

Abstract
Renal medullary carcinoma (RMC) is one of the most aggressive renal cell carcinomas. It predominantly afflicts young adults and adolescents with sickle cell trait and other sickle hemoglobinopathies, and is refractory to targeted and anti-angiogenic therapies used in patients with clear-cell renal cell carcinoma. Platinum-based cytotoxic chemotherapy is the mainstay for RMC treatment. Based on recent advances in the diagnosis, management, and clinical trial development for RMC, a panel of experts met in October 2017 and developed updated consensus recommendations to inform clinicians, researchers, and patients. Because RMC often aggressively recurs while patients are still recovering from nephrectomy, upfront chemotherapy should be considered for the majority of patients, including those with localized disease. After safety and dosing information have been established in adults, phase II and III trials enrolling patients with RMC should allow patients aged 12 years and older to be accrued. Patients with the very rare unclassified renal cell carcinoma with medullary phenotype variant should be included in RMC trials. Medical providers should be aware that RMC can afflict subjects of all races, and not only those of the African descent, and that the presence of sickle cell trait, or of other sickle hemoglobinopathies, can impact drug responses and toxicity.
Keywords: renal medullary carcinoma, sickle hemoglobinopathies, SMARCB1, unclassified renal cell carcinoma with medullary phenotype
Introduction
Originally described in 1995,1 renal medullary carcinoma (RMC) is a rare malignancy that comprises <0.5% of all renal cell carcinomas (RCCs).2 It is almost exclusively found in young adults and adolescents with sickle cell trait and other sickle hemoglobinopathies, and carries a dismal prognosis with < 5% of patients surviving longer than 36 months.2, 3 RMC is resistant to targeted and anti-angiogenic therapies used in patients with clear-cell RCC,3, 4 whereas cytotoxic chemotherapy regimens produced a 29% objective response rate in a multicenter retrospective study.3 In April 2016, a panel of international clinicians and researchers reached an expert consensus on the diagnosis and management of RMC.2 In October 2017, all authors of the present manuscript held a follow-up meeting in Washington, DC to exchange ideas, forge collaborations, and accelerate scientific discoveries for this deadly disease. Based on the feedback received on the original recommendations, the panel of attending experts developed updated proposals on the diagnosis, management, and clinical trial eligibility criteria of patients with RMC. These recommendations were drafted and circulated electronically to all participants who provided further comments and revisions until the following consensus statements were obtained. The major updates since the last meeting are: i) we propose that the term renal cell carcinoma, unclassified, with medullary phenotype (RCCU-MP) should be used for SMARCB1-negative high-grade adenocarcinomas that are histopathologically similar to RMC but occur in individuals without sickle hemoglobinopathies; iii) we recommend that upfront systemic therapy should be considered for most patients with RMC, including those with localized disease; iii) we now provide guidance on developing clinical trials for RMC. We recommend that clinical trials for patients with RMC also include RCCU-MP, particularly if these trials target pathways affected by SMARCB1 loss. Other RCC variants occurring in patients with sickle hemoglobinopathies should be managed as different entities from RMC. Phase II or III trials for patients with RMC / RCCU-MP should routinely allow patients aged 12 years and older to be enrolled. Clinicians and researchers should be particularly aware of the potential interactions that investigational regimens can have with sickle hemoglobinopathies, including sickle cell trait.
