

Abstract
The low molecular weight protein tyrosine phosphatase (LMW-PTP) is a regulator of a number of signaling pathways and has been implicated as a potential target for oncology and diabetes/obesity. There is significant therapeutic interest in developing potent and selective inhibitors to control LMW-PTP activity. We report the discovery of a novel class of LMW-PTP inhibitors derived from SulfoPhenyl Acetic Amide (SPAA), some of which exhibit greater than 50-fold preference for LMW-PTP over a large panel of PTPs. X-ray crystallography reveals that binding of SPAA-based inhibitors induces a striking conformational change in the LMW-PTP active site, leading to the formation of a previously undisclosed hydrophobic pocket to accommodate the α-phenyl ring in the ligand. This induced-fit mechanism is likely a major contributor responsible for the exquisite inhibitor selectivity.
Graphical Abstract
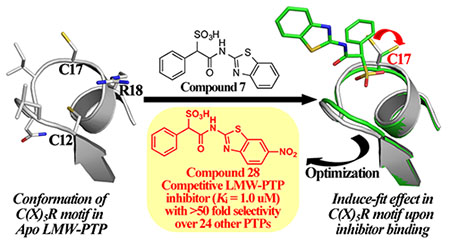
Introduction
As a major posttranslational modification mechanism, protein tyrosine phosphorylation regulates numerous cellular processes including growth, differentiation, migration, and survival.1 Proper level of protein tyrosine phosphorylation, which is dynamically maintained by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs), is essential for normal cellular homeostasis. Perturbation to the balance between PTK and PTP activity induces either excessive or diminished substrate phosphorylation, which is associated with a wide range of pathological conditions such as cancer, diabetes and autoimmune disorders.1,2 Consequently, the ability to selectively modulate signaling pathways mediated by protein tyrosine phosphorylation holds enormous potential for therapeutic intervention, as evidenced by the development of PTK inhibitors for various clinical indications.3 Given the substantial success of PTK inhibitors, there is heightened interest in targeting the opposing PTPs for therapeutic applications.
The PTPs constitute a large family of signaling enzymes (>100) that can be broadly classified into three groups: tyrosine-specific, dual-specific, and the low molecular weight protein tyrosine phosphatase (LMW-PTP).4 The common feature of all PTPs is the active site sequence C(X)5R(S/T), also known as the PTP signature motif or P-loop, which specifically binds the phosphoryl moiety of the substrate. As an enigmatic member of the PTP family, the LMW-PTP, encoded by Acp1, is an 18 kDa cytosolic enzyme found ubiquitously in both prokaryotic and eukaryotic organisms.5–8 LMW-PTP has been shown to either positively or negatively regulate the function of platelet-derived growth factor receptor (PDGFR),9–11 fibroblast growth factor receptor (FGFR),12 epidermal growth factor receptor (EGFR),13 insulin receptor (IR),10,14,15 and is involved in numerous signaling cascades including focal adhesion kinase (FAK),16,17 STAT5,18,19 and Epherin.20,21 However, the mechanism(s) by which these pathways are controlled by LMW-PTP is not yet fully understood.
A number of studies suggest LMW-PTP as a potential oncoprotein. Increased expression of LMW-PTP mRNA and protein levels were observed in malignant samples of breast, colon, bladder and kidney.22,23 More recently, LMW-PTP was reported to enhance the metastatic potential of prostate cancer cells, resulting in worse patient survival.24 LMWPTP is also overexpressed and active in primary colorectal cancer.25 Interestingly, LMW-PTP has also been implicated in the development of type II diabetes and obesity.26–28 For example, LMW-PTP was implicated as a negative regulator of insulin signaling through dephosphorylation of components in the IR pathway.10,14 Diet-induced obese mice treated with LMW-PTP antisense oligonucleotide exhibited decreased plasma insulin, glucose, triglyceride and cholesterol levels, and showed increased glucose and insulin tolerance.15
In view of the emerging recognition of LMW-PTP as a potential target for oncology and diabetes/obesity, there is considerable interest in obtaining small molecule inhibitors of LMW-PTP as potential therapeutics.8,29–37 Moreover, potent and selective inhibitors of LMW-PTP would be invaluable tools for interrogating LMW-PTP biology and therapeutic hypothesis testing. Unfortunately, reported inhibitors lack robust selectivity for LMW-PTP and therefor are not suitable as chemical probes for further mechanistic studies and ultimately, for the development of therapeutic agents targeting LMW-PTP. We describe the identification and characterization of a novel series of LMW-PTP inhibitors, which exhibit greater than 50-fold preference for LMW-PTP over a large panel of PTPs. Crystal structures of LMW-PTP-inhibitor complexes reveal a ligand induced conformational change in LMW-PTP that is responsible for the exquisite inhibitor selectivity.
Results and Discussion
Acquisition of potent and selective LMW-PTP inhibitors.
The PTPs, as a target class, have presented significant difficulties for drug discovery. The high sequence and structure homology within the PTP active site creates a challenge in achieving inhibitor selectivity among closely related family members. In addition, the positive-charged environment of the PTP active site poses unique drug discovery challenges, as most competitive inhibitors by necessity are negatively charged, which complicates drug development due to low cell permeability and bioavailability. Since existing drugs are already endowed with favorable pharmacokinetic properties, we sought to harness the privileged drug space for novel PTP inhibitory scaffolds that can be further optimized into potent and selective PTP inhibitors with improved drug-like properties. To this end, we have recently discovered SulfoPhenyl Acetic Amide (SPAA), a fragment originated from cefsulodin, as a novel pTyr mimetic.38 We found that SPAA inhibited LMW-PTP with an IC50 of 2 mM, as opposed to an IC50 of >50 mM for PTP1B and SHP2. LMW-PTP exists in two isoforms (namely LMW-PTPA and LMW-PTPB, respectively) which differ only by a loop that flanks the catalytic site. This study concerns mostly the LMW-PTPA isoform. In order to increase the potency and selectivity of SPAA for LMW-PTP, we sought to capture additional interactions outside of the active site pocket. To this end, we prepared a SPAA-based focused library by reacting α-sulfophenyl acetic acid with a set of 192 amines of different size, charge, and lipophilicity.38 The library was assembled directly on 96-well plates under standard HBTU amide coupling conditions. Reactions from representative wells were monitored by LC-MS, which indicated a 70-80% product conversion.
Library compounds were screened for inhibitory activity against LMW-PTP at a compound concentration of ~10 μM, and counter-screened against PTP1B and SHP2 to provide preliminary specificity information. Several compounds, 1, 2, 3, 4, 5, 6, 7, 8 (Figure 1), exhibited >50% inhibition against LMW-PTP, but <10% inhibition against PTP1B and SHP2, indicating significant specificity for LMW-PTP. These hits were resynthesized and purified with reversed-phase HPLC, and their IC50 values against LMW-PTP were measured, which are summarized in Figure 1. To our delight, all of the hits display IC50 values less than 10 μM, consistent with our initial screening results. This represents a more than 200-fold gain in binding affinity compared to the parent SPAA. Kinetics studies with compound 7 were carried out to probe its mode of inhibition. Figure 2 showed that 7 is a competitive inhibitor against LMW-PTPA with a Ki of 3.2 ± 0.1 μM. We also determined that compound 7 inhibits the LMW-PTPB isoform with a Ki of 1.7 ± 0.1 μM, indicating that the SPAA-based inhibitors likely inhibit both LMW-PTP isoforms with similar potency. Structure and activity survey revealed that the aniline containing compound 9 and the 2-aminothiazole containing compound 10 have significantly reduced activities, indicating that substituents on aniline (compound 9 vs compounds 1, 2, 3, 4, 5, 8) and the second benzene ring (compound 10 vs compound 7) are required for high affinity binding. The propyl amine containing compound 11 is inactive as expected given that it does not have an aromatic group at the amine terminal, and it was inactive in our initial screening. To gain a more comprehensive picture of how selective these compounds are to the LMW-PTP, we broadened the scope of selectivity profiling by inclusion of an additional 22 phosphatases in addition to PTP1B and SHP2. As shown in Table 1, compounds 3, 5, and 6 exhibit no inhibitory activity against any of the phosphatases in the panel at 200 μM concentration. In particular, compound 3 possesses a greater than 35-fold preference for LMW-PTP over all other PTPs.
Figure 1.
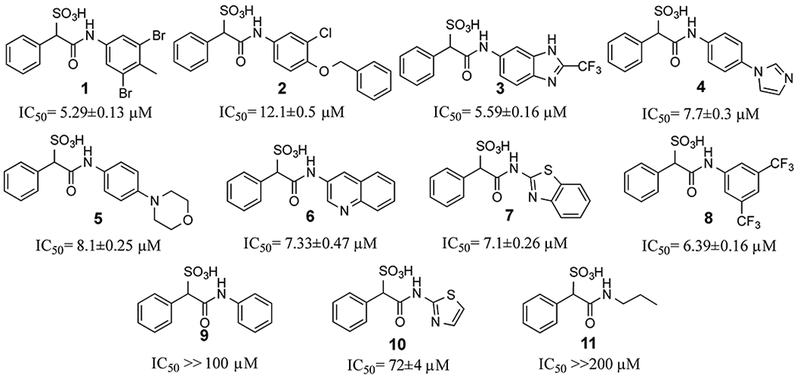
Structure and activity of SPAA-based LMW-PTP inhibitors.
Figure 2.

Lineveaver-Burk plot for compound 7 mediated LMW-PTP inhibition at five different concentrations (0 (●), 5 (μ), 10 (τ), 15 (∇), and 20 (■) μM). pNPP was used as the substrate. The data show that compound 7 inhibits the LMW-PTP catalyzed pNPP hydrolysis in a competitive manner (Ki = 3.2 ± 0.1 μM).
Table 1.
