

Abstract
Membrane proteins represent a considerable fraction of pharmaceutical drug targets. A computational technique to identify ligand binding pockets in these proteins is therefore of great importance. We recently reported such a technique called pMD-membrane that utilizes small molecule probes to detect ligand binding sites and surface hotspots on membrane proteins based on probe-based molecular dynamics simulation. The current work extends pMD-membrane to a diverse set of small organic molecular species that can be used as cosolvents during simulation of membrane proteins. We also describe a projection technique for globally quantifying probe densities on the protein surface and introduce a technique to construct surface topography maps directly from the probe-binding propensity of surface residues. The map reveals surface patterns and geometric features that aid in filtering out high probe density hotspots lacking pocketlike characteristics. We demonstrate the applicability of the extended pMD-membrane and the new analysis tool by exploring the druggability of full-length G12D, G12V, and G13D oncogenic K-Ras mutants bound to a negatively charged lipid bilayer. Using data from 30 pMD-membrane runs conducted in the presence of a 2.8 M cosolvent made up of an equal proportion of seven small organic molecules, we show that our approach robustly identifies known allosteric ligand binding sites and other reactive regions on K-Ras. Our results also show that accessibility of some pockets is modulated by differential membrane interactions.
INTRODUCTION
Identification and characterization of ligand binding sites is an essential step in structure-based drug discovery. This can be achieved computationally by blind docking and related methods such as Ligand Binding Specificity Analysis (LIBSA)1,2 and FTMAP3 or by geometric techniques such as MDpocket.4 Experimental counterparts of these techniques include fragment-based nuclear magnetic resonance (NMR) spectroscopy5,6 and multisolvent crystallography.7 For flexible targets whose ligand-binding site is not readily visible in average experimental structures, probe-based molecular dynamics (pMD) simulation is emerging as the method of choice.8–13 pMD has been applied to a number of soluble proteins (e.g., refs 9–12), but a large number of drug targets are membrane-bound. Examples include surface-bound targets such as Ras GTPases14 and transmembrane proteins such as G protein-coupled receptors (GPCRs).15 Therefore, we recently adapted pMD to be applicable to membrane proteins through the modification of selected pairwise interactions between a probe molecule and lipids.16 Others achieved the same goal by combining grand-canonical Monte Carlo and pMD.17–19 Both approaches have been shown to be effective in sampling interaction of probe molecules with membrane-bound targets. Few advantages of pMD-membrane include ease of implementation and ability to selectively prevent partitioning of probes into the bilayer core, which is important in cases where protein dynamics is coupled to that of the host membrane. The current work expands pMD-membrane to diverse molecular probes as has been done for soluble proteins.9,11,20
Mixed-probe pMD-membrane can potentially allow for a better characterization of the local surface geometry and chemical signature of druggable sites through the analysis of specific functional groups involved in probe-protein interaction. We investigated this concept using seven probe molecules of diverse chemical features (Figure 1): isobutane, acetone, acetamide, acetate, isopropyl alcohol, urea, and dimethyl sulfoxide (DMSO). These probes encompass a wide range of polarities: isobutane is hydrophobic while acetate is charged at pH 7, with the rest being variously polar and carrying key functional groups including methyl, amide, sulfonyl, carboxyl, and hydroxyl moieties. Such diversity allows for selective binding to surface pockets with distinctive chemical signatures. For example, isobutane would ideally detect hydrophobic sites that have low affinity to acetate. Importantly, these compounds represent core fragments of druglike molecules and contain many of their common functional groups.11 Moreover, they are small (only four heavy atoms, 58–78 Da) and can diffuse fast, allowing for efficient sampling of the protein surface in relatively short simulation times.
Figure 1.
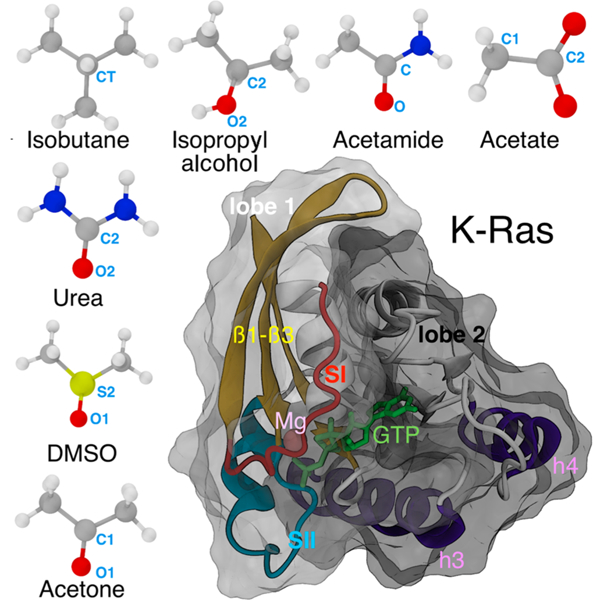
Structure of K-Ras and the small organic probe molecules used in this study. A CPK representation of isobutane, isopropyl alcohol, acetamide, acetate, acetone, DMSO, and urea with carbon, oxygen, nitrogen, sulfur, and hydrogen atoms in gray, red, blue, yellow, and white, respectively. The central labeled atoms (C, C1, C2, CT, and S2) are used for modification of LJ potentials (see Table 1), and the peripheral labeled atoms (any of the terminal carbons in the case of isobutane) are used to define orientation vectors (see Methods). The catalytic domain structure of G12D K-Ras (PDB id: 4DSO) is shown in cartoon with lobe 1 (residues 1–86) and lobe 2 (residues 87–166) highlighted as surface overlays in light gray and black, respectively. The bound GTP (sticks) and Mg (sphere) are also highlighted.
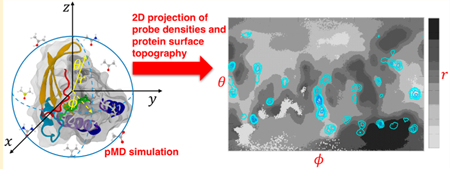
In a typical pMD approach, grid-based spatial mapping of probe occupancies is used to quantify probe densities so that hotspot regions are visualized as isosurfaces corresponding to high probe-occupancy regions.10,20–22 Probe densities can be further processed to estimate probe binding free energies.9,11,12 One challenge with this approach is the difficulty to distinguish probe-binding regions with pocketlike features (grooves or surface depressions) from those that have a high probe binding potential but lack pocketlike features (flat surfaces or protrusions). This may not be a major issue if a detailed prior knowledge of the target ligand binding pockets is available, but it is difficult to interpret probe occupancies if the target surface is not well characterized. An approach that combines probe binding with surface topography of the protein would therefore be desirable. Herein we introduce an integrated analysis technique that characterizes local geometric features of the protein surface as experienced by the probes themselves. This is achieved through a spherical projection technique to globally map probe densities on the protein surface and a surface topography map to reveal surface patterns and local curvatures that are used to filter out hotspots on flat surfaces with no pocketlike geometry. Projection of probe densities on such a two-dimensional (2D) map eliminates the need for multiple views of the protein 3D structure to recognize hotspots with desired geometric features. Our technique thus facilitates comparison of different systems and sites in terms of their potential druggability.
