

Abstract
Deposition of amyloid plaques in limbic and associative cortices is amongst the most recognized histopathologic hallmarks of Alzheimer’s disease. Despite decades of research, there is a lack of consensus over the impact of plaques on neuronal function, with their role in cognitive decline and memory loss undecided. Evidence has emerged suggesting complex and localized axonal pathology around amyloid plaques, with a significant fraction of swellings and dystrophies becoming enriched with putative synaptic vesicles and presynaptic proteins normally colocalized at hotspots of transmitter release. In the absence of hallmark active zone proteins and postsynaptic receptive elements, the axonal swellings surrounding amyloid plaques have been suggested as sites for ectopic release of glutamate, which under reduced clearance can lead to elevated local excitatory drive. Throughout this review, we consider the emerging data suggestive of amyloid plaques as hotspots of compulsive glutamatergic activity. Evidence for local and long-range effects of nonsynaptic glutamate is discussed in the context of circuit dysfunctions and neurodegenerative changes of Alzheimer’s disease.
Keywords: glutamate, axonal dystrophies, ectopic release, paracrine signaling, metabotropic receptors, Alzheimer’s disease
Introduction
Amyloid plaques and neurofibrillary tangles (NFTs) have been long recognized as the most consistent histopathological manifestations of Alzheimer’s disease ( AD). The core constituent of NFTs is hyperphosphorylated axonal protein tau while plaques are composed of pleated sheets of amyloid-β (Aβ) peptide. Despite extensive research, the mechanistic link between these two hallmarks as well as the role of Aβ plaques in the pathobiology of AD remains elusive. Numerous cases of overt dementia with low Aβ load or mild cognitive and emotional changes with high Aβ levels in the brain have been documented (Davies and others 1988; Morris and others 2014; Serrano-Pozo and others 2014). The increasing recognition of multiple physiological effects of Aβ has recently added an extra layer of complexity to interpretation of the amyloid hypothesis of AD. At presynaptic glutamatergic terminals, for instance, both, monomers and oligomers of Aβ influence almost every step of the synaptic vesicle cycle, from voltage-activated calcium influx to transport, docking and fusion of synaptic vesicles, followed by postfusion membrane recovery (Ovsepian and others 2018). The widening acknowledgement of Aβ oligomers as the prime causative of synaptic impairments of AD also calls into question well-recognized disruptive effects of amyloid plaques, with emerging evidence suggesting their neuroprotective role in sequestering toxic Aβ species.
Notwithstanding the conflicting views, there is ample evidence for focal damage caused by amyloid plaques. Localized oxidative stress with mitochondrial dysfunctions, loss of dendritic spines and presynaptic elements, impairments of axonal integrity and focal immune response are just a few to be named (Geddes and Cotman 1989; Mitew and others 2013). Imaging and neurophysiological reports also show plaque-related impairments of calcium homeostasis with alterations in synaptic and neuronal activity, along with the evidence for a considerable variability in the response of brain tissue to amyloid plaques (Busche and others 2008; Busche and Konnerth 2015; Kuchibhotla and others 2008; Ovsepian and others 2017). It emerges that the development of plaques is far from an unpretentious and gradual peptide deposition in the brain but involves complex interactions of accumulating protein mass with surroundings contributed by a range of cellular and molecular players. The highly diverse reaction of brain tissue to amyloid plaques is likely to contribute toward dissociation between the extent of plaque load and cognitive decline in AD. Indeed, the dynamic nature of neurochemical changes combined with the disease progression, distribution of amyloid plaques and affected brain regions could greatly influence the cognitive outcome. Such possibilities have been considered recently, with emerging data pinpointing substantial diversity in plaque-related axonal dystrophies, presenting a range of features and behavior (Adams and Munoz 1993; Dickson and others 1988; Masliah and others 1993; Su and others 1998; Vickers and others 2016a; Vickers and others 2016b). Intriguingly, a substantial proportion of axonal swellings and dystrophies share multiple features with physiologically enlarged presynaptic boutons and axonal varicosities. Like normal axonal varicosities and synaptic terminals, these dystrophies are enriched with presynaptic proteins involved in synaptic vesicle cycle and exocytosis. The majority of dystrophies at plaques is glutamatergic and contains amyloid precursor protein (APP) and proteases catalyzing Aβ production, as well as putative small synaptic vesicles, which typically colocalize with neurotransmitter release sites (Kandalepas and others 2013; Ovsepian and others 2017). Under reduced clearance of glutamate and down-regulation of the astrocyte glutamate transporter EAAT2 in AD (Revett and others 2013; Scimemi and others 2013), the release of this transmitter from dystrophies could lead to its build-up in the extracellular space with activation of extrasynaptic N-methyl-d-aspartate (NMDA) and metabotropic glutamatergic receptors, affecting synaptic transmission and plasticity mechanisms.
