure 5f). The rats injected with lidocaine displayed dysfunction of chewing and eating presumably due to anesthetic effects blocking the motor nerve conductivity. In contrast, the rats injected with CGRP8–37 could eat normally (Supporting Information S10). The food intake was assessed (Figure 5g), and the results showed that the rats injected with CGRP8–37 could eat as much (3.02 ± 0.32 g) as the control group (3.16 ± 0.45 g). Both CGRP8–37 and control groups consumed food amounts approximately 4-fold greater than that the lidocaine group did (0.74 ± 0.46 g). These results demonstrated that targeting CGRP receptors provides a selective analgesic approach without interfering with normal sensation and motor functions, which is highly advantageous compared to traditional sodium channel blockers that tend to eliminate local sensation and benumb peripheral mucles.14,56
Skin Irritation Test.
The safety of the local application of the AMN patch (i.e., MN/CGRP8–37) was evaluated by testing skin irritation following treatment. MN/CGRP8–37 was applied to the dorsal surface of mouse for 20 min once every day for 3 consecutive days. There was no visible irritation observed on the skin treated with MN/CGRP8–37 compared to the untreated skin (Figure 6a). On the fourth day, the skins surrounding the microneedle penetration sites was dissected and prepared for histological examination. Compared to the untreated skin (Figure 6b), there were no overt infiltrated inflammatory cells observed on the skin after repeat insertion of MNs (Figure 6c), indicating the applications of MN/CGRP8– 37 did not induce significant inflammation in the skin.
Figure 6.
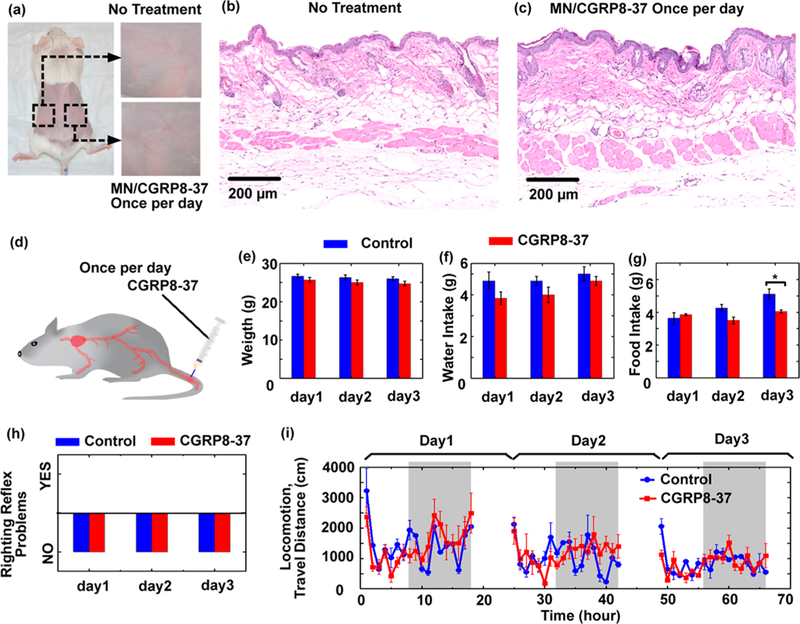
(a-c) MN/CGRP8–37 (i.e., AMN) treatment did not induce over skin irritation. Mice were treated with MN/CGRP8–37 once a day image represents four similar results. (d-i) Evaluation of the risk of systematic exposure to CGRP8–37. (d) Illustration of the experiment on testing neurobehavioral toxicity. CGRP8–37 ( 10mg/kg) was intravenously injected in the blood once per day for up to 3 days. The spontaneous behavior of the mice was assessed after injection every day. (e) The weight, (f) water intake, (g) food intake, and (h) righting reflex were assessed every day after injection. The * indicates statistically significant compared to the control group at the level of p < 0.05 using a Student’s t-test; n = 6 / group. (i) Locomotion was continuously monitored with an automated noninvasive system for 3 days. The gray color indicates the dark phase (night time); n = 6/ group.
Spontaneous Behavior Assessment.
