

Abstract
Phosphoinositide 3-kinase gamma isoform (PI3Kγ) plays a critical role in myeloid-derived cells of the immunosuppressive tumor microenvironment. IPI-549, a recently discovered small molecule selective PI3Kγ inhibitor, is currently under immuno-oncology clinical trials in combination with nivolumab, an anti-PD-1 monoclonal antibody immune checkpoint blocker. The purpose of this study is to investigate whether IPI-549 could reverse P-glycoprotein (P-gp) mediated MDR when combined with chemotherapeutic substrates of P-gp. Cytotoxicity assays showed that IPI-549 reverses P-gp-mediated MDR in SW620/Ad300 and LLC-PK-MDR1 cells. IPI-549 increases the amount of intracellular paclitaxel and inhibits the efflux of paclitaxel out of SW620/Ad300 cells. ABCB1-ATPase assay showed that IPI-549 stimulates the activity of ABCB1-ATPase. IPI-549 does not alter the expression and does not affect the subcellular localization of P-gp in SW620/Ad300 cells. The combination of IPI-549 with paclitaxel showed that IPI-549 potentiates the anti-tumor effects of paclitaxel in P-gp-overexpressing MDR SW620/Ad300 xenograft tumors. With clinical trials beginning to add newly approved immune checkpoint-based immunotherapy into standard-of-care immunogenic chemotherapy to improve patient outcomes, our findings support the rationale of adding IPI-549 to both the chemotherapeutic and immunotherapeutic aspects of cancer combination treatment strategies.
Keywords: ABCB1 transporter, P-glycoprotein, Combination chemotherapy, Multidrug resistance, IPI-549, Immune checkpoint
Graphical Abstract
1. Introduction
In 2018, 1.7 million new cancer cases and 600,000 cancer deaths are projected to occur in the United States, making cancer the second most common cause of death in the U.S. after heart disease [1]. Treatment modalities include surgery, radiation therapy, chemotherapy, immunotherapy, and targeted therapy [2]. To prevent metastasis, systemic chemotherapy is frequently given to patients, but despite initial improvement, relapse frequently occurs due to selective survival of cancer cells expressing the multidrug resistance (MDR) phenotype which remains a significant challenge in treating patients [3,4].
The goal of the chemotherapy is the eradication of tumors and metastatic malignant cells. The heterogeneity of tumors often leads to the development of acquired multidrug resistance (MDR) which attenuates the efficacy of several structurally and mechanistically unrelated chemotherapeutic drugs and often requires treatment to be suspended [5]. At the cellular and molecular level, MDR can be caused by several mechanisms [6]: a) mutations of the drug target; b) decreased drug uptake or decreased influx of drug; c) activation of compensatory cellular survival signaling pathways; d) increased DNA damage repair pathways; and e) increased expression of drug efflux pumps such as the ATP binding cassette (ABC) transporter proteins, implicated in the MDR phenotype [5,6].
The ABC transporter proteins make up a 49 member superfamily of proteins [7]. In cancer cells, MDR is often due to the upregulation of ABC transporter proteins on the cell membrane. P-glycoprotein (P-gp, MDR1, ABCB1) is the most well-studied ABC transporter [8]. Other members of the superfamily most often implicated in MDR to chemotherapy are the multidrug resistance protein-1 (MRP1, ABCC1) [9], and breast cancer resistant protein (BRCP, ABCG2) [10].
“Permeability”-glycoprotein or P-glycoprotein (P-gp), encoded by the MDR1 gene, and also known as ABCB1 [11], is composed of two homologous nucleotide binding domains and two transmembrane domains joined by a linker region [12]. Each transmembrane domain is made of six transmembrane helices which make up a twelve transmembrane helix efflux pump that binds hydrophobic drug substrates [13]. Its hydrophilic region contains the ATP binding site which binds two molecules of ATP. Efflux of a drug substrate leads to hydrolysis of ATP into ADP and inorganic phosphate, allowing the transmembrane domain to bind another substrate to be effluxed. This continuous cycle leads to low intracellular concentrations of substrate drugs and thus survival of MDR cancer cells exposed to drugs of chemotherapy [13]. Anticancer drug substrates of P-gp include the taxanes (paclitaxel, docetaxel), anthracyclines (doxorubicin, daunorubicin), vinca alkaloids (vincristine, vinblastine), epipodophyllotoxins (etoposide, teniposide) and tyrosine kinase inhibitors of EGFR, VEGFR, and Bcr-Abl such as lapatinib, nilotinib, and sunitinib, respectively [14]. In addition to cancer cells, P-gp is highly expressed at the apical surface of epithelial cells, such as in the colon, hepatic bile duct, renal proximal convoluted tubule, pancreatic ductules, adrenal gland, placenta (blood-placenta barrier), testis (blood-testis barrier), and brain capillaries (blood-brain barrier) [15]. Anatomically, P-gp functions as an efflux transporter that limits cellular uptake of drugs from the blood into the brain, and from intestinal lumen into enterocytes. On the other hand, P-gp enhances the elimination of drugs out of the hepatocytes and renal epithelial cells into the bile and urine, respectively [15].
Overexpression of P-gp has been associated with various cancers, including hematological malignancies, breast cancers, acute myeloid leukemia, and solid tumors [16–19]. In order to counteract P-gp-mediated MDR, strategies to develop small molecule drugs which inhibit or block the efflux function of P-gp, referred to as P-gp inhibitors or modulators, or chemosensitizers/reversal agents have been undertaken and have gone through three generations of development [20].
IPI-549 is an investigational first-in-class, small molecule, gamma isoform selective phosphoinositide 3-kinase (PI3Kγ) inhibitor [21,22]. In preclinical studies, inhibition of PI3Kγ by IPI-549 reprogrammed macrophages from an immune-suppressive M2 phenotype to an immune-activating M1 phenotype [22]. The shift of macrophages to the proinflammatory antitumor M1 phenotype enhanced the recruitment, infiltration, and activation of cytotoxic T cells at the tumor site [22]. IPI-549 in combination with anti-CTLA4 or anti-PD-1 immune checkpoint blockers showed synergistic effects in a mouse model [22]. In a phase 1 clinical trial, IPI-549 in combination with nivolumab (anti-PD-1) showed favorable tolerability and signs of clinical activity with immune modulation, and recruiting is currently underway for phase 2 clinical trials [23].
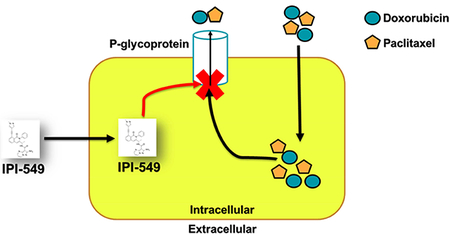
In our own studies, we chose to test whether IPI-549 could act as a chemosensitizing agent to the P-gp-overexpressing MDR phenotype of cancer cells. Most immuno-oncology agents are biological-based therapy in the form of monoclonal antibodies [24]. As a small molecule kinase inhibitor, IPI-549 is an ideal candidate for combination therapy with conventional chemotherapy targeting P-gp-mediated MDR.
2. Materials and Methods
2.1. Reagents
[3H]-paclitaxel (37.9 Ci/mmol) was purchased from Moravek Biochemicals, Inc. (Brea, CA). Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin, and Trypsin/EDTA were purchased from Hyclone, GE Healthcare Life Science (Pittsburgh, PA). Secondary horseradish peroxidase-labeled rabbit anti-mouse IgG was purchased from Cell Signaling Technology (Danvers, MA). 3-(4,5-dimethylthiazol-yl)-2,5- diphenyltetrazolium bromide (MTT), dimethylsulfoxide (DMSO), Triton X-100, propidium iodide and paraformaldehyde were purchased from Sigma-Aldrich (St. Louis, MO). P7965 Monoclonal Anti-P-Glycoprotein (MDR) antibody produced in mouse, monoclonal antibodies BXP-21 to ABCG2, GAPDH, and the secondary horseradish peroxidase-labeled rabbit antimouse IgG were purchased from Sigma-Aldrich (St. Louis, MO). Doxorubicin, vincristine, paclitaxel, colchicine, cisplatin, verapamil, mitoxantrone, and nilotinib were purchased from Sigma-Aldrich (St. Louis, MO). Bovine Serum Albumin (BSA), Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 was purchase from Thermo Scientific (Rockford, IL). IPI-549 was purchased from Chemietek (Indianapolis, IN).