Although the cause of considerable heterogeneity of the localized response to plaques remains unclear, the potential impact of elevated glutamate signaling in AD brain is a matter of great interest and requires careful assessment. The focus of this review centres on plaque-related changes in the brain, with reference to glutamatergic activity. The histochemical, ultrastructural, and molecular evidence is discussed suggesting shared features of normal presynaptic elements and axonal varicosities with dystrophic swellings of AD, which might contribute to the enhanced glutamatergic activity. The emerging focal and extended effects of nonsynaptic glutamate on neurons and networks are considered in the context of the hyperactivity (seizures) of brain circuits and the neurodegenerative process.
Histopathological Evidence for Glutamatergic Impairments at Amyloid Plaques
AD-like proteinaceous deposits in the brain are documented in several neurological diseases, including spongiform encephalopathies, traumatic brain injuries, and neurosyphilis, and occasionally also in the normal ageing brain without a cognitive deficit (Caughey and Lansbury 2003; Fiala 2007). Little is known about their shared features and differences, with future comparative studies anticipated to elucidate the specifics of amyloid plaques of AD. Although multiple mechanisms of biogenesis of Aβ plaques have been considered, factors triggering their formation remain to be determined (Armstrong 1995; Dickson 1997; Fiala 2007; Rifenburg and Perry 1995). Data from mouse models suggests rapid as well as the gradual development of Aβ deposits, which after reaching a certain size enter into a relatively stable phase (Burgold and others 2011; Meyer-Luehmann and others 2008). Immunofluorescence staining with Aβ oligomer-specific antibodies showed that the halo of soluble Aβ surrounding plaque core can extend well beyond the lesion, consistent with diffuse neurochemical changes. Notwithstanding the build-up of a large amount of proteinaceous mass, their emergence is not associated with cell death but deflection of neurons and glial cells to the plaque margins. Axons and presynaptic terminals appear to be particularly sensitive to amyloid lesions, which become swollen and undergo dystrophic changes (Kawai and others 1992; Miyawaki and others 2001; Vickers and others 2016b). The loss of presynaptic elements is especially severe within the plaque core and its immediate vicinity, with dystrophic axonal profiles encircling lesions (Kandalepas and others 2013; Mitew and others 2013; Sadleir and others 2016). With distance from the core, degeneration of glutamatergic elements prevails over 50 to 150 μm ranges, while the loss of GABAergic terminals is restricted to the actual boundaries of lesions. In general, three types of plaque-related axonal changes have been documented: (1) degenerative dystrophies with positively labeled NFT cytoskeletal pathology, indicative of an advanced stage of damage; (2) regenerative NF positive dystrophies enriched with growth proteins (e.g., GAP43), corresponding to neurites and presynaptic terminals undergoing sprouting; and (3) axonal dystrophies with swollen appearance containing an array of presynaptic proteins such as SNAP-25, syntaxin, vGluTl, synaptophysin, voltage-gated calcium channels as well as APP and BACE1 protease (Fig. 1 A).
Figure 1.
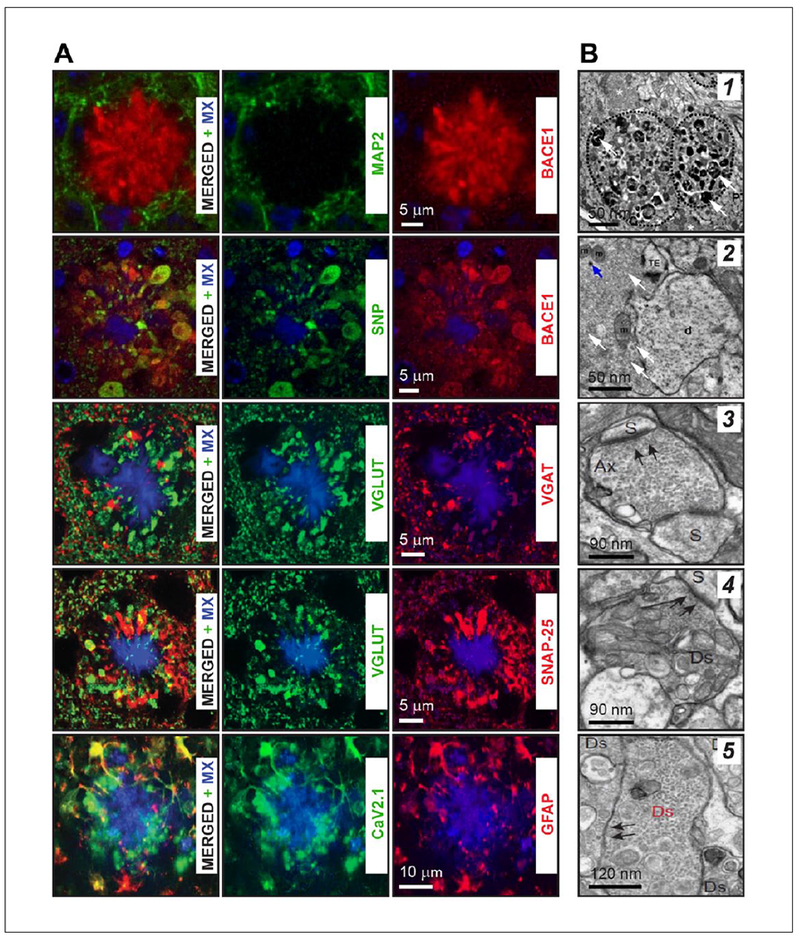