We demonstrate the utility of our extended pMD-membrane and analysis technique by exploring the druggability of three oncogenic K-Ras mutants bound to a negatively charged lipid bilayer. As discussed previously,16 K-Ras is a useful model system not only because it is a major anticancer drug target but also because it adopts multiple membrane orientations, and, in at least one of these, accessibility of a known ligand binding site is partially occluded by the membrane. We show that, for each mutant K-Ras, our approach accurately identifies previously characterized allosteric ligand-binding sites and other reactive surface regions.
METHODS
We simulated full-length G12D, G12V, and G13D oncogenic K-Ras mutants in a negatively charged lipid bilayer and in the presence of seven types of small organic molecules (Figure 1). Each system was simulated in two orientations of the catalytic domain with respect to the bilayer plane.23 Prior to conducting the simulations, however, we had to modify selected force field parameters to avoid probe self-aggregation and interaction with the hydrophobic core of the bilayer.
Modification of Selected Nonbonded Interaction Terms.
As described previously10,16 and detailed in Table 1, the Lennard-Jones (LJ) pairwise nonbonded interactions between selected central atoms of each probe and lipid molecules were modified using the NBFIX term implemented in the CHARMM force field.24 These included interactions between atoms CT, C2, C, C2, C1, S2, and C2 of isobutane, isopropyl alcohol, acetamide, acetate, acetone, DMSO, and urea (Figure 1, atom names based on CGenFF) and CTL2 of lipid molecules. After several trial simulations and building on our previous experience,16 we found that reducing the LJ potential well depth to 0.01 kcal/mol and increasing the minimum interparticle distance to 7–10 Å ensures that interaction of probes with lipid tails is eliminated but not their interaction with the polar bilayer surface (Figure 2B). The LJ interaction between the CT atom pairs of the water-immiscible isobutane was similarly modified to prevent them from self-aggregating.
Table 1.
Summary of Modified LJ Terms between Selected Atoms of Probe and Lipid Molecules
| before modification |
after modification |
|||||
|---|---|---|---|---|---|---|
| interaction sites on molecule 1 | interaction site on molecule 2 | offset from center of mass of molecule 2 (Å) | ||||
| isobutane:CT(CG311) | isobutane:CT(CG311) | 0.38 | 4.00 | 0.0320 | 10.0 | 0.0100 |
| POPC/POPS:Ca(CTL2) | isobutane:CT(CG311) | 0.38 | 4.01 | 0.0432 | 10.0 | 0.0100 |
| POPC/POPS:Ca(CTL2) | isopropyl alcohol:C2(CG311) | 0.38 | 4.01 | 0.0432 | 7.00 | 0.0100 |
| POPC/POPS:Ca(CTL2) | acetamide:C(CG2O1) | 0.08 | 4.00 | 0.0785 | 7.00 | 0.0100 |
| POPC/POPS:Ca(CTL2) | acetate:C2(CG203) | 0.10 | 4.00 | 0.0626 | 7.00 | 0.0100 |
| POPC/POPS:Ca(CTL2) | acetone:C1(CG205) | 0.09 | 4.00 | 0.0710 | 7.00 | 0.0100 |
| POPC/POPS:Ca(CTL2) | DMSO:S2(SG303) | 0.44 | 4.00 | 0.1400 | 7.00 | 0.0100 |
| POPC/POPS:Ca(CTL2) | urea:C2(CG2O6) | 0.09 | 4.00 | 0.0626 | 7.00 | 0.0100 |
An additional interaction site between each probe molecule and the membrane lipids is introduced to repel probe molecules from the core of the lipid bilayer and from each other if needed. In particular, we chose the location of the interaction site for the repulsion between lipid CLT2 atom (column 1; POPC:C1, C3, C11, C12, C22–C28, C33–C39, C211–C217, C310–C315 and POPS:C1, C3, C11, C22–C28, C33–C39, C211–C217, C310–C315) and a selected atom of each probe molecule (column 2). CGenFF force field atom types of the modified interaction sites are shown in round brackets. Ideally, the chosen interaction site should be located at the center of mass of the probe molecule to reduce any preferred orientation or artificial orientational structure in the density of probes close to the bilayer surface. This is not always possible and requires introducing dummy atoms into the topologies of the probe molecules. As an approximate approach, we chose the closest probe atom to the center of mass. The third column summarizes the actual mismatch between the central atom of each probe molecule and its center of mass. The remaining columns tabulate the modifications in the nonbonded LJ parameters used to keep probes away from the bilayer hydrophobic core or from each other.
Figure 2.
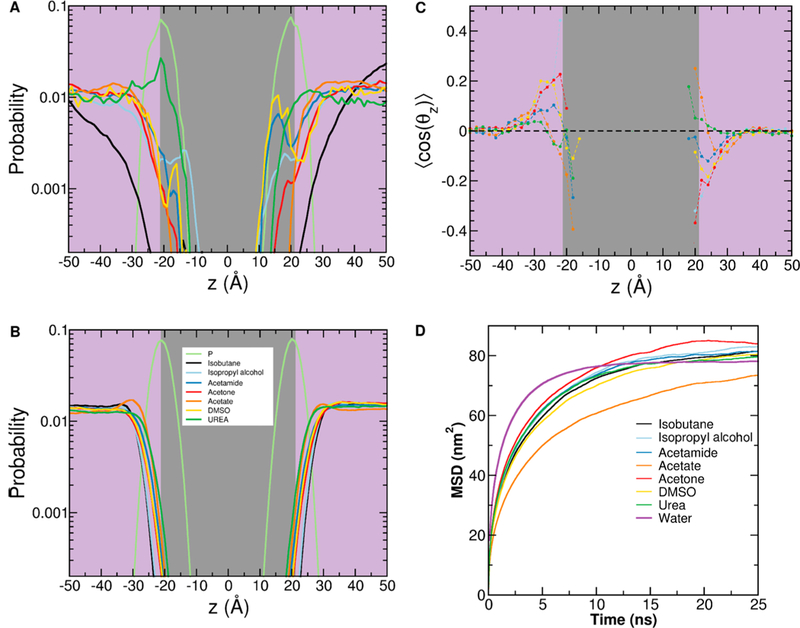
Simulation of membrane-bound K-Ras in mixed solvent. Shown are the density profiles of the lipid phosphate group and the probes, with the purple panel indicating bulk solvent and the gray panel indicating the bilayer core. The profiles are from simulations without (A) and with (B) the force field modifications described in Table 1 and Methods. (C) Ensemble-averaged orientation of each probe type with respect to the bilayer normal (see Methods). (D) Mean square displacement (MSD) of each probe type and water showing the overall similarity in diffusivity.
System Preparation.
