

Abstract
Background and aims:
Atherosclerosis is a widespread and complicated disease involving phenotypic modulation and transdifferentiation of vascular smooth muscle cells (SMCs), the predominant cells in aorta, as well as changes in endothelial cells and infiltrating monocytes. Alterations in DNA methylation are likely to play central roles in these phenotypic changes, just as they do in normal differentiation and cancer.
Methods:
We examined genome-wide DNA methylation changes in atherosclerotic aorta using more stringent criteria for differentially methylated regions (DMRs) than in previous studies and compared these DMRs to tissue-specific epigenetic features.
Results:
We found that disease-linked hypermethylated DMRs account for 85% of the total atherosclerosis-associated DMRs and often overlap aorta-associated enhancer chromatin. These hypermethylated DMRs were associated with functionally different sets of genes than were atherosclerosis-linked hypomethylated DMRs. The extent and nature of the DMRs could not be explained as direct effects of monocyte/macrophage infiltration. Among the known atherosclerosis- and contractile SMC-related genes that exhibited hypermethylated DMRs at aorta enhancer chromatin were ACTA2 (aorta α2 smooth muscle actin), ELN (elastin), MYOCD (myocardin), C9orf3 (miR-23b and miR-27b host gene), and MYH11 (smooth muscle myosin). Our analyses also suggest a role in atherosclerosis for developmental transcription factor genes having little or no previous association with atherosclerosis, such as, NR2F2 (COUP-TFII) and TBX18.
Conclusions:
We provide evidence for atherosclerosis-linked DNA methylation changes in aorta SMCs that might help minimize or reverse the standard contractile character of many of these cells by down-modulating aorta SMC-related enhancers, thereby facilitating pro-atherosclerotic phenotypic changes in many SMCs.
Keywords: atherosclerosis, DNA methylation, chromatin, smooth muscle cells, enhancers, super- enhancer, contractile phenotype
Graphical Abstract
Total atherosclerosis-linked or tissue-specific normal aorta differentially methylated regions (DMRs): Hypermethylated vs. hypomethylated DMRs
1. Introduction
Smooth muscle cells (SMCs), the predominant cells in aorta, can display remarkable phenotypic plasticity, as seen in atherosclerosis. Atherosclerosis involves initial damage to the single-cell endothelial layer of the intima, abnormal retention of lipids, a chronic inflammatory response, and vascular remodeling involving SMCs from the multilayered media [1]. Monocytes infiltrate the intima and many differentiate into macrophages and lipid-loaded macrophage-like foam cells. In addition, there is the full or partial loss of the normal contractile phenotype of many SMCs to give a constellation of states called the synthetic SMC phenotype [2] in which SMCs become more rounded and mobile, begin to actively proliferate, lose their ability to contract, and increase synthesis of certain extracellular proteins. Phenotypic modulation of SMCs during development of atherosclerosis can be protective or disease-promoting [3, 4].
Despite the importance of epigenetics to cell differentiation, there have been only a small number of reports about atherosclerosis-related epigenetic changes, and some of these studies focused on endothelial cells or non-aorta atherosclerosis [5]. A comparison of atherosclerotic human femoral artery samples with mammary artery samples as controls indicated disease-related DNA hypomethylation of gene and promoter regions [6]; however, non-disease related tissue-specific differences might be major contributors to the observed differential methylation. The most comprehensive studies of DNA methylation changes in human aorta examined pairs of atherosclerotic aorta and control aorta from the same individual using a microarray assay (15 sample pairs) or whole-genome bisulfite-sequencing (WGBS or bisulfite-seq; 1 pair of samples) [7, 8]. Grade-independent differentially methylated sites (54625 in >3000 genes) were predominantly disease-hypermethylated. However, in contrast to WGBS, microarray studies lack the power to resolve differentially methylated regions (DMRs) in most non-promoter regions, e.g., enhancers.
We re-evaluated the DNA epigenetics of atherosclerotic aorta using available WGBS data from [7, 9] and much more stringent criteria for calculating DMRs to reflect mostly changes in SMCs. We also compared atherosclerosis-DMRs to normal tissue-specific DMRs, genome-wide chromatin modifications, and transcription profiles [9, 10] to elucidate biological associations of the disease DMRs. In addition, we examined individual gene neighborhoods of interest because composite epigenetic analyses of genome-wide data can miss important insights into the likely functionality of DMRs [11]. We discovered a very strong preference for atherosclerosis-linked DNA hypermethylation at aorta-associated enhancer-like regions, including in genes well known to be associated with atherosclerosis.