IC50 of selected compounds against LMW-PTP and 24 other PTPs with pNPP as a substrate.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 28 | |
|---|---|---|---|---|---|---|---|---|---|
| LMW-PTP | 5.3 | 12.1 | 5.6 | 7.7 | 8.1 | 7.3 | 7.1 | 6.4 | 2.1 |
| SHP2 | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| PTP1B | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| TC-PTP | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| SHP1 | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| LYP | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| HePTP | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | 50.0 | 94.0 | >>100 |
| Meg2 | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| PTPH1 | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| FAP-1 | >>200 | 73.5 | >>200 | >>100 | >>200 | >>200 | 34.0 | 76.0 | >>100 |
| PEST | 72.3 | 69.9 | >>200 | >>100 | >>200 | >>200 | 43.5 | 66.9 | >>100 |
| CD45 | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| LAR | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| PTPα | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| PTPβ | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| PTPγ | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| PTPε | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| PTPσ | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| PTPμ | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| MKP3 | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| VHR | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| VHZ | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| CDC14A | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
| SSu72 | >>200 | 66.6 | >>200 | >>100 | >>200 | >>200 | 69.9 | 83.0 | >>100 |
| PP5 | >>200 | >>200 | >>200 | >>100 | >>200 | >>200 | >>200 | >>200 | >>100 |
Structural basis of LMW-PTP inhibition by the SPAA-based inhibitors.
To elucidate the structural basis of LMW-PTP inhibition and to aid the design of more potent and selective LMW-PTP inhibitors, we sought to determine the crystal structures of LMW-PTP in complex with the SPAA-based inhibitors. We initially obtained LMW-PTP crystals under previously published conditions including 100 mM MES buffer, 30% PEGME 5,000 and 0.2 M ammonium sulfate39 in the presence of 1 mM compound 7. The structure was determined by molecular replacement, using the published LMW-PTP structure as the search model (PDBID:5PNT). However, compound 7 was nowhere to be found. Instead, a MES molecule occupied the active site (Figure 3A). MES is a week inhibitor of LMW-PTP, with an IC50 greater than 10 mM. The sulfonic acid moiety mimics the interaction of the PO4 group of a substrate within the phosphatase active site. The rest of the molecule interacts with the surrounding residues by van der Waals interactions. We then systematically screened alternate crystallization conditions without MES and finalized 25-30% PEGME 5,000 in 100 mM Bis-Tris, pH 6.0-6.5 for optimal crystal growth of the LMW-PTP•7 complexes. Again, the structure of LMW-PTP•7 was determined by molecular replacement, and compound 7 was unambiguously identified around the C(X)5R(S/T) PTP signature motif (Figure 3B). For comparison, we also crystallized and solved the structure of LMW-PTP in complex with compound 9, which has an IC50 > 100 μM, under the same conditions (Figure 3C). Finally, by soaking the LMW-PTP•MES crystals in 100 mM Bis-Tris buffer, we obtained the crystal structure of apo LMW-PTP (Figure 3D). In the apo LMW-PTP structure, the MES molecule is replaced by three water molecules, which are well organized to form hydrogen bonds with the amides of the P-loop residues. The data collection and refinement statistics for all four crystal structures are summarized in Table 2. The overall LMW-PTP structures are very similar and the root mean square derivation (rmsd) for all Cα atoms is 0.341 Å between LMW-PTP•7 and LMW-PTP•MES, 0.529 Å between LMW-PTP•7 and LMW-PTP•9, and 0.630 between LMW-PTP•7 and the apo enzyme.
Figure 3.
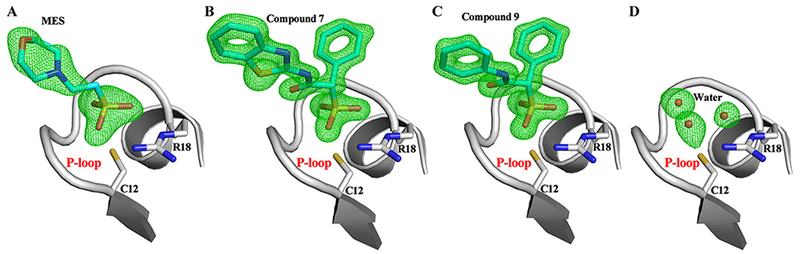
Small molecule ligand or water is identified at the active site of LMW-PTP. Unbiased Fo-Fc difference map of (A) MES, (B) Compound 7, (C) Compound 9, and (D) three waters contoured at +3σ level is shown in green mesh. Small molecules are shown in stick (carbon in cyan), waters are shown in sphere, and the catalytic Cys12 and Arg18 in LMW-PTP are shown in stick (carbon in gray).
Table 2.
Data collection and structure refinement statistics.
| LMW-PTP•7 | LMW-PTP•9 | LMW-PTP•MES | LMW-PTP | |
|---|---|---|---|---|
| Data Collection | ||||
| Space group | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions | ||||
| a (Å) | 34.801 | 34.190 | 33.690 | 31.957 |
| b (Å) | 53.963 | 54.573 | 54.826 | 55.453 |
| c (Å) | 97.330 | 97.515 | 95.191 | 96.326 |
| α=β=γ (deg) | 90.0 | 90.0 | 90.0 | 90.0 |
| Resolution (Å) | 50.00-1.50 | 50.00-1.45 | 50.00-1.91 | 25.00-2.05 |
| Total observations | 182980 | 233460 | 94514 | 75930 |
| Unique observations | 28910 | 33262 | 14276 | 11281 |
| Completeness (%) | 95.5 (74.5) | 99.9 (100.0) | 99.7 (100.0) | 99.9 (99.6) |
| Redundancy | 6.3 (4.5) | 7.0 (6.7) | 6.6 (6.6) | 6.7 (6.4) |
| Rmerge (%) | 5.9 (60.7) | 7.6 (79.9) | 15.8 (79.4) | 10.2 (80.2) |
| <I/δ(I)> | 27.9 (2.0) | 24.4 (1.9) | 27.7 (2.9) | 25.5 (2.6) |
| Structure Refinement | ||||
| Resolution (Å) | 27.81-1.50 | 21.63-1.45 | 27.46-1.91 | 24.03-2.05 |
| No. of reflections used | 28731 | 33197 | 14231 | 11197 |
| No. of protein atoms | 1280 | 1257 | 1225 | 1238 |
| No. of inhibitor atoms | 23 | 32 | 12 | n.a. |
| No. of water | 228 | 268 | 121 | 69 |
| Rwork/Rfree | 0.167/0.194 | 0.164/0.177 | 0.185/0.228 | 0.193/0.266 |
| srms deviations | ||||
| Bond length (Å) | 0.006 | 0.006 | 0.008 | 0.008 |
| Bond angel (deg) | 1.175 | 1.095 | 1.989 | 1.066 |
| Average B factors (Å2) | ||||
| Protein | 18.43 | 15.90 | 32.29 | 36.02 |
| Water | 32.90 | 29.39 | 37.93 | 37.57 |
| Inhibitor | 12.49 | 12.10 | 30.48 | n.a. |
| All atoms | 20.59 | 18.24 | 32.79 | 36.10 |
The data were collected from a single crystal. Values in parentheses are for highest-resolution shell.
Although the overall three-dimensional structures are quite similar between LMW-PTP•7 and LMW-PTP•MES, superimposition of both structures revealed a localized conformational change in the active site. In the LMW-PTP•MES complex, the side chain dihedral angle χ1 (∠C-CA-CB-SG) of Cys17, which is located in the P-loop, is 167.2 degree, while in the presence of compound 7, the side chain of Cys17 is pushed 114.3 degree away by the α-phenyl ring in 7, adapting a new χ1 of 52.9 degree (Figure 4A). In addition, the movement of the Cys17 side chain also shifts the P-loop downward by 1.4 Å from its original position (Figure 4A). Interestingly, the conformation of the side chain of Cys17 in the apo form of LMW-PTP is identical to that observed in LMW-PTP•MES structure (Figure 4B), while the side chain of Cys17 exists in the same conformation in both LMW-PTP•9 and LMW-PTP•7 structures (Figure 4C). The net result of this SPAA ligand induced conformational change is the creation of a hydrophobic cavity constituted by residues Ile16, Cys17, Pro130, Tyr131, Phe138 and the side-chain aliphatic atoms of Asp56, which is well suited for the α-phenyl ring in compound 7 and 9 (Figure 4D). To the best of our knowledge, no other PTPs undergo this structural change in the active site upon ligand binding.
Figure 4.
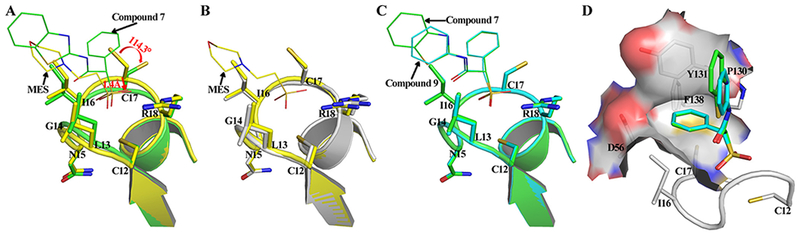
Compound 7 or 9 binding with LMW-PTP induces localized conformation changes leading to the formation of a new hydrophobic cavity within active site pocket. The structure superimposition of (A) LMW-PTP•MES (yellow) with LMW-PTP•7 (green), (B) LMW-PTP•MES (yellow) with LMW-PTP apo (gray), (C) LMW-PTP•7 (yellow) with LMW-PTP•9 (cyan) was performed, the small molecules are shown in thin stick, and local conformation around active site is shown in ribbon diagram with p-loop residues shown in stick. (D) New hydrophobic cavity induced by Compound 7 (carbon in green) or 9 (carbon in cyan) binding accommodates the α-phenyl ring in compound 7 or 9 very well. Residues constituting the cavity are shown in stick underneath the surface.
To better understand the unique preference of SPAA for LMW-PTP, we compared the active site features of LMW-PTP with those of PTP1B, a representative of the classical PTPs. As shown in Figure 5A, sequence alignment and structure superimposition of the signature motif C(X)5R(S/T) of LMW-PTP•7 with that of the PTP1B structure (PDBID: 1PXH) revealed that: 1) although the backbones of the two signature motifs align very well but only 3 of 7 residues are conserved between LMW-PTP and PTP1B, making the active site pockets rather different in shape and electrostatic potential between the two enzymes; 2) Cys17 plays a key role in the induce-fit process to create a hydrophobic cavity accommodating the α-phenyl ring of compound 7¸ but the corresponding residue in PTP1B (and other classical PTPs) is Gly220, which is incapable of doing the same; 3) a severe steric clash exists between the α-phenyl ring and Gln262, and another between the benzothiazole and Tyr46. Since the conserved Gln262 and Tyr46 come from the adjacent active site Q-loop and pY-loop of PTP1B, steric clashes may exist whether the WPD-loop is in the closed (Figure 5B) or open conformation (Figure 5C). Collectively, the active site specificity manifested by the induced-fit mechanism is unique for LMW-PTP, and likely responsible, in large part, for SPAA’s preference to the LMW-PTP over PTP1B, SHP2 and other PTPs as well.
