

Abstract
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are associated with loss of nuclear TDP-43. Here we identify that TDP-43 regulates expression of the neuronal growth-associated factor stathmin-2. Lowered TDP-43 levels, which reduce its binding to sites within the first intron of stathmin-2 pre-mRNA, uncover a cryptic polyadenylation site whose utilization produces a truncated, non-functional mRNA. Reduced stathmin-2 expression is found in neurons trans-differentiated from patient fibroblasts expressing an ALS-causing TDP-43 mutation, in motor cortex and spinal motor neurons from sporadic ALS patients and familial ALS patients with expansion in C9orf72, and in induced pluripotent stem cell (iPSC)-derived motor neurons depleted of TDP-43. Remarkably, while reduction in TDP-43 is shown to inhibit axonal regeneration of iPSC-derived motor neurons, rescue of stathmin-2 expression restores axonal regenerative capacity. Thus, premature polyadenylation-mediated reduction in stathmin-2 is a hallmark of ALS/FTD that functionally links reduced nuclear TDP-43 function to enhanced neuronal vulnerability.
Introduction
Amyotrophic lateral sclerosis (ALS) is a late-onset neurodegenerative disease of the motor system, characterized by selective and progressive loss of motor neurons, eventually leading to paralysis and death within 2–5 years 1. Ninety percent of ALS cases are not associated with a family history and referred to as sporadic ALS. The remaining ten percent are caused by inherited mutations in a variety of genes, including the most frequent genetic cause of ALS represented by GGGGCC repeat expansions in the C9orf72 gene 2,3. Notably, many of the genes whose mutation is linked to ALS also cause or contribute to another age-dependent neurodegenerative disorder frontotemporal dementia (FTD). A landmark contribution to understanding cellular mechanisms of ALS and FTD came from the discovery of cytoplasmic accumulation and nuclear loss of the RNA binding protein TDP-43 from affected neurons in most instances of ALS, as well as in approximately 45% of patients with FTD 4,5. Over 40 dominantly inherited mutations in the gene encoding TDP-43 have subsequently been identified in familial ALS patients 6, implicating TDP-43 dysfunction in the vast majority of ALS cases.
TDP-43 is involved in fundamental RNA processing activities including RNA transcription, splicing, and transport 7–10. TDP-43 binds to thousands of pre-mRNA/mRNA targets, with high affinity for GU-rich sequences, including autoregulation of its own mRNA via binding to the 3’ untranslated region (3’ UTR) 8,9,11. Reduction in TDP-43 from an otherwise normal adult nervous system alters the splicing or expression levels of more than 1500 RNAs, including long intron-containing transcripts 8,12. Considering the central role of TDP-43 in RNA metabolism, it is anticipated that loss of nuclear TDP-43 in affected neurons of ALS and FTD patients drives processing alterations of multiple RNAs 8,12. However, a direct link to neuronal degeneration from reduction or mutation in TDP-43 remains elusive.
Microtubules are key components of the neuronal cytoskeleton, essential for maintaining the stability, shape and proper function of the neuron 13. An intrinsic property of microtubules is their “dynamic instability” 14, i.e., the ability to switch between rapid polymerization and rapid shrinkage (known as catastrophe) in a process that is largely dampened by microtubule-associated proteins 15. One of these (encoded by the STMN2 gene) is the neuronal stathmin-2 (also known as SCG10), which is thought to play an important role in neurite outgrowth 16, most likely by promoting microtubule dynamics in axonal growth cones 17,18. The four proteins of the stathmin family, each encoded by a different gene, affect microtubule dynamics by either directly promoting catastrophe 19 or sequestering tubulin to prevent assembly 20. Stathmin-2, the only essential mammalian stathmin 20, contains two tubulin-binding domains and a conserved N-terminal membrane-anchoring domain containing two palmitoylation sites 16,20.
Stathmin-2 is anterogradely transported along axons 21 and has been implicated as an essential component for axonal regeneration 20. Upon axonal injury, stathmin-2 is upregulated and recruited to growth cones of regenerating axons 22. Moreover, stathmin-2 has been proposed as an axonal-maintenance factor whose loss accelerates a neuronal degeneration program 21. In Drosophila, expression of a mutant stathmin causes retraction of motor neurons from innervated neuromuscular junctions 23.
We now identify that the mRNA encoding stathmin-2 is significantly lost after TDP-43 depletion and in neurons trans-differentiated from multiple patient fibroblasts, each carrying an ALS-causing mutation in TDP-43. Mechanistically, TDP-43 disruption is shown to drive premature polyadenylation and aberrant splicing in intron one of stathmin-2 pre-mRNA, producing a non-functional mRNA. Aberrant polyadenylation/splicing of stathmin-2 pre-mRNA is also consistently found in spinal motor neurons and motor cortex of sporadic ALS and C9orf72 ALS patients, supporting stathmin-2 loss of function as a key driver of motor neuron degeneration. Indeed, suppression of TDP-43 or stathmin-2 in iPSC-derived motor neurons leads to inhibition of axonal regeneration after induced damage. Importantly, restoration of stathmin-2 levels rescues axonal regeneration ability in the absence of TDP-43, evidence supporting rescue of stathmin-2 levels as a potential therapeutic approach in neurodegenerative diseases – especially ALS and FTD – affected by TDP-43 proteinopathy.
Results
TDP-43 depletion or disease-causing mutation suppresses stathmin-2
Two independent strategies were undertaken to manipulate TDP-43 function in a human neuronal cell line: 1) depletion of TDP-43 by siRNA and 2) genome editing with CRISPR-Cas9 to introduce an ALS-causing mutation into both endogenous TDP-43 gene loci. The human neuronal cell line SH-SY5Y was chosen as a model as it has been shown to maintain a diploid karyotype and acquire substantial neuronal character when differentiated 24. siRNA treatment reduced TDP-43 mRNA levels by >80% when compared to cells treated with a control siRNA (Fig. 1a). The consequences of lowering TDP-43 were then examined on a genome-wide basis. Libraries of RNAs were prepared, sequenced, and mapped to the human genome. This approach confirmed reduction of TDP-43 to 1/4th of its initial level (Fig. 1b). Amid 299 and 219 mRNAs that were down or up regulated, respectively (with fold changes >1.5, false discovery rate [FDR] <0.05) (Supplementary Table 1), the most affected mRNA (with >85% reduction - Fig. 1b and Supplementary Fig. 1a) was the one encoding the neuronal growth-associated protein stathmin-2 16,18. Reduction in stathmin-2 mRNA upon TDP-43 depletion was confirmed by quantitative PCR (qPCR) (Fig. 1c) and an 8-fold reduction of the 22 kD stathmin-2 protein was identified by immunoblotting (Fig. 1d and Supplementary Fig. 1b).
Figure 1. TDP-43 depletion or genome editing in a human neuronal cell line identifies significant reduction in stathmin-2 expression levels.

(a) Quantitative real-time PCR analysis confirming siRNA-mediated reduction of TDP-43 mRNA levels in SH-SY5Y cells. Expression of TRFC and GAPDH mRNAs were used as endogenous controls. Cells were treated with siControl (white bar, black dots, mean=1) or siTDP-43 (gray bar, red dots, mean=0.19) for 96 hours in three biologically independent experiments (two-tailed t-test, P=0.0012, n=3), error bars represent SEM, **P<0.01. (b) Volcano-plot showing differentially expressed genes in SH-SY5Y cells depleted of TDP-43 by siRNA treatment. Genes with significant changes in mRNA levels are represented by red dots (n=3 biologically independent experiments, fold change >1.5 and FDR<0.05) by DESeq2 51. Increased red dot size represents increased statistical significance (measured by FDR<0.05). RNA-seq analysis identified 518 misregulated genes and confirmed 4 fold reduction in TDP-43 mRNA levels. Stathmin-2 mRNA showed the strongest reduction (6.5 fold) upon TDP-43 depletion. Up and down regulated genes’ counts are indicated. Expression values were determined as transcripts per kilobase per million mapped reads (TPM). (c) Quantitative real-time PCR analysis confirming reduction of stathmin-2 mRNA expression levels (mean=0.13) in SH-SY5Y cells treated with siRNA targeting TDP-43 compared to cells treated with siControl (mean = 1) for 96 hours in three biologically independent experiments (two-tailed t-test, P=0.0005, n=3), error bars represent SEM. ***P<0.001 (d) Immunoblotting of TDP-43 and stathmin-2 in SH-SY5Y cells treated with siControl or siTDP43 for 96 hours. α-tubulin served as a loading control. Three biological replicates are shown. (e) Representative Immunofluorescence of TDP-43 (green) and lamin-B (red) in SH-SY5Y lines expressing wild type or mutant TDP-43 by genome-editing. Genotypes are indicated, experiment was reproduced 3 times independently with similar results. For unprocessed blots, see Supplementary Fig. 10. (f) Volcano plot depicting 950 differentially expressed genes identified by genome-wide RNA-seq. Significant changes in mRNA levels between SH-SY5YWT and SH-SY5YN352S/N352S lines are represented by red dots (n=2 biologically independent experiments, fold change > 1.5, FDR<0.05) by DESeq2 51. Increased red dot size represents increased statistical significance (measured by FDR<0.05). Expression values were calculated as transcripts per kilobase per million mapped reads (TPM). (g) Quantitative real-time PCR analysis confirming significant reduction in stathmin-2 mRNA levels between isogenic SH-SY5YWT/WT (white bar, black dots, mean=1) and SH-SY5YN352S/N352S (black bar, red dots, mean=0.58) lines. Expression of TRFC and GAPDH mRNAs were used as endogenous controls (two-tailed t-test, P=0.004, n=3 independent biological experiments), error bars represent SEM. **P<0.01. (h) Expression changes of 42 overlapping genes from figures 1b and 1f are plotted.