2. Materials and methods
For the atherosclerotic and control aorta samples from the same individual (88 yo female, athero aorta A, aortic arch, and control aorta A, thoracic aorta), the bisulfite-seq data from Zaina et al. [7] were used. These data were supplemented with two additional control aorta bisulfite-seq profiles from a 34 yo male and a 30 yo female (control aortas B and C, respectively; Roadmap Epigenetics Project [9, 12]). The atherosclerotic aorta sample included the muscular tissue underneath the plaque [7], which should consist predominantly of SMCs [13]. Determination of DMRs using the Uniform Product distribution [14], mapping them, and comparing them to normal epigenetic and transcriptomic databases are described in an expanded Methods section in a companion article in Data in Brief [15]. In Fig. 1, the gene subregions relative to the transcription start site (TSS) or end site (TES) are as follows: promoter, TSS −2 kb to TSS +0.2 kb; gene body, TSS +0.2 kb to TES +2 kb; intergenic , other positions.
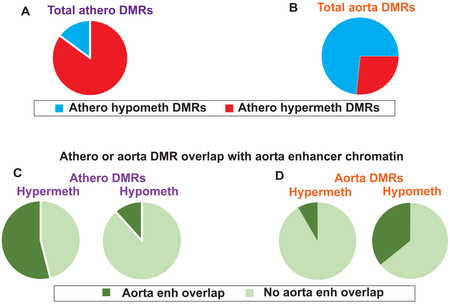
Fig. 1. Atherosclerosis-associated and normal aorta DMRs: genome-wide associations.

(A) Pie charts showing that athero DMRs were mostly athero hypermeth DMRs. (B) Athero hypermeth DMRs, but not athero hypometh DMRs, were greatly enriched in aorta enhancer chromatin. (C) The distribution of athero DMRs among gene regions. (D) Bar graphs showing the strong overlap of athero DMRs with aorta DMRs of the opposite directionality (athero hypermeth with aorta hypometh and vice versa) and athero DMRs overlap with monocyte DMRs of the same directionality. (E-G), parallel to (A-C) but for tissue-specific aorta DMRs (control aorta vs. heart/skeletal muscle/lung/adipose/monocytes), which indicated that the athero hypermeth DMRs are most similar to the aorta hypometh DMRs and vice versa. Enh, enhancer-type chromatin.
3. Results
3.1. Atherosclerosis-associated hypermethylated DMRs were predominant over hypomethylated DMRs and often overlapped aorta enhancers
Previously, Zaina and coworkers [7, 16] using one matched set of atherosclerotic aorta (grade VII, including underlying muscle) and uninvolved aorta from the same individual (athero aorta A and control aorta A) identified 54,625 DMRs with ≥5 consecutive and consistently differentially methylated CpG sites from WGBS data (Fisher exact test, p<0.05) [7]. We re-determined atherosclerosis-associated DMRs using much more stringent conditions and included WGBS data from two additional control aortas [9, 16] (controls B and C). We required an absolute percent methylation difference (PMD) of >20%, a length of >250 bp and gaps <200 bp as well as ≥5 differentially methylated sites per DMR and that the DMRs were significant in both the paired samples (athero aorta A vs. control aorta A) and the atherosclerotic aorta vs. three control aortas. With our criteria, there were only 9220 DMRs associated with 4535 genes (see Data in Brief Table 1 [15]). Zaina et al. also did a microarray analysis of 15 atherosclerotic/normal aorta pairs [7]. Of their 892 genes reported to have differentially methylated sites, 454 were associated with athero DMRs ascertained by our stringent criteria. Moreover, 15 out of 18 sites in nine genes (HOXA2, HOXA9, HOXA11-AS, HOXC4, HOXC11, PDGFA, PLAT, PRRX1 and PXDN) that Zaina et al. verified as significantly athero hypermethylated or hypomethylated by pyrosequencing [7] overlapped DMRs in our study.
We found that 85% of all the athero DMRs were hypermethylated (athero hypermeth DMRs) and 15% hypomethylated (athero hypometh DMRs; Fig. 1A). About 44% of the genes with athero hypometh DMRs were also associated with athero hypermeth DMRs. This result might be explained, in part, by previous reports of tissue-specific DNA hypomethylation and hypermethylation being positively associated with DNA upregulation depending on the gene region, chromatin, and cell context [11]. We sorted the hypermethylated DMRs by PMD >40% or PMD = 20 to 40% (1032 and 6819 DMRs, respectively) and the hypomethylated DMRs by PMD <−40% or PMD −20 to −40% (75 and 1294 DMRs, respectively; see Data in Brief Tables 1a-1d [15]). Leukocyte infiltration or clonal expansion of atypical SMCs into the plaque region during atherosclerosis [17, 18] are unlikely to produce athero DMRs defined by a 20% absolute PMD threshold, and this is especially unlikely for DMRs with absolute PMDs of >40%.
We also compared the two control aorta samples from the 30 and 34 yo individuals with the control aorta from the 88 yo atherosclerotic patient. Unlike the atherosclerotic vs. the control aorta DMRs from the 88 yo patient for which hypermethylation predominated, the 88 yo control vs. 30 - 34 yo controls had more hypometh DMRs (5868 DMRs) than hypermeth DMRs (4212 DMRs). These DMRs could be due to several factors, including biological differences and technical variation because the bisulfite sequencing was performed in different labs with different sequencing depths. However, this finding argues against age-related changes accounting for the predominance of hypermethylation over hypomethylation in the athero DMRs.