Initial structures for G12D K-Ras in membrane orientation states OS1 and OS2 were obtained from a previous study.23 For G12V, we mutated Asp to Val at position 12 in the G12D structure. Similarly, for G13D, we mutated Asp to Gly at position 12 and Gly to Asp at position 13. In each case, the C-terminal cysteine residue was farnesylated and carboxyl methylated, while the N-terminus was protonated. All other side chains were set to their standard protonation state at neutral pH with His neutral, Asp and Glu negatively charged, and Lys and Arg positively charged. The resulting structure of each mutant in each orientation was placed in a 112 × 112 × 120 Å3 box containing 28,000 TIP3P water molecules and an anionic lipid bilayer consisting of 320 POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) and 96 POPS (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine) lipids. A minimum of 14 Å buffer between the edges of the box and protein/membrane atoms was used to prevent protein interaction with its periodic images. To this system we added a total of 1,400 probe molecules, i.e. 200 molecules of each probe type. The probes were distributed/oriented randomly in the aqueous phase with their heavy atoms at least 4 Å away from any protein, membrane, and other probe heavy atom. We assigned a negative charge on acetate and left all other probes neutral taking into account their pKa values, which is about 4.7 for acetate and >12 for the others except for urea, which is neither basic nor acidic in solution and was therefore uncharged. Notice that in each system we maintained a fixed water:total probe mole fraction of 20:1. This corresponds to a 0.4 M concentration for each probe type, which is dilute when compared with the relatively high probe concentration that was necessary for probes to adequately sample various protein surfaces within short MD simulation times.11,16,22 However, using a higher total probe concentration would likely affect the structure and dynamics of the protein due to increased competition between probe-protein and intraprotein interactions, as well as other nonspecific interactions and crowding effects.25 More broadly, while high probe concentration better saturates the protein surface and thus improves detection of binding sites, it will also increase the signal-to-noise ratio and consequently complicate interpretation of the data.21 As a trade-off, we decided to have a system that is significantly dilute in terms of individual probe types but sufficiently concentrated in terms of the total cosolvent to water ratio.
Molecular Dynamics Simulation.
After adding Na+ and Cl- ions to the systems described above to maintain overall charge neutrality and a physiologic ionic strength of 0.15 M, the resulting ∼157,000 atom systems were simulated with the GPU-enabled NAMD2.9.26 We conducted five 60 ns-long independent simulations for each mutant at each orientation, i.e., a total of 30 simulations and an aggregate production simulation time of 1.8 μs. This allowed us to adequately sample local conformational flexibilities of the protein and diflusional relaxation of the probes at the relatively low concentration of 0.4 M per probe (versus >2 M in our previous study16). The simulations were carried out with the CHARMM36 force field with cMAP dihedral correction27 for proteins and lipids28 and the CHARMM general force field (CGenFF) for the small organic molecules.29 Each system was energy minimized by 5,000 steps of conjugate gradient minimization with the Cα atoms restrained to their initial position by a harmonic restraint of force constant k = 4 kcal mol–1 Å–2. Simulated annealing was used to homogenize the probes, ions, and water molecules around the protein and the bilayer. This entailed application of a harmonic restraint with k = 4 kcal mol–1 Å–2 on the protein and lipid heavy atoms while incrementing the temperature by 30 K every 10 ps until 600 K, followed by cooling by 10 K every 10 ps until a final temperature of 310 K was achieved. The resulting system was equilibrated for 5 ns while progressively reducing k to zero. Production runs commenced from the five equilibrated systems of each K-Ras mutant in the two membrane orientation states.
During each simulation, a 2 fs time step was used with SHAKE applied to covalent bonds involving hydrogen atoms. The isothermal–isobaric (NPT) ensemble and periodic boundary conditions were used. Temperature was kept constant at 310 K using Langevin dynamics with a damping coefficient of 10 ps–1 and pressure at 1.0 atm using the Nose-Hoover Langevin piston method with a piston period of 200 fs and decay time interval of 100 fs. Short-range nonbonded interactions were truncated at 10 Å and smoothly switched off between 10 and 12 Å, with a 14 Å cutoff used for pair list updates. Long-range electrostatic interactions were computed using the Particle Mesh Ewald (PME) method30 with a grid density of one grid point per Å. In total, 30 simulations were conducted with an aggregate equilibration and production simulation time of 2.0 μs. Coordinates were saved every 2 ps, yielding a concatenated trajectory of ∼300 ns or 150,000 frames per system.
Probe-Derived Surface Topography Map (PSTM) and Two-Dimensional Projection of Probe Densities.
A major challenge in ligand binding site identification using pMD is to distinguish between interaction spots that are likely to be pocketlike from those that are not suitable for ligand binding.12,21 As shown in our previous work,12 additional independent analysis using structure-based approaches such as SiteMAP31 or MDPocket4 can help alleviate this problem. However, it would be desirable to directly use probe binding propensities to achieve this goal. Here we introduce a novel technique that allows for the identification of probe-binding regions that are amenable for ligand binding directly from protein-probe interactions.
The starting point for our technique is a polar coordinate system whose origin is set to the center of mass of the protein; the protein is assumed to be relatively rigid and approximately spherical. If the coordinate system axes are fixed with respect to the protein principal axes, spherical coordinates of a given probe heavy atom can be defined by r, θ, and ϕ. We define r as the distance of a probe atom from the center of mass of the protein, which in our test case is the catalytic domain of K-Ras, while θ and ϕ are the azimuthal and polar angles. A 2D protein surface topography map (PSTM) is then constructed on a Δϕ° × Δθ° (ϕ, θ) grid spanning the whole protein surface by calculating the average distance of surface-bound probe atoms from the center of the protein()and their spherical angular location (i + 0.5)Δϕ, (j + 0.5)Δθ. A probe is considered surface-bound if any of its heavy atoms are within 4.5 Å from any heavy atom of the protein. Our approach shares some similarities to a recent work by Koromylsova et al.,32 but there are also important differences in the way in which the surface is defined. Specifically, whereas Koromylsova et al.32 use geometric surface descriptors and conventional protein surface calculation methods,33 we utilize probe-protein interactions to map surface depressions and protrusions.
To visualize probe densities, we used a technique similar to the one used in PSTM construction to create a probe-based protein surface density map (PPSD). In this technique, the location of the surface-interacting probe is projected onto a discretized enclosing sphere and binned to a Δϕ° × Δθ° (ϕ, θ) grid that spans the whole protein surface, as follows
where is the approximate molar concentration of probe type p at the surface of the protein corresponding to a surface element around the angular location ((i + 0.5) Δϕ, (j + 0.5) Δθ). The two indices i and j range from 0 to and, respectively. nprobes(p,m) is the total number of heavy atoms of probe p at the mth trajectory frame that are within 4.5 Å from any heavy atom of the catalytic domain of K-Ras. nframes is the total number of frames in the concatenated trajectory. Δθ and Δ ϕ are the discretized grid sizes of the azimuthal and polar angles in radians, which in the current work were set to 0.0174 × 0.0174 (a degree equivalent of 1° × 1°). Such a high-resolution mapping was possible because we used a high sampling frequency (coordinates were saved every 2 ps) and a relatively high probe bulk concentration (0.4 M). C0 is the standard concentration, is the Kronecker delta function, and is the integer floor function. ϕlm and θlm are the angular and azimuthal angles of the lth heavy atom of a probe molecule that is in contact with the protein at the mth frame. and are approximated by the average and standard deviation of the distance of probe atoms from the center of the protein, with the probes located in the corresponding surface element within 4.5 Å from any heavy atom of the catalytic domain. Since can be overestimated due to the lower limit choice of and considering all probe atoms as independent species, we normalized by the number of heavy atoms in probe type p Finally, to reduce cluttering in the density maps, surface elements that are not sampled at least 1% of the time or exhibit large r fluctuations were assigned a density of zero. Correspondingly, probe surface densities were smoothed out to reduce the noise in their profile using a standard image noise filter achieved by averaging each surface element with the surrounding neighbor elements using
As shown in the Results/Discussion section, combining PSTM and PPSD proved to be effective in delineating ligand binding pockets by filtering out other high probe density surfaces that do not possess pocketlike characteristics, such as interfaces of oligomerization34–36 or crystal packing.21 Note that our approach is most suitable for globular proteins with approximately comparable principal moments. For the catalytic domain of our test case K-Ras, the ratio of the three eigenvalues of the moment of inertia tensors is 1.00:0.90:0.84, indicating a near spherical geometry. In principle, it is possible to use a similar projection for targets that deviate from globularity but whose surface is homomorphic to a sphere by carefully choosing a coordinate system that ensures all relevant regions are close to the equator. Furthermore, PSTM is periodic (i.e., regions with ϕ = –180° and ϕ = 180° are adjacent), and regions closer to the poles (i.e., θ > 170° or θ < 10°) are substantially smaller than those close to the equator (θ ∼ 90°). Therefore, care is required while interpreting data in these regions. Finally, PSTM can also be derived from probe-free MD simulations using water molecules as probes. In this case, however, instances of probe-induced structural changes that may be occasionally important are not likely to be captured.