Using a CRISPR-Cas9 site-selective nuclease, we next genetically engineered SH-SY5Y cells to express a familial ALS-causing mutation (asparagine substituted to serine at amino acid 352 – TDP-43N352S) from both endogenous TDP-43 alleles (Supplementary Fig. 2a). Immunofluorescence imaging confirmed that mutant TDP-43 protein remained mostly nuclear, similar to wild type TDP-43 expressed in the original SH-SY5Y line (Fig. 1e and Supplementary Fig. 2b). Transcriptional profiling (i.e., RNA-seq) of the wild type and mutant lines revealed that introduction of the disease-causing TDP-43 mutation led to altered expression of 950 mRNAs (451 downregulated and 499 upregulated; fold change >1.5, FDR <0.05) (Supplementary Table 2), including moderate reduction in TDP-43 (Fig. 1f). In addition, previously described alternative splicing changes linked to TDP-43 loss of function were observed in TDP-43 mutant-expressing cells (Supplementary Fig. 2c), consistent with both loss of normal TDP-43 function and a gain of aberrant function, as seen previously in mice 25–27.
Analysis of RNAs from isogenic wild type and mutant lines (using qPCR) confirmed a 1.7 fold reduction of stathmin-2 mRNA (Fig. 1g), while immunoblotting revealed more than two fold reduction of stathmin-2 protein in TDP-43N352S mutant cells (Supplementary Fig. 2d). A search for gene expression changes that overlapped between cells with TDP-43 loss of function (Fig. 1b) and TDP-43N352S mutant cells (Fig. 1f) identified stathmin-2 mRNA (and 18 additional RNAs) to be downregulated, together with another 23 that were upregulated (Fig. 1h).
Reduced stathmin-2 in TDP-43 mutant human neurons
We then tested whether stathmin-2 expression was reduced in human neurons directly converted from ALS patients’ fibroblasts. To this end, we used eight fibroblast lines obtained from an extended family that included four carriers heterozygous for the ALS-linked mutant TDP-43N352S and four individuals without the mutation (Supplementary Fig. 3a and Supplementary Table 3). Overall stathmin-2 mRNA levels were already reduced (by 50%) (Supplementary Fig. 3b) in the mutant fibroblast lines relative to controls despite primarily nuclear localization of TDP-43 protein both in control and ALS patient fibroblasts (Supplementary Fig. 3c-d). Fibroblasts from all eight lines were directly induced into neurons (herein referred to as iNeurons) 28 by expression of the neuronal-specific transcription activator Brn2 and reduction of the RNA-binding protein PTB (Fig. 2a).
Figure 2. Reduced stathmin-2 levels in human neurons produced by direct conversion from ALS patient fibroblasts expressing mutant TDP-43.
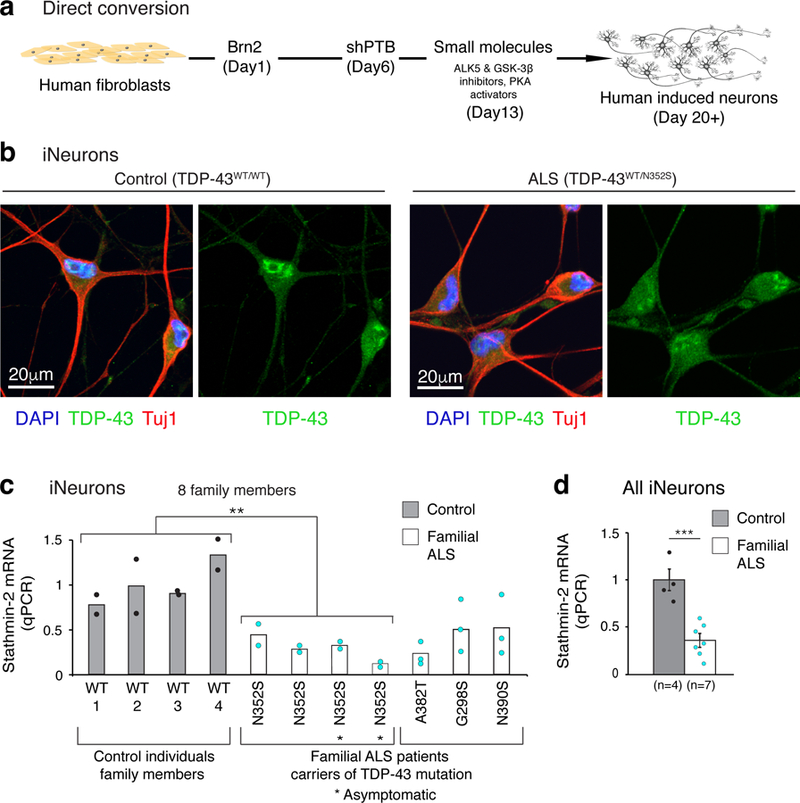
(a) Summary of the direct conversion strategy adopted from Xue et al. 2013 28. (b) Induced neurons (iNeurons) converted from fibroblasts of control and ALS patient. Immunofluorescence staining at day 20 of differentiation revealed partial cytoplasmic mislocalization of TDP-43 (green) in ALS iNeurons. Neuron-specific Class III β-tubulin (Tuj1) (red) was used as a neuronal marker, cell nuclei are visualized by DAPI staining (blue). Experiment was repeated independently with similar results from 3 control individuals and 3 familial ALS donor lines (with mutant TDP-43). (c) qPCR analysis of stathmin-2 mRNA levels in iNeurons from control (grey bars, black dots) and familial ALS patients with different TDP-43 mutations (white bars, turquoise dots). Plotted are 2 biologically independent experiments per each individual from an extended family that included 4 carriers heterozygous for the ALS-linked mutant TDP-43N352S (n=4, mean=0.3) and 4 individuals without the mutation (n=4, mean=1). Two-tailed t-test, P = 0.008. Also plotted are three biologically independent experiments of three additional ALS patients (non-family members) carriers of the indicated mutations. (d) Summary of stathmin-2 expression level measured by qPCR in iNeurons from controls (n=4, gray, mean = 1) and familial ALS patients (n=7, white, mean=0.36). P=0.0005, two-tailed t-test, SEM. **P<0.01***, ***P<0.001.
TDP-43 was almost exclusively nuclear in wild type and mutant fibroblasts (Supplementary Fig. 3c-d). When converted into iNeurons, a higher proportion of wild type TDP-43 accumulated in the cytoplasm and this relocalization was enhanced in iNeurons expressing the N352S mutation (Fig. 2b and Supplementary Fig. 3e). Importantly, stathmin-2 mRNA was significantly downregulated (more than 3 fold) in all four sets of TDP-43N352S iNeurons relative to the four iNeuron lines derived from fibroblasts of healthy family members (p<0.01, n=4) (Fig. 2c).
We next tested iNeurons generated from familial ALS patient fibroblasts carrying three other mutations: glycine to serine at position 298 (TDP-43G298S), alanine to threonine at position 382 (TDP-43A382T), and asparagine to serine at position 390 (TDP-43N390S). Stathmin-2 mRNA was reduced (relative to iNeurons from non-ALS individuals) in all of these familial ALS iNeurons (Fig. 2c-d). These data demonstrate that, similar to reduction of TDP-43, ALS-linked mutations in TDP-43 suppress stathmin-2 expression level in human iNeurons.