We examined chromatin state segmentation analysis profiles, which use genome-wide histone modifications typically associated with promoter, enhancer, repressor, or actively transcribed chromatin [9]. Surprisingly, 53% of the athero hypermeth DMRs overlapped ≥50 bp of enhancer-type (enh) chromatin in normal aorta, and 64% of these DMRs at enh chromatin were in gene bodies (Figs. 1B, 2A, 3A and 4A). Only 12% of athero hypometh DMRs overlapped aorta enh chromatin (Fig. 1B).
Fig. 2. Atherosclerosis-associated hypermethylation in the aorta-related ELN super-enhancer and hypomethylation in leukocyte-associated PTPN6.

(A) ELN displayed an aorta-specific super-enhancer (dotted box) that overlapped many athero hypermeth DMRs (red) and aorta-specific (tissue-specific) hypometh DMRs (blue; chr7:73,381,618-73,486,319). Bisulfite-seq is plotted as average % methylation at CpGs; blue horizontal bars, regions that display significant hypomethylation relative to the rest of the same genome [12]. (B) PTPN6 displayed monocyte hypometh DMRs (chr12:7,055,252-7,071,384). Gray highlighting, monocyte DMRs with little or no athero hypomethylation. All tracks are aligned and derived from the UCSC Genome Browser with hg19 coordinates. Ctl, control; PBMC, peripheral mononuclear blood cells; brain, prefrontal cortex; heart, left ventricle; expr, expression; str, strong; wk, weak; biv, bivalent; prom, promoter; enh, enhancer; txn chromatin, chromatin with histone marks of active transcription; repr, repressed.
Fig. 3. MYOCD and SRF TF genes were associated with atherosclerosis-linked DNA hypermethylation but the hypermethylation linked to SRF was within the adjacent aorta-repressed PTK7 gene.

(A) MYOCD was linked to athero hypermeth DMRs (chr17:12,527,074-12,600,390). (B) Aorta-specific hypomethylation overlapping athero hypermeth DMRs in PTK7 gene (repressed in aorta) adjacent to SRF (broadly expressed; chr6:43,082,821-43,149,411). (Eight additional athero hypermeth DMRs in this gene region are shown in Data in Brief Table 1 [15]).
Fig. 4. Strong atherosclerosis-associated DMRs: C9orf3, the SMC-related miR-23b host gene, and NR2F2, a master developmental TF gene.

Some of athero DMRs in (A) C9orf3 at chr9:97,478,939-97,923,541 and (B) near NR2F2 (COUP-TFII; chr15:96,862,542-96,914,426) were strong DMRs (PMD >40% or <−40%, respectively; yellow highlighting with a black dot underneath). PBMC have repressed chromatin throughout this gene neighborhood [11]. In (B), dotted line, hypomethylated ovary super-enhancer; green, yellow and pink highlighting, the region whose hypermethylation is positively associated with NR2F2 expression.
3.2. Atherosclerosis-associated hypermethylated DMRs and hypomethylated DMRs are enriched in functionally different sets of genes
The functional associations [19] of genes linked to hypometh or hypermeth athero DMRs were very different (see Data in Brief Table 2a [15]). For example, actin-related gene ontology (GO) terms were highly over-represented among athero hypermeth DMRs but not among athero hypometh DMRs. Integrin signaling and focal adhesion were highly favored GO terms among genes linked to athero hypermeth DMRs (but not athero hypometh DMRs) and are related to the actin/integrin/extracellular matrix signaling pathway [20]. The strongest GO association of athero hypometh (but not hypermeth) DMRs was with genes encoding sequence-specific DNA binding proteins.
Athero hypermeth DMRs often overlapped not just enh chromatin, but also super-enhancers. Superenhancers are especially long and strong enhancers and can be defined as regions of >3 kb enriched in histone H3 lysine-27 acetylation (H3K27ac) [21-23]. A functional analysis [24] of genes associated with athero hypermeth DMRs and super-enhancers revealed, again, high enrichment in GO terms related to actin-binding (see Data in Brief Table 3 [15]).
3.3. Genome-wide analyses of atherosclerosis-associated DMRs indicate partial acquisition of a leukocyte-like epigenetic profile by the atherosclerotic aorta
We identified aorta tissue-specific DMRs and monocyte-specific DMRs for comparison to athero DMRs using WGBS datasets [9] for normal aorta vs. those of monocytes, skeletal muscle (SkM), heart, lung and adipose tissues or of monocytes vs. those of aorta, SkM, heart, lung and adipose tissues. About 77% of athero hypermeth DMRs overlapped aorta hypometh DMRs, and 61% of athero hypometh DMRs overlapped aorta hypermeth DMRs (Fig. 1D). Aorta hypometh DMRs were much more plentiful than aorta hypermeth DMRs and much more likely to overlap enhancers (Fig. 1E and F). These findings suggest frequent atherosclerosis-linked increases in DNA methylation targeted to aorta hypometh DMRs at aorta enhancers.