RESULTS AND DISCUSSION
The current work extends a previously reported probe-based MD technique16 for ligand binding site identification in membrane proteins. In what follows we describe the probe molecules used in this study and modifications to selected LJ pairwise nonbonded interaction terms to minimize undesirable probe—probe and probe-lipid interactions. We then discuss the application of a novel analysis technique to facilitate the exploration of interaction hotspots from probe-binding propensities and surface topography. Finally, we demonstrate the usefulness of our approach to identify allosteric ligand binding pockets and other interaction hotspots on the surface of three oncogenic K-Ras mutants bound to an anionic bilayer in two different orientations.
Selection of Probe Molecules.
The use of multiple probe types spanning a large chemical space would allow for a better exploration of reactive surfaces on target proteins. This is because different probes can have different propensities for interaction with hotspot residues, which could potentially lead to new pockets or help differentiate “true” binding sites from spurious ones. Therefore, we tested the performance of mixed-probe pMD-membrane relative to its single-probe (isopropyl alcohol-only) counterpart using six additional probes that represent features commonly found in marketed drugs11 (Figure 1). In addition to these, isopropylamine and benzene are also frequently found in marketed drugs,10,11 but our attempt to include them was unsuccessful. The reasons were the electrostatic attraction between the oppositely charged isopropylamine and POPS, and the lack of an atom close to the center of mass of the benzene ring suitable for modification to prevent self- and lipid-interactions, respectively. The latter could be solved using the dummy atom approach of MacKerell and colleagues,10 and the long-range isopropylamine-lipid electrostatic interaction can be reduced by introducing repulsion with POPS head groups or increasing the nonbonded cutoff beyond 12 Å to utilize the CTL2 atom sites. However, the seven probe types listed in Figure 1 are sufficient for the purposes of this work.
Modification of Selected Pairwise Interactions Prevents Probe Self-Aggregation and Bilayer Partitioning.
We recently found that isopropyl alcohol spontaneously partitions into a POPC/POPS bilayer within a few nanoseconds and thereby causes an increase in area per lipid and a concomitant decrease in bilayer thickness.16 We observed similar effects in the current mixed probe simulations where most probe types partition into the hydrophobic core of the bilayer within several nanoseconds (Figure 2A). This can alter membrane fluidity and therefore protein—lipid interactions, which is consistent with previous observations that adsorption of small organic molecules into membranes affects structure and lipid mixing.37–39 In addition, the hydrophobic isobutane is self-aggregating, which reduces its effective bulk concentration and availability for interaction with the protein. To remove these undesired effects, we modified LJ parameters involving selected atoms of probes and lipids (see Table 1 for details; also refs 10 and 16). The result was that each probe species is effectively excluded from the bilayer hydrophobic core but interacts with the headgroup region (Figure 2B). This enables the probes to contact the protein from bulk as well as the membrane side without affecting the bilayer structure. Simulations after these modifications yielded average areas per lipid of ∼ 60.3 ± 0.7 Å2 and bilayer thickness of 41.0 ± 0.4 Å, values very close to those from probe-free simulations.16 Similarly, self-aggregation of isobutane was completely eliminated (not shown).
To check the response of the probes to the charge density at the bilayer surface, we calculated an orientation order parameter where θz is the angle between the membrane normal and a vector from the central to a selected terminal heavy atom of a probe (see Figure 1). Figure 2C shows that that each probe type adopts a specific orientation at the membrane surface; differences in orientation among probes indicate variation in their interaction with lipid head groups. However, the probes differ little in diffusivity as can be seen from the mean square displacement curves in Figure 2D, reflecting their similar molecular weight (58–78 Da). The exception is the charged acetate, which has a slower diffusion than the rest likely due to its favorable interaction with water making it effectively larger in size. Importantly, that the probes are only slightly less diffusive than water shows that their dynamics can be effectively sampled within reasonably short simulation times. Others have arrived at a similar conclusion based on slightly different arguments.9–11,22
Probe-Based Surface Topography Map (PSTM) Facilitates Ligand Binding Site Identification.
As mentioned in Methods, analyzing the dynamic interactions of molecular fragments with hotspot residues in a quantitative manner can be challenging.12,16,21 One of the challenges is the difficulty in distinguishing between bona fide ligand binding sites from other reactive surface patches that are not suitable for ligand binding or are nonspecific. Another challenge is related to data reduction and visualization. While a grid-based analysis of probe occupancy is useful in quantifying the distribution of probe densities on the 3D structure of the target protein10,12,21 (Figure 3C) visualization of the results is complicated by the large number of interaction spots that are irrelevant for ligand binding.12 Our PSTM method overcomes both of these challenges by projecting probe number densities onto a polar coordinate system describing the protein surface topography as a 2D map. The resulting global view of topography and probe densities provides for an easy-to-read yet detailed picture of probe distributions and geometry of ligand binding pockets. This allowed us to simultaneously estimate probe concentrations and localize pocketlike surface depressions surrounded by elevations, as well as valleys too big to be pocketlike.
Figure 3.