2.2. Cell lines and cell culture
The human colon cancer cell line SW620 and its doxorubicin-selected SW620/Ad300 cell line overexpressing ABCB1 were kindly provided by Drs. Susan E. Bates and Robert Robey (NIH, Bethesda, MD). The LLC-PK1 porcine kidney epithelial cell line and the transfected LLCPK-MDR1 cell line, transfected with human ABCB1 cDNA selected under 2 mg/mL G418 antibiotic, were kindly provided by Dr. Michael M. Gottesman (NIH, Bethesda, MD). All cell lines were grown as adherent monolayers in poly-D-lysine coated flasks with DMEM supplemented with 10% FBS or bovine calf serum and 1% penicillin/streptomycin in a humidified incubator containing 5% CO2 at 37 °C.
2.3. Animals
Male athymic mice (NCRNU-M sp/sp) weighing 13–15 grams and age of 4 weeks were purchased from Taconic Farms. All the animals were maintained on an alternating 12 h light/dark cycle with free access to water and rodent chow ad libitum. The mice were maintained at the St. John’s University Animal Care Facility and monitored for tumor growth by palpitation and visual examination. The St. John’s University Institutional Animal Care and Use Committee (IACUC) approved the protocol, and the research was conducted in compliance with the Animal Welfare Act and other federal statutes.
2.4. Cytotoxicity assay
Modified MTT colorimetric assay was used to measure the changes in cytotoxicities of anticancer drugs with or without inhibitors. The protocol used in this study is identical to that previously described [25, 26]. Briefly, 4000–5000 cells were seeded in 160 μL media per well in 96-well plates in triplicate overnight. The cells were then incubated in various concentrations of IPI-549 (20 μL). After a 2 h pre-incubation with IPI-549, increasing concentrations of chemotherapeutic drugs were added to each well (20 μL). After 72 h, 20 μL of MTT solution (4 mg/mL) was added to each well. The plates were then incubated at 37°C for an additional 4 h to allow viable cells to change the yellow MTT into dark blue formazan crystals. The MTT/medium solution was then aspirated without disturbing the cells, and 100 μL of DMSO was added to each well. The plates were placed on a shaking table to mix the formazan into solution. Finally, the absorbance was determined at 570 nm with an Opsys microplate reader (Dynex Technologies, Chantilly, VA). The degree of resistance was calculated by dividing the IC50 values of the MDR cells with or without IPI-549, verapamil (positive control for ABCB1), or nilotinib (positive control for ABCG2) by that of the parental cells without IPI-549, verapamil, or nilotinib. Verapamil and nilotinib were used at non-toxic concentrations of 10 μM and 5 μM, respectively.
2.5. [3H]-labeled substrate intracellular accumulation and efflux assay
[3H]-labeled paclitaxel accumulation and efflux assays were performed on SW620 and SW620/Ad300 cells in the presence or absence of IPI-549 (5 and 10 μM) or verapamil (10 μM) following our previously established protocols [27, 28]. Confluent cells were trypsinized and four aliquots of each cell line were resuspended in DMEM in four centrifuge tubes. The suspended cells were preincubated with or without reversal agents for 2 h at 37°C in DMEM. To measure drug accumulation, the cells were subsequently incubated in 0.1 μM [3H]-paclitaxel for 2 h in the presence or absence of reversal agents at 37°C. The cells were then washed three times in ice cold PBS, pelleted, and lysed in 5 mL of scintillation fluid. Radioactivity was measured in a Packard TRI-CARB 1900CA liquid scintillation analyzer from Packard Instrument Company, Inc. (Downers Grove, IL).
To study the efflux of ABCB1, the same procedure as the accumulation assay was followed. However, after three washes in ice cold PBS, the suspended cells were incubated at 37°C in fresh DMEM with reversal agents. Aliquots of the cells were taken at 0, 30, 60, and 120 minutes, washed three times in ice cold PBS, lysed in 5 mL of scintillation fluid, and radioactivity was measured.
2.6. ABCB1-ATPase assay
ABCB1-containing crude membrane were prepared from High Five insect cells as previously described [29–35]. IPI-549-induced ABCB1-ATPase activity was determined as previously described [29–35]. Briefly, the membrane vesicles (10 μg of protein/reaction) were incubated in ATPase buffer (50 mmol/L MES-Tris, pH 6.8, 50 mmol/L KCl, 5 mmol/L sodium azide, 1 mmol/L EGTA, 1 mmol/L ouabain, 2 mmol/L dithiothreitol, and 10 mmol/L MgCl2) at 37°C for 5 min with or without 0.3 mmol/L vanadate, then incubated in 0–40 μM IPI-549 at 37°C for 3 minutes. The ABCB1-ATPase reaction was induced by the addition of 5 mmol/L Mg-ATP. After incubation at 37°C for 20 min, the reactions were stopped by loading 0.1 mL of 5% SDS solution. The amount of Pi released was quantified at 800 nm using a Bio-Rad SmartSpec Plus Spectrophotometer.
2.7. Preparation of total cell lysates and western blotting
SW620/Ad300 cells were treated with IPI-549 (10 μM) for up to 72 h. Samples were taken at 0, 24, 48, and 72 h and blotted using our previously established protocols [27, 28].
2.8. Immunofluorescence of P-gp
SW620/Ad300 cells were seeded in 24-well plates with coverslips and were allowed to grow overnight, followed by treatment with 10 μM IPI-549 for 72 h. Fluorescence microscopy was performed as previously described [29–35] and images were captured with an EVOS FL Auto 2 Cell Imaging System fluorescent microscope in the laboratory of Dr. Vikas Dukhande (St. John’s University, Queens, NY).
2.9. Induced-Fit Docking analysis
The structure of IPI-549 was input using the entry editor of Maestro v11.1 followed by energy minimization using OPLS3 force field by Macromodel v11.5 (Schrödinger, LLC, New York, NY, USA, 2017). The structure was then prepared by LigPrep v4.1 (Schrödinger, LLC, New York, NY, USA, 2017). The human ABCB1 homology model based on the refined mouse ABCB1 protein (PDB ID: 4M1M) was kindly provided by S. Aller. The receptor docking grid with length of 25 Å was set up by selecting all identified drug interacting amino acid residues as centroid [36, 37]. Flexible docking simulation on the binding of IPI-549 with human ABCB1 homology model was performed using the XP (extra precision) mode of Glide v7.4 (Schrödinger, LLC, New York, NY, USA, 2017). The IFD protocol was run with defaulted parameters, and the Glide gscore, which indicates the approximate ligand binding free energy [38] were calculated and expressed as kcal/mol [36, 37].
2.10. MDR xenograft tumor model
SW620 (4 X 106) and SW620/Ad300 (5 X 106) cells were injected subcutaneously at the flank near the armpits of athymic nude mice. When the subcutaneous tumors were approximately 0.5 × 0.5 cm in size (day 0), the mice were randomized into four treatment groups. The vehicle used to deliver the IPI-549 and paclitaxel by intraperitoneal (i.p.) injection was ethanol/Cremophor ELP/saline (10%/10%/80%). Group one received the vehicle only; group two received vehicle plus 3 mg/kg IPI-549; group three received vehicle plus 15 mg/kg paclitaxel; group four, the combination group, received vehicle plus 3 mg/kg IPI-549 one h prior to administration of vehicle plus 15 mg/kg paclitaxel. The drug doses were administered every 3 days with a total of 4 doses. Tumor volume was measured using calipers, and body weights were recorded prior to each dosing [39, 40]. The body weights and tumor sizes of the mice were monitored every third day to assess tumor progression. The two perpendicular diameters (termed A and B) were recorded every third day and tumor volume was estimated according to the following formula published previously [39, 40]: V = π/6 × [(A + B)/2]. The mice were euthanized by carbon dioxide inhalation after four treatments. Tumor tissue was excised and weighed.
The ratio of growth inhibition (IR) for tumor weight (IRW) and tumor volume (IRV, at the end of 18-days treatments) were estimated according to the formulas given below [39]: IRV(%) = (1- Mean tumor volume of the experimental group/Mean tumor volume of the control group) X 100 IRW(%) = (1- Mean tumor weight of the experimental group/Mean tumor weight of the control group) X 100
2.11. Statistical analysis
All experiments were repeated at least three times and the differences were determined using the two-tailed Student’s t-test and statistical significance was determined at p < 0.05.