Figure 5.
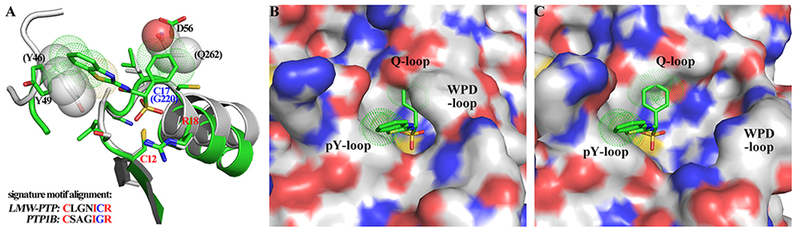
The binding mode between compound 7 and LMW-PTP is unique and can hardly be adopted by the classical PTPs like PTP1B. (A) Sequence alignment and structure superimposition of the signature motif of LMW-PTP and PTP1B (PDBID: 1PXH). The conserved residues are shown and labeled in red; the residue number in PTP1B is shown in parenthesis; the steric conflicts are presented by solid spheres (atoms in PTP1B) and dotted spheres (atoms in compound 7). These conflicts may exist whether PTP1B is in (B) WPD-closed (PDBID: 1PXH) or (C) WPD-open (PDBID: 2HNP) conformation.
In addition to revealing a novel induced-fit mechanism for LMW-PTP inhibition by the SPAA-containing inhibitors, the structure of LMW-PTP•7 also uncovers the basis for its potency: the sulfonic acid moiety is in close proximity to the P-loop and is stabilized by six hydrogen bonds with main chain amides of Leu13, Gly14, Ile16, Cys17, Arg18 and the side chain amide of Arg18 (Figure 6) with a distance ranging from 2.8 to 3.2 Å. In addition, the amide NH of compound 7 forms a hydrogen bond with carboxylate O of the general acid Asp129 with a distance of 3.0 Å. This is a new interaction that was never observed in previously reported LMW-PTP inhibitors. Moreover, a water molecule bridges another two hydrogen bonds between the N atom within thiazole and the carboxylate O of Asp129. Besides the hydrogen bond interactions mentioned above, compound 7 is also involved in a network of π-π interactions with LMW-PTP (Figure 6). π-π interaction has long been acknowledged as an integral force in maintaining protein stability40,41 as well as in differential recognition of medicinal agents containing aromatic substituents by their protein targets.42 In the unliganded state, the phenyl ring of Tyr131 is parallel to that of Tyr49 on the opposite side of the empty active site, with a distance of ~9.9 Å between the two aromatic rings. When compound 7 is bound in the active site, the benzothiazole ring of 7 is sandwiched between the phenyl rings of Tyr49 and Tyr131. The distance of the closest atoms between the phenyl ring of Tyr49 and the benzothiazole ring is ~3.5 Å. Similarly, the distance of the closest atoms between the phenyl ring of Tyr131 and the benzothiazole ring is ~3.8 Å. Hence, the benzothiazole ring falls within the London dispersion distance with both of these tyrosine residues.43 The position and orientation of the Tyr131 side chain is constrained by Tyr132 through an edge to face π-π interaction, with the distance of ~4.0 Å (mass center to mass center) (Figure 6). Tyr131 not only forms a π-π interaction with the phenyl ring of benzothiazole moiety, it also forms an edge to face π-π interaction with the α-phenyl ring of 7, with a distance of ~4.2 Å (Figure 6). Taken together, these π-π interactions form a network involving three tyrosine residues from LMW-PTP (Tyr49, Tyr131 and Tyr132) and the benzothiazole and α-phenyl ring from compound 7. These interactions, in addition to those mentioned above, likely play a major role for tight binding of compound 7.
Figure 6.
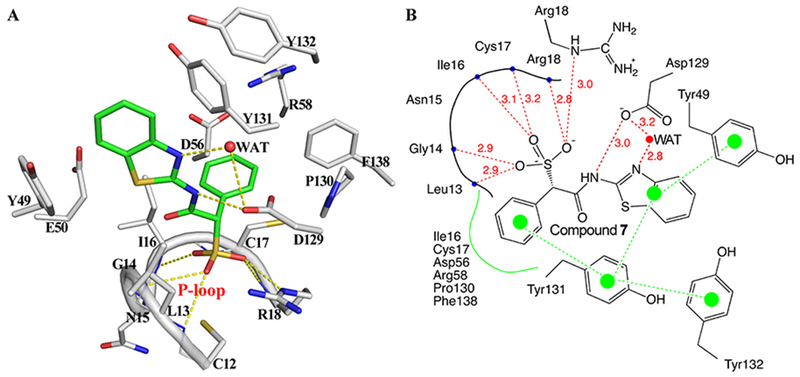
Interaction details for Compound 7 with LMW-PTP. (A) Interaction details represented in 3D space. All residues within 5 Å distance of Compound 7 (carbon in green) are shown in stick (carbon in gray), hydrogen-bonds are shown in yellow dash line. (B) 2D interaction diagram. Hydrogen-bonds are indicated by red dash line with distance labeled. Hydrophobic and π-π interactions are highlighted in green color.
In order to understand why installation of a double aromatic ring to the SPAA moiety generated much better inhibitors for LMW-PTP than did the single ring counterparts (Figure 1), we also determined the structure of LMW-PTP•9. Superimposition of the LMW-PTP•9 structure to that of LMW-PTP•7 revealed that the sulfonic acid moiety and the α-phenyl ring of compound 9 are located at exactly the same positions in the active site as the corresponding groups in compound 7 (Figure 4C). However, the aniline ring of compound 9 overlaps with only half of the double aromatic benzothiazole ring of compound 7. Although it still forms a π-π interaction with Tyr131, the distance between the aniline ring and Tyr49 increases to about 4.5 Å, significantly longer than what is required for optimal London dispersion interaction. Moreover, the nonpolar benzene ring also eliminates the water-bridged hydrogen bonds interaction with Asp129. Thus, the reduction in π-π interactions and water-bridged polar interactions between LMW-PTP and the single benzene ring at the acetamide position may account for the >15-fold decrease in inhibitory activity for compound 9 versus compound 7. Compared to compound 7, the thiazole group in 10 may still retain the water-bridged hydrogen bond interactions but likely loses the π-π interactions due to the absence of the terminal benzene ring. This may explains why compound 10 is superior to compound 9 even though it has a smaller single aromatic ring, suggesting that appropriately positioned polar atoms in the first aromatic ring will benefit binding affinity.
Initial optimization of compound 7.
Based on the structural insights, we carried out an initial structure-activity relationship study in order to lay the foundation to develop more potent and selective LMW-PTP inhibitors. From the crystal structures, the α-phenyl ring is situated in a hydrophobic cavity, bordered by residues Ile16, Cys17, Pro130, Tyr131, Phe138 and the side-chain aliphatic atoms of Asp56. We explored whether installation of a small substituent on the α-benzene ring could capture additional interactions with the enzyme, leading to increased binding affinity (Figure 7). Substituents to enhance hydrophobic interactions included ortho-chloro (compound 13), ortho-methyl (compound 14), meta-methyl (compound 16), para-methyl (compound 20), and para-bromo (compound 21); and substituents aimed for gaining potential hydrogen-bonding interactions included ortho-fluoro (compound 12), meta-fluoro (compound 15), meta-nitrile (compound 17), meta-nitro (compound 18), and meta-trifluoromethyl (compound 19). We also introduced electron donating substituents (compound 14, 16, 20) and electron withdrawing substituents (compounds 17, 18, 19) to ascertain whether perturbation of electronic properties would influence potential π-π interactions and thereby inhibitor activity. Unfortunately, we found that modifications to the α-phenyl ring led to significant diminished inhibitory activity toward LMW-PTP, indicating that the ligand binding induced hydrophobic pocket can only accommodate a naked benzene ring.
Figure 7.
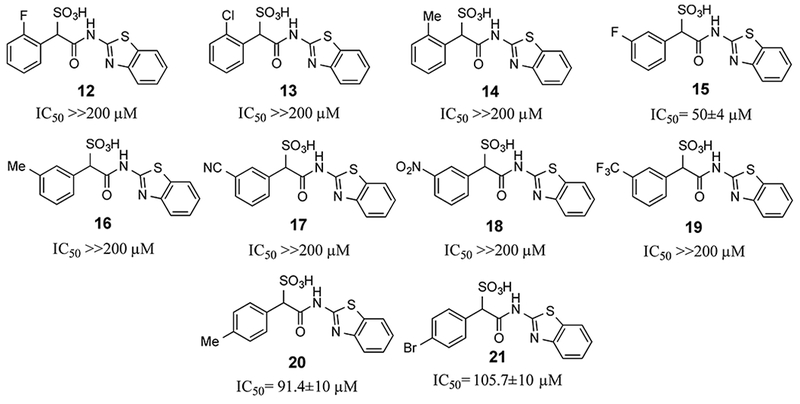
Structure and activity of SPAA inhibitors with various substituents on the α-benzene ring.
We next explored substituents on the benzothiazole ring in compound 7. Analysis of the LMW-PTP•7 crystal structure showed that the amide NH of 7 forms a hydrogen bond with Asp129, and the benzothiazole moiety forms strong π-π stacking interactions with Tyr49, Tyr131, and Tyr132. We reasoned that by installing small substituents with appropriate electronic properties and polarities at the benzene ring of benzothiazole, we could enhance the hydrogen bonding interaction between the amide NH and Asp129, and/or hydrophobic and polar interactions between the benzothiazole moiety and Tyr 49, Tyr131, and Tyr132. Hence we designed and synthesized a series of compounds with electron donating groups (compounds 23, 24, 25, 29, 31), electron withdrawing groups (compounds 26, 28, 32), hydrophobic groups (22, 23, 25, 27, 29), and polar groups (compounds 24, 26, 28, 30, 31, 32). As summarized in Figure 8, most derivatives displayed slightly increased activity over parent compound 7. Specifically, compound 28 with a nitro group at the 6 position and compound 32 with carboxylate group at the 6 position are 3.5- and 2.5-fold more active than 7, with a measured IC50 at 2.1 ± 0.2 μM and 3.1 ± 0.1 μM, respectively. Nitro and carboxylate groups are both electron withdrawing which make the NH amide bond more acidic, which likely enhances its hydrogen bonding interaction with Asp129. In addition, the nitro and carboxylate groups are in close proximity to Tyr49, to make a new hydrogen-bonding interaction with the hydroxyl group of Tyr49. Kinetics studies revealed that compound 28 is also a competitive inhibitor for LMW-PTP with a Ki at 1.0 ± 0.1 μM. More importantly, compound 28 exhibited at least over 50-fold selectivity for LMW-PTP over the panel of 24 phosphatases (Table 1), making compound 28 the most potent and selective LMW-PTP inhibitor reported to date. Deletion of the sulfonic acid in 28 completely abolishes compound 33’s ability to inhibit LMW-PTP, consistent with the importance of sulfonic acid for inhibition.