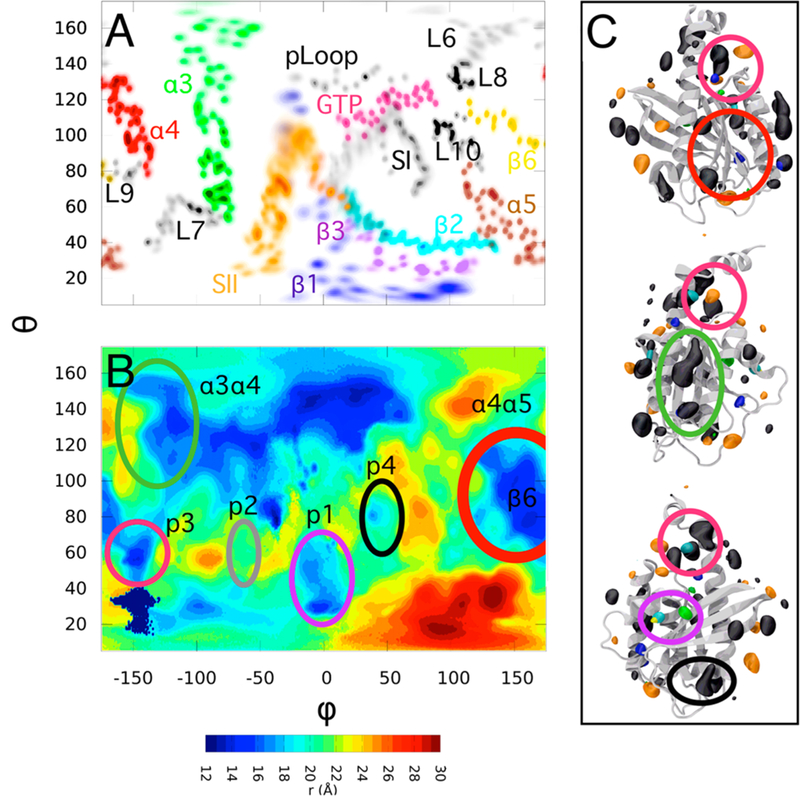
Probe-derived surface topography map (PSTM) of K-Ras. (A) Annotated projection of K-Ras on the surface of an enclosing sphere to link secondary structure elements to their location on the surface map. (B) PSTM of G12D K-Ras in OS2 with ligand binding pockets and other reactive regions circled in the same color as in the corresponding regions on the 3D structure shown in (C). For the images in (C) a recursive filtering has been applied to reduce noise and smooth the probe density isosurfaces (see Figure S1). Pocket p1 spans the region at the intersection of β1-β3 and SII, p2 is between SII and α3, p3 is surrounded by L7, L9, and α5, and p4 is located behind SI and above β2.
Figure 3 shows an example of a probe-derived 2D surface map for G12D K-Ras in one of its two membrane orientation states (OS2). To connect the probe-derived surface features with the secondary structure elements we have (i) annotated key regions and secondary structures on the 2D map (Figure 3A), (ii) highlighted the density of probes derived from grid analysis on the 3D structure using the VolMap plugin in VMD40 (Figure 3C) and (iii) labeled key regions on each panel by previously described ligand binding pockets p1 to p4,12 as well as other reactive surfaces (see below). These annotations illustrate the relationship between different regions of the protein and the PSTM, a 2D topography map derived from the radial location of probes interacting with surface residues. The resulting global view of the protein in terms of surface depressions (Figure 3B) is easier to read than cluttered probe occupancies on the 3D structure (Figure 3C). We note that the generation of such a PSTM is underpinned by two important considerations. One is the choice of the coordinate system. For example, to generate the data shown in Figure 3A,B we first defined a coordinate system whose origin and axes correspond to the center of mass and the three principal moments of inertia axes of the catalytic domain of K-Ras. Then we removed translational and rotational degrees of freedom of the protein by aligning each MD snapshot to a reference structure using Cα atoms of residues 1–24) 41–56) and 76–166. This resulted in a feature-rich global surface topography (Figure 3B). The second factor is the idea that true ligand-binding pockets will appear as local depressions relative to the surrounding topography. This is the case for our test system K-Ras since all of the previously characterized ligand-binding sites (labeled p1, p2, p3; and p4) are visible in the PSTM. The map also delineates the geometry of each pocket and its location: p1 is made up of residues on switch 2, β1, and β3; p2 is between α2 and α3; p3 involves L7 and α5; and p4 lies between switch 2 and β2. The map also captures other reactive surfaces including the two flat surfaces spanning α4, β6; and α5 on the one hand and α3 and α4 on the other. These surfaces appear as extended shallow grooves and hence are unlikely to be suitable for drug binding. Instead, they participate in membrane binding23,41 and self-interaction35,36 (see Figure 7 in ref 12, Figures 2 and 4 in ref 23, and Figure 1B in ref 36). Finally the nucleotide-binding pocket appears as a deep depression, while the effector-binding region centered on β2 is a bulging hotspot. In sum( PSTM implicitly accounts for the dynamics of the protein as it utilizes the ensemble of probe-protein contacts to map the surface, wherein surface depressions and protrusions can be used to localize a potential ligand-binding site. PSTM thus offers an effective means by which to analyze, visualize, and interpret probe occupancies derived from pMD simulations.
figure 7.
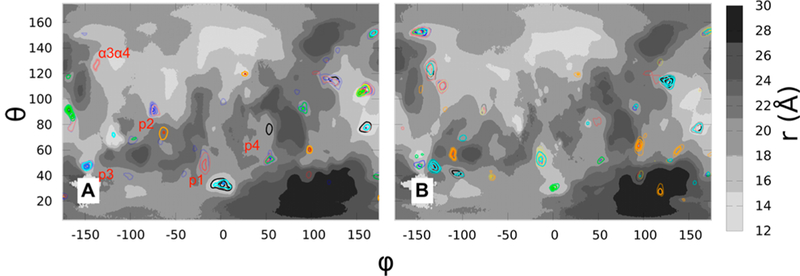
Probe density maps superimposed on the protein topography map of G12D K-Ras in (A) OS1 and (B) OS2. Densities of isobutane, isopropyl alcohol, acetamide, acetate, acetone, DMSO, and urea are shown in black, cyan, blue, orange, red, yellow, and green at the same contour level of
Figure 4.
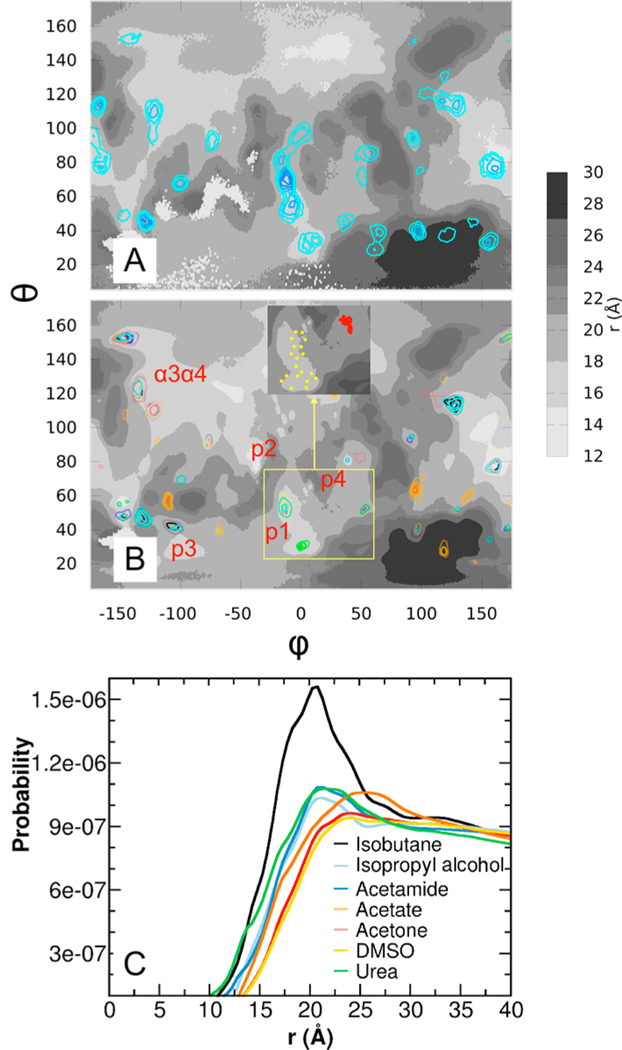
Probe binding analysis. Density of probes in the single-probe (A) and mixed-probe simulations (B) projected onto the respective PSTM of G12D K-Ras catalytic domain in OS2. A contour level of was used in both maps, where c0 = 2.8 M and 0.4 M is the bulk concentration of probes in the single- and mixedprobe simulations, respectively, and s is the calculated molar concentration at the surface. The inset in (B) shows projection of the coordinates of two Ras-bound ligands on the PSTM: 2-(1H-indol-3-ylmethyl)-1H-imidazo[4,5-c]pyridine (PDB ID 4EPV) that binds at p1 (yellow) and S-adenosylmethionine (4YNM) that targets p4 (red). (C) Probability density profile of the seven probe molecules as a function of distance from the center of mass of the catalytic domain of K-Ras. The probability, p(r), of finding a probe between r and r+dr shown in part C was normalized using
Comparison of Hotspots Detected by Single- and Mixed-Probe Simulations.
