

Abstract
Advances in genomic technologies have led to a wealth of information identifying genetic polymorphisms in membrane transporters, specifically how these polymorphisms affect drug disposition and response. This review describes the current perspective of the International Transporter Consortium (ITC) on clinically important polymorphisms in membrane transporters. ITC suggests that in addition to previously recommended polymorphisms in ABCG2 (BCRP) and SLCO1B1 (OATP1B1), polymorphisms in the emerging transporter, SLC22A1 (OCT1), be considered during drug development. Collectively, polymorphisms in these transporters are important determinants of interindividual differences in the levels, toxicities, and response to many drugs.
Keywords: Pharmacogenomics, organic cation transporter, organic anion transporter, breast cancer resistance protein, organic anion transporting polypeptide, next generation sequencing, genomewide association study
Introduction
Historically, pharmacogenetic research has involved candidate gene studies focused largely on associations between common polymorphisms in drug metabolizing enzymes (e.g., CYP2D6 and CYP2C19) and drug disposition (mostly), toxicity (sometimes) and/or efficacy (rarely). Enormous technological advances in sequencing and genotyping have ushered in a new era in human genetics and pharmacogenetics research. Genomewide genotyping and sequencing approaches are widely being used to discover variants in any gene that underlie various phenotypes. As a result, both common and rare variants in many genes, including membrane transporter genes, have been identified as major determinants of variation in drug disposition, response and toxicity. In this State of the Art Review, we describe the current knowledge of genetic polymorphisms and their effects on drug response in two major superfamilies of transporters: the Solute Carrier Superfamily (SLC) and the ATP-Binding Cassette superfamily (ABC). In particular, we focus on transporters that have been recognized by the International Transporter Consortium (ITC)1 and regulatory authorities as important determinants of drug disposition and drug-drug interactions (DDI) (see Table 1). In a previous publication from the ITC, we highlighted associations between common polymorphisms in two transporters, BCRP (ABCG2) and OATP1B1 (SLCO1B1), and variation in therapeutic drug response, drug toxicity or drug disposition, and recommended that polymorphisms in these two transporters be considered during drug development2. Since that review, studies have continued to be published demonstrating significant associations between various pharmacogenomics traits and polymorphisms in ABCG2 and SLCO1B1. In addition, a number of recent studies have demonstrated that genetic variants in the Organic Cation Transporter 1 (OCT1, SLC22A1) are associated with the disposition and/or effects of a number of prescription drugs3–5. It is now not uncommon for regulatory agencies to expect data regarding the pharmacogenetic impact of genetic variation in these transporters in relevant drug development programs. Therefore, this review summarizes associations between polymorphisms in ABCG2, SLCO1B1 and SLC22A1 and pharmacogenomic phenotypes, focusing on clinical evidence and implications (Figure 1). In addition, associations between genetic variants in other transporters and clinical drug phenotypes are described with some emphasis on transporters that are known to mediate clinical DDIs (Figure 1). Notably, the review focuses on germline (heritable) variants, and does not include mutations that may arise in somatic cells such as tumors. Following our summaries of clinical associations of germline variants in transporters, we describe functional genomic studies of heritable transporter polymorphisms. The review ends with a description of recent discoveries in transporter polymorphisms, which have been made using genomewide association (GWA) and massively parallel sequencing approaches. Comments on the use of pharmacogenomics data to support modeling and simulation as well as future directions are included.
Table 1.
List of URLs to the guidelines and guidance from Food and Drug Administration (FDA), European Medical Agency (EMA), Pharmaceuticals and Medical Devices Agency (PMDA) and International Council for Harmonization (ICH), which are mentioned in this review.
Figure 1.
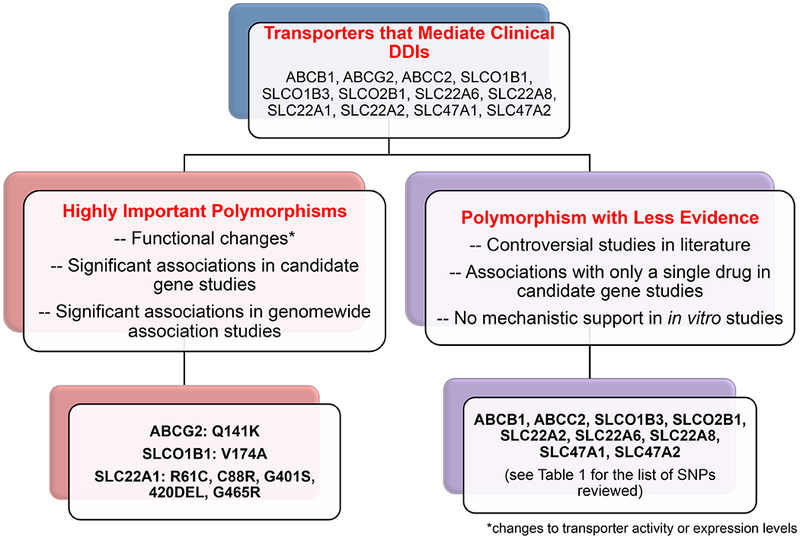
An overview of selecting highly important polymorphisms in transporters that mediate clinical drug-drug interactions.
1. Highly Important Transporter Polymorphisms in ABCG2 (BCRP), SLCO1B1 (OATP1B1) and SLC22A1 (OCT1)
In the previous ITC publication, polymorphisms in ABCG2 and SLCO1B1 were considered highly significant because they met the following strict criteria: (i) At least one genomewide significant association (p<5×10−8) between a polymorphism in the transporter and a pharmacologic trait for one or more drugs. The trait could reflect drug disposition, therapeutic response or drug toxicity; (ii) Significant associations in multiple candidate gene studies with pharmacologic traits; and (iii) In vitro evidence that the polymorphism(s) exhibited functionally important effects on transporter activity or expression levels. In this review, these criteria again support the selection of functionally significant polymorphisms in ABCG2 and SLCO1B1. In this updated publication, we considered transporters in which compelling evidence for an association with a drug response phenotype existed in multiple candidate gene studies (without GWA evidence). OCT1 (SLC22A1) represented a transporter that met this criterion. In particular, many candidate gene studies indicate an important role for polymorphisms in OCT1 as modulators of drug disposition and response (Figure 1). In addition, although none of the SLC22A1 polymorphisms have been associated with a pharmacogenomics trait in GWAS, SLC22A1 polymorphisms have been associated with levels of endogenous metabolites, and other traits in several GWAS (see GWAS Catalog).
Highly Important Polymorphisms in ABCG2 and SLCO1B1:
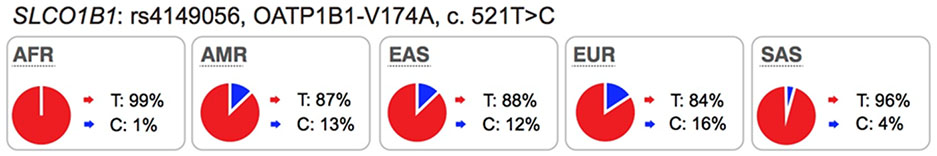
Since publication of the previous manuscript2, new reports continue to validate associations between pharmacologic traits and polymorphisms in ABCG2 and SLCO1B1. To date, a total of eight GWAS have reported genomewide level significant associations (p<5×10−8) between polymorphisms in ABCG2 and SLCO1B1 and drug disposition or response (Table S1). Significant associations have been described for drugs used in the treatment of lipid disorders (statins), gout (allopurinol), cancer (methotrexate) and cardiovascular disorders (ticagrelor). Among the eight GWAS, four are related to drug disposition and four are related to therapeutic or adverse drug response. All of the drugs were known substrates of the transporters with the exception of allopurinol. Allopurinol was discovered to be a substrate of ABCG2, through functional studies motivated by GWAS finding6. Future studies are required to replicate the associations between SLCO1B1 genetic polymorphisms and the active ticagrelor metabolite, AR-C124910XX, and to determine whether the metabolite is also a substrate of OATP1B17. In general, the nonsynonymous polymorphism in SLCO1B1 (rs4149056) which results in an amino acid substitution (Val174Ala) is of most interest, and has been either directly associated with the pharmacologic trait or is in linkage disequilibrium with another variant that has been associated with the trait (Table S1). For ABCG2, multiple polymorphisms have been associated with various traits, though typically, the missense polymorphism rs2231142, resulting in a substitution of lysine for glutamine (Q141K) is in linkage disequilibrium with the associated polymorphisms (Table S1). Collectively, the studies summarized in Table S1 continue to support the ITC recommendation that genetic polymorphisms in SLCO1B1 and ABCG2 should be monitored during drug development. It is worth mentioning that the Clinical Pharmacogenetics Implementation Consortium (CPIC) has included polymorphisms in SLCO1B1 and ABCG2 as important determinants of drug disposition and response (https://cpicpgx.org/genes-drugs/). Among the gene/drug pairs, CPIC has published extensive evidence from the literature to support simvastatin dosing based on SLCO1B1 genotype (https://cpicpgx.org/guidelines/guideline-for-simvastatin-and-slco1b1/).