TDP-43 represses premature polyadenylation in stathmin-2 pre-mRNA
The stathmin-2 gene is annotated to contain 5 constitutive exons (Refseq ID: NM_001199214.1) plus a proposed alternative exon between exons 4 and 5 whose use was undetectable (using RNA-seq) under any condition in our SH-SY5Y cells (Fig. 3a). Reduction or mutation in TDP-43, however, induced a new spliced exon, with RNA-seq reads mapping within intron one (Fig. 3a, red arrow). This new exon, herein called “exon 2a”, was absent in wild type cells, but appeared either when TDP-43 was depleted or when endogenous TDP-43 was edited to carry the N352S mutation. Prominent utilization of this new exon was observed when TDP-43 was reduced, in line with >6 fold suppression of RNAs containing exons 2 through 5 (Fig. 3a, upper panel). This was accompanied by the corresponding loss of stathmin-2 protein (Fig. 1d and Supplementary Fig. 1b).
Figure 3. TDP-43 regulates stathmin-2 mRNA levels by repressing premature polyadenylation.

(a) RNA-seq reads mapped to the genomic region of stathmin-2 reveal incorporation of a new exon, originated from intron 1, into the mature stathmin-2 mRNA. Red arrows indicate the intronic region of aberrant splicing (exon 2a) in SH-SY5Y cells upon TDP-43 depletion (upper panels) or expression of mutant TDP-43 (lower panels). Experiment was repeated independently three times (TDP-43 depletion) or twice (expression of mutant TDP-43) with similar results. (b) Representative RT-PCR (left, experiment was repeated 3 times independently with similar results) and qPCR (right, n=3 biologically independent experiments) analyses confirmed expression of the new spliced mRNA isoform containing exon 2a upon TDP-43 depletion (mean=8.7, P=0.00078) or mutation (mean=3.8, P=0.0001). The location of primers in exons 1 and 2a is shown. RT-PCR of the CENP-A transcript was used as loading control for RT-PCR, TFRC and GAPDH were used as qPCR normalizers (***p<0.001, two-tailed t-test, SEM). For uncropped gel images, see Supplementary Fig. 10. (c) Schematic representation of stathmin-2 pre-mRNA (upper panel) and alternative RNA isoforms (lower panels) in normal cells (i) or cells with TDP-43 deficiency (ii). Constitutive exons are represented by black boxes, exon 2a is in red and the thin red box represents the newly acquired 3’UTR. Light grey boxes represent 3’ or 5’ UTRs of normal stathmin-2 mRNA. (d) Sequence of exon 2a is shown in red, including the embedded in-frame UAG codon generating a premature stop codon. Highlighted are potential TDP-43 binding sites located 127 upstream of the alternative polyadenylation signal. (e) 3’ end sequencing by reverse transcription using oligo(dT)-VN primers confirmed exon 2a as the terminal exon of short stathmin-2 mRNA. (f) Genome browser track obtained from cross-linking and immuno-precipitation (iCLIP) for TDP-43 in human SH-SY5Y cells 9, revealed TDP-43 physical binding to exon 2a, located in intron 1 of stathmin-2 pre-mRNA.
Altered splicing and ligation of exon 1 to exon 2a in SH-SY5Y cells after TDP-43 depletion or in the presence of a TDP-43 mutation was then confirmed by RT-PCR and qPCR (Fig. 3b). However, while fusion between exon 1 and exon 2a was consistently observed (Fig. 3b), no RNAs containing exon 2a ligated to the downstream exon 2 were identified using primers targeting the flanking exons 1 and 2 (Supplementary Fig. 4a-c). To test the possibility that incorporation of exon 2a drives alternative polyadenylation within what is normally intron one (Fig. 3c), we used anchored oligo(dT) primers for reverse transcription followed by PCR and sequencing. This analysis confirmed polyadenylation of the stathmin-2 RNA containing exon 2a as the terminal exon (Fig. 3d-e).
A polyadenylation signal “AUUAAA” 29 was identified 24 nucleotides upstream to the polyadenylation site (Fig. 3d). This cryptic polyadenylation sequence in the human stathmin-2 gene is conserved among most primates, but is notably absent in mouse or rat (Supplementary Fig. 4d). qPCR analysis on nascent stathmin-2 pre-mRNAs identified reduction in full-length stathmin-2 transcripts in SH-SY5Y cells expressing mutant TDP-43 (Supplementary Fig. 4e), consistent with increased levels of truncated exon 2a-containing RNAs (Fig. 3b).
The prematurely polyadenylated RNA includes 227 nucleotides originating from exon 2a (hg38; chr8:79,616,822–79,617,048) with its predicted 16 amino acid translation product initiating at the normal AUG codon in exon 1 and ending at 11 codons into exon 2a (Fig. 3c-d). Three “GUGUGU” hexamers marking potential binding sites of TDP-43 (as we and others have previously described 8,9) were identified in a region spanning 30 nucleotides downstream of the 3’ splice site that produces exon 2a (Fig. 3d). Indeed, analysis of ultraviolet cross-linking and immuno-precipitation (iCLIP) data for TDP-43 in human SH-SY5Y cells 9, confirmed TDP-43 binding (shown by mapped reads) to the stathmin-2 pre-mRNA at a single region containing the three GUGUGU sequences (Fig. 3f), thus confirming physical interaction with TDP-43 protein. Altogether, we conclude that reduction in TDP-43 causes de-repression and efficient use of a cryptic polyadenylation site in what normally is intron 1, which results in truncation of the stathmin-2 pre-mRNA and suppression of functional stathmin-2 expression.
Stathmin-2 mRNA is prominently expressed in motor neurons
Comparison of published evidence of the abundance of stathmin-2 mRNA in ribosome-bound RNAs across specific cell types of the murine central nervous system 30 revealed 25 and 15 fold enrichment of translated stathmin-2 mRNAs in motor neurons relative to astrocytes and oligodendrocytes, respectively (Supplementary Fig. 5a). Remarkably, further analysis of these data 31 identified stathmin-2 to be among the 25 most abundant actively translated mRNAs isolated from adult motor neurons in mice. Similarly, our analysis of RNA sequencing from laser capture microdissected (LCM) human lumbar spinal motor neurons from seven healthy controls 31 determined that in human spinal motor neurons stathmin-2 is the 20th most abundant mRNA, only slightly less abundant that the mRNAs encoding the three neurofilament subunits NF-M, NF-L and NF-H (Supplementary Fig. 5b). Further, of the four stathmin genes, only stathmin-2 was enriched within human motor neurons (Supplementary Fig. 5c).
Suppression of stathmin-2 by premature polyadenylation is a hallmark of ALS
Analysis of the RNA sequencing reads mapping to the stathmin-2 gene from lumbar motor neurons captured by laser microdissection revealed a clear segregation between controls and sporadic ALS cases. RNAs from all analyzed sporadic ALS patients (n=13) contained inclusion of exon 2a (Fig. 4a and Supplementary Fig. 6). In contrast, RNAs including exon 2a were absent in mRNA from all tested (n=7) non-ALS individuals. Importantly, just as we had determined in TDP-43 mutant expressing SH-SY5Y cells and patient-derived iNeurons (Fig. 1 and 2), overall stathmin-2 mRNA levels in sporadic ALS patients were significantly decreased relative to those in motor neurons from non-ALS individuals (Fig. 4b). By contrast, stathmin-1 mRNA expression was low in both healthy and ALS spinal motor neurons (Fig. 4b). Additionally, analysis of RNAs extracted from anterior horns of thoracic spinal cord from 13 sporadic ALS patients and three familial ALS patients carrying GGGGCC expansion in C9orf72 revealed aberrant processing of stathmin-2 mRNA in all, but not in RNAs from eight healthy individuals or three familial ALS patients with SOD1 mutations (Fig. 4c and Supplementary Fig. 7a). Consistent with the latter, mice developing fatal motor neuron disease from expressing an ALS-linked mutation in SOD1 retained normal stathmin-2 mRNA levels despite apparent motor neuron damage 30, demonstrating that stathmin-2 expression is not affected by motor neuron degeneration per se (Supplementary Fig. 5a). Recognizing that TDP-43 pathology is found in sporadic and familial ALS linked to C9orf72 expansion 32 but not SOD1-mediated ALS (Supplementary Fig. 8) 33, these results are consistent with premature polyadenylation of stathmin-2 mRNA being triggered by TDP-43 dysfunction and the corresponding suppression of stathmin-2 expression in both thoracic (Fig. 4c and Supplementary Fig. 7a) and lumbar (Fig. 4a) motor neurons.