3. Results
3.1. IPI-549 sensitizes ABCB1-overexpressing cells to ABCB1 chemotherapeutic substrates, but not cisplatin.
To determine a non-toxic concentration of IPI-549 to be used in reversal studies, cytotoxicity assays were performed on all cell lines. Results showed that 10 μM IPI-549 allowed more than 80% survival on parental human colon cancer cell line SW620 and doxorubicin-resistant cell line SW620/Ad300 (Figure 1B). Results also showed that 5 μM IPI-549 allowed more than 80% survival in LLC-PK1, transfected LLC-PK-MDR1 (Figure 1C), parental human colon cancer cells S1, and mitoxantrone-resistant S1-M1–80 cells (Figure 1D).
Figure 1. Chemical structure of IPI-549 and cytotoxicity of IPI-549 on parental and drugresistant cells.

Chemical structure of IPI-549 (A). Cell survival (%) percentage was measured after treatment with IPI-549 for 72 h on parental and drug-resistant cells: SW620 and SW620/AD300 cells (B), LLC-PK1 and LLC-PK-MDR1 cells (C), S1 and S1-M1–80 cells (D). Points with error bars represent the mean ± SD for independent determinations in triplicate. The above figures are representative of three independent experiments.
To determine if IPI-549 could reverse ABCB1-mediated MDR, modified MTT assays were performed combining IPI-549 with chemotherapeutic ABCB1 substrates. SW620/Ad300 cells without ABCB1 inhibition (IPI-549 or verapamil) showed resistance folds of 65.8, 104.8, 82.0, and 48.0 to doxorubicin, vincristine, paclitaxel, and colchicine, respectively, as compared to SW620 cells (Table 1). IPI-549 at 5 and 10 μM significantly sensitized SW620/Ad300 cells to ABCB1 substrates (Table 1). Similar reductions in IC50 values to ABCB1 substrates were observed in transfected LLC-PK-MDR1 cells on exposure to 2.5 and 5 μM IPI-549 (Table 2). Verapamil was used as a positive control ABCB1 reversal agent, while cisplatin was used as a negative control since it is not a substrate of ABCB1. These results suggest that IPI-549 is able to re-sensitize ABCB1-overexpressing cells to ABCB1 chemotherapeutic substrates in both drug-selected and transfected cell lines.
Table 1. The effect of IPI-549 on the resistance fold of MDR ABCBl-overexpressing cells.
| Drug | SW620 IC50 ± SD (μM) | RFa | SW620/Ad300 IC50 ± SD (μM) | RFa |
|---|---|---|---|---|
| Doxorubicin | 0.0912 ± 0.001 | 1.0 | 6.0037 ± 0.107 | 65.7 |
| +IPI-549 5 μM | 0.0924 ± 0.005 | 1.0 | 0.5607 ± 0.005 | 6.1* |
| +IPI-549 10 μM | 0.0809 ± 0.003 | 1.0 | 0.0935 ± 0.009 | 1.0* |
| +Verapamil 10 μM | 0.0878 ± 0.001 | 1.0 | 0.0793± 0.007 | 0.9* |
| Vincristine | 0.0075 ± 0.001 | 1.0 | 0.7858 ± 0.072 | 104.7 |
| +IPI-549 5 μM | 0.0064 ± 0.003 | 1.0 | 0.0212 ± 0.002 | 2.8* |
| +IPI-549 10 μM | 0.0072 ± 0.004 | 1.0 | 0.0098 ± 0.001 | 1.3* |
| +Verapamil 10 μM | 0.0068 ± 0.001 | 1.0 | 0.0083 ± 0.001 | 1.1* |
| Paclitaxel | 0.0076 ± 0.006 | 1.0 | 0.7133 ± 0.052 | 82.0 |
| +IPI-549 5 μM | 0.0072 ± 0.001 | 1.0 | 0.0582 ± 0.003 | 6.6* |
| +IPI-549 10 μM | 0.0078 ± 0.002 | 1.0 | 0.0067 ± 0.003 | 0.7* |
| +Verapamil 10 μM | 0.0068 ± 0.001 | 1.0 | 0.0080 ± 0.005 | 0.9* |
| Colchicine | 0.0069 ± 0.003 | 1.0 | 0.8114 ± 0.010 | 47.9 |
| +IPI-549 5 μM | 0.0069 ± 0.003 | 1.0 | 0.0682 ± 0.002 | 4.0* |
| +IPI-549 10 μM | 0.0066 ± 0.002 | 1.0 | 0.0228 ± 0.003 | 1.3* |
| +Verapamil 10 μM | 0.0072 ± 0.002 | 1.0 | 0.0921 ± 0.006 | 5.4* |
| Cisplatin | 15.291 ± 0.079 | 1.0 | 28.100 ± 0.083 | 1.8 |
| +IPI-549 5 μM | 28.733 ± 0.023 | 1.0 | 29.794 ± 0.049 | 1.9 |
| +IPI-549 10 μM | 20.835 ± 0.065 | 1.0 | 22.807 ± 0.059 | 1.4 |
| +Verapamil 10 μM | 19.934 ± 0.028 | 1.0 | 29.350 ± 0.061 | 1.9 |
IC50 values are represented as a mean ± SD of three independent experiments performed in triplicate.
Resistance fold (RF) was the IC50 values of the SW620/Ad300 cells with and without IPI-549 or verapamil divided by the IC50 value of SW620 cells without reversing agents.
P < 0.01 versus the control group.
Table 2. The effect of IPI-549 on the resistance fold of ABCBl-transfected cells.
| Drug | LLC-PK1 IC50 ± SD (μM) | RFa | LLC-PK-MDR1 IC50 ± SD (μM) | RFa |
|---|---|---|---|---|
| Doxorubicin | 0.0808 ± 0.002 | 1.0 | 1.7064 ± 0.030 | 21.1 |
| +IPI-549 5 μM | 0.0873 ± 0.003 | 1.0 | 0.2365 ± 0.035 | 2.9* |
| +IPI-549 10 μM | 0.0847 ± 0.001 | 1.0 | 0.1383 ± 0.011 | 1.7* |
| +Verapamil 10 μM | 0.0817 ± 0.002 | 1.0 | 0.2276 ± 0.034 | 2.8* |
| Vincristine | 0.0298 ± 0.007 | 1.0 | 0.8584 ± 0.023 | 28.7 |
| +IPI-549 5 μM | 0.0269 ± 0.003 | 1.0 | 0.0750 ± 0.002 | 2.5* |
| +IPI-549 10 μM | 0.0213 ± 0.001 | 1.0 | 0.0317 ± 0.001 | 1.1* |
| +Verapamil 10 μM | 0.0245 ± 0.003 | 1.0 | 0.0201 ± 0.001 | 0.6* |
| Paclitaxel | 0.0906 ± 0.006 | 1.0 | 8.0263 ± 0.029 | 93.1 |
| +IPI-549 5 μM | 0.0979 ± 0.003 | 1.0 | 0.3169 ± 0.025 | 3.3* |
| +IPI-549 10 μM | 0.0965 ± 0.003 | 1.0 | 0.0605 ± 0.006 | 0.7* |
| +Verapamil 10 μM | 0.0947 ± 0.003 | 1.0 | 0.0757 ± 0.001 | 0.8* |
| Colchicine | 0.0765 ± 0.001 | 1.0 | 0.9360 ± 0.041 | 24.5 |
| +IPI-549 5 μM | 0.0896 ± 0.002 | 1.0 | 0.6686 ± 0.042 | 9.1* |
| +IPI-549 10 μM | 0.0850 ± 0.003 | 1.0 | 0.0866 ± 0.003 | 1.1* |
| +Verapamil 10 μM | 0.0846 ± 0.004 | 1.0 | 0.2959 ± 0.021 | 5.2* |
| Cisplatin | 20.628 ± 0.035 | 1.0 | 27.087 ± 0.083 | 1.3 |
| +IPI-549 5 μM | 20.235 ± 0.018 | 1.0 | 21.565 ± 0.015 | 1.0 |
| +IPI-549 10 μM | 20.923 ± 0.016 | 1.0 | 37.754 ± 0.010 | 1.8 |
| +Verapamil 10 μM | 20.244 ± 0.023 | 1.0 | 26.888 ± 0.039 | 1.3 |
IC50 values are represented as a mean ± SD of three independent experiments performed in triplicate.
Resistance fold (RF) was the IC50 values of the LLC-PK-MDR1 cells with and without IPI-549 or verapamil divided by the IC50 value of LLC-PK1 cells without reversing agents.
P < 0.01 versus the control group.