Figure 8.

Structures of compound 7 derivatives and their inhibitory activities against LMW-PTP.
Cellular activity of compound 28.
To assess the effects of 28 in insulin signaling pathway, we pretreated HepG2 cells with 28 for 2 h and then stimulated the cells with 50 nM insulin for 30 min. As shown in Figure 9A, compound 28 enhanced AKT phosphorylation in a dose-dependent manner relative to the vehicle DMSO. To ensure that the cellular activity of compound 28 is due to inhibition of LMW-PTP and not nonspecific effects, a structurally related but inactive analog of compound 28, compound 33 (IC50 >> 100 μM for LMW-PTP, Figure 8), was also evaluated. As expected, at 16 μM compound 33 exerted no effect on insulin induced AKT phosphorylation while at the same concentration compound 28 increased AKT phosphorylation by 260% (Figure 9B). The results indicate that compound 28 promotes insulin induced AKT phosphorylation through specifically inhibiting LMW-PTP activity.
Figure 9.

Cellular effect of compound 28 on insulin induced AKT phosphorylation in HepG2 cells. (A), compound 28 dose dependently enhanced AKT phosphorylation. (B), compound 33, a structurally related but inactive analog, had no effect on AKT phosphorylation. The bands were quantified by Image J, and the ratios of pAKT/AKT were normalized to the vehicle control.
In summary, we have developed a novel class of LMW-PTP inhibitors based on SPAA by using a focused library synthesis approach. High throughput screening and kinetic studies of the library components led to the identification of a number of hits with excellent potency and selectivity for LMW-PTP. Subsequent crystallography studies disclosed that sulfonic acid is in close proximity with the active site PTP signature motif and stabilized by multiple hydrogen bonds. The structure also revealed a striking ligand-induced conformational change in LMW-PTP that is essential for inhibitor specificity. Moreover, the structure also identified a triad of aromatic residues Tyr49, Tyr131, and Tyr132 that contribute to the potency and specificity of the inhibitors by forming strong π-π stacking interactions with the aromatic moieties in the ligand. Structure-guided optimization of compound 7 resulted in compound 28, which displayed a Ki value of 1.0 ± 0.1 μM for LMW-PTP and an unprecedented >50 fold selectivity for LMW-PTP over 24 other PTPs, marking it the first truly specific LMW-PTP inhibitor. Our study provides a solid foundation upon which more potent and selective LMW-PTP inhibitors can be designed. These compounds are also expected to serve as useful tools to study LMW-PTP’s biological function, and are excellent starting points for developing novel therapeutics for cancer and type 2 diabetes.
Experimental Section
Materials and General Procedures.
p-Nitrophenyl phosphate (pNPP) was purchased from Fluke. Crystallization reagents were from Hampton Research. DNA primers were from Genwiz Inc. and other reagents were obtained from Fisher or Sigma. Reagents were used as purchased from Sigma-Aldrich and Fisher Scientific. 1H and 13C NMR spectra were obtained on a BrukerAvance II 500 MHz NMR spectrometer with TMS or residual solvent as standard. Mass spectra were obtained using an Agilent Technologies 6130 quadrupole LC/MS. HPLC purification was carried out on a Waters Delta 600 equipped with a Sunfire Prep C18 OBD column (30 mm/150 mm, 5 μm) with methanol water (both containing 0.1% TFA) as mobile phase (gradient: 50-100% methanol, flow 10 mL/min). The purity of all final tested compounds was established to be>95% by Agilent Technologies 6130 quadrupole LC/MS by using methanol water (both containing 0.1% TFA) as the mobile phase (gradient: 0-100% methanol, flow 1.0 mL/min), with UV monitoring at the fixed wavelength of 254 nm.
Library Synthesis.
To all wells of 96-well plates were added sulfophenylacetic acid (20 μL, 20 mM in DMF), HBTU (20 μL, 20 mM in DMF), HOBt (20 μL, 20 mM in DMF), DIEA (20 μL, 60 mM in DMF), and corresponding 192 amines (He et al., 2015) from storage plates (1.2 μL, 0.5 mM in DMF), the reaction plates were placed at rt for 1 day. The reaction wells from aniline were monitored by LC-MS to show that reactions occurred well in great conversions. Thus a library of 192 compounds was obtained with estimated concentration at 4 mM (assuming 80% product yield), which was screened against LMW-PTP and other PTPs. All compounds were prepared as racemic mixtures.
Representative Procedure for the Synthesis of Products.
To α-Sulfophenyl acetyl chloride (0.234 g, 1 mmol) and DIEA (0.522 mL, 3 mmol) in DMF (2 mL) was added propyl amine (0.09 mL, 1.1mmol), the mixture was stirred at rt for 1 h. After quenching with water, it was subjected to HPLC purification, and product 11 was obtained as colorless oil (93% yield, >95% purity).
Characterizations of Compounds 1 to 33.
1:1H NMR (500 MHz, DMSO) δ 10.44 (s, 1H), 7.89 (s, 2H), 7.59-7.58 (m, 2H), 7.28-7.26 (m, 3H), 4.78 (s, 1H), 2.43 (s, 3H); 13C NMR (500 MHz, DMSO) δ 166.5, 139.0, 134.8, 130.6, 130.0, 127.4, 127.0, 124.2, 121.8, 71.6, 22.6. ESI-HRMS Cacld. for C15H12Br2NO4S (M-H+): 459.8859; found 459.8866.
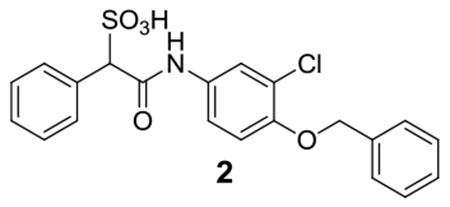
2:1H NMR (500 MHz, DMSO) δ 10.29 (s, 1H), 7.83 (d, J = 1.7 Hz, 1H), 7.57-7.56 (m, 2H), 7.46-7.17 (m, 10H), 5.15 (s, 2H), 4.73 (s, 1 H); 13C NMR (DMSO) δ 165.9, 149.4, 136.7, 135.1, 133.2, 129.9, 128.5, 127.9, 127.5, 127.4, 126.9, 121.3, 120.5, 118.7, 114.7, 71.7, 70.3. ESI-HRMS Cacld.for C21H17ClNO5S (M-H+): 430.0521, found 430.0519.
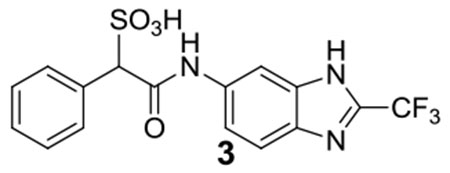
3: 1H NMR (500 MHz, DMSO) δ 10.48 (s, 1H), 8.23 (s, 1H), 7.67 (d, J = 8.7 Hz, 1H), 7.61 (d, J = 7.3 Hz, 2H), 7.35-7.25 (m, 4H), 4.82 (s, 1H); 13C NMR (DMSO) δ 166.1, 139.92, 139.6, 136.4, 136.2, 135.5, 135.2, 129.9, 127.4, 127.0, 120.1, 118.2, 118.0, 116.9, 104.1, 71.8. ESI-HRMS Cacld.for C16H11F3N3O4S (M-H+): 398.0428; found 398.0434.
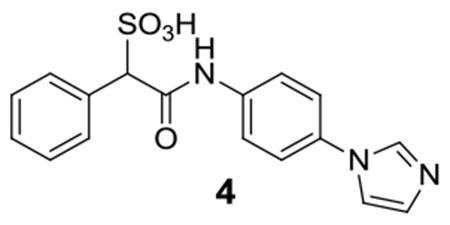
4:1H NMR (500 MHz, DMSO) δ 10.60 (s, 1H), 9.65 (t, J = 1.3 Hz, 1H), 8.26 (t, J = 1.7 Hz, 1H), 7.90 (t, J = 1.6 Hz, 1H), 7.84-7.83 (m, 2H), 7.76-7.73 (m, 2H), 7.59-7.58 (m, 2H), 7.30-7.24 (m, 3H), 4.80 (s, 1H); 13C NMR (DMSO) δ 166.5, 140.2, 135.1, 134.3, 130.0, 129.6, 127.3, 127.0, 122.6, 120.8, 120.7, 119.7, 71.8. ESI-HRMS Cacld. for C17H14N3O4S (M-H+): 356.0711; found 356.0706.
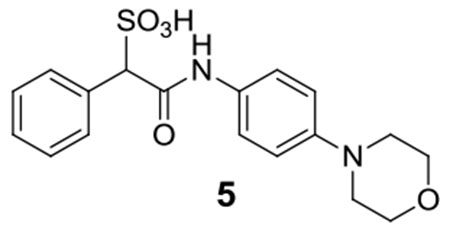
5:1H NMR (500 MHz, DMSO) δ 10.43 (s, 1H), 7.64-7.58 (m, 4H), 7.41 (d, J = 8.4 Hz, 2H), 7.30-7.25 (m, 3H), 7.40-7.34 (m, 3H), 4.82 (s, 1H), 3.90 (t, J = 4.9 Hz, 4H), 3.43 (s, 4H); 13C NMR (DMSO) δ 166.1, 135.0, 129.9, 127.4, 127.0, 120.0, 71.6, 64.5, 52.8. ESI-HRMS Cacld. for C18H19N2O5S (M-H+): 375.1020; found 375.1020.