Definition of RMC and its distinguishing features compared with other rare forms of RCC
RMC is a high-grade adenocarcinoma with a predilection for the renal medullary anatomical region and putatively arising from a cell within the distal nephron structures, including potentially the loop of Henle, distal convoluted tubules, connecting tubules or the collecting duct system.5, 6 The most recent 2012 International Society of Urological Pathology Vancouver classification of RCC recognized RMC as a distinct histologic classification but did not explicitly address whether the presence of a sickle hemoglobinopathy should be an obligate diagnostic criterion.7 We propose that RMC is defined by two features. First, is the loss of protein expression of the tumor suppressor gene SMARCB1, also known as INI1, hSNF5, or BAF47.8, 9 Second, is the presence of a sickle hemoglobinopathy, such as sickle cell trait, sickle cell disease, sickle cell beta thalassemia, or hemoglobin SC disease.3, 10 The most common mechanisms of SMARCB1 inactivation in RMC are deletions and inactivating translocations.11, 12 A recently proposed model of RMC pathogenesis postulates that these SMARCB1 deletions and translocations can occur via the deregulation of low fidelity DNA repair pathways due to regional ischemia induced by red blood cell sickling in the renal inner medulla, most commonly in the right kidney.13
We propose that the provisional term RCCU-MP can be used to describe the very rare cases of SMARCB1-negative high-grade adenocarcinomas arising from the distal nephron in patients without sickle cell trait or other sickle hemoglobinopathies.14 SMARCB1 loss by immunohistochemistry is essential to establish the diagnosis of RMC or RCCU-MP. However, this immunostain is often not available in resource-limited settings or not ordered by the pathologist due to the rarity of this disease. Given the clinical importance of distinguishing RMC and RCCU-MP from other malignancies, it is the strong recommendation of this panel that pathologists and clinicians consider either the routine incorporation of SMARCB1 immunohistochemistry and/or secondary review of the pathology at specialized centers in all patients with a suspicion of RMC or RCCU-MP. The differential diagnosis of high-grade adenocarcinomas arising from the distal nephron is very narrow; a recent article by Ohe et al. provides an excellent overview of the clinicopathological features and morphological patterns that should raise the suspicion for RMC, particularly in patients with sickle cell trait or other sickle hemoglobinopathies.6 More specifically, reticular/yolk sac tumor-like patterns are considerably more common in RMC (85% of cases) compared with collecting duct carcinoma (CDC; 8% of cases) or fumarate hydratase (FH)-deficient RCC (3% of cases).6 The tubulocystic patterns seen in 66% of FH-deficient RCC cases are generally not found in RMC.6 Furthermore, sickled erythrocytes are noted within the tumor specimen and the adjacent kidney tissue in all RMC cases.1, 6
Because many individuals are unaware that they carry the sickle cell trait,15 hemoglobin electrophoresis should be ordered in all suspected RMC or RCCU-MP cases with unknown sickle cell status, regardless of race. However, not all high-grade adenocarcinomas that arise from the distal nephron of patients with sickle cell trait, or other sickle hemoglobinopathies, are RMC. We caution medical providers that this anchoring bias16 can lead to misdiagnosis. Individuals with sickle hemoglobinopathies are at the same risk as those without for developing RCC other than RMC. For example, young individuals with sickle cell trait can develop FH-deficient RCC or CDC with retained SMARCB1 (Tannir, Rao, and Msaouel, unpublished data). Given the major role that SMARCB1 loss plays in RMC biology,17 and in the absence of other biological evidence (e.g., the loss of other subunits of the BAF and PBAF complexes to which SMARCB1 belongs18), such RCC cases should be considered distinct from RMC.
A separate RCC variant arising from the renal medulla has been reported in 3 patients with sickle cell trait, and characteristically harbors a t(2;10)(p23;q22) fusion of anaplastic lymphoma kinase (ALK) with vinculin (VCL) while demonstrating intact SMARCB1 expression.19–21 Although the very small number of cases reported thus far precludes definitive conclusions, VCL-ALK RCC appears to be relatively indolent (median ki-67 of 5%), and to be associated with favorable prognosis.19–21 Given its distinct histopathology, clinical behavior, and molecular characteristics, VCL-ALK RCC should be managed as a different entity from RMC.
Diagnosis and management recommendations
RMC most commonly presents as a unilateral, solid, hypoenhancing mass (mean maximum dimension of 6.6 cm) arising from the renal medulla and abutting the pelvicalyceal system.22 It demonstrates a predilection for the right kidney in ~70% of cases.3, 23 Hematuria and flank pain are the most common presenting symptoms in 66% of cases, and 50% will have constitutional symptoms such as unintentional weight loss or, less commonly, night sweats.3 Diagnosis should initially be established by biopsy of the primary tumor or the most accessible metastatic lesion. When possible, fresh frozen biopsy samples should be stored for research purposes (Figure 1).