3.2. IPI-549 partially sensitizes ABCG2-overexpressing cells to ABCG2 chemotherapeutic substrates.
To determine if IPI-549 could reverse ABCG2-mediated MDR, modified MTT assays were performed combining IPI-549 with chemotherapeutic ABCG2 substrates on mitoxantrone-resistant S1-M1–80 and parental S1 cells (Table 3). S1-M1–80 cells showed resistance fold to mitoxantrone of 1032.8 compared to S1 cells. IPI-549 at 2.5 and 5 μM partially decreased the resistance fold of S1-M1–80 cells to mitoxantrone. Nilotinib at 5 μM was used as a positive control ABCG2 reversal agent (Table 3). The results suggest that IPI-549 partially reverses resistance to mitoxantrone in ABCG2-overexpressing cells.
Table 3. The effect of IPI-549 on the resistance fold of MDR ABCG2-overexpressing cells.
| Drug | S1 IC50 ± SD (μM) | RFa | S1-M1–80 IC50 ± SD (μM) | RFa |
|---|---|---|---|---|
| Mitoxantrone | 0.0089 ± 0.001 | 1.0 | 9.1993 ± 0.027 | 1032.8 |
| +IPI-549 2.5 μM | 0.0073 ± 0.015 | 1.0 | 6.1125 ± 0.002 | 686.2 |
| +IPI-549 5 μM | 0.0084 ± 0.075 | 1.0 | 5.0815 ± 0.029 | 570.4 |
| +Nilotinib 5 μM | 0.0073 ± 0.007 | 1.0 | 0.0830 ± 0.005 | 9.32* |
IC50 values are represented as a mean ± SD of three independent experiments performed in triplicate.
Resistance fold (RF) was the IC50 values of the S1-M1–80 cells with and without IPI- 549 or verapamil divided by the IC50 value of S1 cells without reversing agents.
P < 0.01 versus the control group.
3.3. IPI-549 increases the accumulation and inhibits the efflux of [3H]-paclitaxel in ABCB1-overexpressing cells.
To investigate the mechanism by which IPI-549 sensitizes ABCB1-overexpressing cells to ABCB1 chemotherapeutic substrates, we examined the effect of IPI-549 on the accumulation of [3H]-paclitaxel in ABCB1-overexpressing cells. Intracellular [3H]-paclitaxel was measured in ABCB1-overexpressing cells in the presence or absence of IPI-549 or verapamil (Figure 2A).The intracellular concentration of [3H]-paclitaxel was significantly increased with 5 and 10 μM IPI-549 in SW620/Ad300 cells (Figure 2A). The intracellular concentration of [3H]-paclitaxel was also significantly increased with 10 μM verapamil in SW620/Ad300 cells (Figure 2A). The results suggest that IPI-549 increases the intracellular accumulation of [3H]-paclitaxel in SW620/Ad300 cells in a concentration-dependent manner. Neither IPI-549 nor verapamil significantly altered the intracellular levels of [3H]-paclitaxel in parental SW620 cells.
Figure 2. Effect of IPI-549 on the accumulation and efflux of [3H]-paclitaxel and the effect of IPI-549 on ABCB1-ATPase activity.

The effect of IPI-549 on accumulation of [3H]-paclitaxel in SW620 and SW620/AD300 cells (A) and effect of IPI-549 on the efflux of [3H]-paclitaxel on SW620 (B) and SW620/Ad300 cells (C). A time course (0, 30, 60, 120 min) versus percentage of intracellular [3H]-paclitaxel remaining (%) was plotted. Columns are the mean of triplicate determinations; the error bars represent the SD, ** P<0.01, * P<0.05 versus the control group without the reversal agent. Verapamil 10 μM was used as positive control. Effect of IPI-549 on ABCB1-ATPase activity (D). Crude membranes (10 μg protein/reaction) from High Five cells expressing ABCB1 were incubated with increasing concentrations of IPI-549 (0–40 μM).The inset shows stimulation of ATP hydrolysis at concentration of 0–10 μM IPI-549. The mean values are plotted and error bars depict SD.
To test if the increased accumulation of [3H]-paclitaxel is due to inhibition of efflux activity by IPI-549, the efflux assay was performed (Figure 2B and 2C). The remaining amount of intracellular [3H]-paclitaxel in SW620/Ad300 cells (Figure 2C) was significantly lower compared to the parental SW620 cells (Figure 2B) due to the active transport of [3H]-paclitaxel out of the cell by the ABCB1 transporter. The remaining intracellular [3H]-paclitaxel was significantly increased with 5 and 10 μM IPI-549 in SW620/Ad300 cells after 120 minutes (Figure 2C). These results were comparable to the decrease in efflux of [3H]-paclitaxel by verapamil at 10 μM.
3.4. IPI-549 stimulates ABCB1-ATPase activity.
The drug efflux function of ABCB1 is coupled to ATP hydrolysis which can be stimulated in the presence of ABCB1 substrates. To assess the effect of IPI-549 on ABCB1-ATPase activity, the hydrolysis of ATP by ABCB1-ATPase was measured in the presence of 0–40 μM IPI-549 (Figure 2D). IPI-549 stimulated ABCB1-mediated ATPase activity in a concentration-dependent manner with a maximal stimulation of 4-fold basal activity at 40 μM IPI-549. At the 10 μM IPI-549 concentration used in this study, the inset in Figure 4 shows that the concentration required for 50% stimulation is 4.1 μM IPI-549. This suggests that IPI-549 interacts with the ABCB1 substrate binding site acting as a competitive substrate and stimulating ABCB1-ATPase activity.
Figure 4. Induced Fit Docking (IFD) analysis of IPI-549 to ABCB1.

The best-scored binding pose of IPI-549 within human homology ABCB1 (Glide gscore: −14.602 kcal/mol) predicted by IFD computation is shown in (A). The location of the IPI-549 molecule as a ball and stick model is shown within the ABCB1 internal cavity, with the atoms colored as carbon–orange, hydrogen– white, oxygen–red, nitrogen–blue. Amino acids that have hydrogen bonding or π-π stacking interactions with IPI-549 are shown and depicted as sticks with the same color scheme as above except that carbon atoms are represented in grey. Only polar hydrogens are shown. Dotted yellow lines indicate hydrogen-bonding interactions, while dotted blue lines indicate π -π stacking interactions. Values of the relevant distances are given in Å. (B) The two-dimensional ligand−receptor interaction diagram of IPI-549 and human ABCB1. The amino acids within 4 Å are shown as colored bubbles, blue indicates polar residues, and green indicates hydrophobic residues. Grey circles indicate solvent exposure. Hydrogen bonds are shown by the purple arrow, and π-π stacking aromatic interactions are shown by green lines.
3.5. Effects of IPI-549 on the expression level and subcellular localization of ABCB1
Immunoblot analysis indicated a band with an approximate molecular weight of 170 kDa in the SW620/Ad300 cell lysate and no such band in the parental SW620 cell lysate, suggesting the presence of ABCB1 protein. To confirm that the reversal of resistance in SW620/Ad300 cells was not due to a decreased expression of ABCB1 caused by IPI-549, SW620/Ad300 cells were incubated 72 h under 10 μM IPI-549. There was no significant change in expression of ABCB1 in the SW620/Ad300 cells over 72 h of exposure to 10 μM IPI-549 (Figure 3A and 3B). Analysis of the immunofluorescence imaging of ABCB1 after exposure to 10 μM IPI-549 over 72 h showed no change in subcellular localization of ABCB1 from the cellular membrane, consistent with the role of ABCB1 as a membrane-bound efflux pump (Figure 3C).
Figure 3. Effect of IPI-549 on the expression and subcellular localization of ABCB1 on MDR SW620/Ad300 cells.

SW620/Ad300 cells were treated with 10 μM IPI-549 for 0, 24, 48, and 72 h. Equal amounts (80 μg) of cell lysate were loaded into each well and subjected to western blot analysis (A). Bar graphs of ABCB1 blot analysis were performed with ImageJ software of the National Institutes of Health (B). The differences were not statistically significant (P > 0.05). The effect of 10 μM IPI-549 on the subcellular localization of ABCB1 on SW620/Ad300 (C). Nuclear staining was performed with propidium iodide (PI); ABCB1 staining was performed with green fluorescence protein (GFP)-labeled ABCB1 antibody.
3.6. IPI-549 Docking analysis to human homology ABCB1
To understand the binding mechanism of IPI-549 to the human homology ABCB1 model at the molecular level, Induced Fit Docking (IFD) studies were performed on the ABCB1 substrate binding domains. The best-scored binding pose of IPI-549 within human homology ABCB1 gave a Glide gscore of −14.602 kcal/mol predicted by IFD computation as shown in Figure 4A and 4B.