Highly Important Polymorphisms in SLC22A1:
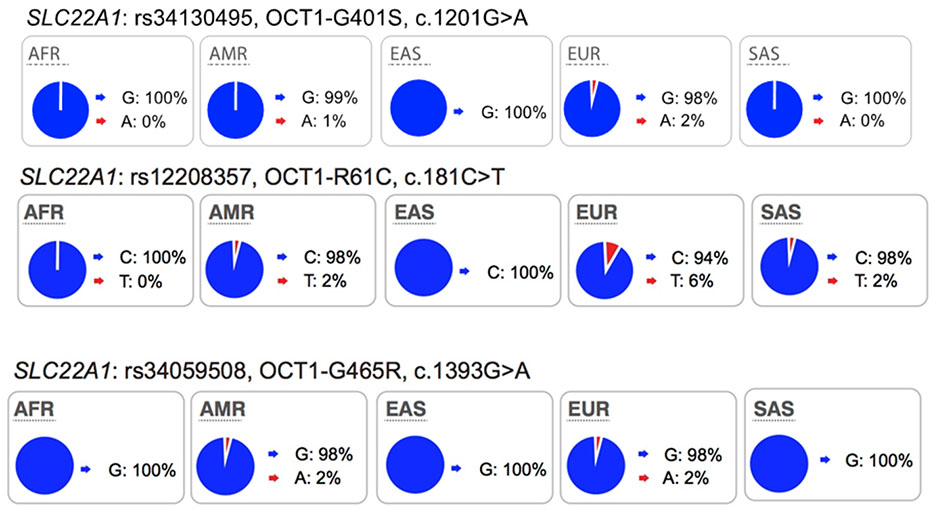
During the third workshop of the ITC, SLC22A1 was discussed as being increasingly important in drug development due to a high level of evidence from a number of clinical pharmacogenomic studies (Table S2). In this issue, Zamek-Gliszczynski et al. and Chu et al. respectively highlight the importance of SLC22A1 as an emerging transporter and provide also information on endogenous biomarkers associated with its polymorphisms. In particular, these studies reported significant associations between SLC22A1 polymorphisms and drug disposition, response and toxicity. SLC22A1 is abundantly expressed in the liver, and found on the sinusoidal membrane of hepatocytes. Polymorphisms in the transporter have been well-characterized in in vitro uptake studies, and many polymorphisms lead to functional changes in protein expression and transport activity (Table S3). Importantly, a number of clinical studies have been published describing significant associations between SLC22A1 polymorphisms and drug pharmacokinetics and pharmacodynamics effects (Table S2). Many of these associations have focused on the anti-diabetic drug, metformin, and include metformin disposition8–11, response12,13 and adverse effects14,15. However, studies of the effects of SLC22A1 polymorphisms on metformin pharmacokinetics and pharmacodynamics have been inconsistent. For example, while significant associations of non-synonymous SLC22A1 polymorphisms with metformin plasma concentrations were observed in several pharmacogenomics studies 8–11,16, other studies failed to observe such effects in either healthy subjects or patients with type 2 diabetes17–19. Recently, using 11C-metformin positron emission tomography (PET)/computed tomography (CT), Sundelin et al. showed that individuals who are carriers of the OCT1 reduced function variants, Met420Del and Arg61Cys, have decreased concentrations of metformin in the liver without changes in systemic plasma levels compared with individuals with reference SLC22A110. Together these studies suggest that hepatic distribution of metformin is decreased in carriers of SLC22A1 reduced function variants and that plasma levels of metformin do not reflect hepatic exposure. These findings are in contrast to minor changes in the liver exposure and pharmacodynamics of statins in the case of SLCO1B1 polymorphisms20. Renal clearance is also a significant contributor to metformin elimination unlike statins where hepatic elimination is the predominant route. Therefore, metformin liver and plasma exposure will be affected by multiple mechanisms and consequences of reduced uptake into liver on systemic plasma levels would not be similar to statins21 (also see Guo et al, Advancing Predictions of Tissue and Intracellular Drug Concentrations Using In Vitro, Imaging and PBPK Modeling Approaches, this Clin Pharmacol Ther issue).
Associations between SLC22A1 polymorphisms and drug levels or response to prescription drugs other than metformin have been described (Table S2). These studies have demonstrated significant associations between reduced function non-synonymous variants of SLC22A1 and the anti-migraine drug, sumatriptan22, the opiate analgesic drugs or their metabolites, morphine4 and O-desmethyltramadol23, and the anti-nausea drug, ondansetron5. In these studies, volunteers were classified based on the number of active SLC22A1 alleles that they harbor, e.g., zero, one or two active alleles (Table S2). Some studies identified significant correlations between systemic plasma levels of drug and genetic polymorphisms in SLC22A1. For example, the exposure to fenoterol, a β2-adrenergic receptor agonist used in the treatment of asthma, was inversely correlated with the number of active SLC22A1 alleles24, i.e. higher levels of fenoterol were associated with zero active alleles of SLC22A1. In addition, drug exposure was correlated with effect, that is, individuals with less active SLC22A1 alleles and higher systemic concentrations had a greater increase in heart rate, an adverse effect of β2-adrenergic receptor agonists24. Studies with SLC22A1 polymorphisms have used a different study design than studies focused on polymorphisms of ABCG2 and SLCO1B1. In particular, reduced function polymorphisms are combined for SLC22A1 and the number of active alleles is associated with a pharmacologic trait4,9,23, whereas for SLCO1B1 and ABCG2, most studies have focused on associations between a single polymorphism and a pharmacologic trait.
Similar to ABCG2 and SLCO1B1, the studies of SLC22A1 polymorphisms have important implications for evaluation of potential DDI risk. For example, a perpetrator drug that inhibits OCT1 at clinically relevant concentrations may phenocopy the effect of the SLC22A1 reduced function polymorphisms. For example, an OCT1 inhibitor may increase fenoterol levels resulting in increased heart rate effects of the drug. To our knowledge, to date no approved drugs have been found to inhibit OCT1 clinically (see Zamek-Gliszczynski et al. in this issue).
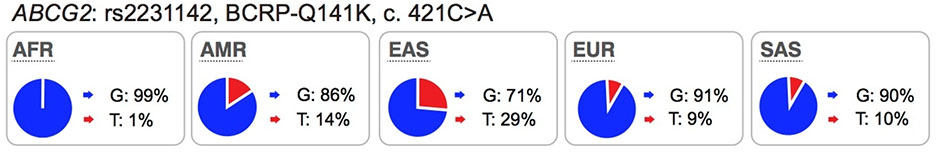
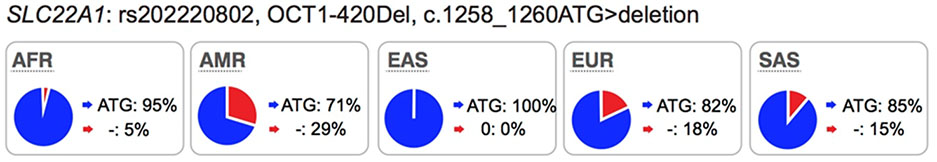
Collectively, review of the available data in the literature suggests that in addition to ABCG2 and SLCO1B1, polymorphisms in SLC22A1 be considered during drug development, particularly for drugs, which are OCT1 substrates. We suggest that drug developers consider conducting pharmacogenomic studies for new drugs that are substrates of OCT1, especially for drugs that have narrow therapeutic indices. Further, we suggest that the non-synonymous SNPs in SLC22A1, Arg61Cys, Cys88Arg, Gly401Ser, Met420del, and Gly465Arg, are given the highest priorities and that the data analysis may consider individuals with 0, 1 or 2 of any of these non-synonymous SNPs of SLC22A1, similar to published studies4,9,23 (Table S2). The allele frequencies in the ethnic population should be considered in the design of the study (see Table S3 for allele frequencies of non-synonymous OCT1 SNPs in various ethnic groups). In some ethnic groups, allele frequencies of reduced function polymorphisms are very low, and therefore, carriers of these alleles are uncommon, making pharmacogenomic studies exceedingly difficult. Pharmacogenomic studies need to focus on ethnic groups that have higher allele frequencies of reduced function polymorphisms of OCT1 such as individuals of European or Hispanic ancestry (Table 2).
Table 2.
Highlights of the in vitro and in vivo effects of key polymorphisms in twelve transporters recommended by FDA or EMA as important transporters in drug development. The major and minor allele frequencies reported in the figure below are alleles on the forward strand, except for alleles in ABCG2 and ABCB1 genes.
| Transporter variants and allele frequencies in different populations1 | Supporting evidence | In vitro effect | *In vivo effect |
|---|---|---|---|
 |
KO mice: Yes DDI: Yes GWAS: Yes |
Reduced expression levels and reduced transporter activity. | Increased plasma levels of drug substrate (e.g. atorvastatin, tenofovir) due to reduced efflux of drug from the intestine. Increased efficacy of drug substrate (e.g. rosuvastatin) due to reduced efflux of drug in the liver. |
 |
KO mice: Yes DDI: Yes GWAS: Yes |
Reduced transporter activity (see Supplemental Table 1) | Increased plasma levels of substrates (e.g. pravastatin, atorvastatin). Increased toxicity of drug substrates (e.g. simvastatin, cerivastatin) due to increased plasma levels. |
|
KO mice: Yes DDI: Yes GWAS: Yes (metabolite levels) |
Reduced transporter activity and some with reduced protein expressions on the membrane (see Supplemental Table 1) | Increased plasma levels of drug, which could lead to increased drug toxicity (see Table 2). |
 |
KO mice: Yes DDI: Yes GWAS: Yes (serum creatinine and metabolite levels) |
Contradictory results in in vitro studies. Some groups showed reduced uptake, and some showed increased or no effect (see Supplemental Table 1) | Contradictory results in literature about the effect of the variant with metformin disposition. |
|
SLC22A6 and SLC22A8 No polymorphisms in these genes that are significantly associated with pharmacogenomic traits in multiple studies. |
KO mice: Yes DDI: Yes GWAS: No |
A few reduced function missense variants and with low allele frequency (see Supplemental Table 1). | No reported in vivo effect of OAT1 reduced function variant with pharmacogenomic trait. |
| KO mice: Yes DDI: Yes GWAS: No |
Only one pharmacogenomics study related to cefotaxime clearance. | ||
 |
KO mice: Yes DDI: Yes GWAS: Yes (serum creatinine) |
C-allele showed reduced luciferase activity and expression levels. No known function of rs2289669. |
Promoter variant, rs2252281 (C-allele) and intronic variant, rs2289669 (A-allele) associated with better metformin response in some studies. |
 |
KO mice: **Available but no data DDI: Yes GWAS: No |
A-allele showed increased luciferase activity and expression levels. | Promoter variant, rs12943590 (A-allele) associated with poor response to metformin in some studies. |
 |
KO mice: Yes (Oatp1a/1b knockout) DDI: Yes GWAS: Yes |
The haplotype, T334-G699, has higher transporter activity compared to G334-A699. | 334T>G and 699G>A associated with increased toxicity and increased drug levels (e.g. mycophenolate mofetil, paclitaxel). |
 |
KO mice: **Available but no data DDI: Yes GWAS: No |
Both variants have similar activity as the reference allele. | Some effect on drug disposition but not reproducible (e.g. montelukast). |
 |
KO mice: Yes DDI: Yes GWAS: No |
T-allele of Ile1145Ile showed reduced expression levels. T-allele of Ala893Ser showed decreased efflux of substrates. A-allele of Ala893Thr showed increased efflux of substrates. |
Controversial and conflicting results with both variants (e.g. fexofenadine, tacrolimus) (see more examples in PharmGKB or ClinVar) |
 |
KO mice: Yes DDI: Yes GWAS: No |
T-allele is associated with higher luciferase activity. | Controversial and conflicting results (e.g. tenofovir, atorvastatin, methotrexate) (see more examples in PharmGKB or ClinVar) |
Population according to 1000 Genomes Project only. AFR: African; AMR: Ad Mixed American; EUR: European; EAS: East Asian; SAS: South Asian
See resources from ClinVar (https://www.ncbi.nih.gov/clinvar/) and PharmGKB (https://www.pharmgkb.org). Both resources provide collection of in vitro and in vivo data of the SNP with clinical phenotypes, including drug response traits.