Figure 1:
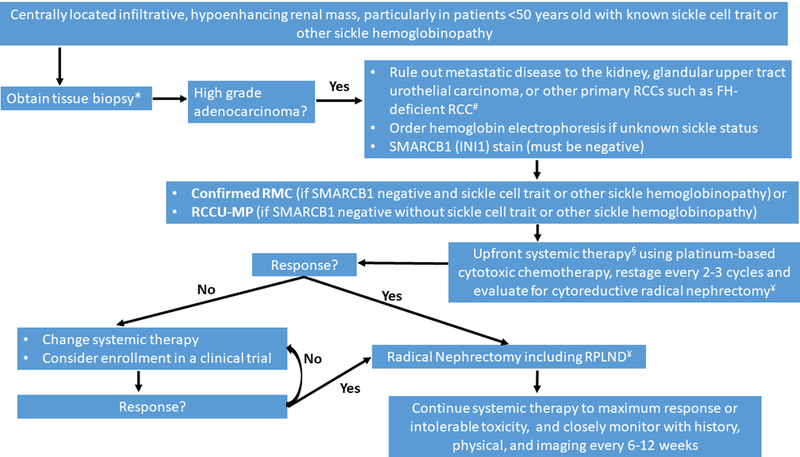
Diagnosis and management recommendations for renal medullary carcinoma (RMC) and renal cell carcinoma, unclassified with medullary phenotype (RCCU-MP). FH-deficient RCC: fumarate hydratase-deficient renal cell carcinoma; RCC: renal cell carcinoma; RPLND: retroperitoneal lymph node dissection
*Whenever possible, fresh frozen tissue should be saved for research use.
#RMC tumor tissues always contain sickled erythrocytes, often demonstrate reticular/yolk sac tumor-like patterns, and lack the tubulocystic patterns seen in FH-deficient RCCs. Secondary review of the pathology at specialized centers is strongly encouraged.
§Upfront surgery can be considered for isolated tumors ≤4 cm in greatest dimension, confined to the kidney. These patients should be followed closely post-operatively with history, physical, and imaging.
¥Cytoreductive radical nephrectomy should be considered if feasible based on response to systemic therapy, performance status, and surgical evaluation.
Up to 94% of patients with RMC present with nodal and/or visceral metastases.3 With few exceptions, even patients presenting with localized disease will quickly develop distant metastases, often within weeks. It is common for patients to develop early recurrence while still recovering from nephrectomy resulting in rapid deterioration of performance status and compromising the ability to receive systemic therapy. We therefore recommend that upfront systemic therapy (platinum-based cytotoxic chemotherapy or clinical trial) should be considered for the majority of patients with newly diagnosed RMC, including those with localized disease at presentation (Figure 1). Upfront surgery can however be chosen for the very rare cases of isolated RMC tumors ≤4 cm in greatest dimension, limited to the kidney. Given the high risk of recurrence, these patients should be very closely monitored post-operatively.
Cytoreductive nephrectomy was associated with improved overall survival compared with systemic therapy alone (16.4 versus 7.0 months) in a retrospective multicenter analysis,3 and should be considered based on response to upfront systemic therapy, performance status, and surgical evaluation. Because of the high risk for rapid recurrence, postoperative systemic therapy should be considered in all patients following definitive or cytoreductive nephrectomy. No clinical trials have demonstrated survival benefit from nephrectomy and adjuvant or neoadjuvant systemic therapy, but the application of chemotherapy and surgery in these settings is based on the combined experience of this expert panel. If nephrectomy is not feasible, embolization can effectively palliate symptomatic hematuria when necessary.
The most commonly used systemic therapies are platinum-based cytotoxic regimens.3, 24 No particular cytotoxic chemotherapy combination has shown superiority over others. However, even relatively low-intensity regimens can occasionally produce gratifying responses. Durable complete responses were noted in two out of 22 (9%) patients with metastatic RMC treated with carboplatin plus paclitaxel,3 with one patient currently disease-free six years and the other four years from initial diagnosis. Radical nephrectomy is preferred over partial nephrectomy due to the infiltrative nature and anatomical location of RMC.