To directly compare mixed- and single-probe pMD-membrane, we kept the total concentration of probes exactly the same in the two approaches. Then we analyzed the previously reported single-probe (isopropyl alcohol) simulation of G12D16 with PSTM and PPSD representations. Figure 4A,B shows overlays of probe densities on PSTMs for each probe type at contour levels of ≤ where c0 = 2.8 M and 0.4 M is the bulk concentration of probes in the single- and mixed-probe simulations, respectively. One can see that the single- and mixed-probe runs yielded qualitatively similar distributions identifying similar regions as reactive surfaces. These include pockets p1, p3, and p4 (see ref 12) as well as surfaces α3α4 and α4α5. Thus, at least for Ras, isopropyl alcohol-only pMD is probably sufficient for the purpose of binding site identification consistent with the fact that isopropyl alcohol exists in 55% of all drugs and 57% of approved drugs.11 In addition the majority of the hotspot regions in the isopropyl alcohol-only simulation are more saturated (i.e., sampled better) than in the mixed-probe simulation. This is true even when considering the sum of the densities in the latter (not shown). A major reason is that the overall propensity of acetone and DMSO to interact with K-Ras is significantly smaller than that of isopropyl alcohol (see below and Table S2). A second reason could be the 7-fold higher relative concentration of isopropyl alcohol in the single-probe simulation. Although probe-occupancy may not necessarily scale linearly with concentration, concentration will likely affect sampling efficiency and saturation of some sites. Third, saturation may be affected by competition among probes in the mixed-probe system. However it is important to emphasize that the relative reactivity of probes is strictly system specific, and so our observations on K-Ras described above should not be taken as universally applicable.
The mixed-probe run performed better in detecting certain reactive regions. This includes pocket p312 and the reactive surface α3α423,36 (Figure 4). In addition the negatively charged acetate persistently interacts with the polycationic membrane-interacting C-terminus of K-Ras. These examples demonstrate the significance of probe chemical diversity for detecting ligand-binding sites that might be missed in a single-probe approach. To further illustrate this point, we show in Figure 4C the probability distribution of each probe type with respect to their distance from the center of mass of the catalytic domain of G12D K-Ras. All of the profiles level off and converge around 25 Å, followed by a slow decline due perhaps to asymmetry induced by the bilayer. It is clear, however, that the hydrophobic isobutane has higher overall tendency to interact with K-Ras, followed by isopropyl alcohol, acetamide, and urea; acetone and DMSO have the least preference. As an additional simple measure of the reactivity of different probe types with surface residues, we calculated the average total number of contacts of each probe with the protein (Table S1). One can see that, for example, isobutane and urea are about twice more likely to be found on the protein surface than acetone or DMSO. The data also highlights a relationship between probe identity and interaction with the protein and thereby a link between overall surface saturation and probe binding affinity. Taken together, our results show that, for K-Ras, single-probe pMD-membrane performs remarkably well in detecting ligand-binding sites, but the mixed-probe approach offers additional insights about surface patches that bind to multiple probe types. One application of the latter information is that orientations and positions of the different probes can guide the design of larger, selective and more potent ligands.
Test of Concept PStM/PPSD Analysis of Membrane-Bound K-Ras.
The GTPase K-Ras is a molecular switch that mediates signal transduction from cell surface receptors to the nucleolus to regulate cell survival, proliferation, growth, and development.42 Its biological activity requires attachment to the inner surface of the plasma membrane.43 K-Ras interconverts between GTP-bound on and GDP-bound off conformational states that differ in the structure of switch SI (residues 25–40) and SII (residues 57–75)44 (see Figure 1). Mutations that stabilize the on state can lead to cancer, including colon, lung, and pancreatic cancer.45,46 Therefore, K-Ras is currently one of the most important anticancer drug targets.47,48 The catalytic domain of K-Ras adopts multiple orientations on the membrane surface.23,41 These include orientation states OS1 where helices 3 and 4 are in contact with the membrane and OS2 where parts of β1–3 and helix 2 interact with the membrane (Figure 5A,B). As discussed in the SI, probe binding did not induce significant changes in the structure or membrane orientation (Figures S2–S5), and the mutants exhibit similar structural (Figures S2–4) and orientational dynamics (Figure S5) within the simulation times. Therefore, any potential difference in probe binding among the different K-Ras mutants or orientation states has to arise from small local differences rather than large-scale conformational reorganizations.
Figure 5.
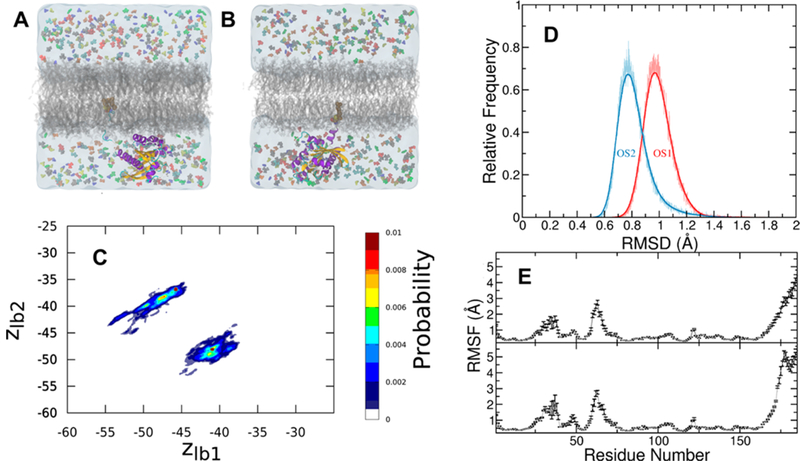
Protein conformational dynamics in the presence of probes. (A, B) K-Ras in orientation state 1 (OS1) in which helices 3 and 4 are in contact with the bilayer surface (A) and orientation state 2 (OS2) in which the central β sheet β1−3 and helix 2 are in contact with the membrane (B). The protein is shown in cartoon colored in purple (helices), yellow (strands), and cyan/white (turns). Farnesyl in vdW spheres is inserted into the core of the bilayer (gray). Water is in transparent light blue shading, and the probes are in differently colored spheres. Sodium, magnesium, and chloride ions as well as GTP are omitted for clarity. (B) Membrane orientation dynamics of G12D K-Ras catalytic domain measured by the vertical distance of lobe 1 and lobe 2 from the bilayer center. (D) Distribution of ensemble-averaged backbone root-mean square deviation (RSMD) of the K-Ras catalytic domain from a combined trajectory of all simulations in OS1 (red) and OS2 (blue). (E) Backbone root-mean square fluctuations (RMSF) of G12D K-Ras in OS1 (bottom) and OS2 (top).