Available but no data: Knockout mice are available from International the Mouse Phenotyping Consortium (IMPC), www.mousephenotype.org but no published data about the mice.
2. Polymorphisms in Other Drug Transporters with Less Evidence
In addition to SLCO1B1, SLC22A1 and ABCG2, other important membrane drug transporters recommended by the FDA, PMDA and EMA drug-drug interaction guidance/guidelines for study during drug development include SLC22A2 (OCT2), SLC22A6 (OAT1), SLC22A8 (OAT3), SLCO1B3 (OATP1B3), SLC47A1 (MATE1), SLC47A2 (MATE2K) and ABCB1 (P-gp) (see Table 1 for the list of URLs to regulatory guidances and guidelines mentioned in this review). Below we briefly summarize candidate gene studies of these transporters. Table 2 gives an overview of the key polymorphisms highlighted below. For lists of publications about these polymorphisms, see the following publicly available resources, PharmGKB, ClinVar, Human Longevity Inc. Open Search and other (see Figure 2).
Figure 2.
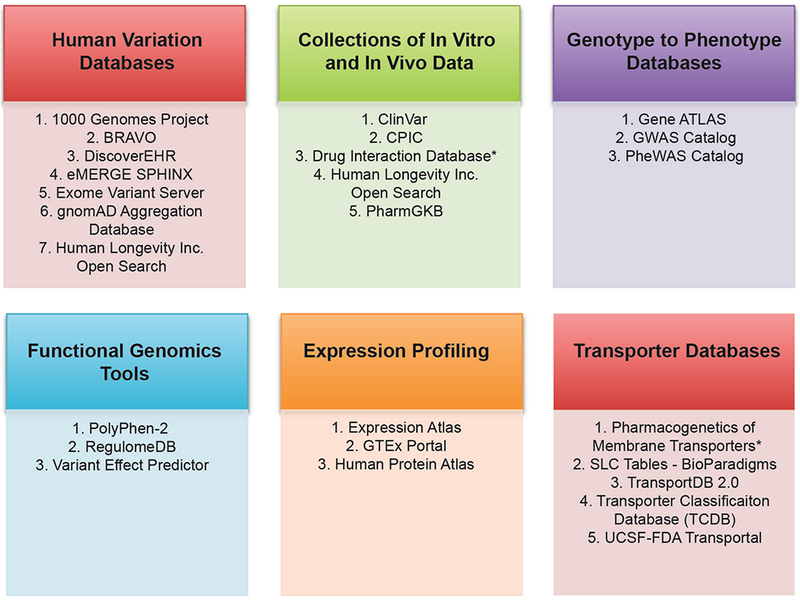
Commonly used databases for pharmacogenomics and membrane transporter research. The figure shows different categories of databases. The asterisk (*) shows that the database is not publicly available. See Table S4 for links to each of the databases, as well as for descriptions of the database categories.
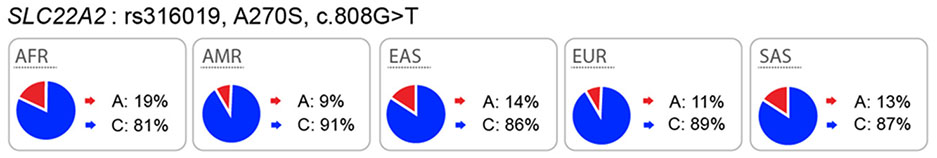
Polymorphisms in SLC22A2 (OCT2):
OCT2 mediates the uptake of many drugs including antidiabetic drugs, platinum anti-cancer drugs, and histamine H2 receptor blockers, across the basolateral membrane of tubular epithelial cells25. In vitro studies of SLC22A2 genetic polymorphisms have been controversial. For example, some studies have shown that genetic variants of SLC22A2 result in a significantly reduced uptake of metformin, lamivudine and other substrates26–29, whereas other studies have failed to show effects of SLC22A2 polymorphisms on substrate uptake30,31. Clinical data suggest that SLC22A2 variants are associated with metformin disposition and response30,32–34, cisplatin toxicity35, as well as with levels of a variety of endogenous metabolites in humans36. However, here again, clinical studies have been inconclusive as various studies report opposite associations between these genetic variants in SLC22A2 and clinical drug levels28,30,34.
Polymorphisms in SLC22A6 (OAT1) and SLC22A8 (OAT3):
OAT1 and OAT3 are renal transporters involved in the elimination and disposition of important drugs including statins, diuretics, nonsteroidal anti-inflammatory drugs, and anti-microbial drugs (see UCSF-FDA Transportal, http://transportal.compbio.ucsf.edu/). Data indicate that the coding regions of SLC22A6 and SLC22A8 have low genetic and functional diversity and suggest that coding region variants of these transporters may not contribute substantially to inter-individual differences in renal elimination of xenobiotics37–39. Despite clinical evidence from DDI studies, which demonstrate that inhibition of these transporters significantly changes the levels of many endogenous metabolites (e.g. taurine, creatinine, indoxyl sulfate, bile acid conjugates40–42) or victim drugs43,44, there are few significant associations of genetic polymorphisms in SLC22A6 or SLC22A8 with drug levels or response. In fact, to our knowledge, only one study showed a significant association (p<0.05) of an SLC22A8 Asian-specific non-synonymous variant, Ile305Phe (rs11568482) with reduced renal and secretory clearance of the antibiotic, cefotaxime45. However, the effect of this variant has not been evaluated in an independent study.
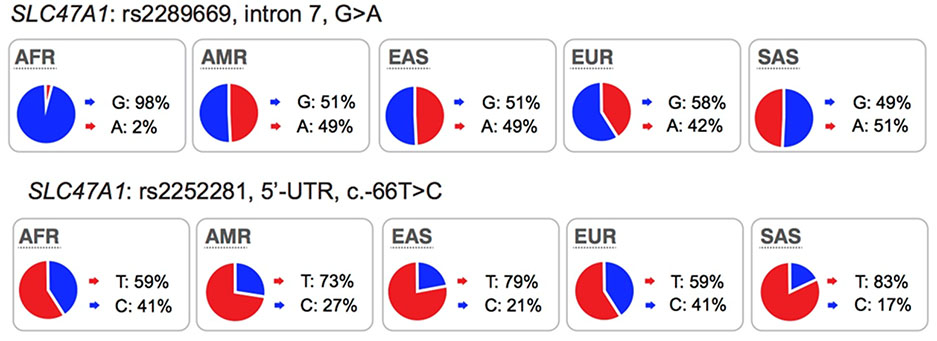
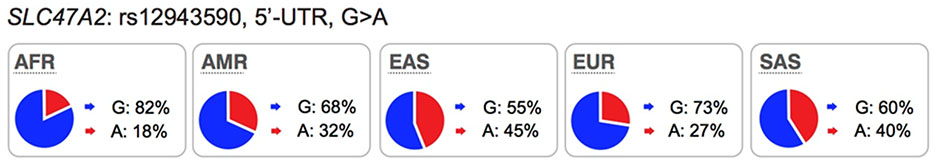
Polymorphisms in SLC47A1 (MATE1) and SLC47A2 (MATE2):
MATE1 is expressed on the canalicular membrane of hepatocytes and on the apical membrane of the renal proximal tubule cell where it serves to secrete drugs into the urine46. MATE2 is expressed specifically on the apical membrane of the proximal tubule and works in concert with MATE1 in renal drug secretion47. These transporters are known to transport many drugs including metformin, oxaliplatin and cimetidine (see UCSF-FDA Transportal). Although in clinical studies, MATE1/MATE2 inhibitors produced significant differences in endogenous metabolite levels48 and in the disposition of MATE1/MATE2 substrates49,50, evidence indicating that genetic polymorphisms in the transporters can phenocopy the effects of clinical MATE inhibitors is inconsistent. For example, an intronic variant in SLC47A1 (rs2289669), without known function, is associated with metformin response in several studies12,18 but not in other studies17. Similarly, the functional variants in SLC47A1 (5’UTR, rs2252281) and SLC47A2 (5’UTR, rs12943590) also showed significant associations with metformin disposition and response in a small cohort of approximately 200 patients with type 2 diabetes51,52 but were not associated with response to metformin in a large cohort of patients with type 2 diabetes17. Considering that changes in metformin pharmacokinetic and pharmacodynamics as a result of transporter modulation are complex, these findings would need to be confirmed with other drug probes. Recent studies have shown that there are ethnic differences metformin response. In particular, patients from African American ancestry have better response to metformin than patients from European ancestry53,54. Further, the pharmacogenomic studies involving SLC47A1 or SLC47A2 have been carried out primarily in individuals of European ancestries. Thus, further studies are needed to determine the effects of polymorphisms in the MATEs on the disposition and response to metformin in other populations.
Polymorphisms in SLCO2B1 (OATP2B1):
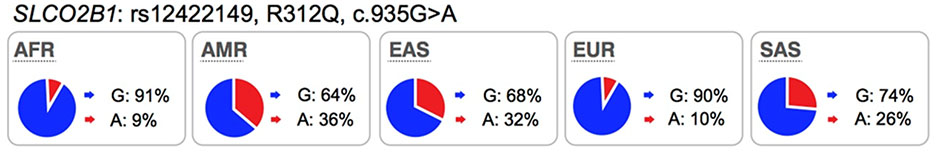
OATP2B1 is expressed in all tissues (GTEx Portal), including the tissues of interest for xenobiotic absorption and disposition, such as brain (luminal membrane of endothelial cells of the blood brain barrier), small intestine, and liver (basolateral/sinusoidal membrane of hepatocytes)1. Further, the transporter is expressed in many tumors such as tumors originating in breast and prostate55. OATP2B1 mediates the transport of structurally diverse organic anions (sulfobromophthalein, methotrexate, pemetrexed, folic acid, reduced folates) (see UCSF-FDA Transportal). Emerging data suggest that SNPs within SLCO2B1 associate with altered protein function and clinical efficacy and/or safety. Several studies have evaluated the impact of an SLCO2B1 non-synonymous SNP, rs12422149 (Arg312Gln) on drug response. The results show that the OATP2B1-Arg312 is associated with increased transport of dehydroepiandrosterone (DHEAS) and increased resistance to androgen-deprivation therapy (ADT)56 as well as lower overall survival rates in patients with prostate cancer56,57. Another independent study showed that OATP2B1-Arg312 is significantly associated with shorter time to progression in prostate cancer patients treated with androgen deprivation58. Furthermore, the nonsynonymous polymorphism, OATP2B1-Gln312, was associated with poorer response to rosuvastatin59. However, the effect of this SLCO2B1 non-synonymous variant was not significant in the disposition or response for other substrates, e.g. montelukast60,61. Several clinical studies have shown that apple juice or grapefruit juice can inhibit OATP2B1 and hence reduce absorption (reduce plasma AUC levels by >1.5 fold) of several drugs that are substrates of OATP2B1, such as fexofenadine, atorvastatin, and celiprolol62,63. More recently, a drug in early development, ronacaleret, unexpectedly reduced plasma levels of rosuvastatin, a substrate of OATP2B164 by 50%. These interaction studies suggest that, similar to the inhibition effects of juice or drugs on OATP2B1 transport function, SLCO2B1 reduced function variants may play a role in modulating drug disposition (Table S3).