The IPI-549 core structure was majorly stabilized in a hydrophobic cavity lined by a number of aromatic and hydrophobic residues, including Leu65, Met69, Phe72, Phe303, Ile306, Tyr307, Tyr310, Phe335, Phe336, Leu339, Ile340, Phe343, Leu724, Phe728, Ala729, Phe732, Tyr953, Phe957, Leu975, Phe978, Phe983, Met 986, Ala987, and Val 991 (Figure 7B). It was predicted that the carbonyl oxygen of the carboxamide group in IPI-549 was involved in hydrogen bonding interactions with the hydroxyl group on the side chain of Tyr307 (-CO…HOGln725, 1.75 Å), and the amino group on the side chain of Gln725 (-CO…H2N-Gln725, 1.82 Å), respectively. Another hydrogen bonding interaction was found between the 2-amino group on the pyrazole ring of IPI-549 and the carbonyl oxygen on the side chain of Gln990 (-NH2…OCGln990, 2.07 Å).
Figure 7.

The effect of vehicle, IPI-549, paclitaxel, and combination (IPI-549 + paclitaxel) on the body weight (A), white blood cell count (B) and platelet count (C) in athymic mice at the end of the 15-day treatment period. The differences were not statistically significant (P > 05).
Several π-π stacking interactions were predicted between IPI-549 and several aromatic residues in the human homology ABCB1 drug binding pocket. The 1-methyl-pyrazole ring in the IPI-549 structure was engaged in two π-π stacking interactions with the phenyl ring of Phe336 and Phe983, respectively. The benzene ring of the 1,2 dihydroisoquinoline group in IPI-549 formed a π-π stacking interaction with the phenyl ring of Phe343. The 2-phenyl ring attached to the 1,2 dihydroisoquinoline group in IPI-549 also formed a π-π stacking interaction with Phe728.
The overall docking pose indicates significant hydrophobic, electrostatic, and aromatic interactions between IPI-549 and human homology ABCB1.
3.7. IPI-549 potentiates the anti-tumor activity of paclitaxel in an ABCB1-overexpressing tumor xenograft model.
After evaluating the in vitro reversal effects of IPI-549 on ABCB1-overexpressing SW620/Ad300 colon cancer cells, we further translated these findings in an in vivo tumor xenograft model (Figures 5 and 6).
Figure 5. Effect of IPI-549 and paclitaxel on the growth of parental SW620 tumors in athymic mice.

The images of excised SW620 tumors implanted subcutaneously in athymic NCR mice (n=6) treated with vehicle, IPI-549, paclitaxel, and the combination of IPI-549 and paclitaxel (A). Changes in tumor volume over time following the implantation (B). Data points represent the mean tumor volumes for each treatment group. The mean weight of the excised SW620 tumors from the mice treated with vehicle, IPI-549, paclitaxel, and the combination of IPI-549 and paclitaxel at the end of the 15-day treatment period (C). Error bars, SD. * p < 0.05 versus the vehicle group.
Figure 6. Effect of IPI-549 and paclitaxel on the growth of MDR SW620/Ad300 tumors in athymic mice.

The images of excised SW620/Ad300 tumors implanted subcutaneously in athymic NCR mice (n=6) treated with vehicle, IPI-549, paclitaxel, and the combination of IPI-549 and paclitaxel (A). Changes in tumor volume over time following the implantation (B). Data points represent the mean tumor volumes for each treatment group. The mean weight of the excised SW620/Ad300 tumors from the mice treated with vehicle, IPI-549, paclitaxel, and the combination of IPI-549 and paclitaxel at the end of the 15-day treatment period (C). Error bars, SD. * p < 0.05 versus the vehicle group. # p < 0.05 versus the paclitaxel group
IPI-549 alone caused no significant growth inhibition of SW620 tumors (17% IRV and 27% IRW) or SW620/Ad300 tumors (24% IRV and 17% IRW) (Table 4). Treatment with paclitaxel alone caused significant growth inhibition in SW620 tumors (58% IRV and 68% IRW) while the combination of IPI-549 with paclitaxel slightly enhanced paclitaxel’s effects (71% IRV and 69% IRW) (Table 4). The parental SW620 tumors remained sensitive to paclitaxel with or without IPI-549. On the MDR SW620/Ad300 tumors, paclitaxel alone showed less anti-tumor effect (42% IRV and 37 %IRW) compared to the combination of IPI-549 with paclitaxel (74% IRV and 72% IRW). This showed that IPI-549 potentiates the anti-tumor effect of paclitaxel on MDR SW620/Ad300 tumors. There was no significant change in mouse body weight (Figure 7A), white blood cell count (Figure 7B) or platelet count (Figure 7C) in all groups after the 15-day treatment period.
Table 4. The ratios of growth inhibition (IR) for tumor volume (IRV) and tumor weight (IRW) of SW620 and SW620/Ad300 xenograft tumors at the end of the 15-day treatment period.
| SW620 tumors | SW620 tumors | SW620/Ad300 tumors | SW620/Ad300 tumors | |
|---|---|---|---|---|
| IRVa(%) | IRWb(%) | IRVa(%) | IRWb(%) | |
| IPI-549 | 17 | 27 | 24 | 17 |
| Paclitaxel | 58* | 68* | 42 | 37 |
| Combination | 71* | 69* | 74*# | 72*# |
IRV and IRW are calculated as compared with the vehicle-only control group as described in the Materials and Methods section.
IRV and IRW are calculated as compared with the vehicle-only control group as described in the Materials and Methods section.
Significant (P < 0.05) as compared with vehicle-only group
Significant (P < 0.05) as compared with paclitaxel-only group
4. Discussion
The targeting of immune cells with monoclonal immune checkpoint blocker (ICB) antibodies to stimulate an immune response or to inhibit immune suppression has shown the most promise with six monoclonal ICB antibodies approved by the FDA targeting immune checkpoints for treating cancer patients [41]. Currently FDA-approved ICBs are ipilimumab (anti-CTLA-4), pembrolizumab (anti-PD-1), nivolumab (anti-PD-1), atezolizumab (anti-PD-L1), avelumab (anti-PD-L1), and durvalumab (anti-PD-L1) [41]. But the low response rate of patients to ICB as monotherapy has led to clinical trials combining immunogenic chemotherapeutic agents with ICB-based immunotherapy [42].
Aside from its cytotoxic effect on cancer cells, chemotherapy is also being utilized for priming the immune system for a more robust immune attack on tumor cells [42].Preconditioning the immunosuppressive tumor microenvironment (TME) with single low-dose doxorubicin or paclitaxel, both of which are substrates of P-gp, has been shown to enhance adoptive T cell transfer therapy in a mouse model, while reducing the numbers of immunosuppressive myeloid-derived suppressor cells (MDSC) and regulatory T lymphocytes (Tregs), and increasing the number of tumor-infiltrating cytotoxic T lymphocytes (CTLs) [43]. Tumors lacking T cell infiltration, referred to as “cold” tumors, are often not responsive to ICB therapy. As an agent inducing immunogenic cell death (ICD) [44], doxorubicin was used to prime a microenvironment lacking T cells, converting it into a “hot” tumor displaying antitumor T cell immunity and responsiveness to combined ICB therapy of anti-PD-1 and anti-CTLA-4 monoclonal antibodies [45]. Because of its ICD inducing ability, doxorubicin at low concentrations is being tested in combination with ICB nivolumab in an adaptive phase 2 randomized trial of nivolumab after induction of ICD by doxorubicin in the treatment of triple-negative breast cancer (TNBC) [46] (TONIC trial; NCT02499367). There is also an ongoing randomized phase 3 trial of avelumab, an anti-PD-L1 monoclonal antibody, in combination with pegylated liposomal doxorubicin in patients with platinum-resistant/refractory recurrent epithelial ovarian cancer (NCT02580058) [47].
Paclitaxel is also being tested in combination with immunotherapy because of its immunomodulating effect of promoting proinflammatory cytokine secretion and enhancing the priming and lytic activity of CD8+ T cells [48]. Paclitaxel administered prior to surgery in breast cancer patients was shown to increase tumor-infiltrating lymphocytes which correlated with clinical response due to the induction of antitumor T cells [49]. In breast cancer patients immediately after surgery, the sequential dosing of doxorubicin, cyclophosphamide, and paclitaxel caused increased T cell and NK proliferation and immune-mediated tumor cell lysis [50]. The increased influx of tumor-infiltrating lymphocytes was correlated with positive responses to neoadjuvant anthracycline/taxane-based chemotherapy in breast cancer patients [51]. Paclitaxel in combination with anti-PD-1 antibody has been shown to enhance tumor regression, prolong mouse survival by NF-κB signal activation, and increase the number of tumor-infiltrating lymphocytes [52].