Figure 4. Abnormal stathmin-2 mRNA processing is a disease hallmark in affected spinal motor neurons of sporadic ALS patients.

(a) RNA sequencing of control and sporadic ALS laser-captured spinal motor neurons reveals robust signature of exon 2a incorporation in stathmin-2 mRNA in sporadic ALS samples but not in non-ALS aged controls. Lower diagram shows the genomic region of stathmin-2 and mapped RNA reads of exon 2a are indicated by a red arrow. Data analyzed from from Krach et al., 2018 31. (b) Stathmin-2 mRNA expression is suppressed in spinal motor neurons of sporadic ALS patients (gray bar, n=13, mean = 423 TPM, P=0.03) relative to non-ALS aged controls (white bar, n=7, mean=971 TPM, P=0.019). Two-tailed t-test, SEM. RNA sequencing data was analyzed from Krach et al., 2018 31. Stathmin-1 mRNA expression is shown to the right (control mean=25, ALS patients mean = 44.5). Transcripts per million counts (TPM) are represented, two-tailed t-test. P=0.02. *P<0.05, SEM. (c-d) RT-PCR using primers targeted to exon 1 and exon 2a confirms expression of truncated stathmin-2 isoform in (c) thoracic spinal cord or (d) motor cortex from sporadic and C9orf72 ALS patients, but not in spinal cords of mutant SOD1 carriers. For uncropped gel images, see Supplementary Fig. 10 (e-f) Lumbar spinal cord and motor cortex sections isolated from control individuals and sporadic ALS patients were hybridized with locked nucleic acid (LNA) probes targeting intron one of stathmin-2 pre-mRNA (for the truncated RNA) or exon 5 of stathmin-2 pre-mRNA. Signal is in blue, counterstain is nuclear fast red. (g) Table summarizing human samples with expression of stathmin-2 truncated RNA, identified by RT-PCR or in-situ hybridization. Data from supplementary Fig. 7 is included.
Extension of similar analyses to the motor cortex identified altered processing of stathmin-2 mRNA to include exon 2a in 2/2 C9orf72 samples and 6/9 sporadic ALS samples (Fig. 4d). For ALS cases that were also diagnosed with frontotemporal dementia (FTD), premature polyadenylation was seen in frontal cortex-derived RNAs in 3 of 4 sporadic ALS/FTD patients, and one of one C9orf72 expansion patient (Supplementary Fig. 7b).
Lastly, chromogenic in situ hybridization (CISH) was used to test on a cell by cell basis for stable accumulation of exon 2a-containing stathmin-2 RNAs in spinal cord and motor cortex samples from five control individuals and four sporadic ALS patients (Fig. 4e-g and Supplementary Fig. 7c,d). In agreement with prior analyses of RNAs derived by laser microdissection (Fig. 4a) or from bulk tissues (Fig. 4c,d), RNA probes hybridizing to stathmin-2 exon 2a were increased in both nuclear and cytoplasmic compartments of spinal cord neurons (97% of sporadic ALS motor neurons versus 0% of control; n=69 and 41, respectively) and in neurons from layers 3 and 5 of motor cortex from all ALS-derived samples, with 100% of scored sporadic ALS cortical neurons staining positive for exon 2a (n=50) versus 0% of control cortical neurons (n=50). Low basal levels of hybridization (presumably corresponding to the newly transcribed stathmin-2 pre-mRNA) were found in tissues from non-ALS individuals, with an intensity only slightly above that of a scrambled sequence control probe.
A probe hybridizing to exon five of the stathmin-2 mRNA revealed an apparent reduction in affected regions of sporadic ALS, with only 35% of sALS lumbar motor neurons showing weakly positive staining for STMN2 exon five versus 70% of control patient motor neurons (n=65 and 61, respectively). The reduction of exon five staining was even more substantial in the motor cortex where 0% of scored neurons showed a positive signal, compared with a strong signal in 100% of non-ALS cortical neurons (Fig. 4e,f and Supplementary Fig. 7c,d).
Impaired axonal regeneration upon TDP-43 loss in human iPSC-derived motor neurons is alleviated by restoring stathmin-2
To determine the functional consequence of TDP-43-mediated reduction in stathmin-2, induced pluripotent stem cells (iPSC) were generated using a non-integrating approach, validated to retain a normal karyotype (Supplementary Fig. 9a) and expression of pluripotent stem cell markers (Supplementary Fig. 9b-d), and differentiated into motor neurons (Supplementary Fig. 9e). Motor neuron precursors (expressing Hb9) appeared within 21 days (Supplementary Fig. 9f) and by 28 days developed Map2 positive dendrites and long axons accumulating neurofilaments (Supplementary Fig. 9g-h). At day 29, motor neurons were treated with antisense oligonucleotides (ASOs) that direct catalytic degradation of TDP-43 mRNAs through the action of endogenous RNase H. TDP-43 mRNA levels were reduced in a dose-dependent manner (Fig. 5a).
Figure 5: Impaired axonal regeneration upon TDP-43 loss in human iPSC-derived motor neurons is alleviated by stathmin-2 restoration.

(a) Quantitative real-time PCR analysis confirming ASO-mediated reduction of TDP-43 mRNA (gray, dose-dependent mean values are 0.71, 0.6, 0.43, respectively) or stathmin-2 mRNA (red, dose-dependent mean values are 0.67, 0.31, 0.09, respectively) levels in human iPSC-derived motor neurons. Control bar (white, mean=1) represents 3 biological replicates, expression of TRFC mRNA was used as endogenous control. Error bars represent SEM. (b) RT-PCR analysis confirmed altered splicing and ligation of exon 1 and exon 2a upon treatment with ASOs targeting TDP-43, in iPSC-derived motor neurons. Three ASO concentrations were tested and a dose dependent effect on expression of stathmin-2 short RNA is shown. Experiment was reproduced five times independently with similar results. For uncropped gel images, see Supplementary Fig. 10. (c) Quantitative real-time PCR analysis confirming ASO-mediated reduction of stathmin-2 mRNA (red, dose-dependent mean values are 0.23, 0.12, 0.07, respectively) or TDP-43 mRNA (gray, dose-dependent mean values are 0.99, 0.95, 1.04, respectively) levels in human iPSC-derived motor neurons. Control bar (mean=1) represents 3 biological replicates, expression of TRFC mRNA was used as endogenous control. Error bars represent SEM. (d) Immunoblotting of TDP-43 and Stathmin-2 in iPSC-derived motor neurons treated with mouse Malat-1 ASO as control (n=2 biologically independent experiments), TDP-43 ASO (n=2 biologically independent experiments) or stathmin-2 ASO (n=2 biologically independent experiments) for 20 days. α-Tubulin served as a loading control. Stathmin-2 protein was profoundly suppressed following TDP-43 ASO or stathmin-2 ASO treatments. Experiment was repeated independently five times with similar results. For uncropped blots, see Supplementary Fig. 10. (e) Schematic illustration of motor neurons in microfluidic chambers before and after axotomy. Motor neuron precursors were plated at the somatic/proximal compartment and axons reached into the distal/axonal compartment during a maturation period of another eight days. One dose of ASOs was added every 12 days to the somatic compartment. After 20 days of culturing with ASOs, aspiration-based axotomy was performed at the distal compartment of each chamber leading to a complete removal of axons in that compartment. Recovery was allowed for 24 hours. (f) Timeline of iPSC-derived motor neuron maturation, ASO treatment and axotomy is shown. (g,i,k,m,o) Representative confocal immunofluorescence images of microgrooves and distal compartments, 24 hours post axotomy. Insets show high-magnification of regenerating axons’ growth cones. Axonal regeneration and growth cones are observed by immunofluorescence of stathmin-2 (green) and NF-H (red) in the terminals of motor neurons treated with control ASOs (g,h) but not in motor neurons treated with stathmin-2 (i,j) or TDP-43 (k,l) ASOs. (m,n,o,p) iPSCs-derived motor neurons treated with ASOs against stathmin-2 (m,n) or TDP43 (o,p) for 20 days and transduced with stathmin-2 expressing lentivirus for the last 96 hours showed axonal regeneration with growth cones in all axonal terminals. (h,j,l,n,p) Quantifications of 300 axons per condition are shown.