Functional analyses also revealed associations of athero (disease-related) hypermeth DMRs and aorta (normal tissue-specific) hypometh DMRs. For example, the DNA sequence binding motif assigned to serum response factor (SRF), which plays a critical role in the development of vascular SMC [25], was strongly enriched at promoters for both athero hypermeth DMR genes and aorta hypometh DMR genes (see Data in Brief Table 2a [15]). Another example is that both athero hypometh DMRs and aorta hypermeth DMRs were very highly enriched at homeobox transcription factor (TF) genes.
In contrast, monocyte DMRs (but not SkM or heart DMRs) often overlapped athero DMRs with the same directionality, namely, monocyte hypermeth DMRs with athero hypermeth DMRs and monocyte hypometh DMRs with athero hypometh DMRs (Fig. 1D). While this finding might reflect the contribution of infiltrating monocytes to the observed athero DMRs, such a conclusion is refuted by the extent of DNA methylation differences observed among strong athero DMRs (absolute value of PMD >40%). Moreover, there was no significant enrichment for specifically leukocyte-related GO terms among strong athero DMRs (see Data in Brief Table 2B [15]). Lastly, epigenetic and transcriptomic profiles of many individual gene regions were inconsistent with athero DMRs being explained by monocyte (or any type of leukocyte) contamination. For example, the leukocyte-associated genes PTPN6 , CD79B, and SH3BP2 displayed monocyte hypometh DMRs that were also hypomethylated in other leukocyte cells types but mostly highly methylated in atherosclerotic aorta (Fig. 2B, gray highlighting, and see Data in Brief Figs. 1 and 2 [15]). All these findings argue against monocyte/macrophage infiltration explaining the observed athero DMRs.
3.4. Some of the hypermethylated DMRs are linked to key SMC genes previously implicated in atherosclerosis
Seven out of 14 synthetic phenotype markers and 12 of 18 contractile phenotype markers for SMCs [26] displayed athero DMRs (see Data in Brief Table 1 [15]). In addition, many of the 270 genes (see Data in Brief Table 4 [15]) that have been related to atherosclerosis [27] were associated with athero DMRs. ELN, which has 41 references linking it to the disease [27] and encodes an extracellular protein that stabilizes arteries, displayed multiple athero hypermeth DMRs. ELN is expressed at extremely high levels specifically in aorta, coronary artery and tibial artery (see Data in Brief Table 5 [15]). Its athero hypermeth DMRs were embedded in 103-kb super-enhancer, and most overlapped tissue-specific aorta hypometh DMRs (Fig. 2A, long dotted box). Super-enhancers generally exhibit overall depletion in DNA methylation and are associated with very high gene expression [21, 22, 28]. ACTA2, MYH11 and MYH10, which encode either actins or actin-binding myosins and were previously implicated in atherosclerosis (see Data in Brief Table 4 [15]), also exhibited multiple athero hypermeth DMRs that overlapped aorta super-enhancers or standard enhancers (see Data in Brief Figs. 3-5 [15]).
Among the transcription- or translation-regulatory genes linked to athero hypermeth DMRs and implicated in atherosclerosis are MYOCD, SRF, SMAD3 and C9orf3 (host gene for miR-23b and miR-27b; Figs. 3 and 4A). SMAD3, a broadly expressed gene, displayed athero hypermeth DMRs that might be due to disease-related decreases in its aorta-specific pattern of enh chromatin or to tissue-specific isoform usage (see Data in Brief Fig. 6 [15]). SMAD3 has been genetically linked to control of COL4A1 and COL4A2 [29], which also displayed multiple athero hypermeth DMRs.
MYOCD, a critical and specific TF for SMCs and heart, exhibited four athero hypermeth DMRs located within the gene (Fig. 3A and data not shown). Seven more were in or adjacent to the upstream non-coding RNA (ncRNA) gene LINC00670 (see Data in Brief Table S1a and b [15]), which is expressed preferentially in arteries (see Data in Brief Table 5 [15]). MYOCD is a transcriptional co-activator of the broadly expressed TF SRF [30]. Although the SRF gene did not display DMRs, PTK7, which is upstream of SRF, had three athero hypermeth DMRs that overlap aorta hypometh DMRs (Fig. 3B). Because PTK7 has very low expression in aorta, it is likely the aorta hypometh DMRs upregulate SRF rather than PTK7 and so hypermethylation at these PTK7 regions in atherosclerotic aorta could be down-regulate SRF.