Nanoparticle albumin bound (nab)-paclitaxel has become the preferred form for combining with immunotherapy since it does not require immune suppressive corticosteroid pretreatment for hypersensitivity reactions caused by paclitaxel [53], thus preserving the patient’s immunity. In a phase 1b clinical trial, nab-paclitaxel combined with atezolizumab, an anti PD-L1 monoclonal antibody, caused marked improvement in the objective response rate (ORR) in patients with metastatic TNBC [54]. It has led to a large phase 3 double-blinded randomized clinical trial (IMpassion130) of nab-paclitaxel combined with atezolizumab as a first-line therapy in patients of metastatic TNBC (NCT02425891) [55].
Similar phase 3 randomized trials are underway in which atezolizumab is being tested in combination with carboplatin/nab-paclitaxel or carboplatin/paclitaxel for patients with stage IV untreated non-squamous NSCLC (IMpower131, NCT02367794) [55]. Numerous other phase 1, 2, and 3 trials are underway combining paclitaxel or nab-paclitaxel with immune checkpoint inhibitors nivolumab (anti-PD1), pembrolizumab (anti-PD1), atezolizumab (anti-PD-L1), durvalumab (anti-PD-L1), and ipilimumab (anti-CTLA-4) monoclonal antibodies for patients with advanced/metastatic NSCLC, metastatic breast cancer, metastatic prostate cancer, metastatic or recurrent TNBC, and various other metastatic solid tumors [55].
Pharmacological inhibition of PI3Kγ by IPI-549 has been shown to stimulate and prolong NF-κB activation while inhibiting C/EBPβ activation, thus restoring cytotoxic T cell activation [21, 22]. In a further study, IPI-549 alone was shown to inhibit tumor growth in breast cancer, melanoma, colon cancer, and lung cancer tumor models while reducing lung metastasis [22]. IPI-549 switched the activity of immune suppressive M2 macrophages into a more inflammatory anti-tumor M1 phenotype which correlated with an increase in cytotoxic T lymphocyte infiltration at the tumor site and increased expression of granzyme B [22]. When IPI-549 was combined with ICB nivolumab in ICB-resistant breast cancer, melanoma, and lung cancer models there was an increased M1/M2 ratio and improved effector T cell function [22]. In testing IPI-549 in combination with double ICB therapy of anti-CTLA-4 combined with antiPD-1, there was complete remission in 30% of breast cancer models and remission of 80% in melanoma models [22].
In a phase 1/1b clinical trial (NCT02637531), IPI-549 was tested as monotherapy and in combination with nivolumab in patients with advanced solid tumors who had received prior treatment. Two patients demonstrated preliminary partial responses, one patient with adrenocortical carcinoma and one patient with gallbladder carcinoma [23]. Patient enrollment is currently underway for a phase 2 trial combining IPI-549 with nivolumab [23].
In our study, we have demonstrated that IPI-549 acts as a chemosensitizing agent to the P-gp-overexpressing MDR phenotype of cancer cells. In our cell viability assay, IPI-549 potentiated the cytotoxicity of ABCB1 substrate drugs in ABCB1-overexpressing cancer cell lines and ABCB1-transfected cell lines. IPI-549 significantly increased the intracellular concentration of [3H]-paclitaxel in SW620/Ad300 cells and inhibited the efflux of [3H]-paclitaxel out of SW620/Ad300 cells. IPI-549 caused no significant change in expression level of ABCB1 on western blot, and there was no detectable change in subcellular localization of ABCB1 as evaluated by immunofluorescence staining with ABCB1 remaining at the extracellular membrane. The overall docking analysis showed significant hydrophobic, electrostatic, and aromatic interactions between IPI-549 and human homology ABCB1 in the transmembrane domain of P-gp.
Our in vivo study showed that IPI-549 potentiates the anti-tumor effect of paclitaxel with a notable decrease of tumor sizes and weights in the combination group (IPI-549 + paclitaxel) in the MDR SW620/Ad300 tumors. IPI-549 alone had no significant effect on slowing the growth of parental SW620 and MDR SW620/Ad300 tumors (Table 4). Compared to SW620 tumors, SW620/Ad300 tumors were resistant to paclitaxel alone due to the overexpression of ABCB1 (Table 4). Although SW620/Ad300 tumor growth was not significantly controlled by either IPI-549 or paclitaxel alone, the growth of SW620/Ad300 tumors was controlled by the combination of IPI-549 and paclitaxel (Table 4). These results showed that IPI-549 potentiates the anti-tumor effect of paclitaxel on ABCB1-overexpressing MDR SW620/Ad300 tumors.
Doxorubicin and paclitaxel, chemotherapeutic substrates of P-gp, are two of the most widely used agents in the treatment of cancer and both have immunogenic properties [42, 56, 57] for which they are being tested in combination with ICB-based immunotherapy [46, 47]. Therefore, combining IPI-549 with doxorubicin can possibly decrease the dose of doxorubicin necessary for inducing immunogenic cell death [56, 57] in MDR cancer cells, avoiding doxorubicin-induced cardiotoxicity [58], and stimulating tumor infiltrating lymphocytes and responsiveness to ICB therapy [59]. Likewise, combining IPI-549 with doxorubicin or paclitaxel can have synergistic effects in promoting an anti-tumor microenvironment by chemotherapeutic depletion of immunosuppressive MDSC [60] and Tregs [61]. De Henau et al. has shown that PI3Kγ inhibition by IPI-549 can be used to overcome resistance to immune checkpoint blocker (ICB) therapy [22]. Our preclinical study has shown that IPI-549 acts as a sensitizing agent to the MDR P-gp-overexpressing phenotype which develops in cancer cells after extended chemotherapeutic drug exposure. With clinical trials beginning to integrate ICB immunotherapy into standard-of-care immunogenic chemotherapy to improve patient outcomes [55, 59, 62, 63], our findings support the rationale of adding IPI-549 to both the chemotherapeutic and immunotherapeutic aspects of cancer combination treatment strategies.
Highlights.
IPI-549 sensitizes multidrug resistant cancer cells to P-glycoprotein chemotherapeutic substrate drugs.
IPI-549 increases the intracellular concentration and inhibits the efflux of paclitaxel from P-glycoprotein overexpressing cancer cells.
IPI-549 stimulates ABCB1-ATPase activity as a competitive substrate of Pglycoprotein.
IPI-549 potentiates the anti-tumor effect of paclitaxel on P-glycoprotein-mediated multidrug resistant cancer cells in a mouse tumor xenograft model.
5. Acknowledgements
We are thankful for Drs. Susan E. Bates and Robert W. Robey (NIH, MD) for providing the SW620, SW620/Ad300, S1, and S1-M1-80 cells. We thank Dr. Micheal M. Gottesman (NIH, Bethesda, MD) for providing the LLC-PK1 and LLC-PK-MDR1 cells. We thank Dr. Tanaji T. Talele (St. John’s University, Queens, New York) for providing the computational resources of induced-fit docking analysis. We thank Dr. Vikas Dukhande (St. John’s University, Queens, NY) for the use of his EVOS FL Auto 2 Cell Imaging System fluorescent microscope.
6. Funding
This work was supported by funds from NIH (No. 1R15CA143701 and No. 1R15GM116043-01) and St. John’s University Research Seed Grant (No. 579-1110-7002) to Z. Chen.