Therapeutic Advances
Given the low cure rates with cytotoxic chemotherapy regimens,3 patients with RMC should be considered for clinical trials investigating novel treatment strategies, whenever possible.2 Preclinical data showed that the loss of SMARCB1 promotes cancer cell growth via deregulated chromatin remodeling mechanisms, including the aberrant activation of the histone methyltransferase EZH2, thus making this pathway an attractive therapeutic target for patients with RMC.25, 26 The first clinical trial (NCT02601950) testing the use of the EZH2 inhibitor tazemetostat in 14 patients with RMC and 1 patient with RCCU-MP was recently completed (Tannir et al. manuscript under preparation). In a first-in-human, multicenter, open-label, phase I trial in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumors, tazemetostat was well-tolerated with the most common adverse events being asthenia (55% of cases), anemia (22%), anorexia (22%), muscle spasms (22%), nausea (20%), and vomiting (19%). Furthermore, a durable complete response was noted in 1/7 (14.3%) patients with SMARCB1-negative malignant rhabdoid tumor, and a second patient (14.3%) had a partial response. In addition, prolonged stable disease was noted in 2/3 (66%) patients with SMARCB1-negative epithelioid sarcoma.27 However, enrollment in tazemetostat clinical trials in the United States is currently on hold following a safety report of a pediatric patient with advanced poorly differentiated chordoma who developed secondary T-cell lymphoma. Studies in mice have indeed suggested that Ezh2 inactivation can lead to hematologic malignancies28, 29, and patients on EZH2 inhibitors should therefore be closely monitored for these complications.
SMARCB1 inactivation has also been shown to induce significant upregulation of protein anabolism which can render cells susceptible to disruption of their proteostatic machinery.30 Accordingly, one case report noted a durable (>24 months) complete response to single-agent bortezomib in a patient with RMC.31 However, this result was not replicated in other patients with RMC who received single-agent bortezomib following progression on cytotoxic chemotherapy.3, 32 Nevertheless, the combination of bortezomib with cytotoxic chemotherapy achieved durable responses in two pediatric patients with RMC,33 and therefore combining proteasome inhibition with other therapies should be further investigated. A phase II clinical trial (NCT03587662) evaluating the combination of the second-generation proteasome inhibitor ixazomib with the nucleoside analog gemcitabine and the topoisomerase II inhibitor doxorubicin in patients with RMC began accrual in August 2018 and includes patients ≥12 years old . The use of topoisomerase II inhibitors in patients with RMC is supported by whole-genome expression and immunohistochemistry analyses showing overexpression of topoisomerase II in these tumors.34, 35
There are currently three active clinical trials that specifically include patients with RMC (Table 1). All three trials are exploring the use of immune checkpoint inhibitors in patients with RMC, based on case reports demonstrating a complete response using the programmed death-1 (PD-1) inhibitor nivolumab in one out of three patients with RMC.36, 37 Emerging insights into the biology of RMC and of other malignancies driven by SMARCB1 loss30, 38 will guide the development of targeted therapeutic strategies. RMC trials should systematically collect tissue samples suitable for biobanking and future research. In addition, we recommend that all RMC trials include patients with RCCU-MP given that these malignancies are histopathologically similar to RMC, demonstrate an analogously aggressive clinical behavior, and are molecularly defined by the loss of SMARCB1.14
Table 1.
Ongoing clinical trials for patients with RMC.
| Trial identifier | Trial Title | Phase | Treatment | Mechanism of Action | Primary Endpoint |
N | Eligible ages |
Comments |
|---|---|---|---|---|---|---|---|---|
| NCT02721732 | Study for the Evaluation of Efficacy of Pembrolizumab (MK- 3475) in Patients With Rare Tumors |
II | Pembrolizumab | PD-1 inhibitor | NPR | Up to 25 patients |
≥18 years old |
NPR is defined as the percentage of patients who are alive and progression-free at 27 weeks as assessed by RECIST 1.1 criteria |
|
NCT02496208 |
Cabozantinib-s-malate and Nivolumab With or Without Ipilimumab in Treating Patients With Metastatic Genitourinary Tumors |
I |
Cabozantinib + nivolumab +/− ipilimumab |
Cabozantinib: multitargeted TKI Nivolumab: PD-1 inhibitor Ipilimumab: CTLA-4 inhibitor |
DLT and RP2D |
Up to 135 Patients in all cohorts |
≥18 years old |
Includes an expansion portion |
|
NCT03274258 |
A phase 2 trial of immunotherapy in patients with carcinomas arising from the renal medulla |
II |
Nivolumab + ipilimumab or Nivolumab + NKTR214 |
Nivolumab: PD-1 inhibitor Ipilimumab: CTLA-4 inhibitor NKTR214: CD122 agonist |
ORR |
Up to 30 Patients for each combinat ion |