Although CGRP8– 37 was delivered by MN patch in this study, avoiding systemic exposure, it is possible some CGRP8–37 entered circulating blood. The potential systemic side effects of such exposure were assessed by monitoring general neurobehavior following intravenous injections to the mouse tail (Figure 6d). A high dose of CGRP8–37 (10 mg/kg) was injected once a day for 3 consecutive days. The concentrations of CGRP8–37 in the plasma (estimated around 100 μM, Supporting Information S11) should be significantly higher than the local concentrations of CGRP8–37 (<10 μM) transdermally delivered by MNs or SC injections. Following each daily injection, the health conditions and spontaneous behaviors of the mice were assessed, including the body weight, water and food intake, clinical observations, righting reflex (a measure of unconsciousness and rolling response), 57,58 and locomotion. There were no significant differences between the CGRP8–37 group and the control group in body weight and water intake for all 3 days and food intake for the first 2 days (Figure 6e-g). Only food intake showed a small yet significant difference on the third day, where the group treated with CGRP8–37 showed less food intake than the control group. However, the amount of food intake was still within the normal range for mice (4–5 g per day). The mice from all groups appeared clinically normal, and none of the mice from either group showed any righting reflex defects (Figure 6h), indicating normal consciousness and rolling response. Locomotion was continuously monitored using the SmartCage, an automated noninvasive monitoring system for 3 days (Figure 6h).59 The CGRP8–37 group displayed similar travel distances as the control group, with increased activities in the dark phase corresponding to the normal active behaviors of mice, especially in the first day when the mice were placed in fresh homecages and in a new environment. These results indicate that the systemic exposure of high dose of CGRP8–37 did not alter the animals’ general health state and behaviors, suggesting the safe nature of this peptide CGRP antagonist (Supporting Information S12).
CONCLUSION
AMN patches transdermally delivered CGRP antagonist peptide in a painless manner, producing effective and safe analgesia on neuropathic pain models including SNI, diabetic neuropathy, and neurogenic inflammatory pain induced by UV radiation in rats. Effective analgesia was selectively achieved without disrupting normal pain sensation and motor functions. These differential actions result from the high specificity of the CGRP antagonist peptide on blocking the overactivation of the CGRP receptors that causes nociceptive hypersensitivity under pathological conditions. Peptide drugs are generally safe with regards to off-target effects, owing to their high selectivity against their targets and highly efficient peptide degradation into recyclable amino acids by proteinases (or peptidases). In contrast, small molecular drugs often suffer from systematic side effects and detoxification issues.30,31 However, there are many challenges to peptide delivery including limited absorption in the gastrointestinal tract and duration of action in vivo. In our studies, we took the advantages of peptide drugs while overcoming these challenges by using the MN transdermal delivery technology.32,60 The present study demonstrates that peptide-bearing AMN can be safely applied without inducing skin irritation or undesirable systemic side effects, presenting an important technical advance toward alternative treatments for neuropathic pain. This new approach may potentially lead to a reduction in the use of opioid analgesics and open alternative opportunities for pharmaceuticals to use peptide-bearing MNs for other clinical applications.
METHODS
AMN Patch Fabrication and Application.
AMN patches were fabricated by a two-step centrifugation casting method using a polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning, USA) mold containing cone cavities (44 cone cavities, each 0.8 mm in depth and ~440 μm in base diameter; TheraJect, Inc., CA, USA). For MN patched loaded with CGRP8–37 (MN/CGRP8–37), in the first step, ~2 mg CGRP8–37 (VTHRLAGLLSRSGGVVKDNFVPTNVGSEAFNH2, synthesized by GenScript, NJ, USA) was dissolved in 500 μL 8% (w/w) sodium carboxylmethyl cellulose (SCMC, Sigma-Aldrich, MO, USA) solution, and then 50 μL of this solution was added to the mold and centrifuged at 4000 rpm for 5 min to drive the solution into the cavities. The solution out of the cavities was removed after centrifugation and collected for reuse. After air-drying overnight, another 200 μL solution containing 8% (w/w) SCMC alone was poured on the mold and centrifuged at 4000 rpm for 1 min. The solution was dried overnight to form an MN patch, and the patch was peeled off from the mold.
For MN patches loaded with dextran labeled with Rhodamine B, 0.5 mg dextran-Rhodamine B (MW ~ 3000 Da; Nanocs, USA) was dissolved in 500 μL 8% (w/w) SCMC solution, and this solution was used to prepare the MN tips. For MN patches loaded with lidocaine (MN/lidocaine), 75 mg lidocaine hydrochloride (Sigma-Aldrich) was dissolved in 500 μL 8% (w/w) SCMC solution, and this solution was used to prepare the MN tips. The rest of the procedures were similar to the preparation of the AMN patches. After fabrication, AMN patches were stored at 4 °C (in a freezer) until application. Typically, the AMN patches were stored in 24-well plates sealed with parafilm. These plates were generally further stored in a plastic tight head secondary container to protect from moisture. Upon use, MNs were gently inserted into the skin of animals and were held on the skin with adhesive tape. Twenty min after insertion, the MN patches were removed, and the pain behavior test was performed.
MN Characterization.
For scanning electron microscope (SEM) imaging, MN patches were sputter-coated with gold-palladium using DESK V HP (Denton Vacuum, NJ, USA) and then imaged with FEI/ Philips XL30 FEG ESEM. For confocal fluorescent microscopy, MN patches were imaged with a Zeiss LSM 700 Laser Scanning Confocal.