Polymorphisms in SLCO1B3 (OATP1B3):
OATP1B3 is predominantly expressed in hepatocytes (basolateral/sinusoidal membrane)1. The transporter mediates the transmembrane flux of bile acids, cholesterol, hormones and their metabolites as well as drugs such as HMG-CoA reductase inhibitors, anti-hypertensive drugs, anti-cancer drugs, HIV protease inhibitors and others (see UCSF-FDA Transportal). An assessment of polymorphisms (SNPs) in SLCO1B3 revealed that a number of variants were associated with anti-cancer drug and immunosuppressant drug response or toxicity (see https://www.pharmgkb.org/gene/PA35844/variantAnnotation). For example, patients who are carriers of the T allele of rs4149117 (OATP1B3-Ser112Ala, c.334T>G), were 19% less sensitive to thrombocytopenia from carboplatin/paclitaxel treatment, than the homozygotes for the G allele65. SLCO1B3 699GG (rs7311358, OATP1B3-Met233Ile, c.699G>A) and 344TT (rs4149117) genotypes are also associated with non-response to imatinib in patients with chronic myeloid leukemia66. Collectively, studies suggest that OATP1B3 may be an important polymorphic transporter for new and approved drugs.
Polymorphisms in ABCB1 (P-gp, MDR1):
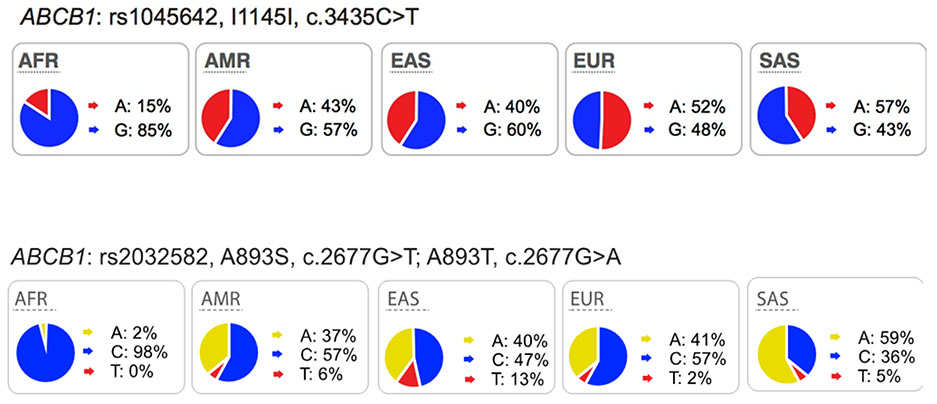
The multi-drug resistance-1 (MDR1) protein belongs to ATP-binding cassette (ABC) family and encodes the efflux pump, P-glycoprotein (P-gp). P-gp, expressed in tumors, mediates drug resistance to many anti-cancer drugs67 and its roles in the intestine and blood-brain barrier have been extensively studied using different experimental methodologies such as knockout mice68, imaging probes69 and clinical studies of genetic polymorphisms70. The most highly studied polymorphisms in ABCB1 are the synonymous variant, C3435T (Ile1145Ile, rs1045642) and the non-synonymous variant, G2677T, Ala893Ser (rs2032582). These common polymorphisms are found in all ethnic groups. Functional studies showed that the C3435T variant is associated with lower expression levels of P-gp in the duodenum71 and placenta72 and that these lower expression levels have been correlated with increased digoxin plasma concentrations71. The ABCB1 non-synonymous variant, Ala893Ser, has been shown to cause reduced function and increased accumulation of substrates inside cells73,74. However, controversial and conflicting data of these polymorphisms were reviewed previously75,76 (also see resources: https://www.pharmgkb.org/search?connections&gaSearch=rs2032582&query=rs2032582 and https://www.pharmgkb.org/search?connections&gaSearch=rs1045642&query=rs1045642).
Polymorphisms in ABCC2 (MRP2):
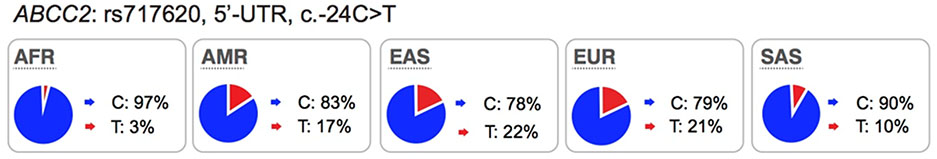
The multi-drug resistance associated protein-2 (MRP2) belongs to the ABC family. This transporter is highly expressed in the liver and kidney, and serves to efflux drugs from the cells into the bile and urine, respectively. Rare mutations in ABCC2 are known to cause a Mendelian disorder, Dubin-Johnson syndrome, which causes conjugated hyperbilirubinemia (https://www.omim.org/entry/601107). Many drugs and glucuronide-conjugated drug metabolites are known substrates of MRP2, e.g. methotrexate, the irinotecan inactive metabolite, SN-38 glucuronide, and etoposide (see UCSF-FDA Transportal). The most highly studied polymorphism in ABCC2 is the promoter variant, c.−24C>T (rs717620). This promoter variant is associated with lower expression levels of ABCC277 (Table S3). Several candidate gene studies have identified significant associations of the variant with drug levels78,79, toxicity80,81 and response82 of anti-cancer drugs, such as methotrexate and irinotecan. The effect of this variant on drug response shows contradictory results, which may be due to small sample sizes or differences in the populations studied (see PharmGKB for clinical studies which are significant or not significantly associated with this promoter variant).
In summary, though there is some evidence that SNPs in the transporters in this section contribute to variation in drug response and toxicity, more experiments are clearly needed. For SNPs in SLC22A2 (OCT2), SLCO2B1 (OATP2B1), SLC47A1 (MATE1), ABCB1 (P-gp), and ABCC2 (MRP2), reports in the literature have been conflicting, and sample sizes have generally been small. For variants of SLCO1B3 (OATP1B3) and SLC22A6 (OAT1), only a few studies have been published. Clarification with further studies and studies of larger sample sizes will greatly enhance our understanding of the clinical implications of polymorphisms in these transporters.
3. Functional Genomics of Drug Transporters
From a mechanistic view, polymorphisms in membrane transporter genes can modulate transport function by affecting either the activity of the transporter or expression level of the transporter protein. Polymorphisms can affect transporter activity through several mechanisms. For example, polymorphisms in the binding site of the transporter may directly affect the binding and de-binding of substrates and may act in a substrate-dependent fashion (e.g. SLC47A2 missense variant, Gly393Arg)51. In contrast, missense variants not found in the binding site may affect the expression level of the transporter protein in the plasma membrane by altering trafficking to and from the plasma membrane or protein stability (e.g. SLC47A2 missense variant, Pro162Leu)51. Noncoding region variants as well as synonymous variants (and some non-synonymous variants) may affect the mRNA expression level of the transporter by affecting rates of transcription or RNA stability. In this section, we provide a list of functionally characterized coding and promoter variants of twelve major transporters that play a role in drug absorption, disposition and toxicity, along with their in vitro effects on protein function or expression (Table S3). The majority of the SNPs were discovered in large sequencing projects of multiple genes, and a large research project focused on membrane transporter polymorphisms, the Pharmacogenomics of Membrane Transporters project83. More recently, genetic polymorphisms across the genome have been discovered through multiple sequencing efforts, and frequently accessed databases now post genetic variants in all genes: Exome Variant Server and eMERGE SPHINX (see more examples in Figure 2 and Table S4). Below, we highlight key examples in which the genetic polymorphism leads to functional changes.
Transporter variants that cause changes to transporter activity or protein expression:
Nonsynonymous transporter variants have been characterized using site-directed mutagenesis in mammalian cells (e.g. HEK293, CHO, HeLa) or X. laevis oocytes (see references in Table S3). For transporter variants that retain some functional activity, kinetic studies have been performed to determine the Michaelis-Menten constants (Km and Vmax values) of the variant transporters for various substrates. For some transporters, further studies to determine the molecular mechanisms responsible for changes in kinetic parameters have been performed including structural modeling, in situ docking of substrates in the binding pocket of the variant transporter, and measurement of the transporter protein level on the cell membrane. A glance at the table (Table S3) shows 180 non-synonymous variants that have been characterized in cellular assays of which ~50% exhibited reduced transporter function and/or expression levels on the membrane compared to the reference allele. Many of these reduced function variants are substrate-dependent. Generalizable findings for functional studies of drug transporter variants are as follows:
Several well-characterized xenobiotic transporters have common reduced function nonsynonymous variants found at allele frequencies greater than 10% in particular ethnic populations. These include: OATP1B1-V174A (rs4149056), BCRP-Q141K (rs2231142), and OCT1–420del (rs202220802). These variants have been associated with changes in efficacy and safety for a number of drugs and are considered in this manuscript as highly significant transporter polymorphisms (see Table S1 and S2).
Mechanisms responsible for reduced function are more frequently related to reduced protein expression levels on the cell surface membrane (e.g., the OATP1B1-V174A and BCRP-Q141K) rather than direct effects on substrate binding and translocation.
Less common and rare nonsynonymous variants (minor allele frequency (MAF) < 1%) are more likely to exhibit significant reductions in their uptake kinetics than more common variants.
Most of the twelve transporters have at least one less common nonsynonymous variant (1% - 5% in at least one population) that affects transporter kinetic parameters and many of these are present in one ethnic group only. Examples are: OCT1-G401S (European), OCT2-K432Q (African), MATE2-P162L (African), OAT1-R50H (African), OAT3-I305F (Asian) and OATP1B1-G488A (African).
The majority of less common reduced-function transporter variants have not been associated with drug disposition or response in clinical studies, presumably due to their low allele frequencies and the difficulty of obtaining sufficient numbers of study subjects who harbor the variants.
Collectively, these finding suggest that further studies should be conducted assessing the associations of less common and rare variants in transporters and drug response, or combinations of less common and rare variants in individual transporters (as in the case of OCT1).