ASO-induced reduction in TDP-43 mRNA (up to 55%) was consistently accompanied by a greater (up to 12 fold) dose-dependent reduction in stathmin-2 mRNAs, coupled with a corresponding increase in RNAs containing exon 1 ligated to exon 2a (Fig. 5b). Dose-dependent suppression (up to 15 fold) of stathmin-2 expression in iPSC-derived motor neurons (without affecting TDP-43 levels) was also achieved with an ASO targeted to the 3’ UTR of full length stathmin-2 mRNA (Fig. 5c). Immunoblotting confirmed that within 12 days of treatment with ASOs targeting either TDP-43 or stathmin-2 mRNAs there was nearly complete loss of the 22kD stathmin-2 protein (Fig. 5d). Motor neuronal viability was not affected under these conditions (Supplementary Fig. 9i).
To assess the functional consequences of stathmin-2 depletion in the axonal regeneration process (Fig. 5e), motor neuron precursors were seeded into the proximal somatic compartment of a microfluidic device. Over a nine days maturation period, axons extended through microgroove-embedded channels (830 μm in length) that exclude neuronal cell bodies but allow axonal extension into the distal compartment. Matured motor neurons were treated for 20 days with ASOs added to the somatic compartment to reduce synthesis and accumulation of either TDP-43 or stathmin-2 (Fig. 5f). Axons were maintained after loss of either protein. After axons in the distal compartment were mechanically axotomized, axonal regrowth of motor neurons treated with non-targeting, control ASOs initiated within 24 hours (visualized by NF-H immunostaining in red - Fig. 5g-h). Stathmin-2 appeared in a punctate pattern along these regenerating axons and accumulated in the growth cones (Fig. 5g), consistent with a role in promoting axonal regrowth. Indeed, following ASO-mediated reduction in stathmin-2 (Fig. 5i,j) or TDP-43 (Fig. 5k,l), axonal regeneration after axotomy was almost completely suppressed, with 10% (Fig. 5j) and 13% (Fig. 5l) recovery rates relative to those treated with control ASOs, respectively. Together, these data demonstrate increased vulnerability of motor neurons upon reduced accumulation of the neuronal-growth factor stathmin-2.
We next tested if the failure to regenerate after axotomy of iPSC-derived human motor neurons following depletion of either stathmin-2 or TDP-43 could be rescued by restoration of stathmin-2 expression. To do this, after a 16 day ASO-mediated suppression of stathmin-2 or TDP-43, iPSC-derived motor neurons were transduced with a lentivirus carrying a stathmin-2-encoding gene whose RNA did not contain the cryptic polyadenylation site found in the endogenously encoded stathmin-2 gene. Despite a sustained reduction in stathmin-2, subsequent restoration of stathmin-2 expression almost completely rescued regeneration after axotomy (Fig. 5m,n). More remarkably, although TDP-43 affects the levels or splicing of many RNAs (Fig. 1b and ref. 8,9), restoration of stathmin-2 alone was sufficient to rescue regeneration after axotomy of TDP-43 depleted motor neurons (Fig. 5o,p).
Discussion
Using three independent approaches, we have identified that reduction or mutation in TDP-43 induces aberrant processing of stathmin-2 pre-mRNA, leading to efficient usage of a cryptic polyadenylation site that truncates mRNAs encoding stathmin-2, thereby suppressing stathmin-2 synthesis. Our data strongly support that under normal conditions TDP-43 acts to maintain stathmin-2 expression by repressing this premature polyadenylation within intron one of the stathmin-2 pre-mRNA. Loss of nuclear TDP-43 is a nearly universal pathological hallmark in ALS, and notably, we observed efficient premature polyadenylation of stathmin-2 in motor neurons of all tested sporadic ALS patients and in patients with repeat expansion in C9orf72, the most common inherited cause of ALS and FTD. In contrast, altered processing of stathmin-2 was not found in SOD1 ALS patients who do not exhibit TDP-43 pathology. Thus, our data offer strong support that loss of stathmin-2 RNA by premature polyadenylation and subsequent reduced levels of stathmin-2 protein in affected neurons are hallmarks of sporadic and C9orf72-expansion mediated ALS and FTD.
Thousands of mRNA targets affected by TDP-43 dysfunction have been identified in multiple cell lines and in vivo 8,9,11,25. Notably, reduced TDP-43 has been linked to the abnormal inclusion of a set of cryptic exons that may lead to nonsense-mediated decay and reduced levels of transcripts in different cell types and tissues from mouse and human 10,34–36. To that, we have established here that TDP-43 also acts in the normal situation to suppress polyadenylation within exon 2a in stathmin-2 pre-mRNA. Reduced TDP-43 in human motor neurons is accompanied by nearly complete loss of expression of functional stathmin-2. Recognizing that splicing typically occurs co-transcriptionally 37, it is likely that the premature polyadenylation in stathmin-2 mRNA is the driving event in stathmin-2 suppression, with premature polyadenylation inducing the splicing machinery to use a previously cryptic 3’ site to resolve splicing initiated at the normal 5’ splice site in intron one.
We note that abnormal processing of stathmin-2 is not recapitulated in mice expressing TDP-43 transgenes or in TDP-43 deficient mice, as the cryptic polyadenylation signal and three GU-rich sequences in intron one of the human stathmin-2 gene are not found in the corresponding mouse intron (Supplementary Fig 4d). Consistently, stathmin-2 pre-mRNA is not bound by TDP-43 in the murine nervous system and stathmin-2 mRNA level is not altered upon ASO-mediated reduction of TDP-43 in wild-type mice 8. By mining TDP-43 iCLIP data 9, we have identified physical interaction of TDP-43 with exon 2a of human stathmin-2 pre-mRNA. This strongly supports that in healthy neurons TDP-43 binding represses premature polyadenylation of stathmin-2 pre-mRNA. TDP-43 loss of function during disease relieves this inhibition, producing exon 2a-containing, truncated stathmin-2 mRNAs as seen in all sporadic and C9ORF72-mediated ALS examples tested here.
Our evidence supports a broader role for TDP-43 in modulating polyadenylation within the nervous system, similar to the role previously shown for the neuronal factor Nova 38, as well as additional RNA-binding proteins 39,40 including the ALS-associated protein FUS 12,41. Since computational analysis has recently identified extensive alternative polyadenylation in cerebellum of C9orf72 ALS and sporadic ALS subjects 42, as well as in HEK293 cells with TDP-43 suppression 43, the global impact of TDP-43 on alternative polyadenylation in human motor neurons should now be determined.
Impaired microtubule dynamics has been widely implicated in neurodegeneration, and microtubule-stabilizing agents have been proposed as a therapeutic approach 44. While disruption of TDP-43 nuclear function affects the processing of numerous RNA targets, our data provide a link between increased motor neuron vulnerability in ALS and suppression of stathmin-2, a tubulin binding protein previously established to affect microtubule dynamics 18. The family of vertebrate stathmins includes stathmins 1–4 that are likely to serve both complementary and distinct functions in the nervous system 20. Each stathmin possesses two conserved tubulin-binding domains that are capable of interacting with two α/β tubulin heterodimers 20.
Individual stathmins may have unique functional properties. Stathmins-2 and 4 are highly expressed in neurons 16, with only stathmin-2 mRNA enriched in motor neurons (Supplementary Fig. 5b, c). Increased expression of stathmin-1 and stathmin-2 has been reported in cellular and mouse models of spinal muscular atrophy (SMA) 45 and in motor neurons of SMA-like mice 45 and ALS mice expressing mutant superoxide dismutase 1 (SOD1) 46,47. Stathmin-2 has been proposed as a neuronal regeneration marker 22 (with expression similar to the axonal growth associated protein GAP-43 48) and we have shown here that axonal regeneration after reduction in TDP-43 can be restored by maintaining stathmin-2 levels (Fig. 5). In contrast, reduction in stathmin-1, which we show to be expressed in motor neurons at 1/30th the level of stathmin-2 (Fig. 4b), has been reported to rescue axonal pathology in motor neurons in pmn mice that have mutation in the TBCE chaperone for folding tubulin 49.
The stathmin-2 loss we have identified in motor neurons from ALS patients is strongly expected to alter microtubule dynamics and function, thereby contributing to denervation from neuromuscular junctions and/or axonal degeneration. This scenario is supported by evidence of destabilized neuromuscular junctions and impaired synapse stability in Drosophila expressing mutant stathmin 23. Likewise, a paralytic phenotype was shown in Drosophila following stathmin silencing 50. The high level of stathmin-2 shown here to accumulate in normal human spinal motor neurons highlights the potentially deleterious impact from its profound reduction upon reduced nuclear TDP-43 function in the neuronal population preferentially affected in ALS.