The amount of CGRP8–37 in the AMN patch was determined with Calcitonin Gene Related Peptide (CGRP) - Enzyme Immunoassay (EIA) Kit (Phoenix Pharmaceuticals, Inc., CA, USA). This EIA kit has 100% cross-reactivity with CGRP8–37. The MN tips were immersed in 1 mL DI water to allow the tips to be dissolved. The patch substrate was immersed into another 1 mL DI water to dissolve. The CGRP8–37 concentration of each solution was measured with the EIA according to the standard protocol of the product.
The stability of CGRP8–37 in MN patches was determined with EIA assay. In order to get higher signals for quantification, a higher amount of CGRP8–37 (~10 μg) was loaded in each MNs or aqueous sample, and the CGRP8–37 was loaded homogeneously in the whole MN patch rather than localized in the MN tips in order to avoid the variation of peptide loading during MN fabrication.
The preparation method and storage conditions for different samples were as follow. “Before MN fabrication”: 10.0 μg CGRP8–37 was dissolved in 100 μL DI water. “After MN fabrication”: 10.0 μg CGRP8–37 was dissolved in 100 ^L 8% SCMC solution, and the solution was poured to the mold and dried overnight to form MN patch. “In MNs at 4 °C, 1week”: the MN samples containing ~10.0 μg CGRP8–37 were stored at 4 °C for 1 week. “In MNs, heated to 90 °C “: the MN samples containing ~10.0 μg CGRP8–37 were heated up to 90 °C for 40 min to destabilize the CGRP8–37. This was used as a negative control to confirm that the EIA can be used to determine the effective amount of CGRP8–37. “MNs w/o peptide”: the MN was fabricated without loading CGRP8–37. This was used as a negative control to confirm that the EIA is specific to CGRP8–37 rather than SCMC. Each sample was diluted with another 900 μL DI water, and the CGRP8–37 concentration of each solution was measured with the EIA according to the standard protocol of the product.
For the dextran-Rhodamine B releasing experiment, MN patches loaded with dextran-Rhodamine B were applied to the dorsal skin of rats for 20 min and then removed. After 2 or 6 h, the treated sites were dissected. Frozen sections were imaged by confocal microscopy (Olympus FV-1000) to determine the distribution of the released dextran-Rhodamine B.
Experimental Animals.
Male Sprague-Dawley rats weighing 300–350 g and male C57BL/6 mice weighing 25–35 g were used (Envigo, former Harlan Laboratories, IN, USA). Rats or mice were housed in a climate-controlled room under a 12 h/12 h light/dark cycle and provided food and water ad libitum. The Animal Care and Use Committees of Stanford University and AfaSci approved all surgical and testing procedures prior to initiation of studies. For all surgical procedures, deep anesthesia was maintained throughout surgery with 2% isoflurane. All incisions were closed in layers with 4–0 silk sutures.
Thermal Hyperalgesia Testing.
For the thermal pain threshold determination on rat hindpaw, the Hargreaves test was used with the a paw thermal stimulator system (Department of Anesthesiology, University of California, San Diego, CA, USA), which applies a high-intensity beam of light directed to the hindpaw to induce pain. The time it takes for the animal to withdraw its hindpaw (withdrawal latency) was measured. A cutoff of 20 s is employed to avoid excessive tissue injury. For the thermal test on cheeks, thermal stimulation was provided by a custom-built infrared laser stimulator (Lasmed, Inc. Mountain View, CA, USA), which uses a fiberoptic to allow precise, hand-held positioning of the beam. The beam itself was collimated, allowing for approximately 5 cm working distance, while producing a reliable 3 mm spot size. This stimulator has been used successfully in animal and human behavioral and electrophysiological experiments. After habituation, withdrawal latencies from low-intensity laser heat were assessed (typical latencies ~10 s). Responses of the injured as well as the uninjured cheeks were assessed by measuring withdrawal latencies in response to stimulation of the middle surface of cheeks.
Mechanical Allodynia and Hyperalgesia Testing.
Mechanical allodynia was measured using von Frey monofilaments (Bioseb, Chaville, France). Individual rats were subjected to habituation within a cylinder on a suspended metal mesh; mechanical withdrawal threshold to the application of a von Frey probe to the foot was measured using the up-down method. An ascending series of von Frey hairs of logarithmically incremental force (3.2, 5.2, 8.3, 15, 29, 44, 64, 94, and 160 mN) were applied to sites in the middle (tibial nerve distribution) aspect of the plantar surface of the affected hindpaw. Each von Frey hair was applied to the test area for about 2–3 s, with a 1–2 min interval between stimuli. If the animal showed no response to the highest von Frey hair (160 mN), a von Frey threshold of 260 mN, corresponding to the next log increment in potential von Frey probes, was assigned as the threshold.
Spared Nerve Injury (SNI) Model.