≥18 years old |
Any number of prior therapies is allowed Initial cohorts will be treated with nivolumab + ipilimumab. Futility and toxicity will be assessed in every 10 patients. If excessive toxicity and/or insufficient efficacy then will switch to different combination immunotherapy (nivolumab + NKTR214) |
|
NCT03587662 |
Phase II Trial of Ixazomib combined with Gemcitabine and Doxorubicin in Patients With Renal Medullary Carcinoma |
II |
Ixazomib + gemcitabine + doxorubicin |
Ixazomib: proteasome inhibitor Gemcitabine: inhibits DNA synthesis Doxorubicin: inhibits topoisomerase II and generates free radicals |
DCR at 28 weeks and ORR (joint primary endpoint) |
Up to 30 patients |
≥12 years old |
Any number of prior therapies is allowed. The co-primary endpoints will be compared with the historical ORR and 28- week DCR of 29% and 14%, respectively. |
CTLA-4: cytotoxic T lymphocyte antigen-4; DCR: disease control rate; DLT: dose-limiting toxicity; NPR: non-progression rate; PD-1: programmed death-1; ORR: objective response rate; RP2D: recommended phase II dose; TKI: tyrosine kinase inhibitor
RMC typically manifests in young adults and children as young as 9 years old.3 Phase I trials of pediatric patients typically find the maximum-tolerated doses (MTDs) and safety profiles to be close to those found in trials of adult patients for the majority of new agents studied, prompting current pediatric phase I trials to limit the study number of dose levels and doses outside 80–160% of the adult MTD and/or recommended phase II dose.39 Due to the similarity in drug metabolism and excretion between adults and postpubertal adolescents, the American Society of Clinical Oncology-Friends of Cancer Research Minimum Age Working Group recently recommended that, once safety and dosing information is available for adults, phase II and III trials spanning pediatric and adult patient populations should routinely enroll patients aged 12 years and older.40 To facilitate the development of novel therapeutic strategies for pediatric patients with RMC, we accordingly recommend that adult clinical trials include pediatric oncology investigators and pediatric clinical trial networks, and allow phase II or III trials to simultaneously enroll adult and pediatric cohorts 12 years old or older.
RMC clinical trials can benefit from emerging insights into the pharmacogenomics and pharmacokinetic effects of race and ethnicity on drug responses or toxicity.41 Therefore, researchers should be aware of such variations. Furthermore, clinicians and researchers designing clinical trial protocols for RMC patients should take into account the presence of sickle cell trait or other sickle hemoglobinopathies in this population. For example, although they are generally tolerated in patients with sickle cell trait,42 granulocyte colony-stimulating factors can cause severe adverse effects in patients with sickle cell disease or other sickle hemoglobinopathies of similar severity.43
Insights gained from the study of rare tumors can help us understand and treat the more common forms of cancer. We therefore encourage industry partners to design trials that either specifically target patients with RMC or ensure that these patients are included as a specific patient cohort, under the guidance of the considerations outlined above. In addition, the FDA Office of Orphan Products Development has developed a list of specific incentives for industry to develop products for rare diseases such as RMC.44
Increasing awareness and advocacy
Moving forward, there is a need to increase awareness of RMC, particularly in areas with high prevalence of sickle cell trait and/or other sickle hemoglobinopathies. Social media efforts such as RMC Support and the Chris “CJ” Johnson Foundation have been invaluable towards this goal. We are in the process of establishing an RMC Alliance (upcoming website at http://RMCAlliance.org), bringing together expert clinicians, researchers, and patient advocates, to better understand how and why RMC occurs, and to develop better strategies to screen for, diagnose, and treat this deadly disease. An important initial goal of this alliance is to establish a RMC registry to define the true incidence of this disease.
Acknowledgments:
The authors would like to thank KCCure for supporting this meeting, as well as Cora Connor, Ritchie Johnson, and Wander Powell for raising awareness and advocating for patients with renal medullary carcinoma. We are grateful to the Children’s National Health System for hosting this meeting and to Ashley Haston for providing administrative assistance. We have received support from the Chris “CJ” Johnson Foundation Inc., R.M.C. Inc., the Kidney Cancer Association, NIH grant T32 CA009666 (PM), R00CA187565 (HCH), the Conquer Cancer Foundation Young Investigator Award (PM), the Gabrielle’s Angel Foundation (HCH), and CPRIT grant RR170036 (HCH).
Footnotes
Conflicts of interest:
The authors declare no potential conflicts of interest.