The athero hypermeth DMRs associated with C9orf3 were especially plentiful (Fig. 4A; 23 DMRs, some of which had PMD >40%, dotted yellow highlighting). This is the host gene for six miRNAs including miR-23b, which helps prevent SMC phenotypic switching in cultured cells [31]. Analysis of chromatin data and isoform-specific GTEx data [10] suggests an alternative promoter is used for transcription in aorta and many other tissues (black box, chromatin state segmentation; Fig. 4A). The cluster of athero hypermeth DMRs immediately downstream of this promoter could down-modulate its activity.
3.5. Unusual atherosclerosis-associated DMRs associated with genes encoding developmental transcription factors: TBX and HOX family genes and NR2F2
Many of the genes with unusually dense clusters of DMRs or especially strong DMRs encode early developmental TFs, including homeobox (HOX) genes, T-box (TBX) TF genes, and NR2F2 (COUP-TFII), a gene specifying a TF involved in many types of embryonic organogenesis including vein formation [32] (see Data in Brief Table 1 [15]). Adjacent to NR2F2 were clusters of hypometh DMRs and of hypermeth DMRs and some overlapped two isoforms of a long ncRNA gene (Fig. 4B). This ncRNA gene and an antisense ncRNA gene upstream of NR2F2 have post-natal tissue-specific expression patterns very similar to those NR2F2 itself (Fig. 4B; see Data in Brief Table 5 [15]). The athero hypermeth DMRs downstream of NR2F2 displayed very low methylation in leukocytes (Fig. 4B, yellow highlighting), where NR2F2 is repressed [11], and unusually high methylation in ovary (Fig. 4B, pink highlighting), where it is most highly expressed. This suggests that high methylation in this ncRNA gene region favors NR2F2 expression. Atherosclerotic aorta also had DNA hypermethylation in this NR2F2-far downstream region (Fig. 4B, green highlighting), which together with the athero hypometh DMRs closer to the gene might indicate upregulation of NR2F2 in atherosclerotic aorta.
Seven of the 13 TBX family genes exhibited athero hypometh DMRs (see Data in Brief Tables 3 and 4 [15]). TBX5 (Fig. 5B) and TBX20 (see Data in Brief Fig. 11 [15]) displayed athero hypometh DMRs in their 5’ gene regions even though they are repressed in normal aorta. Some HOX genes that are silent in aorta displayed athero DMRs (see Data In Brief Figs. 7-10 [15]). In contrast, TBX18 had an athero hypometh DMR in the promoter-upstream region (Fig. 5A) and is expressed moderately in normal aorta. Therefore, while some of the athero DMRs associated with TBX or HOX family genes might affect transcription in cis, others are unlikely to do so because those genes are tightly repressed in the majority of adult tissues, including control aorta.
Fig. 5. Developmental TF genes, TBX18 and TBX5, displayed atherosclerosis-associated hypomethylated DMRs even though TBX5 is not expressed in aorta.

(A) TBX18 (chr6:85,441,091-85,490,593). (B) TBX5 (chr12:114,825,763-114,855,728). Yellow highlighting as in Fig. 4. CpG density, CG dinucleotide positions to show regions with low CpG content that, therefore, cannot display CpG methylation.
4. Discussion
In this report we present evidence that many of the athero hypermeth DMRs and the much less frequent hypometh DMRs might facilitate gene expression changes that could drive SMC phenotypic modulation and pathogenesis and that the athero hypermeth DMRs often occur at aorta enhancers. Our threshold of a 20% methylation difference for scoring a disease-associated DMR resulted in DMRs that should mostly reflect epigenetic alterations in SMCs, the dominant cell type in aorta. Nevertheless, we found a large overlap of the disease-linked DMRs with tissue-specific monocyte DMRs. This is probably due to phenotypic conversion and transdifferentiation of SMCs. Such leukocyte-like conversions can occur even while the cells still express markers of the contractile phenotype, although usually at a subnormal levels [3, 17, 33]. Similarly, we found that for athero DMRs that overlapped monocyte DMRs, the associated genes usually acquired only some of the monocyte hypo- or hypermethylation. This was even the case for genes that were preferentially expressed in monocytes and other leukocytes.
Athero hypermeth DMRs overlapped tissue-specific aorta hypometh DMRs even more frequently than they overlapped monocyte hypermeth DMRs. The over-representation of athero hypermeth DMRs at aorta hypometh DMRs could be explained by these athero DMRs frequently forming in regions of aorta enhancer-type chromatin. This suggests that de novo DNA methylation during atherogenesis is targeted preferentially to cis-acting regulatory regions, especially aorta enhancers. Given the magnitude of methylation changes that we found, the de novo DNA methylation probably occurred mostly in SMCs in the medial layer of atherosclerotic aorta. Chapelle et al. [18] demonstrated that clonal expansion of SMCs from the media is associated with their migration to the neointima and to the plaque. However, many of the SMCs left behind in the media, especially those close to the plaque, lost the elongated morphology characteristic of the contractile phenotype and displayed strong decreases in expression of the contractile marker ACTA2, which was among the genes that we found to have athero hypermeth DMRs. Epigenetic change in a considerable fraction of the medial SMCs would explain how we observed >40% DNA methylation increases or decreases in many DNA regions in atherosclerotic aorta.