Of the three branches of the sciatic nerve, the tibial and the common peroneal nerves were cut and ligated, while leaving the sural nerve intact. Deeply anesthetized animals were placed on a heated surgical bed. The posterolateral aspect of the left thigh was shaved and prepared with iodine surgical prep. An incision was made through the skin and underlying biceps femoris muscle just deep enough to expose the sciatic nerve and its trifurcation into tibial, common peroneal, and sural nerves. The tibial and common peroneal branches were then ligated and cut distal to the point of ligation, taking care not to touch the sural nerve. The intact, or ‘spared,’ sural nerve normally conveys pain sensation from the lateral aspect of the hindpaws. The incision was then sutured in layers. In sham animals, the procedure was the same, except that the nerves were only exposed and not ligated. Animals were allowed to recover from surgery for approximately 2 weeks before continuation of the study. WT Sprague-Dawley rats each underwent SNI on their left hind leg. Thermal and mechanical pain thresholds of the ipsilateral hindpaws were measured using the Hargreaves thermal stimulator and von Frey hair monofilaments (up-down method). The baseline thermal and mechanical thresholds of the rats prior to surgery were assessed. Five weeks post-surgery, the thermal and mechanical thresholds of each rat were reassessed, confirming the presence of hyperalgesia and mechanical allodynia in each rat. These pain conditions lasted for 6–8 weeks, modeling chronic neuropathic pain. Each rat (n = 8 per group) received MN/CGRP8–37 (~1.4 μg/patch), MN patch without drugs (MN/blank), local subcutaneous injection of CGRP 8–37 (~1.6 μg in 50 μL, dissolved in 2% DMSO in 0.5% % hydroxyl propyl cellulose, HPC), lidocaine (1.5%, 50 μL), vehicle (50 μL, 2% DMSO in 0.5% HPC), or intraperitoneal (IP) administration of gabapentin (100 mg/kg, Sigma-Aldrich, MO, USA). Thermal and mechanical thresholds were assessed every hour for up to 5 h after treatment. Each rat’s thermal pain threshold was represented by its withdrawal latency (s), while mechanical pain threshold measurements were expressed as a percentage of the rat’s individual normal nociception prior to SNI surgery (baseline).
Diabetic Neuropathy Model.
Prior to STZ administration, each rat’s baselines of thermal and mechanical thresholds on their individual hindpaws were assessed using the Hargreaves thermal pain test and von Frey mechanical pain test, respectively. In a naϊve rat, the average paw withdrawal latency using the Hargreaves thermal stimulator was approximately 15 s. Once these baseline values were recorded, a dose of STZ (50–55 mg/kg, IP, Sigma-Aldrich) was administered to each rat (n = 6 per group). Two weeks post-STZ administration, the rats’ average withdrawal latency dropped by 45% (withdrawal latency around 8.5 s), confirming the development of neuropathic pain. In addition, each rats’ blood glucose levels were measured using a basic glucose level monitor and verified hyperglycemia status, confirming that the diabetic model was established. MN/patch or MN/blank was patched on the rat hindpaw for 20 min. After removing the patches, thermal and mechanical thresholds were assessed in 1 h.
UVB/Paw Model.
Baseline thermal and mechanical pain thresholds of each rat’s left hindpaws were measured prior to UVB exposure. Each rat received 1200 mJ/cm2 of UVB (over a 25-s span) on its left hindpaw using a fiberglass applicator of the UV Curing Device (Dymax Bluewave 200). Twenty-four h post-UVB exposure, the thermal and mechanical pain thresholds of each rat were reassessed; the presence of hyperalgesia and mechanical allodynia in each rat was confirmed. MN/patch or MN/blank was applied to the left hindpaw, remaining on the hindpaw for approximately 20 min. After treatment, the thermal and mechanical thresholds were measured once every hour for 5 consecutive hours.
UVB/Cheek Model.
When the skin of a rat’s cheek was stretched, the circular (8 mm in diameter) fiberglass applicator of the UV Curing Device was applied to the center of the rat’s depilated left cheek. To produce an appropriate inflammation, a total dose of 1200 mJ/cm2 UVB was administered to one rat cheek for 25 s. The same procedure was performed on the rat’s depilated right cheek. Upon completion of UVB radiation, all rats were returned to their home cages and were assessed for thermal pain 24 h later. At this period, persistent UVB inflammation could be induced. The rats were lightly anesthetized with urethane (800 mg/kg, IP), and heat-induced withdrawal latencies of the cheeks (an indicator of C fiber stimulation responses) were measured. MN/CGRP8–37, MN/lidocaine, or MN/blank was applied to the left cheeks for 20 min. After removing patches, cheek withdrawal latencies induced by continuous and noxious heat were assessed at different time points for up to 3 h post-treatment. Likewise, the same application process and test were carried out on the untreated right cheeks as a negative control. For the sham group, the rats’ left cheeks were not exposed to UVB, but the right cheeks were exposed and used as a control.