To this, our data provide evidence that reduction in stathmin-2, as the consequence of neuronal damage or stress that leads to diminished nuclear TDP-43 function, is itself sufficient to inhibit regeneration of motor axons. Restoring the regenerative capability to iPSC-derived motor neurons depleted of TDP-43 simply by restoring stathmin-2 levels offers a new insight into how TDP-43 dysfunction drives increased vulnerability. Perhaps most importantly, our evidence supports development of therapeutic strategies for ALS, FTD and other neurodegenerative diseases affected by TDP-43 proteinopathy through restoration of stathmin-2 to increase sustainability of affected neurons.
Experimental Procedures
RNA extraction and cDNA synthesis
For RNA extraction from cultured cells and neurons, direct lysis was applied using Trizol reagent (Invitrogen) and RNA was purified according to the manufacturer’s instructions. Human tissue samples were first homogenized in Trizol, using a mechanical tissue homogenizer, and RNA was extracted accordingly.
RT-PCR and real-time PCR
For cDNA synthesis, 1 μg total RNA was reverse transcribed using the high-capacity reverse transcription kit (ABI) or superscript III (Invitrogen) according to the manufactures’ instructions. RT-PCR reactions were performed using Q5 High-Fidelity DNA polymerase (NEB) in a T100 thermocycler PCR machine (Bio-Rad). For splicing analyses, RT-PCR products were separated on 2% polyacrylamide gels and then incubated with SYBR gold (Invitrogen) for imaging and analysis. Quantification of alternative splicing band intensities were determined using ImageJ software and an average of 3 biological replicates was plotted. Quantitative real-time PCR was carried out in triplicates, using iTaq Universal SYBR green (Bio-Rad) in a CFX384 real-time PCR machine. mRNA expression of transferrin receptor protein-1 (TFRC) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as endogenous control genes, as indicated. All primers are listed in Supplementary Table 4.
RNA sequencing
Total RNA was extracted from SH-SY5Y cells as indicated above. Subsequently, 1 μg of total RNA was used for mRNA libraries preparation using the TruSeq RNA kit (Illumina). For the TDP-43 knockdown experiment, 3 biological replicates were sequenced per each condition (siControl/siTDP-43). For TDP-43 genome-editing experiments, 2 biological replicates were sequenced per each condition (TDP-43WT/WT, TDP-43N352S/N352S). Real-time qPCR validations were done with at least 3 replicates. RNA sequencing was carried out on an Illumina HiSeq 4000 platform with a median of 46M reads per sample. 50bp single-end FASTQ files were obtained using the Illumina demultiplexing pipeline. STAR 54 and RSEM 55 were used to align the reads to the human reference sequence HG38 and to calculate the raw counts and transcripts per million (TPM) values for genes, respectively. Genes differentially expressed between sample groups were identified by DESeq2 51.
Genome editing
Both TDP-43 alleles were genetically modified in SH-SY5Y cells. A single guided RNA targeting TDP-43 was designed (Benchlin webtool) and cloned into pSpCas9–2A-GFP plasmid 56 (px458-Addgene) using the BbSI restriction site. The sequence for guided RNA targeting TDP-43 is: GCGGGTAATAACCAAAACCA. To promote homologous recombination, pSpCas9–2A-GFP-gRNA plasmid was electroporated using the Amaxa Nucleofactor (assay A-023) along with a 180 long single-stranded donor oligonucleotides (IDT), containing the desired ALS-causing mutation and four synonymous single-nucleotide replacements to avoid DNA cleavage recurrence by Cas9. 48 hours following electroporation, cells were collected and dissociated using Accutase. GFP-positive cells were sorted and single-cell seeded into 96 wells plates using the SH800S Sony cell sorter. Individual clones were expanded and DNA was extracted for PCR amplification of TDP-43 genomic locus. Replacement of AAT to AGT (c.1055A>G) was confirmed by Sanger sequencing. The entire TDP-43 coding region and all exon-intron junctions (> 500 base-pairs length of each intronic region) were then sequenced to verify the absence of any additional DNA alteration. qPCR analysis was performed with a wild type isogenic SH-SY5Y cell line that had undergone the mutagenesis attempt but not acquired the TDP-43 mutation during the original screening progress.
Immunoblotting
Total-cells extracts were collected in radioimmunoprecipitation lysis buffer (RIPA buffer). Proteins concentrations were determined by Bradford assay (Bio-Rad) and equal amounts of total protein were boiled in SDS sample buffer for 10 minutes before running in 10% acrylamide gel. Proteins were transferred to PVDF, and blocked in 5% milk solution in tris-buffered saline and 0.1% Tween-20 (TBST) for 1 hour before overnight incubation with the following primary antibodies: anti-TDP-43 (1:1000) (ProteinTech, 10782–2-AP), anti-Stathmin-2 (1:2000) (nbp1–49461, NovusBiologicals), anti-Tubulin (1:10,000) (DM1A, Abcam). Immunoblots were washed in TBST and probed with HRP-conjugated secondary antibodies diluted 1:5000 for one hour in room temperature (GE Healthcare), before being exposed to films.
Immunofluorescence and image acquisition
SH-SY5Y cells were grown on poly-D-lysine (Sigma) coated 4-wells glass bottom chambers (Thermo). iNeurons were grown on matrigel coated (Corning, 356230) 4-wells glass bottom chambers (Thermo). At end point, cells were washed one time with phosphate buffer (PBS) and fixed by 4% paraformaldehyde in phosphate buffer (PBS) for 30 minutes at room temperature. Two washes with PBS were followed by cell membrane permeabilization using 0.1% Triton X-100 (Sigma) for 15 minutes at room temperature. After one wash with PBS, cells were incubated for 30 minutes with blocking solution containing 3% bovine serum albumin (Sigma) + 3% donkey serum (Jackson ImmunoResearch) in PBS. Blocking solution was removed and replaced with primary TDP-43 antibody (Proteintech # 10782–2-AP) diluted 1:1000 in fresh blocking solution for overnight incubation at 4°C. Following overnight incubation and 3 washes with PBS, cells were incubated with fluorescently tagged secondary antibody conjugated to Alexa-488 (Thermo Fisher Scientific) diluted 1:500 in blocking solution. Finally, cells were washed 3 times in PBS, the second wash included 10 minutes incubation with DAPI solution (Thermo Fisher Scientific, 100ng/ml) for nuclear staining. The preparation was mounted with ProLong Gold antifade reagent (Thermo Fisher Scientific). Fluorescence images were acquired with Olympus FV1000 confocal microscope, at the microscopy core, the Ludwig institute, UCSD.
Fluorescence quantification from micrographs
Fluorescence quantifications were performed using FIJI image processing software (http://imagej.net). Freehand tool of the software was used to select the subcellular regions such as nuclei or cytoplasm for quantification. Data were plotted in GraphPad Prism software for illustration.
In Situ Hybridization of STMN2 RNA isoforms
Short 5’-Digoxigenin-labeled Locked Nucleic Acid (LNA) probes were designed by QIAGEN (Truncated Exon2A STMN2 RNA isoform probe sequence: TCACACAGAGAGCCAAATTCTT; Normal STMN2 mature mRNA probe sequence to the 3’UTR of STMN2: ATCCTGATATCGCATGATCCAT). For each tissue two serial FFPE tissue sections (7μm) were cut onto charged glass slides. Each of the two sections was hybridized with one of the STMN2 probes using standard ISH protocols and revealed with a commercial anti-DIG antibody and NBT developer kit. Slides were inspected under brightfield microscopy and serial sections of individual neurons were identified and imaged at 40x magnification on a Keyence BZ-X700 fluorescent microscope. For quantification, spinal motor neurons were identified by their large cytoplasm and/or nucleus, the presence of lipofuscin, and their position within Rexed lamina IX of the spinal cord. Neurons of the motor cortex were identified by their shape and relative size and position within cortical layering, with special attention to layers 3 and 5.
Immunohistochemistry for TDP-43 and phosphorylated TDP-43
Formalin-fixed paraffin-embedded sections with 6 µm thickness were deparaffinized with Citrisolv (Decon Lab #1601H) and hydrated with different dilutions of alcohol. Endogenous peroxidase activity was quenched with 0.06% H2O2 in methanol for 15 min. Antigen retrieval was performed in a high pH solution (Vector #H-3301) in a pressure cooker at 125 °C for 20 min. Sections were blocked with 2% FBS (Gibco #10438–026) and 0.2% Triton X-100 (Sigma #T8787) in PBS for 60 min. Following antigen retrieval and blocking, the sections were incubated with primary antibodies at 4 °C overnight as follows: TDP-43 (anti-Rabbit, Proteintech #10782–2-AP, 1:5,000) and phosphorylated TDP-43 (pTDP-43) (pS409/410, anti-Rabbit, Cosmobio #TIP-PTD-P02, 1:1,000). The secondary antibody (ImmPRESS HRP Reagent Kit, anti-Rabbit IgG, Vector #MP-7401) was incubated at room temperature for 60 min, and signals were detected using NovaRED peroxidase substrate kit (Vector #SK-4800) for 2 minutes. Counterstaining was performed with hematoxylin (Ricca chemical #3537–32). TDP-43 and pTDP-43 immunohistochemistry was performed using consecutive sections for each patient.