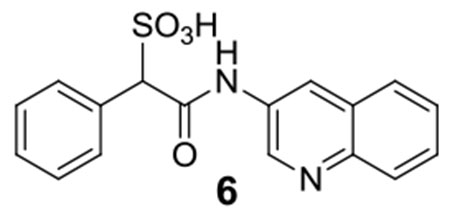
6:1H NMR (500 MHz, DMSO) δ 11.05 (s, 1H), 9.29 (d, J = 2.3 Hz, 1H), 9.01 (d, J = 2.1 Hz, 1H), 8.14 (d, J = 8.1 Hz, 1H), 8.07 (d, J = 8.5 Hz, 1H), 7.85-7.84 (m, 1H), 7.84-7.82 (m, 1H), 7.76-7.63 (m, 2H), 7.32-7.27 (m, 3H), 4.89 (s, 1H); 13C NMR (DMSO) δ 167.2, 141.3, 138.9, 134.7, 133.3, 130.4, 130.0, 128.6, 128.3, 128.2, 127.4, 127.0, 124.2, 99.5, 71.6. ESI-HRMS Cacld. for C17H13N2O4S (M-H+): 341.0602; found 341.0602.

7:1H NMR (500 MHz, DMSO) δ 7.97 (d, J = 7.8 Hz, 1H), 7.75 (d, J = 8.0 Hz, 1H), 7.58 (d, J = 7.1 Hz, 2H), 7.43 (t, J = 7.5 Hz, 1H), 7.33-7.28 (m, 4H), 5.09 (s, 1H); 13C NMR (DMSO) δ 167.0, 157.6, 148.5, 134.3, 131.5, 130.0, 127.6, 127.3, 126.2, 123.6, 121.8, 120.6, 70.3. ESI-HRMS Cacld.for C15H11N2O4S2 (M-H+): 347.0166; found 347.0166.

8:1H NMR (500 MHz, DMSO) δ 10.90 (s, 1H), 8.27 (s, 2H), 7.71 (s, 1H), 7.63-7.61(m, 2H), 7.30-7.24 (s, 5H), 4.85 (s, 1H); 13C NMR (DMSO) δ 167.2, 158.4 (q, J = 38.6 Hz), 141.1, 134.6, 130.8 (q, J = 32.4 Hz), 127.4, 127.1, 123.2 (q, J = 271.0 Hz), 120.0, 115.0 (q, J = 286.2 Hz), 71.8. ESI-HRMS Cacld. for C16H10F6NO4S (M-H+): 426.0240; found 426.0237.
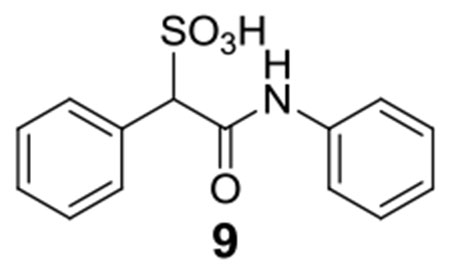
9: 1H NMR (500 MHz, DMSO) δ 10.23 (s, 1H), 7.56-7.54 (m, 4H), 7.30-7.25 (m, 5H), 7.03 (t, J = 7.4 Hz, 1H), 4.76 (s, 1H); 13C NMR (DMSO) δ 166.2, 139.2, 135.2, 130.1, 129.1, 127.8, 127.4, 123.7, 119.3, 72.2. ESI-HRMS Cacld.for C14H13NO4S (M-H+): 292.0638; found 292.0645.
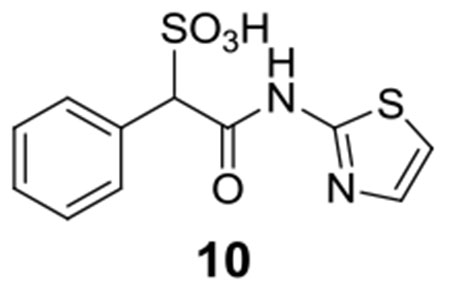
10: 1H NMR (500 MHz, DMSO) δ 12.0 (s, 1H), 7.49-7.23 (m, 7H), 4.93 (s, 1H). ESI-HRMS Cacld. for C11H9N2O4S2 (M-H+): 297.0009; found 296.9999.
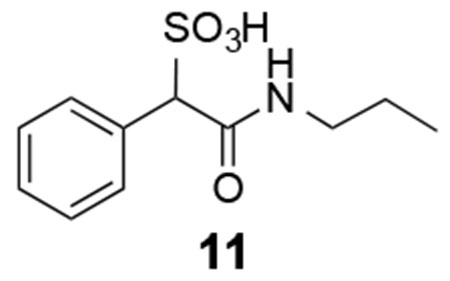
11:1H NMR (500 MHz, CDCl3) δ 8.23 (s, 1H), 7.45-7.44 (m, 2H), 7.27-7.20 (m, 3H), 4.42 (s, 1H), 3.11-3.02 (m, 2 H), 1.46-1.39 (m, 2H), 0.85 (t, J = 7.4 Hz, 3H);13C NMR (500 MHz, CDCl3) δ 167.3, 135.7, 129.6, 127.4, 126.8, 71.5, 40.4, 22.3, 11.4. ESI-HRMS Cacld.for C11H16NO4S (M+H+): m/z 258.0795; found 258.0799.
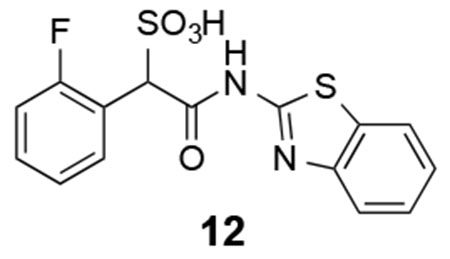
12: 1H NMR (500 MHz, DMSO) δ 12.43 (s, 1H), 8.08-8.05 (m, 1H), 7.97 (d, J = 7.9 Hz, 1H), 7.76 (d, J = 8.1 Hz, 1H), 7.45-7.42 (m, 1H), 7.33-7.29 (m, 2H), 7.19-7.12 (m, 2H), 5.46 (s, 1H). 13C NMR (DMSO) δ 166.3, 160.2 (d, J = 243.9 Hz, 1H), 157.6, 148.5, 131.9 (d, J = 3.3 Hz), 131.5, 129.0 (d, J = 8.1 Hz), 126.1, 123.5, 123.4 (d, J = 3.5 Hz), 121.7, 121.4 (d, J = 13.2 Hz), 114.4 (d, J = 22.4 Hz), 61.6. ESI Cacld. for C15H10FN2O4S2 (M-H+): 365.0071; found 365.0081.
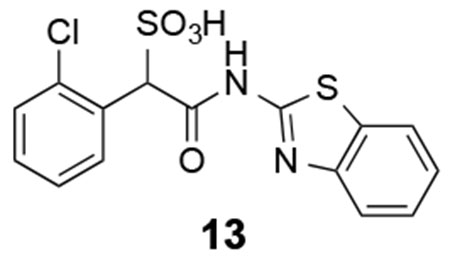
13: 1H NMR (500 MHz, DMSO) δ 8.19 (d, J = 1.8 Hz, 1H), 8.18 (d, J = 1.8 Hz, 1H), 7.97 (d, J = 7.9 Hz, 1H), 7.77-7.41 (m, 2H), 7.35-7.28 (m, 3H), 5.65 (s, 1H). 13C NMR (DMSO) δ 166.2, 157.6, 148.5, 133.9, 132.0, 132.0, 131.5, 128.8, 128.5, 126.2, 126.1, 123.5, 121.7, 120.6, 65.9. ESI Cacld. for C15H10ClN2O4S2 (M-H+): 380.9776; found 380.9773.
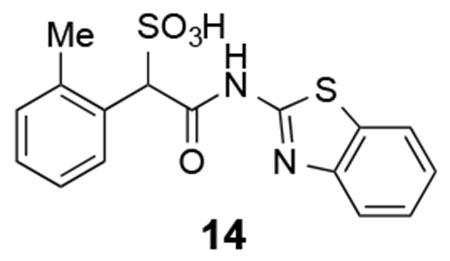
14:1H NMR (500 MHz, DMSO) δ 12.46 (br, 1H), 7.98-7.96 (m, 2H), 7.75 (d, J = 8.1 Hz, 1H), 7.43 (t, J = 7.3 Hz, 1H), 7.30 (t, J = 7.3 Hz, 1H), 7.15-7.14 (m, 3H), 5.34 (s, 1H). 13C NMR (DMSO) δ 167.1, 157.7, 148.5, 137.1, 132.7, 131.4, 129.6, 129.5, 126.9, 126.1, 125.0, 123.5, 121.7, 120.5, 65.7, 19.8. ESI Cacld. for C16H13N2O4S2 (M-H+): 361.0322; found 361.0328.
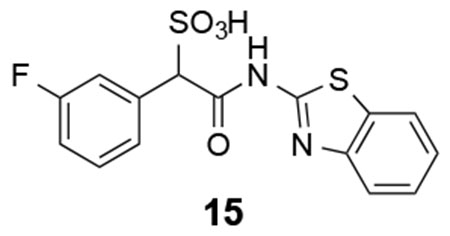
15: 1H NMR (500 MHz, DMSO) δ 12.28 (s, 1H), 7.98 (d, J = 7.7 Hz, 1H), 7.76 (d, J = 7.9 Hz, 1H), 7.51 (d, J = 10.0 Hz, 1H), 7.43 (t, J = 7.4 Hz, 1H), 7.35-7.29 (m, 3H), 7.12 (t, J = 7.1 Hz, 1H), 5.15 (s, 1H); 13C NMR (DMSO) δ 166.6, 161.5 (d, J = 240.0 Hz, 1H), 157.5, 148.5, 136.9 (d, J = 7.9 Hz), 131.5, 129.2 (d, J = 7.9 Hz), 126.2, 126.1, 123.5, 121.7, 120.6, 116.5 (d, J = 22.3 Hz), 114.0 (d, J = 21.0 Hz), 69.6. ESI-HRMS Cacld.for C15H10FN2O4S2 (M-H+): 365.0071; found 365.0071.