Figure 6 displays PSTMs for G12D K-Ras in OS1 and OS2, while Figure S6 compares the two orientations of G12V and G13D. In each case, OS1 and OS2 not only share many common surface features but also have significant differences. For example, p1 is a deep pocket in OS1 but a shallower one in OS2 (Figures 6 and S6). As a result, its relative probe occupancy as measured by the frequency of contact between probes and p1 residues (Table S2 and Figure S7A), or in terms of probe surface densities (Figures 7 and S8), is significantly higher in OS1 than OS2. Pocket p2 is less accessible to probes in all simulations (Figures 7, S6–S8), because opening of this pocket requires a comparatively large reorganization of α2 that typically occurs in the GDP-bound form.44,49,50 Pocket p3 is shallow in OS1 of G13D (Figures S6B,D, S7C, S8B,D and Table S2), and p4 is slightly more accessible to probes in OS1 than OS2 (Figures 6 and S7D and Table S2). There are also some differences beyond the ligand binding pockets. For example, α3α4 is occluded by the membrane in OS1 and so has negligible probe density, but it is accessible to probes in OS2 (Figures 7 and S8). Also, even though α4α5 is solvent exposed in both orientations its probe binding propensity is higher in OS1 (Figures 7 and S8). Comparison of the mutants and orientations in terms of preferences of sites for different probe types provides additional insights. Broadly, almost all probe- binding sites interact with more than one probe type, with different probes typically occupying distinct parts of a site (see variations in the color of the contours within depressions in Figures 7 and S8). For example, p1 consistently binds to isobutane, acetamide, and acetone in OS1, while p3 is predominantly populated by isobutane, isopropyl alcohol, and urea in all systems except for G13D in OS1.
Figure 6.
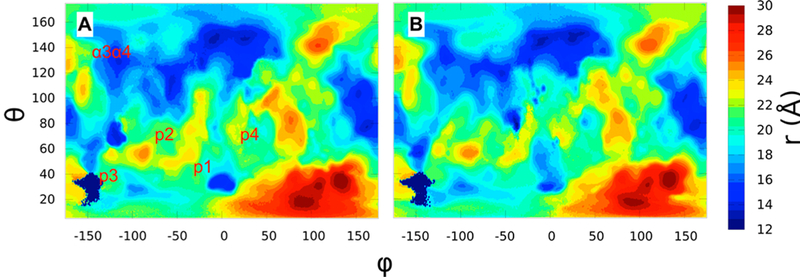
Comparison of protein surface topography maps for G12D K-Ras in (A) OS1 and (B) OS2.
CONCLUSION
MD simulation of proteins in the presence of small organic molecules (pMD) has emerged as a versatile computational solvent mapping method for ligand binding site identification. The basic idea is that the complex kinetics and thermodynamics of protein–ligand interactions underlying the druggability of a site is proportional to its probe-binding propensity. pMD has been successfully applied to many soluble proteins and was recently extended to membrane proteins (pMD-membrane). In the current work we have expanded the scope of pMD-membrane to seven chemically diverse probe types and introduced an approach to filter out spurious hotspots that are not likely to be ligand-binding sites. This was achieved via a Cartesian-to-spherical coordinate system transformation and construction of a 2D topographic map directly from the probe-binding propensity of surface residues. We have demonstrated the robustness of this technique by identifying known druggable sites and other reactive regions on membrane-bound G12D, G12V, and G13D oncogenic K-Ras mutants simulated in two different membrane orientations.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Texas Advanced Computing Center (TACC) and the Extreme Science and Engineering Discovery Environment (XSEDE Project: MCB150054) and the National Institutes of Health General Medical Sciences (Grant No. R01GM100078) and the Cancer Prevention and Research Institute of Texas (CPRIT Grant No. DP150093).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jctc.7b00130.
Text describing the effect probe binding on K-Ras dynamics or membrane binding and convergence of probe-protein interaction; two tables; seven figures (PDF)
AUTHOR INFORMATION
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Hetényi C; van der Spoel D Efficient Docking of Peptides to Proteins without Prior Knowledge of the Binding Site. Protein Sci. 2002, 11, 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hocker HJ; Rambahal N; Gorfe AA LIBSA–A Method for the Determination of Ligand-Binding Preference to Allosteric Sites on Receptor Ensembles. J. Chem. Inf. Model. 2014, 54, 530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ngan CH; Bohnuud T; Mottarella SE; Beglov D; Villar EA; Hall DR; Kozakov D; Vajda S Ftmap: Extended Protein Mapping with User-Selected Probe Molecules. Nucleic Acids Res. 2012, 40, W271–W275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Schmidtke P; Bidon-Chanal A; Luque FJ; Barril X Mdpocket: Open-Source Cavity Detection and Characterization on Molecular Dynamics Trajectories. Bioinformatics 2011, 27, 3276–3285. [DOI] [PubMed] [Google Scholar]
- (5).Congreve M; Chessari G; Tisi D; Woodhead AJ Recent Developments in Fragment-Based Drug Discovery. J. Med. Chem. 2008. 51, 3661–3680. [DOI] [PubMed] [Google Scholar]
- (6).Shuker SB; Hajduk PJ; Meadows RP; Fesik SW Discovering High-Affinity Ligands for Proteins: SAR by NMR. Science 1996, 274, 1531. [DOI] [PubMed] [Google Scholar]
- (7).Mattos C; Ringe D Locating and Characterizing Binding Sites on Proteins. Nat. Biotechnol. 1996, 14, 595–599. [DOI] [PubMed] [Google Scholar]
- (8).Lexa KW; Carlson HA Full Protein Flexibility Is Essential for Proper Hot-Spot Mapping. J. Am. Chem. Soc. 2011, 133, 200–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Seco J; Luque FJ; Barril X Binding Site Detection and Druggability Index from First Principles. J. Med. Chem. 2009, 52, 2363–2371. [DOI] [PubMed] [Google Scholar]
- (10).Guvench O; MacKerell AD Jr Computational Fragment-Based Binding Site Identification by Ligand Competitive Saturation. PLoS Comput. Biol. 2009, 5, e1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bakan A; Nevins N; Lakdawala AS; Bahar I Druggability Assessment of Allosteric Proteins by Dynamics Simulations in the Presence of Probe Molecules. J. Chem. Theory Comput. 2012, 8, 2435– 2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Prakash P; Hancock JF; Gorfe AA Binding Hotspots on K-Ras: Consensus Ligand Binding Sites and Other Reactive Regions from Probe-Based Molecular Dynamics Analysis. Proteins: Struct., Funct., Genet. 2015, 83, 898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Huang D; Caflisch A Small molecule binding to proteins: affinity and binding/unbinding dynamics from atomistic simulations. ChemMedChem 2011, 6, 1578–1580. [DOI] [PubMed] [Google Scholar]
- (14).Gorfe A Mechanisms of Allostery and Membrane Attachment in Ras Gtpases: Implications for Anti-Cancer Drug Discovery. Curr. Med. Chem. 2010, 17, 1–9. [DOI] [PubMed] [Google Scholar]
- (15).Lundstrom K Latest Development in Drug Discovery on G Protein-Coupled Receptors. Curr. Protein Pept. Sci. 2006, 7, 465–470. [DOI] [PubMed] [Google Scholar]