A caveat in this analysis is that the matched atherosclerotic and non-atherosclerotic aorta samples came from different parts of the aorta, aortic arch and thoracic aorta, respectively [7], which have different embryological origins [33]. In addition, the endothelial layers of these two different sections of aorta are subject to different flow dynamics that can alter their epigenetics [34, 35]. The two additional control aortas used for DMR determination were from abdominal aorta or an unspecified portion of aorta. Differences in the embryological origin of curved (atherosclerotic sample) and linear aorta segments (at least two of the three control samples) could result in aorta region-specific DMRs [36-38] that arise during prenatal development but persist in adulthood whether or not they have a postnatal function. This is especially relevant to consideration of the numerous DMRs in HOX genes because of their role in shaping the body-plan early in embryogenesis. However, HOXA6 and HOXA9 were previously shown to be upregulated in atherosclerotic vs. control aorta and to display atherosclerosis-associated hypomethylation at tested CpG sites [7]. Their athero hypometh DMRs might favor transdifferentiation [39] to an osteochondrocytic phenotype (see Data in Brief Fig. 7 and Table S2c [15]).
Among genes associated with athero hypermeth DMRs, there was a strong over-representation of atherosclerosis-relevant GO terms, including actin cytoskeleton organization, smooth muscle contraction, focal adhesion, cell death, and the TGFβ-receptor signaling pathway, which is related to SMC phenotypic switching [40]. ELN, which was associated with many athero hypermeth DMRs, encodes a major structural protein in aorta relevant to atherosclerosis (see Data in Brief Table 4 [15]). The importance of fine-tuned control of ELN levels in aorta is evidenced by the finding that inactivating mutations in a single allele of the gene are linked to supravalvular aortic stenosis syndrome, which includes a congenital narrowing of part of the aorta [41]. We hypothesize that athero hypermeth DMRs in the aorta-specific super-enhancer of ELN down-modulate its enhancer activity and, thereby, decrease the rate of transcription of this gene, given the known link [11, 28, 42] between DNA hypomethylation and enhancers (Fig. 1F).
Other examples of athero hypermeth genes with known relationships to atherosclerosis are COL4A1 and COL4A2, whose protein products are present in subnormal levels in the medial layer of atherosclerotic tissue [29]. Transcription of the genes encoding these proteins is positively regulated by SMAD3, a signaling TF [43], which also is encoded by a gene that exhibited athero hypermeth DMRs (see Data in Brief Fig. 6 [15]). Two other TF-encoding genes whose athero hypermeth DMRs could play a major role in atherosclerosis are MYOCD and SRF, which code for subunits of a MYOCD/SRF TF complex that induces expression of most SMC contractile phenotype marker genes [30], including athero hypermeth ACTA2 and MYH11. These findings illustrate how multifaceted control of gene expression might drive phenotypic change in atherosclerotic aorta SMCs.
Our study also provides epigenetic clues for roles in atherosclerosis of genes that have been little studied in regard to this disease, e.g., NR2F2, the master organogenesis TF and the developmental TF gene, TBX18. TBX18 is implicated in the generation of SMCs during development [40] and was reported to be induced in the endothelial layer of atherosclerotic arteries [44]. NR2F2 is associated with phenotypic switching in vein endothelial cells [45]. These two genes displayed athero hypometh DMRs which suggests disease-linked upregulation of expression because they overlap a promoter or enhancer-type region in aorta.
A possible explanation as to why atherosclerosis-associated DNA hypermethylation greatly exceeds hypomethylation for DMRs comes from recent studies of TET2, a 5-methylcytosine (5mC) dioxygenase. Decreased levels of TET2 in SMC, endothelial cells, and leukocytes have been implicated in atherosclerosis [46-48]. One of the functions of TET2 is participation in a DNA demethylation pathway (5mC → C) through a 5-hydroxymethylcytosine intermediate [49]. Down-regulation of TET2 can limit DNA demethylation resulting in increased genomic 5mC levels by perturbing a dynamic DNA methylation/demethylation cycle that is especially targeted to regulation of enhancer activity [42, 50]. TET2 down-regulation by itself or possibly in conjunction with increased DNA methyltransferase activity might drive predominant DNA methylation gain in atherosclerosis. Importantly, Liu et al. [46] showed that experimentally introduced decreases in TET2 could down-regulate MYOCD, MYH11, and SRF but ascribed this result just to effects of increased promoter methylation. Our finding of frequent atherosclerosis-associated DNA hypermethylation at aorta enhancer-type chromatin, including at these three genes, suggests that enhancer hypermethylation also probably contributes to down-regulating contractile phenotype SMC genes during pathogenesis.