REFERENCES:
- 1.Davis CJ Jr., Mostofi FK, Sesterhenn IA Renal medullary carcinoma. The seventh sickle cell nephropathy. Am J Surg Pathol 1995;19:1–11. [DOI] [PubMed] [Google Scholar]
- 2.Beckermann KE, Sharma D, Chaturvedi S, et al. Renal Medullary Carcinoma: Establishing Standards in Practice. J Oncol Pract 2017;13:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah AY, Karam JA, Malouf GG, et al. Management and outcomes of patients with renal medullary carcinoma: a multicentre collaborative study. BJU Int 2017;120:782–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tannir NM, Plimack E, Ng C, et al. A phase 2 trial of sunitinib in patients with advanced non-clear cell renal cell carcinoma. Eur Urol 2012;62:1013–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kriz W, Kaissling B. Chapter 20 - Structural Organization of the Mammalian Kidney In: Alpern RJ, Moe OW, Caplan M, eds. Seldin and Giebisch’s The Kidney (Fifth Edition): Academic Press; 2013:595–691. [Google Scholar]
- 6.Ohe C, Smith SC, Sirohi D, et al. Reappraisal of Morphologic Differences Between Renal Medullary Carcinoma, Collecting Duct Carcinoma, and Fumarate Hydratase-deficient Renal Cell Carcinoma. Am J Surg Pathol 2018;42:279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Srigley JR, Delahunt B, Eble JN, et al. The International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia. Am J Surg Pathol 2013;37:1469–1489. [DOI] [PubMed] [Google Scholar]
- 8.Margol AS, Judkins AR. Pathology and diagnosis of SMARCB1-deficient tumors. Cancer Genet 2014;207:358–364. [DOI] [PubMed] [Google Scholar]
- 9.Cheng JX, Tretiakova M, Gong C, Mandal S, Krausz T, Taxy JB. Renal medullary carcinoma: rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod Pathol 2008;21:647–652. [DOI] [PubMed] [Google Scholar]
- 10.Dimashkieh H, Choe J, Mutema G. Renal medullary carcinoma: a report of 2 cases and review of the literature. Arch Pathol Lab Med 2003;127:e135–138. [DOI] [PubMed] [Google Scholar]
- 11.Calderaro J, Masliah-Planchon J, Richer W, et al. Balanced Translocations Disrupting SMARCB1 Are Hallmark Recurrent Genetic Alterations in Renal Medullary Carcinomas. Eur Urol 2016;69:1055–1061. [DOI] [PubMed] [Google Scholar]
- 12.Carlo MI, Chaim J, Patil S, et al. Genomic Characterization of Renal Medullary Carcinoma and Treatment Outcomes. Clin Genitourin Cancer 2017;15:e987–e994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Msaouel P, Tannir NM, Walker CL. A Model Linking Sickle Cell Hemoglobinopathies and SMARCB1 Loss in Renal Medullary Carcinoma. Clin Cancer Res 2018. [DOI] [PMC free article] [PubMed]
- 14.Sirohi D, Smith SC, Ohe C, et al. Renal cell carcinoma, unclassified with medullary phenotype: poorly differentiated adenocarcinomas overlapping with renal medullary carcinoma. Hum Pathol 2017;67:134–145. [DOI] [PubMed] [Google Scholar]
- 15.Harrison SE, Walcott CM, Warner TD. Knowledge and Awareness of Sickle Cell Trait Among Young African American Adults. West J Nurs Res 2017;39:1222–1239. [DOI] [PubMed] [Google Scholar]
- 16.Croskerry P The importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med 2003;78:775–780. [DOI] [PubMed] [Google Scholar]
- 17.Kim KH, Roberts CW. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet 2014;207:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hodges C, Kirkland JG, Crabtree GR. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb Perspect Med 2016;6:a026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Debelenko LV, Raimondi SC, Daw N, et al. Renal cell carcinoma with novel VCL-ALK fusion: new representative of ALK-associated tumor spectrum. Mod Pathol 2011;24:430–442. [DOI] [PubMed] [Google Scholar]
- 20.Marino-Enriquez A, Ou WB, Weldon CB, Fletcher JA, Perez-Atayde AR. ALK rearrangement in sickle cell trait-associated renal medullary carcinoma. Genes Chromosomes Cancer 2011;50:146–153. [DOI] [PubMed] [Google Scholar]
- 21.Smith NE, Deyrup AT, Marino-Enriquez A, et al. VCL-ALK renal cell carcinoma in children with sickle-cell trait: the eighth sickle-cell nephropathy? Am J Surg Pathol 2014;38:858–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandberg JK, Mullen EA, Cajaiba MM, et al. Imaging of renal medullary carcinoma in children and young adults: a report from the Children’s Oncology Group. Pediatr Radiol 2017;47:1615–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alvarez O, Rodriguez MM, Jordan L, Sarnaik S. Renal medullary carcinoma and sickle cell trait: A systematic review. Pediatr Blood Cancer 2015;62:1694–1699. [DOI] [PubMed] [Google Scholar]