For the normal nociception model, the procedure was similar to the UVB/Cheek model, except that all the rats’ cheeks were not exposed to UVB radiation.
At each time point, the measured withdraw latency of the left cheek was subtracted by the withdraw latency of the right cheek, and the differences from all time points were averaged as the “Withdrawal Latencies Difference.”
Eating Experiment To Assess the Local Interference with Active Physical Functions.
WT Sprague-Dawley rats were fasted for a full 24 h, but water was freely accessed. After 24 h of fasting, each rat was lightly anesthetized with 2% isoflurane and received a local oral injection of a high dose of CGRP8–37 (~5 μg in 50 μL, dissolved in the vehicle), lidocaine (1.5%, 50 μL, dissolved in the vehicle), or vehicle (50 μL, 0.5% HPC containing 2% DMSO). The injection was made in the oral mucosa directly behind the left temporomandibular joint. After receiving injections, each rat was returned to its homecage for 1 h, then individually placed in fresh homecage (without bedding), and given ~10 min to acclimate to the new environment. Each rat was then given 1 single intact rodent chow biscuit (Rodent Chow 5001, 4.5–5.0 g/biscuit) and permitted exactly 20 min to eat freely. The eating process of each group was recorded with a video camera. Each rat’s biscuit was weighed before and after the 20 min eating test.
Skin Irritation Test.
MN/CGRP8–37 patches were applied to the dorsal skin of mice for 20 min and then removed. Mice were treated on the same area once per day for 3 days. On the fourth day, the treated sites were dissected, fixed, and stained by a standard H&E staining procedure. The slides were scanned and analyzed with NanoZoomer (Hamamatsu, Japan).
Spontaneous Behavior Testing.
Mice were given daily intravenous injections of either CGRP8–37 (10 mg/kg, dissolved in 2% DMSO in 0.5% HPC) or vehicle (2% DMSO in 0.5% HPC) once per day for 3 days. At 15 min post each injection, animals were evaluated for righting reflex and clinical observations. Immediately after clinical observations, the animals were placed in an ordinary rat homecage, which was placed in the SmartCage (AfaSci, Inc. Redwood City, CA, USA) that continuously recorded locomotion (traveling distance, speed and trajectory). The weight of animals, water intake, and food intake were evaluated once a day during each day of the experimental period.
Statistical Analyses.
Rats or mice were randomly assigned to different treatment groups for each experiment. All pain tests and behavioral assessments were conducted in a blinded manner to the experimenter. Data were presented as mean ± SEM, unless specified otherwise. The statistical significance of observed differences was calculated using a two-tailed Student’s t-test when comparing two different groups. A one way ANOVA with a Tukey-Kramer post hoc test was performed when three or more groups were compared. The differences were considered significant for p < 0.05.
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to thank Dr. Sung-Yun Kwon, TheraJect, Inc., Fremont, CA, USA, for the use of equipment and experimental advice. This work was supported by the National Institute of Health grants R44DA026363 and R44 NS086343. X.X. would like to thank the 100 Talents Program of Sun Yat-Sen University (76120–18821104) for support.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.6b06104.
Additional experimental details, calculation methods, supplemental figure and supplemental discussion (PDF)
REFERENCES
- (1).Baron R Mechanisms of Disease: Neuropathic Pain-a Clinical Perspective. Nat. Clin. Pract. Neurol. 2006, 2, 95–106. [DOI] [PubMed] [Google Scholar]
- (2).Dworkin RH; Backonja M; Rowbotham MC; Allen RR; et al. Advances in Neuropathic Pain. Arch. Neurol. 2003, 60, 1524–1534. [DOI] [PubMed] [Google Scholar]
- (3).Backonja M-M Defining Neuropathic Pain. Anesth. Analg. 2003, 97, 785–790. [DOI] [PubMed] [Google Scholar]