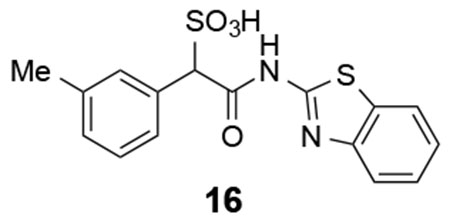
16: 1H NMR (500 MHz, DMSO) δ 7.97 (d, J = 7.9 Hz, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.45-7.36 (m, 3H), 7.30 (t, J = 7.6 Hz, 1H), 7.19 (t, J = 7.6 Hz, 1H), 7.09 (d, J = 7.4 Hz, 1H), 5.02 (s, 1H), 2.30 (s, 3H). 13C NMR (DMSO) δ 167.7, 157.5, 148.4, 136.4, 134.2, 131.4, 130.4, 127.9, 127.5, 127.0, 126.1, 123.5, 121.7, 120.5, 70.3, 21.1. ESI Cacld. for C16H13N2O4S2 (M-H+): 361.0322; found 361.0333.

17: 1H NMR (500 MHz, DMSO) δ 8.06 (s, 1H), 7.98 (d, J = 7.8 Hz, 1H), 7.84 (d, J = 7.9 Hz, 1H), 7.78-7.75 (m, 2H), 7.55 (t, J = 7.8 Hz, 1H), 7.44 (t, J = 7.3 Hz, 1H), 7.31 (d, J = 7.4 Hz, 1H), 5.25 (s, 1H). 13C NMR (DMSO) δ 166.4, 157.5, 148.5, 135.9, 135.1, 133.4, 131.5, 130.9, 128.9, 126.2, 123.6, 121.7, 120.6, 118.9, 110.5, 69.3. ESI Cacld. for C16H10N3O4S2 (M-H+): 372.0118; found 372.0129.
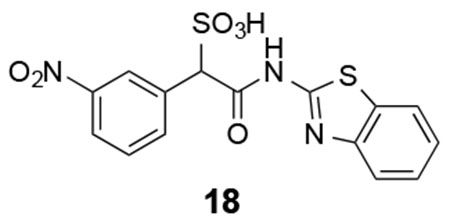
18: 1H NMR (500 MHz, DMSO) δ 8.58 (s, 1H), 8.18 (d, J = 8.0 Hz, 1H), 7.99-7.94 (m, 2H), 7.77 (d, J = 8.1 Hz, 1H), 7.63 (t, J = 7.9 Hz, 1H), 7.44 (t, J = 7.4 Hz, 1H), 7.31 (d, J = 7.4 Hz, 1H), 5.37 (s, 1H). 13C NMR (DMSO) δ 166.3, 157.5, 148.5, 147.2, 137.0, 136.4, 131.5, 129.0, 126.2, 124.6, 123.6, 122.2, 121.7, 120.6, 69.2. ESI Cacld. for C15H10N3O6S2 (M-H+): 392.0017; found 392.0026.

19: 1H NMR (500 MHz, DMSO) δ 8.03 (s, 1H), 7.97 (d, J = 7.9 Hz, 1H), 7.81 (d, J = 7.6 Hz, 1H), 7.76 (d, J = 8.1 Hz, 1H), 7.65 (d, J = 7.7 Hz, 1H), 7.56 (t, J = 7.7 Hz, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.30 (d, J = 7.6 Hz, 1H), 5.29 (s, 1H). 13C NMR (125 MHz, DMSO) δ 166.5, 157.5, 148.5, 135.6, 134.2, 131.5, 128.6, 128.3 (q, J = 31.0 Hz), 126.4 (d, J = 3.6 Hz), 126.2, 124.4 (q, J = 270.4 Hz), 124.0 (d, J = 3.4 Hz), 123.6, 121.7, 120.6, 69.6. ESI Cacld. for C16H10F3N2O4S2 (M-H+): 415.0040; found 415.0046.
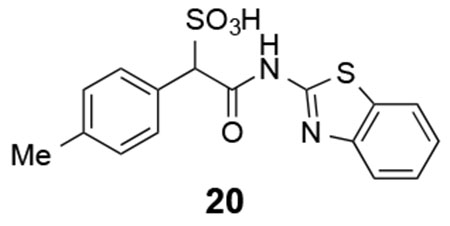
20: 1H NMR (500 MHz, DMSO) δ 7.97 (d, J = 7.8 Hz, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.47-7.42 (m, 3H), 7.30 (t, J = 7.6 Hz, 1H), 7.11 (d, J = 7.8 Hz, 2H), 5.02 (s, 1H), 2.28 (s, 3H). 13C NMR (DMSO) δ 167.2, 157.6, 148.4, 136.4, 131.4, 131.3, 129.7, 128.1, 126.1, 123.5, 121.7, 120.5, 70.0, 20.7. ESI Cacld. for C16H13N2O4S2 (M-H+): 361.0322; found 361.0330.

21: 1H NMR (500 MHz, DMSO) δ 12.28 (s, 1H), 7.97 (d, J = 7.7 Hz, 1H), 7.75 (d, J = 8.0 Hz, 1H), 7.55-7.50 (m, 4H), 7.43 (t, J = 7.5 Hz, 1H), 7.30 (d, J = 7.4 Hz, 1H), 5.12 (s, 1H); 13C NMR (DMSO) δ 166.6, 157.5, 148.5, 133.7, 132.1, 131.5, 130.4, 126.1, 123.5, 121.7, 120.6, 120.6, 69.5. ESI-HRMS Cacld. for C15H10BrN2O4S2 (M-H+): 424.9271; found 424.9270.

22:1H NMR (500 MHz, DMSO) δ 12.57 (s, 1H), 7.92-7.25 (m, 8H), 5.04 (s, 1H). ESI-HRMS Cacld. for C15H10ClN2O4S2 (M-H+): 380.9776; found 380.9777.
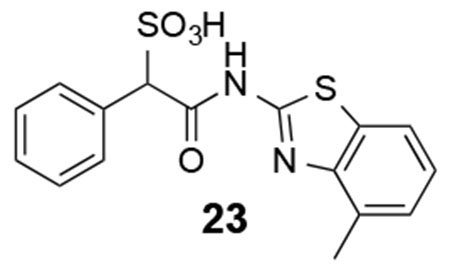
23:1H NMR (500 MHz, DMSO) δ 12.32 (s, 1H), 7.77-7.24 (m, 8H), 5.07 (s, 1H). ESI-HRMS Cacld. for C16H13N2O4S2 (M-H+): 361.0322; found 361.0316.
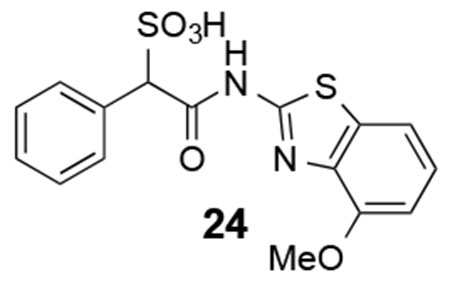
24: 1H NMR (500 MHz, DMSO) δ 12.40 (br, 1H), 7.59-6.98 (m, 8H), 5.10 (s, 1H), 3.91 (br, 3H). ESI-HRMS Cacld. for C16H13N2O5S2 (M-H+): 377.0271; found 377.0266.
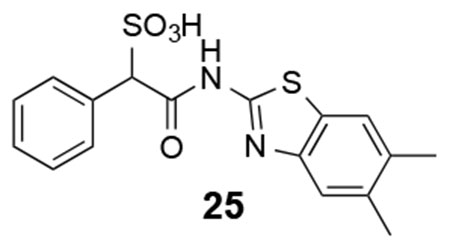
25: 1H NMR (500 MHz, DMSO) δ 12.24 (br, 1H), 7.99-7.29 (m, 7H), 4.94 (s, 1H), 2.30 (6H). ESI-HRMS Cacld. for C17H15N2O4S2 (M-H+): 375.0479; found 375.0473.
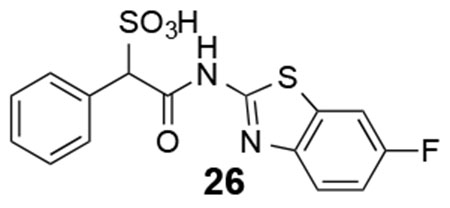
26: 1H NMR (500 MHz, DMSO) δ 12.35 (br, 1H), 7.88-7.28 (m, 8H), 5.07 (s, 1H). ESI-HRMS Cacld. for C15H10FN2O4S2 (M-H+): m/z 365.0071; found 365.0071.
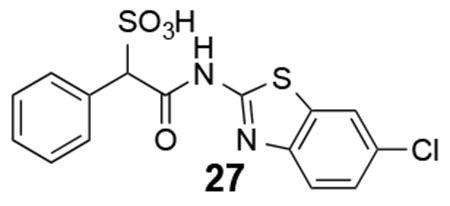
27: 1H NMR (500 MHz, DMSO) δ 12.35 (br, 1H), 8.10 (s. 1H), 7.72-7.27 (m, 7H), 5.05 (s, 1H). ESI-HRMS Cacld. for C15H10ClN2O4S2 (M-H+): 380.9776; found 380.9768.
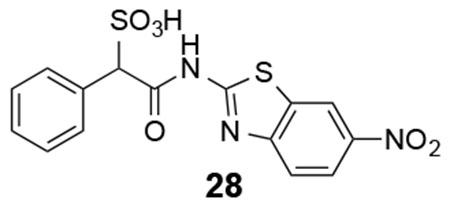
28: 1H NMR (500 MHz, DMSO) δ 12.64 (s, 1H), 9.00 (s. 1H), 8.21-7.24 (m, 7H), 5.09 (s, 1H). ESI-HRMS Cacld. for C15H10N3O6S2 (M-H+): 392.0017; found 392.0014.
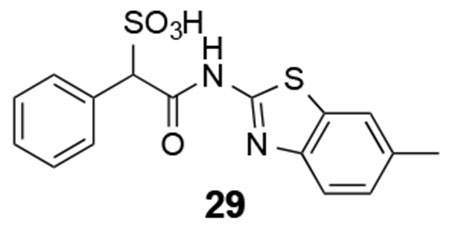
29: 1H NMR (500 MHz, DMSO) δ 12.16 (s, 1H), 7.70-7.23 (m. 8H), 5.00 (s, 1H), 2.35 (s, 3H). ESI-HRMS Cacld. for C16H13N2O4S2 (M-H+): 361.0322; found 361.0315.