Highlights.
Preferential gain of atherosclerosis-linked DNA methylation at aorta enhancers
Hypermethylation at enhancers in some atherosclerosis-downregulated genes
Atherosclerosis-linked hyper- and hypomethylation have different functional associations
Smooth muscle cell phenotype changes explain their leukocyte-like DNA methylation
Important to consider enhancer epigenetic as well as promoter changes in disease
Acknowledgements
We thank Dr. Shin Lin (University of Washington Medical Center, Seattle, Washington) for clinical information about the NIH Roadmap aorta sample (Ctl aorta B).
Financial support
This research was supported in part by grants from the National Institutes of Health (National Center for Advancing Translational Sciences of the National Institutes of Health, award number UL1TR001417, and NS04885) and the Louisiana Cancer Center to ME and by high performance computing resources and services provided by Technology Services at Tulane University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: The authors declare that they have no competing interests.
Conflicts of interests
The authors declare that they do not have anything to disclose regarding conflict of interest with respect to this manuscript.
References
- [1].Tabas I, Bornfeldt KE, Macrophage phenotype and function in different stages of atherosclerosis, Circ. Res, 118 (4) (2016) 653–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Allahverdian S et al. , Smooth muscle cell fate and plasticity in atherosclerosis, Cardiovasc. Res, 114 (4) (2018) 540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Orr AW et al. , Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis, J. Vasc. Res, 47 (2) (2010) 168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Beamish JA et al. , Molecular regulation of contractile smooth muscle cell phenotype: implications for vascular tissue engineering, Tissue Eng. Part B Rev, 16 (5) (2010) 467–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Khyzha N et al. , Epigenetics of atherosclerosis: Emerging mechanisms and methods, Trends Mol. Med, 23 (4) (2017) 332–347. [DOI] [PubMed] [Google Scholar]
- [6].Aavik E et al. , Global DNA methylation analysis of human atherosclerotic plaques reveals extensive genomic hypomethylation and reactivation at imprinted locus 14q32 involving induction of a miRNA cluster, Eur. Heart J, 36 (16) (2015) 993–1000. [DOI] [PubMed] [Google Scholar]
- [7].Zaina S et al. , DNA methylation map of human atherosclerosis, Circ. Cardiovasc. Genet, 7 (5) (2014) 692–700. [DOI] [PubMed] [Google Scholar]
- [8].Valencia-Morales MP et al. , The DNA methylation drift of the atherosclerotic aorta increases with lesion progression, BMC Med. Genomics, 8 (2015) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kundaje A et al. , Integrative analysis of 111 reference human epigenomes, Nature, 518 (7539) (2015) 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].GTEx_Consortium et al. , Genetic effects on gene expression across human tissues, Nature, 550 (7675) (2017) 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Baribault C et al. , Developmentally linked human DNA hypermethylation is associated with down-modulation, repression, and upregulation of transcription, Epigenetics, Epub ahead of print (2018) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Song Q et al. , A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics, PLoS One, 8 (12) (2013) e81148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dubland JA, Francis GA, So Much Cholesterol: the unrecognized importance of smooth muscle cells in atherosclerotic foam cell formation, Curr. Opin. Lipidol, 27 (2) (2016) 155–161. [DOI] [PubMed] [Google Scholar]
- [14].Lacey MR, Baribault C, Ehrlich M, Modeling, simulation and analysis of methylation profiles from reduced representation bisulfite sequencing experiments, Stat. Appl. Genet. Mol. Biol, 12 (6) (2013) 723–742. [DOI] [PubMed] [Google Scholar]
- [15].Lacey M et al. , Data for atherosclerosis differentially methylated regionare often at enhancers, Data in Brief, (2018). [Google Scholar]
- [16].Rosenbloom KR et al. , The UCSC Genome Browser database: 2015 update, Nucleic Acids Res, 43 (Database issue) (2015) D670–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Feil S et al. , Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis, Circ. Res, 115 (7) (2014) 662–667. [DOI] [PubMed] [Google Scholar]
- [18].Chappell J et al. , Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models, Circ. Res, 119 (12) (2016) 1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].McLean CY et al. , GREAT improves functional interpretation of cis-regulatory regions, Nat Biotechnol, 28 (5) (2010) 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Giancotti FG, Ruoslahti E, Integrin signaling, Science, 285 (5430) (1999) 1028–1032. [DOI] [PubMed] [Google Scholar]
- [21].Whyte WA et al. , Master transcription factors and mediator establish super-enhancers at key cell identity genes, Cell, 153 (2) (2013) 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Parker SC et al. , Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants, Proc. Natl. Acad. Sci. U S A, 110 (44) (2013) 17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Khan A, Zhang X, dbSUPER: a database of super-enhancers in mouse and human genome, Nucleic Acids Res, 44 (D1) (2016) D164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Huang DW, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources, Nat. Protoc, 4 (1) (2009) 44–57. [DOI] [PubMed] [Google Scholar]