- (4).Sindrup SH; Jensen TS Efficacy of Pharmacological Treatments of Neuropathic Pain: An Update and Effect Related to Mechanism of Drug Action. Pain 1999, 83, 389–400. [DOI] [PubMed] [Google Scholar]
- (5).MacFarlane BV; Wright a; O’Callaghan J.; Benson H a. Chronic Neuropathic Pain and Its Control by Drugs. Pharmacol. Ther. 1997, 75, 1–19. [DOI] [PubMed] [Google Scholar]
- (6).McQuay HJ; Tramér M; Nye BA; Carroll D; Wiffen PJ; Moore RA A Systematic Review of Antidepressants in Neuropathic Pain. Pain 1996, 68, 217–227. [DOI] [PubMed] [Google Scholar]
- (7).Dieleman JP; Kerklaan J; Huygen FJPM; Bouma PAD; Sturkenboom MCJM Incidence Rates and Treatment of Neuropathic Pain Conditions in the General Population. Pain 2008, 137, 681–688. [DOI] [PubMed] [Google Scholar]
- (8).Jensen TS Anticonvulsants in Neuropathic Pain: Rationale and Clinical Evidence. Eur. J. Pain 2002, 6, 61–68. [DOI] [PubMed] [Google Scholar]
- (9).Saarto T; Wiffen PJ Antidepressants for Neuropathic Pain. Cochrane Database Syst. Rev. 2007, DOI: 10.1002/14651858.CD005454.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hollingshead J; Duhmke R; Cornblath DR, Tramadol for Neuropathic Pain. Cochrane Database Syst. Rev. 2006, 3, CD003726. [DOI] [PubMed] [Google Scholar]
- (11).Rowbotham MC; Twilling L; Davies PS; Reisner L; Taylor K; Mohr D Oral Opioid Therapy for Chronic Peripheral and Central Neuropathic Pain. N. Engl. J. Med. 2003, 348, 1223–1232. [DOI] [PubMed] [Google Scholar]
- (12).Zenz M; Strumpf M; Tryba M Long-Term Oral Opioid Therapy in Patients with Chronic Nonmalignant Pain. J. Pain Symptom Manage. 1992, 7, 69–77. [DOI] [PubMed] [Google Scholar]
- (13).Arner S; Meyerson B.a. Lack of Analgesic Effect of Opioids on Neuropathic and Idiopathic Forms of Pain. Pain 1988, 33, 11–23. [DOI] [PubMed] [Google Scholar]
- (14).Kalso E Sodium Channel Blockers in Neuropathic Pain. Curr. Pharm. Des. 2005, 11, 3005–3011. [DOI] [PubMed] [Google Scholar]
- (15).Brose WG; Cousins MJ Subcutaneous Lidocaine for Treatment of Neuropathic Cancer Pain. Pain 1991, 45, 145–148. [DOI] [PubMed] [Google Scholar]
- (16).Radwan I; Saito S.; Goto F. The Neurotoxicity of Local Anesthetics on Growing Neurons: A Comparative Study of Lidocaine, Bupivacaine, Mepivacaine, and Ropivacaine. Anesth. Analg. 2002, 94, 319–324. [DOI] [PubMed] [Google Scholar]
- (17).Ackerman WE Transient Neurologic Toxicity after Subarachnoid Anesthesia with Hyperbaric 5% Lidocaine. Anesth. Analg. 1993, 77, 1306. [DOI] [PubMed] [Google Scholar]
- (18).Benemei S; Nicoletti P; Capone JG; Geppetti P CGRP Receptors in the Control of Pain and Inflammation. Curr. Opin. Pharmacol. 2009, 9, 9–14. [DOI] [PubMed] [Google Scholar]
- (19).Jang JH; Nam TS; Paik KS; Leem JW Involvement of Peripherally Released Substance P and Calcitonin Gene-Related Peptide in Mediating Mechanical Hyperalgesia in a Traumatic Neuropathy Model of the Rat. Neurosci. Lett. 2004, 360, 129–132. [DOI] [PubMed] [Google Scholar]
- (20).Zimmermann M Pathobiology of Neuropathic Pain. Eur. J. Pharmacol. 2001, 429, 23–37. [DOI] [PubMed] [Google Scholar]
- (21).Doods H; Arndt K; Rudolf K; Just S CGRP Antagonists: Unravelling the Role of CGRP in Migraine. Trends Pharmacol. Sci. 2007, 28, 580–587. [DOI] [PubMed] [Google Scholar]
- (22).Doods H Development of CGRP Antagonists for the Treatment of Migraine. Curr. Opin. Investig. Drugs 2001, 2, 1261–1268. [PubMed] [Google Scholar]
- (23).Degnan AP; Chaturvedula PV; Conway CM; Cook D a; Davis CD.; Denton R.; Han X.; Macci R.; Mathias NR.; Moench P.; et al. Discovery of (R)-4-(8-fluoro-2-oxo-1,2-dihydroquinazolin-3(4H)-yl)-N-(3-(7-methyl-1H-indazol-5-yl)-1-oxo-1-(4-(piperidin-1-yl)piperidin-1-yl)propan-2-yl)piperidine-1-carboxamide (BMS-694153): A potent Antagonist of the Human Calcitonin Gene-Related Peptide Receptor for Migraine with Rapid and Efficient Intranasal Exposure. J. Med. Chem. 2008, 51, 4858–4861. [DOI] [PubMed] [Google Scholar]
- (24).de Prado BM; Russo AF CGRP Receptor Antagonists: A New Frontier of Anti-Migraine Medications. Drug Discovery Today: Ther. Strategies 2006, 3, 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tepper SJ; Stillman MJ Clinical and Preclinical Rationale for CGRP-Receptor Antagonists in the Treatment of Migraine. Headache 2008, 48, 1259–1268. [DOI] [PubMed] [Google Scholar]