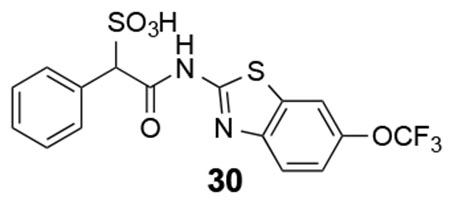
30: 1H NMR (500 MHz, DMSO) δ 12.35 (s, 1H), 8.05-7.25 (m. 8H), 5.04 (s, 1H). ESI-HRMS Cacld. for C16H10F3N2O5S2 (M-H+): 430.9989; found 430.9993.
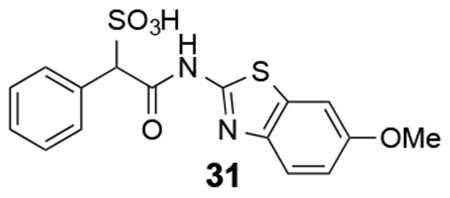
31: 1H NMR (500 MHz, DMSO) δ 12.05 (s, 1H), 7.49-6.93 (m. 8H), 4.94 (s, 1H), 3.70 (s, 3H). ESI-HRMS Cacld. for C16H13N2O5S2 (M-H+): 377.0271; found 377.0268.
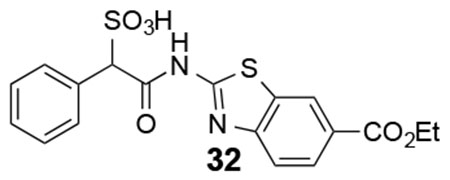
32: 1H NMR (500 MHz, DMSO) δ 12.47 (s, 1H), 8.59 (s, 1H), 7.95-7.25 (m, 8H), 5.10 (s, 1H), 4.26 (br, 2H), 1.27 (br, 3H). ESI-HRMS Cacld. for C18H15N2O6S2 (M-H+): 419.0377; found 419.0374.

33:1H NMR (500 MHz, DMSO) δ 8.70 (d, J = 2.3 Hz, 1H), 8.26 (dd, J = 2.4, 9.0 Hz, 1H), 7.73 (d, J = 9.0 Hz, 1H), 7.38-7.31 (m, 5H); 13C NMR (DMSO) 170.1, 162.4, 153.0, 143.9, 132.5, 129.4, 129.3, 128.0, 121.9, 120.7, 118.1, 43.2. ESI-HRMS Cacld. for C15H10N3O3S (M-H+): 312.0448; found 312.0444.
Protein Expression and Purification.
Expression and purification of the His-tagged LMW-PTP isoform A was performed as described elsewhere.39 LMW-PTPA was expressed in BL21 E coli cells. Protein expression was induced by 0.6 mM IPTG and harvested after overnight incubation at 25 °C. The cells were pelleted by centrifugation at 5,000 rpm and resuspended in 30 ml of 100 mM Tris buffer, pH 7.8, with 500 mM NaCl and 10 mM imidazole. The suspensions were frozen and thawed before physical lysis by a French Press. The bacterial lysate was centrifuged at 15,000 rpm at 4 °C for 30 min. The supernatant was collected and incubated with 2 ml of Ni-NTA resin by end-over-end rotation for 1 h at 4 °C. The beads were washed with the same Tris buffer with 20 mM imidazole to remove nonspecific binding. LMWPTP was eluted with 250 mM imidazole in the same Tris buffer. The protein was concentrated and further purified by going through a Sephadex G75 column. The other PTPs for selectivity studies were expressed and purified from E. coli by the same method used for LMW-PTP. These PTPs included SHP2, PTP1B, TC-PTP, SHP1, LYP, HePTP, Meg2, PTPH1, FAP-1, PTP-PEST, CD45, LAR, PTPα, PTPβ, PTPγ, PTPε, PTPσ, PTPμ, MKP3, VHR, VHZ, CDC14A, SSu72, and PP5.
Crystallization, Data Collection, and Structure Determination.
Protein crystals were first grown in 100 mM MES buffer, 30% PEGME 5,000 and 0.2 M Ammonium sulfate, the same buffer used elsewhere39 in the presence of 1 mM compound 7. Crystallization conditions were systematically scanned against a number of different buffers instead of MES and eventually the optimal condition was finalized for crystal growth of the LMWPTP•7 complex. The buffer grid is 25-30% PEGME 5,000 in 100 mM Bis-Tris, pH 6.0-6.5. The data was collected at the APS 19BM or 19ID beamlines (Argonne, IL). Initial phases of the LMWPTP complexes were determined by molecular replacement with the CCP4 software package. The published LMWPTP structure (5PNT) was used as the initial search model. After one round of refinement by Refmac in the CCP4 package, further refinement was carried out in Phenix.
Determination of Inhibition Constant and IC50.
PTP activity was measured using p-nitrophenyl phosphate (pNPP) as a substrate in 50 mM 3,3-dimethylglutarate (DMG) buffer, pH 7.0 at 25 °C. The reaction was initiated by the addition of enzyme to a reaction solution containing 2-5 mM pNPP with different concentrations of inhibitors. The reactions were incubated at room temperature for 5-10 min and quenched with 5N NaOH. The nonenzymatic hydrolysis of pNPP was corrected by measuring the control without the addition of enzyme. The amount of product p-nitrophenol was determined from the absorbance at 405 nm detected by a Spectra MAX340 microplate spectrophotometer (Molecular Devices) using a molar extinction coefficient of 18,000 M−1cm−1. IC50 was calculated by fitting the data using SigmaPlot. To determine the mode of inhibition, the reactions were initiated by addition of enzyme to the reaction solution containing different concentrations of pNPP with different concentrations of inhibitor. Similarly, Ki was obtained by fitting the data using SigmaPlot Enzyme Kinetics Module. In addition to LMW-PTP, other PTPs evaluated included the receptor-like PTPs, CD45, PTPα, PTPβ, PTPμ, PTPε, LAR, PTPσ and PTPγ, cytosolic PTPs, PTP1B, TC-PTP, Lyp, SHP1, SHP2, PTPH1, HePTP, PTP-Meg2, PTP-FAP1, and PTP-PEST, the dual specificity phosphatase VHR, VHZ, MKP3, CDC14A, RNA Polymerase II CTD Phosphatase, and protein phosphatase 5. The IC50 determination for these PTPs was performed under the same conditions as used for LMW-PTP except that the pNPP concentrations used corresponded to the Km values of the PTPs studied and the enzyme concentrations ranged from 5-20 nM.
Cell culture and immunoblot analysis.
HepG2 cells were cultured at 37 °C and 5% CO2 in Dulbecco’s modified Eagle’s medium (Corning) supplemented with 10% fetal bovine serum (HyClone). The 60-80% confluent HepG2 cells were starved overnight in serum free medium, followed by treatment with vehicle or compound for 2h, and then either left unstimulated or stimulated with 50 nM insulin (Sigma) for 30 min. Cells were lysed and the lysates were electrophoresed on a 10% polyacrylamide gel, transferred to a nitrocellulose membrane, and blotted with anti-phospho-AKT, anti-AKT antibodies (Cell Signaling). The blots were developed by the enhanced chemiluminescence technique using the SuperSignal West Pico Chemiluminescent substrate (Thermo Scientific). The protein levels of phospho-AKT and AKT were quantified by Image J software, and the ratios of pAKT/AKT were normalized to the vehicle control.
Acknowledgement:
This work was supported in part by National Institutes of Health Grants CA69202 and P30CA023168.
Abbreviations footnote:
- LMW-PTP
low molecular weight protein tyrosine phosphatase
- PTP
protein tyrosine phosphatase
- SPAA
SulfoPhenyl Acetic Amide
Footnotes
Ancillary Information
Data deposition: The coordinates for the structures of LMW-PTP•7 (accession number 5KQG), LMW-PTP•9 (accession number 5KQL), LMW-PTP•MES (accession number 5KQM), and the apo-form LMW-PTP (accession number 5KQP) have been deposited in the Protein Data Bank. Authors will release the atomic coordinates and experimental data upon article publication.