Transporter variants that cause changes in transcriptional rates:
Though many noncoding variants in transporter genes are associated with variation in drug response (Table S3), few of these variants have been characterized. A handful of variants in promoter, enhancer or UTR regions of these transporters have been functionally characterized using reporter assays with luminescent probes such as luciferin (19742321). In general, the effect of variants in non-coding regions may be subtle modulating, but not ablating, expression levels. A noncoding region variant in a transporter gene may change the expression level of the mRNA transcript and correspondingly, the transporter protein level. Variants that are associated with changes in transcript levels are termed expression quantitative trait loci (eQTL). Through major efforts of consortia or large research projects, many eQTL databases are available publicly. These databases allow users to look up genetic polymorphisms in any gene, including membrane transporter genes, and determine whether they are significantly associated with mRNA expression levels of the transporter or other genes in various tissues. The most popular eQTL database is the GTEx Portal. In Table S3, we highlighted several SNPs in the promoter and UTR regions of transporters that are known eQTLs and alter luciferase activity in reporter assays.
4. Clinical Study Design and The Use of Modeling and Simulation
Clinical Study Design:
Studies of the clinical effects of transporter polymorphisms can provide enormous insights into the role of a transporter in drug disposition, response and toxicity. Unlike DDI studies, which often involve the use of non-selective inhibitors, studies of transporter polymorphisms provide direct evidence for the pharmacologic role of a particular transporter. However such studies may be challenging due to the low allele frequencies of many transporter polymorphisms (<20%), and the fact that the effects of transporter polymorphisms are largely seen in individuals who are homozygous. Thus, Hardy Weinberg equilibrium predicts that for a variant with an allele frequency of 20%, only 4% of the population will be homozygous. Of course, if allele frequencies are lower, homozygotes will be even rarer; leading to underpowered studies or studies where conclusions are drawn from a handful of patients. Other issues that should be addressed during drug development are when and how to evaluate the effects of genetic polymorphisms on drug disposition or response. As in our previous manuscript2, we recommend that during all phases of drug development, genetic polymorphisms in drug transporters are studied. Furthermore, recent guidances from regulatory groups (e.g., the FDA draft guidance of Clinical Drug Interaction Studies, the FDA guidance of Clinical Pharmacogenomics, the International Council for Harmonization (ICH) Guideline E18 on Genomic Sampling and Management of Genomic Data, and the FDA final guidance of E18 Genomic Sampling and Management of Genomic Data (see URLs in Table 1)) encourage sponsors to routinely collect DNA from all subjects for retrospective analysis of polymorphisms in transporters of interest. In the pre-clinical phases of drug development, it is important to characterize which transporters are important determinants of the drug disposition. For substrates of BCRP or OATP1B1 in which the transporter plays an important role in drug disposition, the ITC recommends that clinical studies are considered to assess the effects of genetic variants on the pharmacokinetics and if possible pharmacodynamics84. Key considerations for pharmacogenomic clinical designs have been reviewed85.
Pharmacogenomic Discovery Studies:
Though there have been a number of candidate gene pharmacogenomics studies focused on transporter polymorphisms, many have been underpowered to make meaningful conclusions. Further, most have focused on more common variants in transporter genes. To enhance reproducibility of results and explore the effects of less common genetic variants, larger sample sizes are needed. In order to accomplish this feat, many investigators are pooling samples to obtain the necessary sample sizes. For example, a recent meta-analysis examining the association between genetic variants in transporter genes and metformin response included up to 7,968 samples17.
In addition to underpowered studies focused on candidate genes, most of the retrospective pharmacogenomic studies have evaluated the effect of transporter variants in isolation, i.e., lack the information on the potential effect of multiple coexisting genetic covariates (e.g., SLCO1B1 and ABCG2 polymorphisms in the same subject) together with demographic factors (e.g., age, ethnicity) on the pharmacokinetics of a drug and/or its DDI sensitivity. The population pharmacokinetic modeling reported recently for simvastatin acid identified combination of genetic and demographic risk factors associated with altered simvastatin exposure and increased myopathy risk that was not solely attributed to rs414905686. In addition, a number of studies have reported differences in transporter activity and plasma exposures of statins between Japanese and Caucasian populations, which appear to extend beyond the SLCO1B1 genotype86,87. However, these findings have been challenged in other studies, which suggest that genetic variants in SLCO1B1 and ABCG2 may explain much of the ethnic differences between plasma exposures of statins88.
Role of Modeling and Simulation:
Modeling and simulation, especially physiologically-based pharmacokinetic (PBPK) models, can be used to understand and project the effects of genetic polymorphisms on pharmacokinetics and pharmacodynamics during drug development, as illustrated in number of literature examples predominantly focusing on OATP1B121,89. Recently, PBPK modeling approach was applied to predict morphine clearance and associated inter-subject variability in children with different OCT1 allelic variants90. Plasma concentrations of many prescription drugs are affected by SLCO1B1 polymorphisms, particularly the OATP1B1-Val174Ala variant (rs4149056, c.521T>C)2. In spite of their strong associations with systemic plasma levels and muscle toxicities of various statins, SLCO1B1 polymorphisms have minimal effect on pharmacodynamics of statins, i.e. their lipid lowering effects20,91. These observations can be explained by understanding that hepatic elimination is the predominant route and that metabolic clearance (simvastatin acid) and biliary efflux (pravastatin) rather than active uptake determine liver exposure of statins21,92,93. In contrast, polymorphisms in efflux transporters on the canalicular membrane such as BCRP (or efflux transporter-mediated DDI) are expected to have a significant effect on the liver exposure84. Verification of these model-derived tissue exposures and associated challenges are discussed in detail in the Guo et al. whitepaper in this Clin Pharmacol Ther issue. A PBPK model-based approach also provides a framework to inform power calculations and guide the design of either pharmacogenetic or DDI studies, as illustrated by Gertz et al.89 where this mechanistic approach was applied to simulate repaglinide exposure in subjects with co-existing CYP2C8*3 and OATP1B1-Val174Ala.
Pharmacogenomic clinical data provide an incredibly valuable dataset for the verification of the PBPK models, as they allow evaluation of the importance of individual transporters/elimination mechanisms in the model. Recent examples illustrate the use of plasma data from different SLCO1B1 variant groups to validate PBPK models developed for repaglinide, simvastatin acid, simeprevir and pitavastatin21,89,94. Following initial verification of the simulated plasma profiles against pharmacogenomic data, PBPK models were subsequently used to predict liver and muscle exposure in subjects with different polymorphism and/or predict DDIs with these clinical probes. Overall, there is less confidence in transporter PBPK models compared to PBPK models for drug metabolizing enzymes because of knowledge gaps in transporter biology and limited experience in determining and modeling the kinetics of transporters (see this issue by Guo Y. et al.). The recently revised FDA draft guidance of In Vitro Metabolism and Transporter-Mediated Drug-Drug Interaction Studies (see Table 1) recommends that clinical data from a wide range of DDI or pharmacogenetic studies be used to verify PBPK models developed for transporter substrates.
5. Future Directions and Conclusions
Future directions will depend on continued use and growth of technology in the discovery of important associations between genetic variants in transporters and drug response, as highlighted below.
Genomewide Association Studies:
Genomewide association studies (GWAS) continue to be a powerful tool for determining the role of transporters in human biology and pharmacology. In fact, in addition to genomewide level significant results between pharmacogenomics traits and polymorphisms in ABCG2 and SLCO1B1, GWAS have revealed associations between polymorphisms in other ABC and SLC transporters and drug response at genomewide level significance (p<5×10−8) (Table S1 and Table S5)95. Surveying the NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/) and Pubmed, we identified eleven publications relevant to pharmacogenomic traits, which described significant associations with polymorphisms in ABC or SLC transporter genes or loci (p<5×10−8). However, with the exception of polymorphisms in ABCG2 and SLCO1B1, associations between polymorphisms in other transporter genes (e.g. SLC17A1, SLC2A2, SLC15A1, SLCO1B3) (Table S5) and pharmacogenomic traits have yet to be replicated in independent cohorts. It is envisioned that some of these associations will be replicated in future studies. GWAS is envisaged to facilitate identification of potential endogenous biomarkers for transporters, as reported recently for OATP1B196, especially considering interest in application of endogenous biomarkers to support evaluation of transporter-mediated DDI risk in early stages of drug development (see Chu et al in this issue for details). In the last few years, there has been growing interest with phenome-wide association studies (PheWAS), in which many phenotypes (disease, traits, drug response, etc) are correlated with a particular single genetic polymorphism97. For example, scanning across all the available GWAS, one could look for new implications of a single polymorphism, such as those in transporters. Such resources are publicly available, e.g. PheWAS Resources, https://phewascatalog.org/
Massively Parallel Sequencing:
In recent years, massively parallel sequencing has been increasingly used for discovering SNPs in pharmacogenes in large and diverse ethnic populations from various regions of the world 98–100. The majority of these studies have focused on CYP enzymes. However some of these studies have included membrane transporter genes known to play a role in drug response, such as SLCO1B1 and ABCG298,99. Leveraging publicly available human variation databases, such as the 1000 Genomes Project, Exome Variant Server, and gnomAD browser (see Figure 2, Table S4) allows an extensive examination of the spectrum of genomic variations across all genes including SLC and ABC transporters. Some advantages of Next Generation Sequencing (NGS) methods are that they are potentially cheaper and analyses are simpler compared to traditional Sanger sequencing. Further, the results generated from NGS allow re-analysis of the raw sequencing data, especially when updated information about new variants or reclassified variants become available in the future. Although NGS has been used for discovering SNPs, its use in the identification of the genetic determinants of drug response and adverse drug response has been very limited. The following are examples of how NGS have been used in the discovery of variants in membrane transporters, which are associated with drug disposition, response or toxicity.
NGS to identify genetic determinants for drug levels (or exposure). Recently, the Niemi laboratory used NGS to sequence candidate genes to identify the genetic basis of inter-individual variation in response to montelukast and clopidogrel as well as concentrations of drug metabolites101,102. These studies were conducted in approximately 190 healthy volunteers, focusing on relevant enzymes and membrane transport proteins. The results showed that individuals who are carriers of uridine diphosphateglucuronosyltransferase (UGT), UGT1A3*2 (rs3821242 and rs6431625) have lower montelukast exposure and greater exposure to montelukast metabolites with very large effect sizes (effect size ≥18% change per copy of minor allele, p<10−9). In general, these SNPs are not readily available in genotyping arrays (see NCBI dbSNP) and thus massive parallel sequencing facilitated the discovery of common SNPs (~40% allele frequencies) associated with montelukast exposure. Other candidate genes, including the transporters, ABCC9 and SLCO1B1 were also associated with montelukast metabolite concentrations.