- (16).Prakash P; Sayyed-Ahmad A; Gorfe AA PMD-Membrane: A Method for Ligand Binding Site Identification in Membrane-Bound Proteins. PLoS Comput. Biol. 2015, 11, e1004469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lakkaraju SK; Yu W; Raman EP; Hershfeld AV; Fang L; Deshpande DA; MacKerell AD Jr, Mapping Functional Group Free Energy Patterns at Protein Occluded Sites: Nuclear Receptors and G-Protein Coupled Receptors. J. Chem. Inf. Model. 2015, 55, 700–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lakkaraju SK; Mbatia H; Hanscom M; Zhao Z; Wu J; Stoica B; MacKerell AD; Faden AI; Xue F Cyclopropyl-Containing Positive Allosteric Modulators of Metabotropic Glutamate Receptor Subtype 5. Bioorg. Med. Chem. Lett. 2015, 25, 2275–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).He X; Lakkaraju SK; Hanscom M; Zhao Z; Wu J; Stoica B; MacKerell AD; Faden AI; Xue F Acyl-2-Aminobenzimida-zoles: A Novel Class of Neuroprotective Agents Targeting Mglur5. Bioorg. Med. Chem. 2015, 23, 2211–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Raman EP; Yu W; Guvench O; MacKerell AD Jr Reproducing Crystal Binding Modes of Ligand Functional Groups Using Site-Identification by Ligand Competitive Saturation (Silcs) Simulations. J. Chem. Inf. Model. 2011, 51, 877–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ung PM; Ghanakota P; Graham SE; Lexa KW; Carlson HA Identifying Binding Hot Spots on Protein Surfaces by Mixed-Solvent Molecular Dynamics: HIV-1 Protease as a Test Case. Biopolymers 2016, 105, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lexa KW; Carlson HA Improving Protocols for Protein Mapping through Proper Comparison to Crystallography Data. J. Chem. Inf. Model. 2013, 53, 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Prakash P; Zhou Y; Liang H; Hancock JF; Gorfe AA Oncogenic K-Ras Binds to an Anionic Membrane in Two Distinct Orientations: A Molecular Dynamics Analysis. Biophys. J. 2016, 110, 1125–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).MacKerell AD; Brooks B; Brooks CL; Nilsson L; Roux B; Won Y; Karplus M CHARMM: The Energy Function and Its Parameterization. Enc. Comput. Chem. 1998, 1, 271–277. [Google Scholar]
- (25).Naddaf L; Sayyed-Ahmad A Intracellular Crowding Effects on the Self-Association of the Bacterial Cell Division Protein Ftsz. Arch. Biochem. Biophys. 2014, 564, 12–19. [DOI] [PubMed] [Google Scholar]
- (26).Phillips JC; et al. Scalable Molecular Dynamics with Namd. J. Comput. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).MacKerell AD; Feig M; Brooks CL Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem. 2004, 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- (28).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD Jr; Pastor RW Update of the Charmm All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Vanommeslaeghe K; et al. Charmm General Force Field: A Force Field for Drug-Like Molecules Compatible with the Charmm All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Darden T; York D; Pedersen L Particle Mesh Ewald: An N-Log (N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- (31).Halgren T New Method for Fast and Accurate Binding-Site Identification and Analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [DOI] [PubMed] [Google Scholar]
- (32).Koromyslova AD; Chugunov AO; Efremov RG Deciphering Fine Molecular Details of Proteins’ Structure and Function with a Protein Surface Topography (PST) Method. J. Chem. Inf. Model. 2014, 54, 1189–1199. [DOI] [PubMed] [Google Scholar]
- (33).Connolly ML Analytical Molecular Surface Calculation. J. Appl Crystallogr. 1983, 16, 548–558. [Google Scholar]
- (34).Sayyed-Ahmad A; Cho K-J; Hancock JF; Gorfe AA Computational Equilibrium Thermodynamic and Kinetic Analysis of K-Ras Dimerization through an Effector Binding Surface Suggests Limited Functional Role. J. Phys. Chem. B 2016, 120, 8547–8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Muratcioglu S; et al. GTP-Dependent K-Ras Dimerization. Structure 2015, 23, 1325–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Prakash P; Sayyed-Ahmad A; Cho K-J; Dolino D; Chen W; Li H; Grant B; Hancock J; Gorfe A Computational and Biochemical Characterization of Two Partially Overlapping Interfaces and Multiple Weak-Affinity K-Ras Dimers. Sci. Rep. 2017, 7, 40109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sayyed-Ahmad A; Lichtenberger LM; Gorfe AA Structure and Dynamics of Cholic Acid and Dodecylphosphocholine- Cholic Acid Aggregates. Langmuir 2010, 26, 13407–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Prakash P; Sayyed-Ahmad A; Zhou Y; Volk DE; Gorenstein DG; Dial E; Lichtenberger LM; Gorfe AA Aggregation Behavior of Ibuprofen, Cholic Acid and Dodecylphosphocholine Micelles. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 3040–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Boggara MB; Krishnamoorti R Partitioning of Nonsteroidal Antiinflammatory Drugs in Lipid Membranes: A Molecular Dynamics Simulation Study. Biophys. J. 2010, 98, 586–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Humphrey W; Dalke A; Schulten K Vmd: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (41).Mazhab-Jafari MT; Marshall CB; Smith MJ; Gasmi-Seabrook GM; Stathopulos PB; Inagaki F; Kay LE; Neel BG; Ikura M Oncogenic and Rasopathy-Associated K-Ras Mutations Relieve Membrane-Dependent Occlusion of the Effector-Binding Site. Proc. Natl. Acad. Sci U. S. A. 2015, 112, 6625–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Karnoub AE; Weinberg RA Ras Oncogenes: Split Personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Hancock JF Ras Proteins: Different Signals from Different Locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–385. [DOI] [PubMed] [Google Scholar]
- (44).Grant BJ; Gorfe AA; McCammon JA Ras Conformational Switching: Simulating Nucleotide-Dependent Conformational Transitions with Accelerated Molecular Dynamics. PLoS Comput. Biol. 2009, 5, e1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bos JL Ras Oncogenes in Human Cancer: A Review. Cancer Res. 1989, 49, 4682–4689. [PubMed] [Google Scholar]
- (46).Eser S; Schnieke A; Schneider G; Saur D Oncogenic Kras Signalling in Pancreatic Cancer. Br. J. Cancer 2014, 111, 817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Vigil D; Cherfils J; Rossman KL; Der CJ Ras Superfamily GEFs and GAPs: Validated and Tractable Targets for Cancer Therapy? Nat. Rev. Cancer 2010, 10, 842–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).McCormick F K-Ras Protein as a Drug Target. J. Mol. Med. 2016, 94, 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Grant BJ; Lukman S; Hocker HJ; Sayyah J; Brown JH; McCammon JA; Gorfe AA Novel Allosteric Sites on Ras for Lead Generation. PLoS One 2011, 6, e25711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Prakash P; Sayyed-Ahmad A; Gorfe AA The Role of Conserved Waters in Conformational Transitions of Q61H K-Ras. PLoS Comput. Biol. 2012, 8, e1002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.