References
- 1.Hunter T Tyrosine phosphorylation: thirty years and counting. Curr. Opin. Cell Biol 2009, 21, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He R; Yu Z-H; Zhang R-Y; Zhang Z-Y Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol. Sin 2014, 35, 1227–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen P; Alessi DR Kinase drug discovery - what’s next in the field? ACS Chem. Biol 2013, 8, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alonso A; Sasin J; Bottini N; Friedberg I; Friedberg I; Osterman A; Godzik A; Hunter T; Dixon J; Mustelin T Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z-Y; Van Etten RL Purification and characterization of a low-molecular-weight acid phosphatase--a phosphotyrosyl-protein phosphatase from bovine heart. Arch. Biochem. Biophys 1990, 282, 39–49. [DOI] [PubMed] [Google Scholar]
- 6.Wang S; Tabernero L; Zhang M; Harms E; Van Etten RL; Stauffacher CV Crystal structures of a low-molecular weight protein tyrosine phosphatase from Saccharomyces cerevisiae and its complex with the substrate p-nitrophenyl phosphate. Biochemistry 2000, 39, 1903–1914. [DOI] [PubMed] [Google Scholar]
- 7.Tolkatchev D; Shaykhutdinov R; Xu P; Plamondon J; Watson DC; Young NM; Ni F Three‐dimensional structure and ligand interactions of the low molecular weight protein tyrosine phosphatase from Campylobacter jejuni. Protein Science 2006, 15, 2381–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maccari R; Ottanà R Low molecular weight phosphotyrosine protein phosphatases as emerging targets for the design of novel therapeutic agents. J. Med. Chem 2012, 55, 2–22. [DOI] [PubMed] [Google Scholar]
- 9.Chiarugi P; Cirri P; Taddei ML; Giannoni E; Fiaschi T; Buricchi F; Camici G; Raugei G; Ramponi G Insight into the role of low molecular weight phosphotyrosine phosphatase (LMW-PTP) on platelet-derived growth factor receptor (PDGF-r) signaling. LMW-PTP controls PDGF-r kinase activity through TYR-857 dephosphorylation. J. Biol. Chem 2002, 277, 37331–37338. [DOI] [PubMed] [Google Scholar]
- 10.Taddei ML; Chiarugi P; Cirri P; Talini D; Camici G; Manao G; Raugei G; Ramponi G LMW-PTP exerts a differential regulation on PDGF- and insulin-mediated signaling. Biochem. Biophys. Res. Commun 2000, 270, 564–569. [DOI] [PubMed] [Google Scholar]
- 11.Shimizu H; Shiota M; Yamada N; Miyazaki K; Ishida N; Kim S; Miyazaki H Low M(r) protein tyrosine phosphatase inhibits growth and migration of vascular smooth muscle cells induced by platelet-derived growth factor. Biochem. Biophys. Res. Commun 2001, 289, 602–607. [DOI] [PubMed] [Google Scholar]
- 12.Park EK; Warner N; Mood K; Pawson T; Daar IO Low-molecular-weight protein tyrosine phosphatase is a positive component of the fibroblast growth factor receptor signaling pathway. Mol. Cell. Biol 2002, 22, 3404–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramponi G; Manao G; Camici G; Cappugi G; Ruggiero M; Bottaro DP The 18 kDa cytosolic acid phosphatase from bovine live has phosphotyrosine phosphatase activity on the autophosphorylated epidermal growth factor receptor. FEBS Lett 1989, 250, 469–473. [DOI] [PubMed] [Google Scholar]
- 14.Chiarugi P; Cirri P; Marra F; Raugei G; Camici G; Manao G; Ramponi G LMW-PTP is a negative regulator of insulin-mediated mitotic and metabolic signalling. Biochem. Biophys. Res. Commun 1997, 238, 676–682. [DOI] [PubMed] [Google Scholar]
- 15.Pandey SK; Yu XX; Watts LM; Michael MD; Sloop KW; Rivard AR; Leedom TA; Manchem VP; Samadzadeh L; McKay RA; Monia BP; Bhanot S Reduction of low molecular weight protein-tyrosine phosphatase expression improves hyperglycemia and insulin sensitivity in obese mice. J. Biol. Chem 2007, 282, 14291–14299. [DOI] [PubMed] [Google Scholar]
- 16.Chiarugi P; Pani G; Giannoni E; Taddei L; Colavitti R; Raugei G; Symons M; Borrello S; Galeotti T; Ramponi G Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J. Cell Biol 2003, 161, 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swanson PA; Kumar A; Samarin S; Vijay-Kumar M; Kundu K; Murthy N; Hansen J; Nusrat A; Neish AS Enteric commensal bacteria potentiate epithelial restitution via reactive oxygen species-mediated inactivation of focal adhesion kinase phosphatases. Proc. Natl. Acad. Sci. USA. 2011, 108, 8803–8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rigacci S; Talini D; Berti A LMW-PTP associates and dephosphorylates STAT5 interacting with its C-terminal domain. Biochem. Biophys. Res. Commun 2003, 312, 360–366. [DOI] [PubMed] [Google Scholar]
- 19.Rigacci S; Guidotti V; Parri M; Berti A Modulation of STAT5 interaction with LMW-PTP during early megakaryocyte differentiation. Biochemistry 2008, 47, 1482–1489. [DOI] [PubMed] [Google Scholar]
- 20.Stein E; Lane AA; Cerretti DP; Schoecklmann HO; Schroff AD; Van Etten RL; Daniel TO Eph receptors discriminate specific ligand oligomers to determine alternative signaling complexes, attachment, and assembly responses. Genes Dev 1998, 12, 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kikawa KD; Vidale DR; Van Etten RL; Kinch MS Regulation of the EphA2 kinase by the low molecular weight tyrosine phosphatase induces transformation. J. Biol. Chem 2002, 277, 39274–39279. [DOI] [PubMed] [Google Scholar]
- 22.Malentacchi F; Marzocchini R; Gelmini S; Orlando C; Serio M; Ramponi G; Raugei G Up-regulated expression of low molecular weight protein tyrosine phosphatases in different human cancers. Biochem. Biophys. Res. Commun 2005, 334, 875–883. [DOI] [PubMed] [Google Scholar]
- 23.Marzocchini R; Malentacchi F; Biagini M; Cirelli D; Luceri C; Caderni G; Raugei G The expression of low molecular weight protein tyrosine phosphatase is up-regulated in 1,2-dimethylhydrazine-induced colon tumours in rats. Int J Cancer 2007, 122, 1675–1678. [DOI] [PubMed] [Google Scholar]
- 24.Ruela-de-Sousa RR; Hoekstra E; Hoogland AM; Souza Queiroz KC; Peppelenbosch MP; Stubbs AP; Pelizzaro-Rocha K; van Leenders GJ; Jenster G; Aoyama H; Ferreira CV; Fuhler GM Low-molecular-weight protein tyrosine phosphatase predicts prostate cancer outcome by increasing the metastatic potential. Eur Urol 2016, 69, 710–719 [DOI] [PubMed] [Google Scholar]
- 25.Hoekstra E; Kodach LL; Das AM; Ruela-de-Sousa RR; Ferreira CV; Hardwick JC; van der Woude CJ; Peppelenbosch MP; Ten Hagen TL; Fuhler GM Low molecular weight protein tyrosine phosphatase (LMWPTP) upregulation mediates malignant potential in colorectal cancer. Oncotarget 2015, 6, 8300–8312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bottini N; MacMurray J; Peters W; Rostamkhani M; Comings DE Association of the acid phosphatase (ACP1) gene with triglyceride levels in obese women. Molecular Genetics and Metabolism 2002, 77, 226–229. [DOI] [PubMed] [Google Scholar]
- 27.Iannaccone U; Bergamaschi A; Magrini A; Marino G; Bottini N; Lucarelli P; Bottini E; Gloria-Bottini F Serum glucose concentration and ACP1 genotype in healthy adult subjects. Metab. Clin. Exp 2005, 54, 891–894. [DOI] [PubMed] [Google Scholar]
- 28.Gloria-Bottini F; Magrini A; Renzo LD; Lorenzo AD; Bergamaschi A; Bottini E Body mass index and acid phosphatase locus 1 in diabetic disorders. Acta Diabetol 2010, 47, 139–143. [DOI] [PubMed] [Google Scholar]
- 29.Zabell AP; Corden S; Helquist P; Stauffacher CV; Wiest O Inhibition studies with rationally designed inhibitors of the human low molecular weight protein tyrosine phosphatase. Bioorg. Med. Chem 2004, 12, 1867–1880. [DOI] [PubMed] [Google Scholar]
- 30.Weitgenant JA; Katsuyama I; Bigi MA; Corden SJ; Markiewicz JT; Zabell APR; Homan KT; Wiest O; Stauffacher CV; Helquist P Synthesis of a 5-azaindole phosphonic acid as a computationally designed inhibitor of the low molecular weight phosphatase HCPTP. Heterocycles 2006, 70, 599–607. [Google Scholar]
- 31.Vidal D; Blobel J; Pérez Y; Thormann M; Pons M Structure-based discovery of new small molecule inhibitors of low molecular weight protein tyrosine phosphatase. Eur. J. Med. Chem 2007, 42, 1102–1108. [DOI] [PubMed] [Google Scholar]
- 32.Maccari R; Ottanà R; Ciurleo R; Paoli P; Manao G; Camici G; Laggner C; Langer T Structure-based optimization of benzoic acids as inhibitors of protein tyrosine phosphatase 1B and low molecular weight protein tyrosine phosphatase. ChemMedChem 2009, 4, 957–962. [DOI] [PubMed] [Google Scholar]
- 33.Ottanà R; Maccari R; Ciurleo R; Paoli P; Jacomelli M; Manao G; Camici G; Laggner C; Langer T 5-Arylidene-2-phenylimino-4-thiazolidinones as PTP1B and LMW-PTP inhibitors. Bioorg. Med. Chem 2009, 17, 1928–1937. [DOI] [PubMed] [Google Scholar]
- 34.Homan KT; Balasubramaniam D; Zabell APR; Wiest O; Helquist P; Stauffacher CV Identification of novel inhibitors for a low molecular weight protein tyrosine phosphatase via virtual screening. Bioorg. Med. Chem 2010, 18, 5449–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forghieri M; Laggner C; Paoli P; Langer T; Manao G; Camici G; Bondioli L; Prati F; Costantino L Synthesis, activity and molecular modeling of a new series of chromones as low molecular weight protein tyrosine phosphatase inhibitors. Bioorg. Med. Chem 2009, 17, 2658–2672. [DOI] [PubMed] [Google Scholar]
- 36.Seiler CL; Richards KA; Jakubowski HV; McIntee EJ Identification of new inhibitors for low molecular weight protein tyrosine phosphatase isoform B. Bioorg. Med. Chem. Lett 2013, 23, 5912–5914 [DOI] [PubMed] [Google Scholar]
- 37.Ottanà R; Maccari R; Amuso S; Wolber G; Schuster D; Herdlinger S; Manao G; Camici G; Paoli P New 4-[(5-arylidene-2-arylimino-4-oxo-3-thiazolidinyl)methyl]benzoic acids active as protein tyrosine phosphatase inhibitors endowed with insulinomimetic effect on mouse C2C12 skeletal muscle cells. Eur. J. Med. Chem 2012, 50, 332–343. [DOI] [PubMed] [Google Scholar]
- 38.He R; Yu Z-H; Zhang R-Y; Wu L; Gunawan A; Lane BS; Shim JS; Zeng L-F; He Y; Chen L; Wells CD; Liu JO; Zhang Z-Y Exploring the existing drug space for novel pTyr mimetic and SHP2 inhibitors. ACS Medicinal Chemistry Letter 2015, 6, 782–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang M; Stauffacher CV; Lin D; Van Etten RL Crystal structure of a human low molecular weight phosphotyrosyl phosphatase. Implications for substrate specificity. J. Biol. Chem 1998, 273, 21714–21720. [DOI] [PubMed] [Google Scholar]
- 40.Burley SK; Petsko GA Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science 1985, 229, 23–28. [DOI] [PubMed] [Google Scholar]
- 41.Burley SK; Petsko GA Amino-aromatic interactions in proteins. FEBS Lett 1986, 203, 139–143. [DOI] [PubMed] [Google Scholar]
- 42.McGaughey GB; Gagné M; Rappé AK pi-Stacking interactions. Alive and well in proteins. J. Biol. Chem 1998, 273, 15458–15463. [DOI] [PubMed] [Google Scholar]
- 43.Riley KE; Hobza P On the importance and origin of aromatic interactions in chemistry and biodisciplines. Acc. Chem. Res 2013, 46, 927–936. [DOI] [PubMed] [Google Scholar]