Cell culture and transfection
SH-SY5Y
The neuroblastoma cells (ATCC) were cultured in DMEM/F12 (Gibco) supplemented with 10% fetal-bovine serum (Omega) and 1% penicillin-streptomycin (Gibco) at 37 °C with 5% CO2. For knockdown experiments, cells were transfected with SMARTpool ON-TARGETplus siRNA targeting TDP-43 (L-012394) or control siRNA pool (D001810–10) (GE Dharmacon) at final concentration of 50nM, for 96 hours in two doses (0, 24h), after complexing with Lipofectamin RNAiMAX (Invitrogen) in Opti-MEM (Gibco) for 20 minutes.
Fibroblasts
Fibroblasts were cultured in DMEM/F12 (Gibco) containing 20% tetracycline-free FBS (Omega) and 1% penicillin-streptomycin (Gibco).
Direct conversion of human fibroblasts into induced neurons
The trans-differentiation assay was performed according to a protocol previously described 57 and is based on expression of neuronal-specific transcriptional factor Brn2 and knockdown of the splicing factor PTB. Briefly, initial lentiviral transduction of fibroblasts by doxycycline-induced Brn2 expressing plasmid was performed. After 16 hours, mediia was replaced and cells were allowed to recover for 24 hours. Cells were then grown under 1 μg/ml puromycin selection (Gibco) for 4 days and remaining cells were transferred into a new culturing dish. After expansion to 40–50% confluency, cells were transduced with shRNA lentivirus designed to suppress PTB mRNA expression. 16 hours later, medium was replaced for 24 hours recovery. After selection with hygromycin B (100µg/ml) for 4 days, remaining cells were seeded for terminal differentiation in matrigel-covered (Corning, 356230) glass chambers (Thermo) for Immunofluorescence or 6 well dish for RNA extraction. 24 hours later, medium was switched to 1:1 DMEMF12:Neurobasal N3 medium plus doxocycline 1 μg/ml (Dox) (Sigma), FGFb 10ng/ml (Invitrogen) and 0.4% B27 (Gibco). The N3 medium contains: 25 μg/ml insulin (Sigma, I9278), 50 μg/ml apo-transferrin (T1147), 30 nM sodium selenite, 20 nM progesterone (Sigma), 100 nM putrescine (Sigma), 0.4% B27 and penicillin/streptomycin (Gibco). Small molecules were added 48 hours later: CHIR99021 1μM (Stemgent, 04–0004-02), SB431542 10μM (Stemgent, 04–0010) and Dbcamp 0.5 μM (Sigma, D0627). 48 hours later, cells were switched to N3 medium containing neuronal growth factors: CNTF (20ng/ml), BDNF (10ng/ml), NT3 (10ng/ml) and GDNF (20ng/ml) purchased from R&D systems, plus 2% FBS, Dox (1μg/ml), FGFb (10ng/ml), 0.4% B27, forskolin 10μM (FSK) (Sigma F6886) and DM 1μM (AMPK Inhibitor, Compound C; Millipore 17126). Medium was changed every other day for 2–4 weeks.
Induced pluripotent cells (iPSC) and motor neurons differentiation
The iPSC line was derived from peripheral blood mononuclear cells donated by a 58 year old healthy Caucasian male, by introducing the episomal DNAs expressing Oct4, Sox2, Klf4, L-Myc, Nanog, and shRNA against p53. The selected clone was fully characterized and demonstrated a normal karyotype with normal self-renewal and differentiation capacity comparable with H9 human embryonic stem cells. NextGen sequencing data shows that no modifications occurred on ALS-related genes. iPS cells were differentiated into motor neurons using the proprietary differentiation protocol patented by iXCells Biotechnologies (Provisional Application # 14359–001-888), summarized in Supplementary Fig. 9.
Antisense oligonucleotide (ASO) treatment
ASOs mediating RNase H-dependent degradation of the TDP-43 mRNA, AAGGCTTCATATTGTACTTT, Ionis Pharmaceuticals), stathmin-2 mRNA, GGTCTTAGTCAAGCTCAGAG, Ionis Pharmaceuticals), or murine Malat-1 as control, GGGTCAGCTGCCAATGCTAG, Ionis Pharmaceuticals), were added to the iPSC-derived motor neurons culture medium at day 29 of maturation. Medium was changed every 3–4 days without addition of ASOs, and RNA was collected after 12 days of treatment (day 41). The MOE-gapmer ASOs are 20 nucleotides in length, wherein the central gap segment comprising ten 2′-deoxyribonucleotides that are flanked on the 5′ and 3′ wings by five 2′-O-methoxyethyl-modified nucleotides. The internucleotide linkages are phosphorothioate, except for the Stathmin-2 ASO which has phosphodiester linkages in the 5’ and 3’ wings of the ASO, and all cytosine residues are 5′-methylcytosines.
Compartmentalized microfluidic devices and axotomy
The master molds to prepare the microfluidic devices were fabricated by photolithography, as previously described (Minteer, 2006), by the Bioengineering Department of the University of California, San Diego, Nano3 Cleanroom Facility. Two compartment devices were molded by soft lithography using Sylgard 182 (Ellsworth Adhesives, Germantown, WI) as previously described 58. Each compartment of the device was 108 μm tall, separated by microgrooves 3.1 μm tall, 16.28 μm wide and 830 μm long. The proximal compartment contained one hole in each side of the compartment 8 mm in diameter, while the distal compartment’s holes were 4 mm in diameter. These characteristics are important to perform an effective vacuum-based axotomy. After curing, the cut devices were bath-sonicated in water, washed in 70% ethanol and sterilized under UV before mounting onto glass coverslips. The devices were coated with matrigel (Corning, 356230) for 1 hour at 37ºC and rinsed with DMEM before plating the cells. Half million iPSC-derived motor neuron precursors were plated in the proximal somatic compartment, and axonal growth to distal compartment was achieved in eight days of maturation. ASOs were then added into the somatic compartment for 20 days of treatment, half media was changed every 4 days and a second dose of ASOs was added at day 12. For aspiration-induced axotomy, media was simultaneously aspirated from one side and added back to the other side of the distal (axonal) compartment, until complete removal of axons from the distal compartment was confirmed.
Lenti-viral transduction
A stathmin-2 open reading frame was cloned into a PL-SIN18-lentiviral vector under transcriptional control of the phosphoglycerate kinase (PGK) promoter. HEK293T cells were used for packaging lentiviruses. Briefly, 0.5 ×106 per well of 293T cells were seeded in a 6-well plate. For lentiviral transfection, 2.5 µg of the lentiviral plasmid (containing stathmin-2 cDNA), 1.25 µg of pMD2.G and 0.625 µg of psPAX2 were co-transfected to each well using Mirus transIT-X2 transfection reagent (Mirus). Culture medium was changed to fresh medium at ~24 hours post transfection. Virus containing supernatants were collected at 48h and 72h after transfection and concentrated using ultracentrifugation. Viral concentrates were stored at −80 °C. Human iPSC-derived motor neurons were cultured to maturation and then infected with lentiviral particles encoding stathmin-2 in the presence of polybrene (Millipore).
Cell viability assay
Human iPSC-derived motor neurons were matured as described in supplementary Fig. 9e, and then treated with control ASOs or ASOs targeting TDP-43 or stathmin-2 for 20 days. Viability of iPSC-derived human motor neurons was examined using LIVE/DEAD Reduced Biohazard Viability/Cytotoxicity Kit (Molecular Probes), according to the manufacturer’s instructions. Quantification of living cells was performed using the Olympus FV1000 confocal microscope.
Human postmortem tissues
Human tissues were obtained using a short postmortem interval acquisition protocol that followed HIPAA-compliant informed consent procedures and were approved by Institutional Review Board (Benaroya Research Institute, Seattle, WA IRB# 10058 and University of California San Diego, San Diego, CA IRB# 120056).
Tissue samples were obtained from patients who met the modified El Escorial criteria for definite ALS (Brooks et al, 2000). Control nervous systems were obtained from non-neurological patients when life support was withdrawn, or from patients on hospice. Autopsies were performed within 10 hours of death, with an average post-mortem interval of 5 hours for the cohort of patients used in this study.