Abbreviations:
- ABC
ATP-binding cassette
- BCRP
breast cancer resistance protein
- CTLA-4
cytotoxic T-lymphocyte associated protein 4
- CTL
cytotoxic T lymphocyte
- DC
dendritic cell
- ICB
immune checkpoint blocker
- ICD
immunogenic cell death
- MDSC
myeloid-derived suppressor cells
- NK
natural killer cells
- NSCLC
non-small cell lung cancer
- PD-1
programmed death protein 1
- PD-L1
programmed death-ligand 1
- P-gp
Permeability-glycoprotein (P-glycoprotein)
- PI3Kγ
phosphoinositide 3-kinase gamma
- Tregs
regulatory T lymphocytes
- TAA
tumor-associated antigens
- TME
tumor microenvironment
- TNBC
triple-negative breast cance
Footnotes
7. Conflict of interests
The authors declare that they have no conflicting interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
8. References
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2018, CA. Cancer J. Clin 68 (2018) 7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- [2].DeVita VT, Rosenberg SA, Two hundred years of cancer research, N. Engl. J. Med 366 (2012) 2207–2214. doi: 10.1056/NEJMra1204479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Thomas H, Coley HM, Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein, Cancer Control J. Moffitt Cancer Cent. 10 (2003) 159–165. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- [4].Zahreddine H, Borden KLB, Mechanisms and insights into drug resistance in cancer, Front. Pharmacol. 4 (2013) 28. doi: 10.3389/fphar.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gottesman MM, Fojo T, Bates SE, Multidrug resistance in cancer: role of ATP-dependent transporters, Nat. Rev. Cancer 2 (2002) 48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- [6].Krishna R, Mayer LD, Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs, Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci 11 (2000) 265–283. [DOI] [PubMed] [Google Scholar]
- [7].Gottesman MM, Pastan I, Biochemistry of multidrug resistance mediated by the multidrug transporter, Annu. Rev. Biochem. 62 (1993) 385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- [8].Gottesman MM, Ludwig J, Xia D, Szakács G, Defeating drug resistance in cancer, Discov. Med 6 (2006) 18–23. [PubMed] [Google Scholar]
- [9].Cole SP, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC, Stewart AJ, Kurz EU, Duncan AM, Deeley RG, Overexpression of a transporter gene in a multidrugresistant human lung cancer cell line, Science. 258 (1992) 1650–1654. [DOI] [PubMed] [Google Scholar]
- [10].Doyle LA, Ross DD, Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2), Oncogene. 22 (2003) 7340–7358. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- [11].Juliano RL, Ling V, A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants, Biochim. Biophys. Acta 455 (1976) 152–162. [DOI] [PubMed] [Google Scholar]
- [12].Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM, Biochemical, cellular, and pharmacological aspects of the multidrug transporter, Annu. Rev. Pharmacol. Toxicol 39 (1999) 361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- [13].Jones PM, George AM, Opening of the ADP-bound active site in the ABC transporter ATPase dimer: evidence for a constant contact, alternating sites model for the catalytic cycle, Proteins. 75 (2009) 387–396. doi: 10.1002/prot.22250. [DOI] [PubMed] [Google Scholar]
- [14].Beretta GL, Cassinelli G, Pennati M, Zuco V, Gatti L, Overcoming ABC transportermediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents, Eur. J. Med. Chem 142 (2017) 271–289. doi: 10.1016/j.ejmech.2017.07.062. [DOI] [PubMed] [Google Scholar]
- [15].Genovese I, Ilari A, Assaraf YG, Fazi F, Colotti G, Not only P-glycoprotein: Amplification of the ABCB1-containing chromosome region 7q21 confers multidrug resistance upon cancer cells by coordinated overexpression of an assortment of resistance-related proteins, Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother 32 (2017) 23–46. doi: 10.1016/j.drup.2017.10.003. [DOI] [PubMed] [Google Scholar]
- [16].Pérez-Sayáns M, Somoza-Martín JM, Barros-Angueira F, Diz PG, Rey JMG, García-García A, Multidrug resistance in oral squamous cell carcinoma: The role of vacuolar ATPases, Cancer Lett. 295 (2010) 135–143. doi: 10.1016/j.canlet.2010.03.019. [DOI] [PubMed] [Google Scholar]
- [17].Verrelle P, Meissonnier F, Fonck Y, Feillel V, Dionet C, Kwiatkowski F, Plagne R, Chassagne J, Clinical relevance of immunohistochemical detection of multidrug resistance Pglycoprotein in breast carcinoma, J. Natl. Cancer Inst 83 (1991) 111–116. [DOI] [PubMed] [Google Scholar]
- [18].Marie JP, P-glycoprotein in adult hematologic malignancies, Hematol. Oncol. Clin. North Am 9 (1995) 239–249. [PubMed] [Google Scholar]
- [19].Leighton JC, Goldstein LJ, P-glycoprotein in adult solid tumors. Expression and prognostic significance, Hematol. Oncol. Clin. North Am 9 (1995) 251–273. [PubMed] [Google Scholar]
- [20].Kathawala RJ, Gupta P, Ashby CR, Chen Z-S, The modulation of ABC transportermediated multidrug resistance in cancer: a review of the past decade, Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother 18 (2015) 1–17. doi: 10.1016/j.drup.2014.11.002. [DOI] [PubMed] [Google Scholar]
- [21].Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, Schmid MC, Pink M, Winkler DG, Rausch M, Palombella VJ, Kutok J, McGovern K, Frazer KA, Wu X, Karin M, Sasik R, Cohen EEW, Varner JA, PI3Kγ is a molecular switch that controls immune suppression, Nature. 539 (2016) 437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, Budhu S, Ghosh A, Pink M, Tchaicha J, Douglas M, Tibbitts T, Sharma S, Proctor J, Kosmider N, White K, Stern H, Soglia J, Adams J, Palombella VJ, McGovern K, Kutok JL, Wolchok JD, Merghoub T, Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells, Nature. 539 (2016) 443–447. doi: 10.1038/nature20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sullivan R, Hong DS, Tolcher AW, Patnaik A, Shapiro G, Chmielowski B, Ribas A, Brail LH, Roberts J, Lee L, O’Connell B, Kutok JL, Mahabhashyam S, Ullmann CD, Postow MA, Wolchok JD, Initial results from first-in-human study of IPI-549, a tumor macrophagetargeting agent, combined with nivolumab in advanced solid tumors., J Clin Oncol 36 (2018) Suppl Abstr 3013. (n.d.). http://abstracts.asco.org/214/AbstView_214_230109.html. [Google Scholar]
- [24].Dempke WCM, Fenchel K, Uciechowski P, Dale SP, Second- and third-generation drugs for immuno-oncology treatment-The more the better?, Eur. J. Cancer Oxf. Engl. 1990 74 (2017) 55–72. doi: 10.1016/j.ejca.2017.01.001. [DOI] [PubMed] [Google Scholar]
- [25].Zhang YK, Zhang XY, Zhang GN, Wang YJ, Xu H, Zhang D, Shukla S, Liu L, Yang DH, Ambudkar SV, Chen ZS, Selective reversal of BCRP-mediated MDR by VEGFR-2 inhibitor ZM323881, Biochem Pharmacol, (2017) 132:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang H, Kathawala RJ, Wang Y-J, Zhang Y-K, Patel A, Shukla S, Robey RW, Talele TT, Ashby CR, Ambudkar SV, Bates SE, Fu L-W, Chen Z-S, Linsitinib (OSI-906) antagonizes ATP-binding cassette subfamily G member 2 and subfamily C member 10-mediated drug resistance, Int. J. Biochem. Cell Biol 51 (2014) 111–119. doi: 10.1016/j.biocel.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang YK, Zhang GN, Wang YJ, Patel BA, Talele TT, Yang DH, Chen ZS, Bafetinib (INNO-406) reverses multidrug resistance by inhibiting the efflux function of ABCB1 and ABCG2 transporters, Sci Rep, 6 (2016) 25694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang H, Wang Y-J, Zhang Y-K, Wang D-S, Kathawala RJ, Patel A, Talele TT, Chen Z-S, Fu L-W, AST1306, a potent EGFR inhibitor, antagonizes ATP-binding cassette subfamily G member 2-mediated multidrug resistance, Cancer Lett. 350 (2014) 61–68. doi: 10.1016/j.canlet.2014.04.008. [DOI] [PubMed] [Google Scholar]
- [29].Ambudkar SV, Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells, Methods Enzymol. 292 (1998) 504–514. [DOI] [PubMed] [Google Scholar]
- [30].Zhang YK, Zhang H, Zhang GN, Wang YJ, Kathawala RJ, Si R, Patel BA, Xu J, Chen ZS, Semi-synthetic ocotillol analogues as selective ABCB1-mediated drug resistance reversal agents, Oncotarget, 6 (2015) 24277–24290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT, Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes, J Med Chem, 49 (2006) 6177–6196. [DOI] [PubMed] [Google Scholar]