- 24.Iacovelli R, Modica D, Palazzo A, Trenta P, Piesco G, Cortesi E. Clinical outcome and prognostic factors in renal medullary carcinoma: A pooled analysis from 18 years of medical literature. Can Urol Assoc J 2015;9:E172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med 2016;22:128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knutson SK, Warholic NM, Wigle TJ, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 2013;110:7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Italiano A, Soria JC, Toulmonde M, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 2018;19:649–659. [DOI] [PubMed] [Google Scholar]
- 28.Simon C, Chagraoui J, Krosl J, et al. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev 2012;26:651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muto T, Sashida G, Oshima M, et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J Exp Med 2013;210:2627–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Genovese G, Carugo A, Tepper J, et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature 2017;542:362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ronnen EA, Kondagunta GV, Motzer RJ. Medullary Renal Cell Carcinoma and Response to Therapy With Bortezomib. Journal of Clinical Oncology 2006;24:e14–e14. [DOI] [PubMed] [Google Scholar]
- 32.Rathmell WK, Monk JP. High-dose-intensity MVAC for Advanced Renal Medullary Carcinoma: Report of Three Cases and Literature Review. Urology 2008;72:659–663. [DOI] [PubMed] [Google Scholar]
- 33.Carden MA, Smith S, Meany H, Yin H, Alazraki A, Rapkin LB. Platinum plus bortezomib for the treatment of pediatric renal medullary carcinoma: Two cases. Pediatr Blood Cancer 2017;64. [DOI] [PubMed] [Google Scholar]
- 34.Schaeffer EM, Guzzo TJ, Furge KA, et al. Renal medullary carcinoma: molecular, pathological and clinical evidence for treatment with topoisomerase-inhibiting therapy. BJU Int 2010;106:62–65. [DOI] [PubMed] [Google Scholar]
- 35.Albadine R, Wang W, Brownlee NA, et al. Topoisomerase II alpha status in renal medullary carcinoma: immuno-expression and gene copy alterations of a potential target of therapy. J Urol 2009;182:735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beckermann KE, Jolly PC, Kim JY, et al. Clinical and immunologic correlates of response to PD-1 blockade in a patient with metastatic renal medullary carcinoma. J Immunother Cancer 2017;5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sodji Q, Klein K, Sravan K, Parikh J. Predictive role of PD-L1 expression in the response of renal Medullary carcinoma to PD-1 inhibition. J Immunother Cancer 2017;5:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Lee RS, Alver BH, et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat Genet 2017;49:289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee DP, Skolnik JM, Adamson PC. Pediatric phase I trials in oncology: an analysis of study conduct efficiency. J Clin Oncol 2005;23:8431–8441. [DOI] [PubMed] [Google Scholar]
- 40.Gore L, Ivy SP, Balis FM, et al. Modernizing Clinical Trial Eligibility: Recommendations of the American Society of Clinical Oncology-Friends of Cancer Research Minimum Age Working Group. J Clin Oncol 2017;35:3781–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Donnell PH, Dolan ME. Cancer pharmacoethnicity: ethnic differences in susceptibility to the effects of chemotherapy. Clin Cancer Res 2009;15:4806–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang EM, Areman EM, David-Ocampo V, et al. Mobilization, collection, and processing of peripheral blood stem cells in individuals with sickle cell trait. Blood 2002;99:850–855. [DOI] [PubMed] [Google Scholar]
- 43.Fitzhugh CD, Hsieh MM, Bolan CD, Saenz C, Tisdale JF. Granulocyte colony-stimulating factor (G-CSF) administration in individuals with sickle cell disease: time for a moratorium? Cytotherapy 2009;11:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Developing Products for Rare Diseases & Conditions: Food & Drug Administration Office of Orphan Products Development.