Information on human samples is provided in Supplementary Table 5.
Statistics
Statistical tests were performed using GraphPad Prism. Student’s t-test (two tails or one tail) were used as indicated in the text. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications. Experiments were not randomized. Data distribution was assumed to be normal but this was not formally tested. Data collection and analysis were not performed blind to the conditions of the experiments.
Reporting summary
Extended information on experimental design is available in the Life Sciences Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
The plasmids for expression of Brn2 and shPTB were kind gifts from X. D. Fu, UCSD. We thank the viral vector core facility at Sanford Burnham Prebys (SBP) Medical Discovery Institute for the kind contribution of the SIN18 vector. We thank B. Ren for providing the Illumina sequencing platform. We thank A. Goginashvili for valued input on the manuscript. We thank J. Nuovo at Phylogeny Inc. (Columbus, OH) for performing the CISH experiment. This work was supported by grants from NINDS/NIH R01-NS27036 to D.W.C, Target ALS (S20A00) and NINDS/NIH (R01-NS087227) to C.L.-T. and R01-NS088578 to J.R. C.L.-T. was recipient of a Career Development grant from the Muscular Dystrophy Association (MDA). Z.M was recipient of EMBO long-term fellowship and is currently supported by the Human Frontiers Science Program (HFSP) long-term fellowship. J.L.-E. was recipient of a Milton Safenowitz postdoctoral fellowship. M.W.B. was supported by the National Institute of General Medical Sciences of the National Institutes of Health Award T32-GM008666. M.A.M, C.F.B., and F.R. are employees and D.W.C. is a consultant for Ionis Pharmaceuticals.
Footnotes
Accession code
All RNA sequencing data generated and analyzed for this study have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE122069.
Data availability statement
The data that support the findings of this study are readily available from the corresponding authors upon reasonable request.
Competing interests
The authors declare no competing interests.
References
- 1.Taylor JP, Brown RH Jr. & Cleveland DW Decoding ALS: from genes to mechanism. Nature 539, 197–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renton AE et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeJesus-Hernandez M et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–56 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neumann M et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–3 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Ling SC, Polymenidou M & Cleveland DW Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lagier-Tourenne C, Polymenidou M & Cleveland DW TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19, R46–64 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alami NH et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polymenidou M et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14, 459–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tollervey JR et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14, 452–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ling JP, Pletnikova O, Troncoso JC & Wong PC TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ayala YM et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 30, 277–88 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lagier-Tourenne C et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15, 1488–97 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kapitein LC & Hoogenraad CC Building the Neuronal Microtubule Cytoskeleton. Neuron 87, 492–506 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Mitchison T & Kirschner M Dynamic instability of microtubule growth. Nature 312, 237–42 (1984). [DOI] [PubMed] [Google Scholar]
- 15.Desai A & Mitchison TJ Microtubule polymerization dynamics. Annu Rev Cell Dev Biol 13, 83–117 (1997). [DOI] [PubMed] [Google Scholar]
- 16.Stein R, Mori N, Matthews K, Lo LC & Anderson DJ The NGF-inducible SCG10 mRNA encodes a novel membrane-bound protein present in growth cones and abundant in developing neurons. Neuron 1, 463–76 (1988). [DOI] [PubMed] [Google Scholar]
- 17.Morii H, Shiraishi-Yamaguchi Y & Mori N SCG10, a microtubule destabilizing factor, stimulates the neurite outgrowth by modulating microtubule dynamics in rat hippocampal primary cultured neurons. J Neurobiol 66, 1101–14 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Riederer BM et al. Regulation of microtubule dynamics by the neuronal growth-associated protein SCG10. Proc Natl Acad Sci U S A 94, 741–5 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belmont LD & Mitchison TJ Identification of a protein that interacts with tubulin dimers and increases the catastrophe rate of microtubules. Cell 84, 623–31 (1996). [DOI] [PubMed] [Google Scholar]
- 20.Chauvin S & Sobel A Neuronal stathmins: a family of phosphoproteins cooperating for neuronal development, plasticity and regeneration. Prog Neurobiol 126, 1–18 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Shin JE et al. SCG10 is a JNK target in the axonal degeneration pathway. Proc Natl Acad Sci U S A 109, E3696–705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shin JE, Geisler S & DiAntonio A Dynamic regulation of SCG10 in regenerating axons after injury. Exp Neurol 252, 1–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graf ER, Heerssen HM, Wright CM, Davis GW & DiAntonio A Stathmin is required for stability of the Drosophila neuromuscular junction. J Neurosci 31, 15026–34 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yusuf M, Leung K, Morris KJ & Volpi EV Comprehensive cytogenomic profile of the in vitro neuronal model SH-SY5Y. Neurogenetics 14, 63–70 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnold ES et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U S A 110, E736–45 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White MA et al. TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat Neurosci 21, 552–563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fratta P et al. Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J (2018). [DOI] [PMC free article] [PubMed]
- 28.Xue Y et al. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 152, 82–96 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tian B & Graber JH Signals for pre-mRNA cleavage and polyadenylation. Wiley Interdiscip Rev RNA 3, 385–96 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun S et al. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc Natl Acad Sci U S A 112, E6993–7002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krach F et al. Transcriptome-pathology correlation identifies interplay between TDP-43 and the expression of its kinase CK1E in sporadic ALS. Acta Neuropathol 136, 405–423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saberi S, Stauffer JE, Schulte DJ & Ravits J Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurol Clin 33, 855–76 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mackenzie IR et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61, 427–34 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Sun M et al. Cryptic exon incorporation occurs in Alzheimer’s brain lacking TDP-43 inclusion but exhibiting nuclear clearance of TDP-43. Acta Neuropathol 133, 923–931 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeong YH et al. Tdp-43 cryptic exons are highly variable between cell types. Mol Neurodegener 12, 13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Humphrey J, Emmett W, Fratta P, Isaacs AM & Plagnol V Quantitative analysis of cryptic splicing associated with TDP-43 depletion. BMC Med Genomics 10, 38 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naftelberg S, Schor IE, Ast G & Kornblihtt AR Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem 84, 165–98 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Licatalosi DD et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456, 464–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elkon R, Ugalde AP & Agami R Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet 14, 496–506 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Tian B & Manley JL Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol 18, 18–30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masuda A et al. Position-specific binding of FUS to nascent RNA regulates mRNA length. Genes Dev 29, 1045–57 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prudencio M et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat Neurosci 18, 1175–82 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rot G et al. High-Resolution RNA Maps Suggest Common Principles of Splicing and Polyadenylation Regulation by TDP-43. Cell Rep 19, 1056–1067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brunden KR, Lee VM, Smith AB 3rd, Trojanowski JQ & Ballatore C Altered microtubule dynamics in neurodegenerative disease: Therapeutic potential of microtubule-stabilizing drugs. Neurobiol Dis (2016). [DOI] [PMC free article] [PubMed]
- 45.Wen HL et al. Stathmin, a microtubule-destabilizing protein, is dysregulated in spinal muscular atrophy. Hum Mol Genet 19, 1766–78 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Strey CW et al. Dysregulation of stathmin, a microtubule-destabilizing protein, and up-regulation of Hsp25, Hsp27, and the antioxidant peroxiredoxin 6 in a mouse model of familial amyotrophic lateral sclerosis. Am J Pathol 165, 1701–18 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bellouze S et al. Stathmin 1/2-triggered microtubule loss mediates Golgi fragmentation in mutant SOD1 motor neurons. Mol Neurodegener 11, 43 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mason MR, Lieberman AR, Grenningloh G & Anderson PN Transcriptional upregulation of SCG10 and CAP-23 is correlated with regeneration of the axons of peripheral and central neurons in vivo. Mol Cell Neurosci 20, 595–615 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Selvaraj BT, Frank N, Bender FL, Asan E & Sendtner M Local axonal function of STAT3 rescues axon degeneration in the pmn model of motoneuron disease. J Cell Biol 199, 437–51 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duncan JE, Lytle NK, Zuniga A & Goldstein LS The Microtubule Regulatory Protein Stathmin Is Required to Maintain the Integrity of Axonal Microtubules in Drosophila. PLoS One 8, e68324 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rabin SJ et al. Sporadic ALS has compartment-specific aberrant exon splicing and altered cell-matrix adhesion biology. Hum Mol Genet 19, 313–28 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ho R et al. ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat Neurosci 19, 1256–67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xue Y et al. Sequential regulatory loops as key gatekeepers for neuronal reprogramming in human cells. Nat Neurosci 19, 807–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taylor AM et al. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods 2, 599–605 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.