- [32].Farid R, Day T, Friesner RA, Pearlstein RA, New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies, Bioorg Med Chem, 14 (2006) 3160–3173. [DOI] [PubMed] [Google Scholar]
- [33].Herling A, Konig M, Bulik S, Holzhutter HG, Enzymatic features of the glucose metabolism in tumor cells, FEBS J, 278 (2011) 2436–2459. [DOI] [PubMed] [Google Scholar]
- [34].Winterfeld S, Ernst S, Borsch M, Gerken U, Kuhn A, Real time observation of single membrane protein insertion events by the Escherichia coli insertase YidC, PLoS One, 8 (2013) e59023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang YJ, Zhang YK, Zhang GN, Al Rihani SB, Wei MN, Gupta P, Zhang XY, Shukla S, Ambudkar SV, Kaddoumi A, Shi Z, Chen ZS, Regorafenib overcomes chemotherapeutic multidrug resistance mediated by ABCB1 transporter in colorectal cancer: In vitro and in vivo study, Cancer Lett, 396 (2017) 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G, Structure of P-glycoprotein reveals a molecular basis for polyspecific drug binding, Science. 323 (2009) 1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Li J, Jaimes KF, Aller SG, Refined structures of mouse P-glycoprotein, Protein Sci. Publ. Protein Soc 23 (2014) 34–46. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT, Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes, J. Med. Chem 49 (2006) 6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- [39].Dai C, Tiwari AK, Wu C-P, Su X-D, Wang S-R, Liu D, Ashby CR, Huang Y, Robey RW, Liang Y, Chen L, Shi C-J, Ambudkar SV, Chen Z-S, Fu L, Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2, Cancer Res. 68 (2008) 7905–7914. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang Y-J, Huang Y, Anreddy N, Zhang G-N, Zhang Y-K, Xie M, Lin D, Yang D-H, Zhang M, Chen Z-S, Tea nanoparticle, a safe and biocompatible nanocarrier, greatly potentiates the anticancer activity of doxorubicin, Oncotarget. 7 (2015) 5877–5891. doi: 10.18632/oncotarget.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Huck BR, Kötzner L, Urbahns K, Small Molecules Drive Big Improvements in Immuno-Oncology Therapies, Angew. Chem. Int. Ed Engl 57 (2018) 4412–4428. doi: 10.1002/anie.201707816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Apetoh L, Ladoire S, Coukos G, Ghiringhelli F, Combining immunotherapy and anticancer agents: the right path to achieve cancer cure?, Ann. Oncol. Off. J. Eur. Soc. Med. Oncol 26 (2015) 1813–1823. doi: 10.1093/annonc/mdv209. [DOI] [PubMed] [Google Scholar]
- [43].Hsu F-T, Chen T-C, Chuang H-Y, Chang Y-F, Hwang J-J, Enhancement of adoptive T cell transfer with single low dose pretreatment of doxorubicin or paclitaxel in mice, Oncotarget. 6 (2015) 44134–44150. doi: 10.18632/oncotarget.6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kroemer G, Galluzzi L, Kepp O, Zitvogel L, Immunogenic cell death in cancer therapy, Annu. Rev. Immunol 31 (2013) 51–72. doi: 10.1146/annurev-immunol-032712-100008 [DOI] [PubMed] [Google Scholar]
- [45].Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, Lin Y-J, Wojtkiewicz G, Iwamoto Y, Mino-Kenudson M, Huynh TG, Hynes RO, Freeman GJ, Kroemer G, Zitvogel L, Weissleder R, Pittet MJ, Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy, Immunity. 44 (2016) 343–354. doi: 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kok Leonie Voorwerk MK, Horlings Hugo, Sikorska Karolina, Vijver Koen van der, Slagter Maarten, Warren Sarah, Ong SuFey, Wiersma Terry, Russell Nicola, Lalezari Ferry, de Maaker Michiel, Kemper Inge, Mandjes Ingrid A., Chalabi Myriam, Sonke Gabe S., Adaptive phase II randomized trial of nivolumab after induction treatment in triple negative breast cancer (TONIC trial): Final response data stage I and first translational data., J Clin Oncol 36 2018. Suppl Abstr 3013. (n.d.). http://abstracts.asco.org/214/AbstView_214_226853.html. [Google Scholar]
- [47].Pujade-Lauraine E, Fujiwara K, Dychter SS, Devgan G, Monk BJ, Avelumab (anti-PDL1) in platinum-resistant/refractory ovarian cancer: JAVELIN Ovarian 200 Phase III study design, Future Oncol. Lond. Engl (2018). doi: 10.2217/fon-2018-0070. [DOI] [PubMed] [Google Scholar]
- [48].Machiels JP, Reilly RT, Emens LA, Ercolini AM, Lei RY, Weintraub D, Okoye FI, Jaffee EM, Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice, Cancer Res. 61 (2001) 3689–3697. [PubMed] [Google Scholar]
- [49].Demaria S, Volm MD, Shapiro RL, Yee HT, Oratz R, Formenti SC, Muggia F, Symmans WF, Development of tumor-infiltrating lymphocytes in breast cancer after neoadjuvant paclitaxel chemotherapy, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 7 (2001) 3025–3030. [PubMed] [Google Scholar]
- [50].Carson WE, Shapiro CL, Crespin TR, Thornton LM, Andersen BL, Cellular immunity in breast cancer patients completing taxane treatment, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 10 (2004) 3401–3409. doi: 10.1158/1078-0432.CCR-1016-03. [DOI] [PubMed] [Google Scholar]
- [51].Denkert C, Loibl S, Noske A, Roller M, Müller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R, Hanusch C, von Törne C, Weichert W, Engels K, Solbach C, Schrader I, Dietel M, von Minckwitz G, Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer, J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 28 (2010) 105–113. doi: 10.1200/JCO.2009.23.7370. [DOI] [PubMed] [Google Scholar]
- [52].Peng J, Hamanishi J, Matsumura N, Abiko K, Murat K, Baba T, Yamaguchi K, Horikawa N, Hosoe Y, Murphy SK, Konishi I, Mandai M, Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-κB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer, Cancer Res. 75 (2015) 5034–5045. doi: 10.1158/0008-5472.CAN-14-3098. [DOI] [PubMed] [Google Scholar]
- [53].Robinson DM, Keating GM, Albumin-bound Paclitaxel: in metastatic breast cancer, Drugs. 66 (2006) 941–948. [DOI] [PubMed] [Google Scholar]
- [54].Adams S, Diamond JR, Hamiltom EP, Pohlmann PR, Tolaney SM, Molinero L, Phase Ib trial of atezolizumab in combination with nab-paclitaxel in patients with metastatic triple-negative breast cancer (mTNBC)., J. Clin. Oncol 34 No 15suppl May 20 2016. 1009–1009. (n.d.). http://ascopubs.org/doi/abs/10.1200/JCO.2016.34.15_suppl.1009. [Google Scholar]
- [55].Soliman HH, nab-Paclitaxel as a potential partner with checkpoint inhibitors in solid tumors, OncoTargets Ther. 10 (2016) 101–112. doi: 10.2147/OTT.S122974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gebremeskel S, Johnston B, Concepts and mechanisms underlying chemotherapy induced immunogenic cell death: impact on clinical studies and considerations for combined therapies, Oncotarget. 6 (2015) 41600–41619. doi: 10.18632/oncotarget.6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Galluzzi L, Senovilla L, Zitvogel L, Kroemer G, The secret ally: immunostimulation by anticancer drugs, Nat. Rev. Drug Discov 11 (2012) 215–233. doi: 10.1038/nrd3626. [DOI] [PubMed] [Google Scholar]
- [58].Volkova M, Russell R, Anthracycline Cardiotoxicity: Prevalence, Pathogenesis and Treatment, Curr. Cardiol. Rev 7 (2011) 214–220. doi: 10.2174/157340311799960645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yan Y, Kumar AB, Finnes H, Markovic SN, Park S, Dronca RS, Dong H, Combining Immune Checkpoint Inhibitors With Conventional Cancer Therapy, Front. Immunol 9 (2018). doi: 10.3389/fimmu.2018.01739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, Katsanis E, Larmonier N, Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer, Cancer Res. 74 (2014) 104–118. doi: 10.1158/0008-5472.CAN-13-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhang L, Dermawan K, Jin M, Liu R, Zheng H, Xu L, Zhang Y, Cai Y, Chu Y, Xiong S, Differential impairment of regulatory T cells rather than effector T cells by paclitaxelbased chemotherapy, Clin. Immunol. Orlando Fla 129 (2008) 219–229. doi: 10.1016/j.clim.2008.07.013. [DOI] [PubMed] [Google Scholar]
- [62].Emens LA, Middleton G, The interplay of immunotherapy and chemotherapy: harnessing potential synergies, Cancer Immunol. Res 3 (2015) 436–443. doi: 10.1158/2326-6066.CIR-15-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Fournier C, Rivera Vargas T, Martin T, Melis A, Apetoh L, Immunotherapeutic properties of chemotherapy, Curr. Opin. Pharmacol 35 (2017) 83–88. doi: 10.1016/j.coph.2017.05.003. [DOI] [PubMed] [Google Scholar]