NGS to identify genetic determinants for non-response to medication. NGS technology was also used to sequence DNA from patients who are extremely resistant to particular medications. For example, Chua et al. 103 performed whole-exome sequencing of DNA samples from 12 patients who exhibited extreme therapeutic resistance to azathioprine or 6-mercaptopurine. The study was designed to focus on genes within the drug disposition pathway, which included SLC and ABC transporters as well as enzymes103. Although the sample size was small, a potential missense variant in the transporter, SLC17A4, had significantly higher allele frequency in the patients (3/24 (12.5%)) compared to population controls from public data sources (101/8600 (1.2%) (p<0.0001).
In addition to its use in the discovery of variants that associate with clinical drug response, disposition and toxicity, NGS technology is also being used in preemptive genotyping in the patient care setting. The design and implementation of preemptive approaches to integrate pharmacogenomic data in the electronic health record has begun in several healthcare institutions since 2012 and may include functional variants in SLCO1B1 and ABCG2 (see examples104,105). Although genotyping using arrays is most commonly employed in preemptive studies, there are recent examples where exome-sequencing with NGS has slowly been adopted in a few healthcare institutions, such as eMERGE-PGx Project106. In these studies, genotype information is deposited in the electronic health record along with some decision support tools, with the goal of assisting clinicians in the use of genomic information to guide choice of therapies and doses.
Drug Development Perspective:
Pharmaceutical industry-leading practices now incorporate pharmacogenomics routinely in drug development to better understand drug safety and response in individual patients. For many years, pharmacogenomic studies largely focused on common polymorphisms in drug metabolizing enzymes. However, proactive strategies now include genotyping patients using genome-wide platforms that are specifically enriched for ADME genes including transporters. With this approach, data are collected during the conduct of the trial (and across many trials in a drug development program) and can be retrospectively analyzed for association with disposition, toxicity and/or efficacy. Costs of collection, genotyping and long term biorepository storage are now in the realm of making this a relatively low-cost endeavor107,108. Furthermore, as more companies adopt such strategies, logistics are enabled by several commercial organizations offering such services. Such practice has resulted in a relatively low-cost approach to improved understanding of pharmacokinetic and pharmacodynamic mechanisms including the role of particular transporters and enzymes in the absorption, disposition, efficacy and toxicity of drugs. While it is not uncommon to identify genetic polymorphisms that are statistically associated with differences in drug disposition, it is also not uncommon that those differences are deemed clinically non-meaningful when considering the therapeutic window of the drug. When clinically meaningful associations exist, it is important to identify those during drug development and understand their effect with respect to other co-existing polymorphisms so that appropriate dosing or exclusion decisions can be made. In addition to improved practices with the industry and better understanding of pharmacologic mechanism, regulators are increasingly expecting and asking for such data.
CONCLUSION
In this review, evidence has been presented that genetic polymorphisms in the emerging transporter, SLC22A1, be considered in addition to previously recommended polymorphisms in ABCG2, and SLCO1B1 during drug development when evaluating mechanisms responsible for inter-individual variation in drug disposition, response and toxicity. Review of clinical and functional genomic studies of other drug transporters including SLCO2B1, SLCO1B3, ABCB1, and ABCC2 suggests that further studies are needed to understand the role of polymorphisms in these transporters in clinical drug disposition and response. The integrated use of PBPK modeling and pharmacogenomic data to support model development and improve our understanding of the consequences of genetic variants in membrane transporters on drug disposition and response is recommended. Future studies using GWAS and massively parallel sequencing approaches in larger cohorts of patients are clearly needed to advance our understanding of transporter polymorphisms and variation in drug disposition and response so that they may be fully integrated into clinicians’ decision-making when choosing doses and therapies.
Supplementary Material
Table S1. Eight genomewide association studies identified significant associations of polymorphisms in ABCG2 or SLCO1B1 with drug disposition or response (p<5×10-8). A number of studies have since replicated the initial findings from the initial GWAS, and the references for each study can be found in Table S1 below.
Table S2. Important non-synonymous variants in SLC22A1 that have reduced transporter activity and altered substrates disposition, response or adverse effects. These non-synonymous variants have been genotyped in several clinical pharmacogenomics studies. In each of these studies, there is a significant correlation with the number of inactive OCT1 alleles with the studied drug disposition (pharmacokinetic effect, PK) and response and toxicity (pharmacodynamics effect, PD). These studies associated the phenotype with the most common functional amino acid substitutions found in Caucasians (Arg61Cys, Cys88Arg, Gly401Ser, Met420del, and Gly465Arg). Allele frequencies of other ethnic groups and functional variants of other coding variants in SLC22A1 are shown Table S3. References for each study can be found below Table S2.
Table S3. Genetic polymorphisms in 12 transporters and their in vitro consequences. Non-synonymous and untranslated region (UTR) polymorphisms with known in vitro functions are described in this table.
Table S4. Commonly used databases for pharmacogenomics research and membrane transporter research.
Table S5. Genomewide association studies of pharmacogenomic studies that have genomewide significance (p<5×10-8) variants in SLC or ABC transporters.
Acknowledgements
The authors would like to acknowledge Dr. Anuradha Ramamoorthy and members of the ITC for their valuable comments on the manuscript.
Funding The authors acknowledge support from the following NIH grants: GM115370 (K.M.G., S.W.Y.), R01 GM117163 (K.M.G., S.W.Y.), R01 DK103729 (K.M.G. and D.J.B.).
Footnotes
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: The contents of this article reflect the views of the authors and should not be construed to represent the FDA’s views or policies.
Conflict of Interest
Drs. Giacomini and Yee are co-founders of Apricity Therapeutics, Inc., which focuses on transporters in drug discovery and development.
References
- 1.International Transporter C et al. Membrane transporters in drug development. Nat Rev Drug Discov 9, 215–236, doi: 10.1038/nrd3028 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giacomini KM et al. International Transporter Consortium commentary on clinically important transporter polymorphisms. Clin Pharmacol Ther 94, 23–26, doi: 10.1038/clpt.2013.12 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Balyan R et al. OCT1 genetic variants are associated with postoperative morphine-related adverse effects in children. Pharmacogenomics 18, 621–629, doi: 10.2217/pgs-2017-0002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tzvetkov MV et al. Morphine is a substrate of the organic cation transporter OCT1 and polymorphisms in OCT1 gene affect morphine pharmacokinetics after codeine administration. Biochem Pharmacol 86, 666–678, doi: 10.1016/j.bcp.2013.06.019 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Tzvetkov MV et al. Effects of OCT1 polymorphisms on the cellular uptake, plasma concentrations and efficacy of the 5-HT(3) antagonists tropisetron and ondansetron. Pharmacogenomics J 12, 22–29, doi: 10.1038/tpj.2010.75 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Wen CC et al. Genome-wide association study identifies ABCG2 (BCRP) as an allopurinol transporter and a determinant of drug response. Clin Pharmacol Ther 97, 518–525, doi: 10.1002/cpt.89 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varenhorst C et al. Effect of genetic variations on ticagrelor plasma levels and clinical outcomes. Eur Heart J 36, 1901–1912, doi: 10.1093/eurheartj/ehv116 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Christensen MM et al. The pharmacogenetics of metformin and its impact on plasma metformin steady-state levels and glycosylated hemoglobin A1c. Pharmacogenet Genomics 21, 837–850, doi: 10.1097/FPC.0b013e32834c0010 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Shu Y et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther 83, 273–280, doi: 10.1038/sj.clpt.6100275 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sundelin E et al. Genetic Polymorphisms in Organic Cation Transporter 1 Attenuates Hepatic Metformin Exposure in Humans. Clin Pharmacol Ther 102, 841–848, doi: 10.1002/cpt.701 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Tzvetkov MV et al. The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin Pharmacol Ther 86, 299–306, doi: 10.1038/clpt.2009.92 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Becker ML et al. Genetic variation in the organic cation transporter 1 is associated with metformin response in patients with diabetes mellitus. Pharmacogenomics J 9, 242–247, doi: 10.1038/tpj.2009.15 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Shu Y et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest 117, 1422–1431, doi: 10.1172/JCI30558 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dujic T et al. Association of Organic Cation Transporter 1 With Intolerance to Metformin in Type 2 Diabetes: A GoDARTS Study. Diabetes 64, 1786–1793, doi: 10.2337/db14-1388 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tarasova L et al. Association of genetic variation in the organic cation transporters OCT1, OCT2 and multidrug and toxin extrusion 1 transporter protein genes with the gastrointestinal side effects and lower BMI in metformin-treated type 2 diabetes patients. Pharmacogenet Genomics 22, 659–666, doi: 10.1097/FPC.0b013e3283561666 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Christensen MM et al. Endogenous glucose production increases in response to metformin treatment in the glycogen-depleted state in humans: a randomised trial. Diabetologia 58, 2494–2502, doi: 10.1007/s00125-015-3733-2 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Dujic T et al. Variants in Pharmacokinetic Transporters and Glycemic Response to Metformin: A Metgen Meta-Analysis. Clin Pharmacol Ther 101, 763–772, doi: 10.1002/cpt.567 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jablonski KA et al. Common variants in 40 genes assessed for diabetes incidence and response to metformin and lifestyle intervention in the diabetes prevention program. Diabetes 59, 2672–2681, doi: 10.2337/db10-0543 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou K et al. Reduced-function SLC22A1 polymorphisms encoding organic cation transporter 1 and glycemic response to metformin: a GoDARTS study. Diabetes 58, 1434–1439, doi: 10.2337/db08-0896 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chasman DI et al. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ Cardiovasc Genet 5, 257–264, doi: 10.1161/CIRCGENETICS.111.961144 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Tsamandouras N et al. Development and Application of a Mechanistic Pharmacokinetic Model for Simvastatin and its Active Metabolite Simvastatin Acid Using an Integrated Population PBPK Approach. Pharm Res 32, 1864–1883, doi: 10.1007/s11095-014-1581-2 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Matthaei J et al. OCT1 mediates hepatic uptake of sumatriptan and loss-of-function OCT1 polymorphisms affect sumatriptan pharmacokinetics. Clin Pharmacol Ther 99, 633–641, doi: 10.1002/cpt.317 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Tzvetkov MV et al. Genetically polymorphic OCT1: another piece in the puzzle of the variable pharmacokinetics and pharmacodynamics of the opioidergic drug tramadol. Clin Pharmacol Ther 90, 143–150, doi: 10.1038/clpt.2011.56 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Tzvetkov MV et al. Increased Systemic Exposure and Stronger Cardiovascular and Metabolic Adverse Reactions to Fenoterol in Individuals with Heritable OCT1 Deficiency. Clin Pharmacol Ther, doi: 10.1002/cpt.812 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Motohashi H & Inui K Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J 15, 581–588, doi: 10.1208/s12248-013-9465-7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi MK & Song IS Genetic variants of organic cation transporter 1 (OCT1) and OCT2 significantly reduce lamivudine uptake. Biopharm Drug Dispos 33, 170–178, doi: 10.1002/bdd.1783 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Kang HJ et al. Identification and functional characterization of genetic variants of human organic cation transporters in a Korean population. Drug Metab Dispos 35, 667–675, doi: 10.1124/dmd.106.013581 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Song IS, Shin HJ & Shin JG Genetic variants of organic cation transporter 2 (OCT2) significantly reduce metformin uptake in oocytes. Xenobiotica 38, 1252–1262, doi: 10.1080/00498250802130039 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Zolk O, Solbach TF, Konig J & Fromm MF Functional characterization of the human organic cation transporter 2 variant p.270Ala>Ser. Drug Metab Dispos 37, 1312–1318, doi: 10.1124/dmd.108.023762 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Chen Y et al. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenet Genomics 19, 497–504, doi: 10.1097/FPC.0b013e32832cc7e9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teft WA et al. Identification and Characterization of Trimethylamine-N-oxide Uptake and Efflux Transporters. Mol Pharm 14, 310–318, doi: 10.1021/acs.molpharmaceut.6b00937 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goswami S et al. A Longitudinal HbA1c Model Elucidates Genes Linked to Disease Progression on Metformin. Clin Pharmacol Ther 100, 537–547, doi: 10.1002/cpt.428 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song IS et al. Genetic variants of the organic cation transporter 2 influence the disposition of metformin. Clin Pharmacol Ther 84, 559–562, doi: 10.1038/clpt.2008.61 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Wang ZJ, Yin OQ, Tomlinson B & Chow MS OCT2 polymorphisms and in-vivo renal functional consequence: studies with metformin and cimetidine. Pharmacogenet Genomics 18, 637–645, doi: 10.1097/FPC.0b013e328302cd41 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Filipski KK, Mathijssen RH, Mikkelsen TS, Schinkel AH & Sparreboom A Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin Pharmacol Ther 86, 396–402, doi: 10.1038/clpt.2009.139 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin SY et al. An atlas of genetic influences on human blood metabolites. Nat Genet 46, 543–550, doi: 10.1038/ng.2982 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bleasby K, Hall LA, Perry JL, Mohrenweiser HW & Pritchard JB Functional consequences of single nucleotide polymorphisms in the human organic anion transporter hOAT1 (SLC22A6). J Pharmacol Exp Ther 314, 923–931, doi: 10.1124/jpet.105.084301 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Erdman AR et al. The human organic anion transporter 3 (OAT3; SLC22A8): genetic variation and functional genomics. Am J Physiol Renal Physiol 290, F905–912, doi: 10.1152/ajprenal.00272.2005 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Fujita T et al. Functional analysis of polymorphisms in the organic anion transporter, SLC22A6 (OAT1). Pharmacogenet Genomics 15, 201–209 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Bush KT, Wu W, Lun C & Nigam SK The drug transporter OAT3 (SLC22A8) and endogenous metabolite communication via the gut-liver-kidney axis. J Biol Chem 292, 15789–15803, doi: 10.1074/jbc.M117.796516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsuruya Y et al. Investigation of Endogenous Compounds Applicable to Drug-Drug Interaction Studies Involving the Renal Organic Anion Transporters, OAT1 and OAT3, in Humans. Drug Metab Dispos 44, 1925–1933, doi: 10.1124/dmd.116.071472 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Wu W, Bush KT & Nigam SK Key Role for the Organic Anion Transporters, OAT1 and OAT3, in the in vivo Handling of Uremic Toxins and Solutes. Sci Rep 7, 4939, doi: 10.1038/s41598-017-04949-2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Imamura Y et al. 6beta-Hydroxycortisol is an endogenous probe for evaluation of drug-drug interactions involving a multispecific renal organic anion transporter, OAT3/SLC22A8, in healthy subjects. Drug Metab Dispos 42, 685–694, doi: 10.1124/dmd.113.055475 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Shen H et al. Discovery and Validation of Pyridoxic Acid and Homovanillic Acid as Novel Endogenous Plasma Biomarkers of Organic Anion Transporter (OAT) 1 and OAT3 in Cynomolgus Monkeys. Drug Metab Dispos, doi: 10.1124/dmd.117.077586 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Yee SW et al. Reduced renal clearance of cefotaxime in asians with a low-frequency polymorphism of OAT3 (SLC22A8). J Pharm Sci 102, 3451–3457, doi: 10.1002/jps.23581 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Otsuka M et al. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci U S A 102, 17923–17928, doi: 10.1073/pnas.0506483102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masuda S et al. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J Am Soc Nephrol 17, 2127–2135, doi: 10.1681/ASN.2006030205 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Kato K et al. Investigation of endogenous compounds for assessing the drug interactions in the urinary excretion involving multidrug and toxin extrusion proteins. Pharm Res 31, 136–147, doi: 10.1007/s11095-013-1144-y (2014). [DOI] [PubMed] [Google Scholar]
- 49.Kusuhara H et al. Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin Pharmacol Ther 89, 837–844, doi: 10.1038/clpt.2011.36 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Muller F et al. N(1)-methylnicotinamide as an endogenous probe for drug interactions by renal cation transporters: studies on the metformin-trimethoprim interaction. Eur J Clin Pharmacol 71, 85–94, doi: 10.1007/s00228-014-1770-2 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Choi JH et al. A common 5’-UTR variant in MATE2-K is associated with poor response to metformin. Clin Pharmacol Ther 90, 674–684, doi: 10.1038/clpt.2011.165 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stocker SL et al. The effect of novel promoter variants in MATE1 and MATE2 on the pharmacokinetics and pharmacodynamics of metformin. Clin Pharmacol Ther 93, 186–194, doi: 10.1038/clpt.2012.210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams LK et al. Differing effects of metformin on glycemic control by race-ethnicity. J Clin Endocrinol Metab 99, 3160–3168, doi: 10.1210/jc.2014-1539 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang C & Zhang R More effective glycaemic control by metformin in African Americans than in Whites in the prediabetic population. Diabetes Metab 41, 173–175, doi: 10.1016/j.diabet.2015.01.003 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Obaidat A, Roth M & Hagenbuch B The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu Rev Pharmacol Toxicol 52, 135–151, doi: 10.1146/annurev-pharmtox-010510-100556 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X et al. Association of SLCO2B1 Genotypes With Time to Progression and Overall Survival in Patients Receiving Androgen-Deprivation Therapy for Prostate Cancer. J Clin Oncol 34, 352–359, doi: 10.1200/JCO.2015.62.5988 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang M et al. SLCO2B1 and SLCO1B3 may determine time to progression for patients receiving androgen deprivation therapy for prostate cancer. J Clin Oncol 29, 2565–2573, doi: 10.1200/JCO.2010.31.2405 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fujimoto N et al. Polymorphisms of the androgen transporting gene SLCO2B1 may influence the castration resistance of prostate cancer and the racial differences in response to androgen deprivation. Prostate Cancer Prostatic Dis 16, 336–340, doi: 10.1038/pcan.2013.23 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Kim TE et al. The Effect of Genetic Polymorphisms in SLCO2B1 on the Lipid-Lowering Efficacy of Rosuvastatin in Healthy Adults with Elevated Low-Density Lipoprotein. Basic Clin Pharmacol Toxicol 121, 195–201, doi: 10.1111/bcpt.12826 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Kim KA, Lee HM, Joo HJ, Park IB & Park JY Effects of polymorphisms of the SLCO2B1 transporter gene on the pharmacokinetics of montelukast in humans. J Clin Pharmacol 53, 1186–1193, doi: 10.1002/jcph.144 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Tapaninen T, Karonen T, Backman JT, Neuvonen PJ & Niemi M SLCO2B1 c.935G>A single nucleotide polymorphism has no effect on the pharmacokinetics of montelukast and aliskiren. Pharmacogenet Genomics 23, 19–24, doi: 10.1097/FPC.0b013e32835bac90 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Akamine Y et al. The change of pharmacokinetics of fexofenadine enantiomers through the single and simultaneous grapefruit juice ingestion. Drug Metab Pharmacokinet 30, 352–357, doi: 10.1016/j.dmpk.2015.06.005 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Ieiri I et al. Microdosing clinical study: pharmacokinetic, pharmacogenomic (SLCO2B1), and interaction (grapefruit juice) profiles of celiprolol following the oral microdose and therapeutic dose. J Clin Pharmacol 52, 1078–1089, doi: 10.1177/0091270011408612 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Johnson M et al. Inhibition of Intestinal OATP2B1 by the Calcium Receptor Antagonist Ronacaleret Results in a Significant Drug-Drug Interaction by Causing a 2-Fold Decrease in Exposure of Rosuvastatin. Drug Metab Dispos 45, 27–34, doi: 10.1124/dmd.116.072397 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Mbatchi LC et al. Polymorphisms in SLCO1B3 and NR1I2 as genetic determinants of hematotoxicity of carboplatin and paclitaxel combination. Pharmacogenomics 16, 1439–1450, doi: 10.2217/pgs.15.84 (2015). [DOI] [PubMed] [Google Scholar]
- 66.de Lima LT et al. Relationship between SLCO1B3 and ABCA3 polymorphisms and imatinib response in chronic myeloid leukemia patients. Hematology 20, 137–142, doi: 10.1179/1607845414Y.0000000181 (2015). [DOI] [PubMed] [Google Scholar]