- (26).Doods H; Hallermayer G; Wu D; Entzeroth M; Rudolf K; Engel W; Eberlein W Pharmacological Profile of BIBN4096BS, the First Selective Small Molecule CGRP Antagonist. Br. J. Pharmacol. 2000, 129, 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yu LC; Hansson P; Brodda-Jansen G; Theodorsson E; Lundeberg T Intrathecal CGRP8–37-Induced Bilateral Increase in Hindpaw Withdrawal Latency in Rats with Unilateral Inflammation. Br. J. Pharmacol. 1996, 117, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yu L; Hansson P; Lundeberg T Brain Res. 1994, 653, 223–230. [DOI] [PubMed] [Google Scholar]
- (29).Bennett AD; Chastain KM; Hulsebosch CE Alleviation of Mechanical and Thermal Allodynia by CGRP(8–37) in a Rodent Model of Chronic Central Pain. Pain 2000, 86, 163–175. [DOI] [PubMed] [Google Scholar]
- (30).Bruno BJ; Miller GD; Lim CS Basics and Recent Advances in Peptide and Protein Drug Delivery. Ther. Delivery 2013, 4, 1443–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Uhlig T; Kyprianou T; Martinelli FG; Oppici CA; Heiligers D; Hills D; Calvo XR; Verhaert P The Emergence of Peptides in the Pharmaceutical Business: From Exploration to Exploitation. EuPa Open Proteomics 2014, 4, 58–69. [Google Scholar]
- (32).Prausnitz MR; Langer R Transdermal Drug Delivery. Nat. Biotechnol 2008, 26, 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kommareddy S; Baudner BC; Oh S; Kwon SY; Singh M; O’Hagan DT Dissolvable microneedle patches for the delivery of cell-culture-derived influenza vaccine antigens. J. Pharm. Sci. 2012, 101, 1021–1027. [DOI] [PubMed] [Google Scholar]
- (34).Mohammed YH; Yamada M; Lin LL; Grice JE; Roberts MS; Raphael AP; Benson H.a.E.; Prow TW. Microneedle Enhanced Delivery of Cosmeceutically Relevant Peptides in Human Skin. PLoS One 2014, 9, e101956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kim YC; Park JH; Prausnitz MR Microneedles for Drug and Vaccine Delivery. Adv. Drug Delivery Rev. 2012, 64, 1547–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Zhang S; Qiu Y; Gao Y Enhanced Delivery of Hydrophilic Peptides in Vitro by Transdermal Microneedle Pretreatment . Acta Pharm. Sin. B 2014, 4, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sullivan SP; Koutsonanos DG; Del Pilar Martin M; Lee JW; Zarnitsyn V; Choi S-O; Murthy N; Compans RW; Skountzou I; Prausnitz MR Dissolving Polymer Microneedle Patches for Influenza Vaccination. Nat. Med. 2010, 16, 915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Demuth PC; Garcia-Beltran WF; Ai-Ling ML; Hammond PT; Irvine DJ Composite Dissolving Microneedles for Coordinated Control of Antigen and Adjuvant Delivery Kinetics in Transcutaneous Vaccination. Adv. Funct. Mater. 2013, 23, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Yu J; Zhang Y; Ye Y; DiSanto R; Sun W; Ranson D; Ligler FS; Buse JB; Gu Z Microneedle-Array Patches Loaded with Hypoxia-Sensitive Vesicles Provide Fast Glucose-Responsive Insulin Delivery. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 8260–8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ye Y; Wang J; Hu Q; Hochu GM; Xin H; Wang C; Gu Z Synergistic Transcutaneous Immunotherapy Enhances Antitumor Immune Responses through Delivery of Checkpoint Inhibitors. ACS Nano 2016, 10, 8956–63. [DOI] [PubMed] [Google Scholar]
- (41).Chen M-C; Lin Z-W; Ling M-H Near-Infrared Light- Activatable Microneedle System for Treating Superficial Tumors by Combination of Chemotherapy and Photothermal Therapy. ACS Nano 2016, 10, 93–101. [DOI] [PubMed] [Google Scholar]
- (42).Chandrasekhar S; Iyer LK; Panchal JP; Topp EM; Cannon JB; Ranade VV Microarrays and Microneedle Arrays for Delivery of Peptides, Proteins, Vaccines and Other Applications. Expert Opin. Drug Delivery 2013, 10, 1155–1170. [DOI] [PubMed] [Google Scholar]