- [25].Xia XD et al. , Myocardin: A novel player in atherosclerosis, Atherosclerosis, 257 (2017) 266–278. [DOI] [PubMed] [Google Scholar]
- [26].Rensen SS, Doevendans PA, van Eys GJ, Regulation and characteristics of vascular smooth muscle cell phenotypic diversity, Neth. Heart J, 15 (3) (2007) 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xi D et al. , Systematic analysis of the molecular mechanism underlying atherosclerosis using a text mining approach, Hum. Genomics, 10 (1) (2016) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ehrlich KC et al. , DNA hypomethylation in intragenic and intergenic enhancer chromatin of muscle-specific genes usually correlates with their expression, Yale J. Biol. Med, 89 (4) (2016) 441–455. [PMC free article] [PubMed] [Google Scholar]
- [29].Steffensen LB, Rasmussen LM, A role for collagen IV in cardiovascular disease?, Am. J. Physiol. Heart Circ. Physiol, (2018). [DOI] [PubMed] [Google Scholar]
- [30].Miano JM, Myocardin in biology and disease, J. Biomed. Res, 29 (1) (2015) 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Iaconetti C et al. , Down-regulation of miR-23b induces phenotypic switching of vascular smooth muscle cells in vitro and in vivo, Cardiovasc. Res, 107 (4) (2015) 522–533. [DOI] [PubMed] [Google Scholar]
- [32].Lindskog H et al. , Molecular identification of venous progenitors in the dorsal aorta reveals an aortic origin for the cardinal vein in mammals, Development, 141 (5) (2014) 1120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Majesky MW, Developmental basis of vascular smooth muscle diversity, Arterioscler. Thromb. Vasc. Biol, 27 (6) (2007) 1248–1258. [DOI] [PubMed] [Google Scholar]
- [34].Xu S et al. , Flow-dependent epigenetic regulation of IGFBP5 expression by H3K27me3 contributes to endothelial anti-inflammatory effects, Theranostics, 8 (11) (2018) 3007–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jiang YZ et al. , Arterial endothelial methylome: differential DNA methylation in athero-susceptible disturbed flow regions in vivo, BMC Genomics, 16 (2015) 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Alexander MR, Owens GK, Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease, Annu. Rev. Physiol, 74 (2012) 13–40. [DOI] [PubMed] [Google Scholar]
- [37].Gomez D, Swiatlowska P, Owens GK, Epigenetic control of smooth muscle cell Identity and lineage memory, Arterioscler. Thromb. Vasc. Biol, 35 (12) (2015) 2508–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kim M, Costello J, DNA methylation: an epigenetic mark of cellular memory, Exp. Mol. Med, 49 (4) (2017) e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Freise C, Bobb V, Querfeld U, Collagen XIV and a related recombinant fragment protect human vascular smooth muscle cells from calcium-/phosphate-induced osteochondrocytic transdifferentiation, Exp. Cell Res, 358 (2) (2017) 242–252. [DOI] [PubMed] [Google Scholar]
- [40].Wang G et al. , Origin and differentiation of vascular smooth muscle cells, J. Physiol, 593 (14) (2015) 3013–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ge X et al. , Modeling supravalvular aortic stenosis syndrome with human induced pluripotent stem cells, Circulation, 126 (14) (2012) 1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hon GC et al. , 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation, Mol. Cell, 56 (2) (2014) 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Turner AW et al. , Functional interaction between COL4A1/COL4A2 and SMAD3 risk loci for coronary artery disease, Atherosclerosis, 242 (2) (2015) 543–552. [DOI] [PubMed] [Google Scholar]
- [44].Volger OL et al. , Distinctive expression of chemokines and transforming growth factor-beta signaling in human arterial endothelium during atherosclerosis, Am. J. Pathol, 171 (1) (2007) 326–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cui X et al. , Venous endothelial marker COUP-TFII tegulates the distinct pathologic potentials of adult arteries and veins, Sci. Rep, 5 (2015) 16193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Liu R et al. , Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity, Circulation, 128 (18) (2013) 2047–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Peng J et al. , Tet methylcytosine dioxygenase 2 inhibits atherosclerosis via upregulation of autophagy in ApoE−/− mice, Oncotarget, 7 (47) (2016) 76423–76436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Fuster JJ et al. , Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice, Science, 355 (6327) (2017) 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wiehle L et al. , Tet1 and Tet2 protect DNA methylation canyons against hypermethylation, Mol. Cell. Biol, 36 (3) (2016) 452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Rasmussen KD, Helin K, Role of TET enzymes in DNA methylation, development, and cancer, Genes Dev, 30 (7) (2016) 733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]