- 67.Schinkel AH & Borst P Multidrug resistance mediated by P-glycoproteins. Semin Cancer Biol 2, 213–226 (1991). [PubMed] [Google Scholar]
- 68.Xie R, Hammarlund-Udenaes M, de Boer AG & de Lange EC The role of P-glycoprotein in blood-brain barrier transport of morphine: transcortical microdialysis studies in mdr1a (−/−) and mdr1a (+/+) mice. Br J Pharmacol 128, 563–568, doi: 10.1038/sj.bjp.0702804 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Muzi M et al. Imaging of cyclosporine inhibition of P-glycoprotein activity using 11C-verapamil in the brain: studies of healthy humans. J Nucl Med 50, 1267–1275, doi: 10.2967/jnumed.108.059162 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim RB et al. Identification of functionally variant MDR1 alleles among European Americans and African Americans. Clin Pharmacol Ther 70, 189–199, doi: 10.1067/mcp.2001.117412 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Hoffmeyer S et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci U S A 97, 3473–3478, doi: 10.1073/pnas.050585397 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hitzl M et al. Variable expression of P-glycoprotein in the human placenta and its association with mutations of the multidrug resistance 1 gene (MDR1, ABCB1). Pharmacogenetics 14, 309–318 (2004). [DOI] [PubMed] [Google Scholar]
- 73.Gow JM, Hodges LM, Chinn LW & Kroetz DL Substrate-dependent effects of human ABCB1 coding polymorphisms. J Pharmacol Exp Ther 325, 435–442, doi: 10.1124/jpet.107.135194 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schaefer M, Roots I & Gerloff T In-vitro transport characteristics discriminate wild-type ABCB1 (MDR1) from ALA893SER and ALA893THR polymorphisms. Pharmacogenet Genomics 16, 855–861, doi: 10.1097/01.fpc.0000230113.03710.34 (2006). [DOI] [PubMed] [Google Scholar]
- 75.Chinn LW & Kroetz DL ABCB1 pharmacogenetics: progress, pitfalls, and promise. Clin Pharmacol Ther 81, 265–269, doi: 10.1038/sj.clpt.6100052 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Hodges LM et al. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharmacogenet Genomics 21, 152–161, doi: 10.1097/FPC.0b013e3283385a1c (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Itoda M et al. Polymorphisms in the ABCC2 (cMOAT/MRP2) gene found in 72 established cell lines derived from Japanese individuals: an association between single nucleotide polymorphisms in the 5’-untranslated region and exon 28. Drug Metab Dispos 30, 363–364 (2002). [DOI] [PubMed] [Google Scholar]
- 78.Liu Y et al. Association of ABCC2 −24C>T polymorphism with high-dose methotrexate plasma concentrations and toxicities in childhood acute lymphoblastic leukemia. PLoS One 9, e82681, doi: 10.1371/journal.pone.0082681 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rau T et al. High-dose methotrexate in pediatric acute lymphoblastic leukemia: impact of ABCC2 polymorphisms on plasma concentrations. Clin Pharmacol Ther 80, 468–476, doi: 10.1016/j.clpt.2006.08.012 (2006). [DOI] [PubMed] [Google Scholar]
- 80.de Jong FA et al. Irinotecan-induced diarrhea: functional significance of the polymorphic ABCC2 transporter protein. Clin Pharmacol Ther 81, 42–49, doi: 10.1038/sj.clpt.6100019 (2007). [DOI] [PubMed] [Google Scholar]
- 81.Innocenti F et al. Comprehensive pharmacogenetic analysis of irinotecan neutropenia and pharmacokinetics. J Clin Oncol 27, 2604–2614, doi: 10.1200/JCO.2008.20.6300 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Akiyama Y et al. Association of ABCC2 genotype with efficacy of first-line FOLFIRI in Japanese patients with advanced colorectal cancer. Drug Metab Pharmacokinet 27, 325–335 (2012). [DOI] [PubMed] [Google Scholar]
- 83.Kroetz DL, Yee SW & Giacomini KM The pharmacogenomics of membrane transporters project: research at the interface of genomics and transporter pharmacology. Clin Pharmacol Ther 87, 109–116, doi: 10.1038/clpt.2009.226 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee CA et al. Breast cancer resistance protein (ABCG2) in clinical pharmacokinetics and drug interactions: practical recommendations for clinical victim and perpetrator drug-drug interaction study design. Drug Metab Dispos 43, 490–509, doi: 10.1124/dmd.114.062174 (2015). [DOI] [PubMed] [Google Scholar]
- 85.Burt T & Dhillon S Pharmacogenomics in early-phase clinical development. Pharmacogenomics 14, 1085–1097, doi: 10.2217/pgs.13.81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tsamandouras N et al. Identification of the effect of multiple polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid using a population-modeling approach. Clin Pharmacol Ther 96, 90–100, doi: 10.1038/clpt.2014.55 (2014). [DOI] [PubMed] [Google Scholar]
- 87.Tomita Y, Maeda K & Sugiyama Y Ethnic variability in the plasma exposures of OATP1B1 substrates such as HMG-CoA reductase inhibitors: a kinetic consideration of its mechanism. Clin Pharmacol Ther 94, 37–51, doi: 10.1038/clpt.2012.221 (2013). [DOI] [PubMed] [Google Scholar]
- 88.Wu HF et al. Rosuvastatin Pharmacokinetics in Asian and White Subjects Wild Type for Both OATP1B1 and BCRP Under Control and Inhibited Conditions. J Pharm Sci 106, 2751–2757, doi: 10.1016/j.xphs.2017.03.027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gertz M, Tsamandouras N, Sall C, Houston JB & Galetin A Reduced physiologically-based pharmacokinetic model of repaglinide: impact of OATP1B1 and CYP2C8 genotype and source of in vitro data on the prediction of drug-drug interaction risk. Pharm Res 31, 2367–2382, doi: 10.1007/s11095-014-1333-3 (2014). [DOI] [PubMed] [Google Scholar]
- 90.Emoto C et al. Characterization of Contributing Factors to Variability in Morphine Clearance Through PBPK Modeling Implemented With OCT1 Transporter. CPT Pharmacometrics Syst Pharmacol 6, 110–119, doi: 10.1002/psp4.12144 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Search Collaborative Group et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med 359, 789–799, doi: 10.1056/NEJMoa0801936 (2008). [DOI] [PubMed] [Google Scholar]
- 92.Watanabe T, Kusuhara H, Maeda K, Shitara Y & Sugiyama Y Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J Pharmacol Exp Ther 328, 652–662, doi: 10.1124/jpet.108.146647 (2009). [DOI] [PubMed] [Google Scholar]
- 93.Patilea-Vrana G & Unadkat JD Transport vs. Metabolism: What Determines the Pharmacokinetics and Pharmacodynamics of Drugs? Insights From the Extended Clearance Model. Clin Pharmacol Ther 100, 413–418, doi: 10.1002/cpt.437 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Duan P, Zhao P & Zhang L Physiologically Based Pharmacokinetic (PBPK) Modeling of Pitavastatin and Atorvastatin to Predict Drug-Drug Interactions (DDIs). Eur J Drug Metab Pharmacokinet 42, 689–705, doi: 10.1007/s13318-016-0383-9 (2017). [DOI] [PubMed] [Google Scholar]
- 95.Giacomini KM et al. Genome-wide association studies of drug response and toxicity: an opportunity for genome medicine. Nat Rev Drug Discov 16, 1, doi: 10.1038/nrd.2016.234 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yee SW et al. Metabolomic and Genome-wide Association Studies Reveal Potential Endogenous Biomarkers for OATP1B1. Clin Pharmacol Ther 100, 524–536, doi: 10.1002/cpt.434 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Denny JC et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol 31, 1102–1110, doi: 10.1038/nbt.2749 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bush WS et al. Genetic variation among 82 pharmacogenes: The PGRNseq data from the eMERGE network. Clin Pharmacol Ther 100, 160–169, doi: 10.1002/cpt.350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Han SM et al. Targeted Next-Generation Sequencing for Comprehensive Genetic Profiling of Pharmacogenes. Clin Pharmacol Ther 101, 396–405, doi: 10.1002/cpt.532 (2017). [DOI] [PubMed] [Google Scholar]
- 100.Zhou Y, Ingelman-Sundberg M & Lauschke VM Worldwide Distribution of Cytochrome P450 Alleles: A Meta-analysis of Population-scale Sequencing Projects. Clin Pharmacol Ther 102, 688–700, doi: 10.1002/cpt.690 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Neuvonen M et al. Effects of Genetic Variants on Carboxylesterase 1 Gene Expression, and Clopidogrel Pharmacokinetics and Antiplatelet Effects. Basic Clin Pharmacol Toxicol, doi: 10.1111/bcpt.12916 (2017). [DOI] [PubMed] [Google Scholar]
- 102.Hirvensalo P et al. Comprehensive Pharmacogenomic Study Reveals an Important Role of UGT1A3 in Montelukast Pharmacokinetics. Clin Pharmacol Ther, doi: 10.1002/cpt.891 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chua EW et al. Exome sequencing and array-based comparative genomic hybridisation analysis of preferential 6-methylmercaptopurine producers. Pharmacogenomics J 15, 414–421, doi: 10.1038/tpj.2015.9 (2015). [DOI] [PubMed] [Google Scholar]
- 104.Bielinski SJ et al. Preemptive genotyping for personalized medicine: design of the right drug, right dose, right time-using genomic data to individualize treatment protocol. Mayo Clin Proc 89, 25–33, doi: 10.1016/j.mayocp.2013.10.021 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Van Driest SL et al. Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin Pharmacol Ther 95, 423–431, doi: 10.1038/clpt.2013.229 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rasmussen-Torvik LJ et al. Design and anticipated outcomes of the eMERGE-PGx project: a multicenter pilot for preemptive pharmacogenomics in electronic health record systems. Clin Pharmacol Ther 96, 482–489, doi: 10.1038/clpt.2014.137 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Burke W, Tarini B, Press NA & Evans JP Genetic screening. Epidemiol Rev 33, 148–164, doi: 10.1093/epirev/mxr008 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Payne K, Gavan SP, Wright SJ & Thompson AJ Cost-effectiveness analyses of genetic and genomic diagnostic tests. Nat Rev Genet, doi: 10.1038/nrg.2017.108 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Eight genomewide association studies identified significant associations of polymorphisms in ABCG2 or SLCO1B1 with drug disposition or response (p<5×10-8). A number of studies have since replicated the initial findings from the initial GWAS, and the references for each study can be found in Table S1 below.
Table S2. Important non-synonymous variants in SLC22A1 that have reduced transporter activity and altered substrates disposition, response or adverse effects. These non-synonymous variants have been genotyped in several clinical pharmacogenomics studies. In each of these studies, there is a significant correlation with the number of inactive OCT1 alleles with the studied drug disposition (pharmacokinetic effect, PK) and response and toxicity (pharmacodynamics effect, PD). These studies associated the phenotype with the most common functional amino acid substitutions found in Caucasians (Arg61Cys, Cys88Arg, Gly401Ser, Met420del, and Gly465Arg). Allele frequencies of other ethnic groups and functional variants of other coding variants in SLC22A1 are shown Table S3. References for each study can be found below Table S2.
Table S3. Genetic polymorphisms in 12 transporters and their in vitro consequences. Non-synonymous and untranslated region (UTR) polymorphisms with known in vitro functions are described in this table.
Table S4. Commonly used databases for pharmacogenomics research and membrane transporter research.
Table S5. Genomewide association studies of pharmacogenomic studies that have genomewide significance (p<5×10-8) variants in SLC or ABC transporters.