- (43).Zhang Y; Brown K; Siebenaler K; Determan A; Dohmeier D; Hansen K Development of Lidocaine-Coated Microneedle Product for Rapid, Safe, and Prolonged Local Analgesic Action. Pharm. Res. 2012, 29, 170–177. [DOI] [PubMed] [Google Scholar]
- (44).Park JH; Allen MG; Prausnitz MR Biodegradable Polymer Microneedles: Fabrication, Mechanics and Transdermal Drug Delivery. J. Controlled Release 2005, 104, 51–66. [DOI] [PubMed] [Google Scholar]
- (45).Chang C; Zhang L Cellulose-Based Hydrogels: Present Status and Application Prospects. Carbohydr. Polym. 2011, 84, 40–53. [Google Scholar]
- (46).Bachy V; Hervouet C; Becker PD; Chorro L; Carlin LM; Herath S; Papagatsias T; Barbaroux J-B; Oh S-J; Benlahrech A; et al. Langerin Negative Dendritic Cells Promote Potent CD8+ T-Cell Priming by Skin Delivery of Live Adenovirus Vaccine Microneedle Arrays. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Grimsholm O; Rantapää-Dahlqvist S; Forsgren S Levels of Gastrin-Releasing Peptide and Substance P in Synovial Fluid and Serum Correlate with Levels of Cytokines in Rheumatoid Arthritis. Arthritis Res. Ther. 2005, 7, R416–R426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Bourquin A-F; Süveges M; Pertin M; Gilliard N; Sardy S; Davison AC; Spahn DR; Decosterd I Assessment and Analysis of Mechanical Allodynia-like Behavior Induced by Spared Nerve Injury (SNI) in the Mouse. Pain 2006, 122, 14e1–14e14. [DOI] [PubMed] [Google Scholar]
- (49).Decosterd I; Woolf CJ Spared Nerve Injury: An Animal Model of Persistent Peripheral Neuropathic Pain. Pain 2000, 87, 149–158. [DOI] [PubMed] [Google Scholar]
- (50).Kim KJ; Yoon YW; Chung JM Comparison of Three Rodent Neuropathic Pain Models. Exp. Brain Res. 1997, 113, 200–206. [DOI] [PubMed] [Google Scholar]
- (51).Jeffrey W; Allen TLY Assessment of Acute Thermal Nociception in Laboratory Animals Pain Research Methods and Protocols, Methods in Molecular Medicine: Luo ZD., Ed.; Humana Press: Totowa, NJ, 2004; Vol. 99, pp 11–23 [DOI] [PubMed] [Google Scholar]
- (52).Abdi S; Lee DH; Chung JM The Anti-Allodynic Effects of Amitriptyline, Gabapentin, and Lidocaine in a Rat Model of Neuropathic Pain. Anesth. Analg. 1998, 87, 1360–1366. [PubMed] [Google Scholar]
- (53).Backonja M; Glanzman RL. Gabapentin Dosing for Neuropathic Pain: Evidence from Randomized, Placebo-Controlled Clinical Trials. Clin. Ther. 2003, 25, 81–104. [DOI] [PubMed] [Google Scholar]
- (54).Argoff CE; Cole BE; Fishbain DA; Irving GA Diabetic Peripheral Neuropathic Pain: Clinical and Quality-of-Life Issues. Mayo Clin. Proc. 2006, 81, S3–S11. [DOI] [PubMed] [Google Scholar]
- (55).Sycha T; Gustorff B; Lehr S; Tanew A; Eichlerl HG; Schmetterer L A Simple Pain Model for the Evaluation of Analgesic Effects of NSAIDs in Healthy Subjects. Br. J. Clin. Pharmacol. 2003, 56, 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Drenjancevic-Peric I; Drenjancevic IH; Ivic D; Sakic-Zdravcevic K Sodium Channels and Nociception - The Effects of Local Anesthetics. Period. Biol. 2009, 111, 215–218. [Google Scholar]
- (57).Danhof M; Levy G Kinetics of Drug Action in Disease States. I. Effect of Infusion Rate on Phenobarbital Concentrations in Serum, Brain and Cerebrospinal Fluid of Normal Rats at Onset of Loss of Righting Reflex. J. Pharmacol. Exp. Ther. 1984, 229, 44–50. [PubMed] [Google Scholar]
- (58).Irifune M; Katayama S; Takarada T; Shimizu Y; Endo C; Takata T; Morita K; Dohi T; Sato T; Kawahara M MK-801 Enhances Gabaculine-Induced Loss of the Righting Reflex in Mice, but Not Immobility. Can. J. Anaesth. 2007, 54, 998–1005. [DOI] [PubMed] [Google Scholar]
- (59).Khroyan TV; Zhang J; Yang L; Zou B; Xie J; Pascual C; Malik A; Xie J; Zaveri NT; Vazquez J; et al. Rodent Motor and Neuropsychological Behaviour Measured in Home Cages Using the Integrated Modular Platform SmartCage. Clin. Exp. Pharmacol. Physiol. 2012, 39, 614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Prausnitz MR; Mitragotri S; Langer R Current Status and Future Potential of Transdermal Drug Delivery. Nat. Rev. Drug Discovery 2004, 3, 115–124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.