Histochemical and ultrastructural profiles of axonal dystrophies surrounding amyloid plaques. (A) Triple staining of brain slices with Methoxy-X04 (MX, blue in all panels) with dendrite-specific MAP2 protein, BACEI, synaptophysin (SNP), vesicular glutamate transporter (VGLUT), vesicular GABA transporter (VGAT), SNARE protein SNAP-25 (SNAP-25), P/Q type high-voltage gated Ca2+ channel (Cav2.1), and glial fibrillary acidic protein (GFAP). These results suggest that peri-plaque dystrophies like presynaptic glutamatergic nerve terminals are strongly enriched with BACEI, SNAP-25, VGLUTI, synaptophysin, and P/Q type high-voltage gated Ca2+ channels. (B) Ultrastructural data for the enrichment of axonal dystrophies with small, putative synaptic vesicles. (I) Low-magnification image of dystrophic neurite near plaque in the stratum lucidum of CA3 area of the mouse hippocampus filled with electron dense material (white arrows) and vesicles with (2) neighboring vesicle rich presynaptic terminal remaining intact (white arrows). (3) Electron micrograph of normal axon terminal forming two synaptic contacts with two dendritic spines (Ax-S). Black arrows point out one of the synaptic contact and post-synaptic density. (4) A dystrophic putative axon (Ds) establishing synaptic contact (black arrows) with a dendritic spine (S). (5) Dystrophic neurite (Ds in red color) enriched with small synaptic vesicles (black arrows), with some profiles of vesicles juxta-posed at surface membrane ready for release. Although no synaptic contact could be identified, at higher magnification numerous close superpositions of synaptic vesicles with the surface membrane in the absence of PSD were observed. Panels are modified with permission from Daschil and others (2013), Kandalepas and others (2013), Ovsepian and others (2017), and Sadleir and others (2016).
According to Vicker, Terry, and others, there are two independent processes that could trigger the formation of axonal dystrophies around plaques (Masliah and others 1993; Vickers and others 2016a). The first results from the reaction of axons to amyloid lesions, which stimulates regenerative changes and major cytoskeleton alterations and sprouting, whereas the second involves swelling of axons and nerve terminals without signs of regenerative alterations. These phenotypical differences imply the possibility of a differential response of axons or various neuron types to lesions. It emerges that long-range glutamatergic projecting axons of pyramidal neurons are particularly responsive to plaques, while nonpyramidal cells generally do not show dystrophic features. The localized damage and loss of some axon terminals at plaques are paralleled with compensatory enlargement of the remaining presynaptic terminals and bouton swelling, perhaps signifying their adaptive remodeling to counter the collapse of local circuits. Importantly, unlike boutons and varicosities of glutamatergic axons presenting canonical sites for release at specialized active zones, amyloid plaque related axonal swellings and dystrophies, while also enriched with generic presynaptic markers, are devoid of active zone proteins (Sadleir and others 2016), an observation implying the absence of specialized secretory sites. Results of ultrastructural analysis are in general agreement with immunohistochemical data, showing remarkable heterogeneity in the content and behavior of dystrophies, with putative synaptic vesicles and multilamellar, electron-dense endosomes and lysosomal intermediates being differentially present (Masliah and others 1991; Nixon 2014) (Fig. 1B). In dystrophies, BACE1 appears mostly on small translucent intracellular compartments, which are reminiscent of a vGlut1 positive small synaptic vesicle, with occasional fusion profiles identified on the axonal membrane (Ovsepian and others 2017). As glutamate and Aβ secretion from neurons rely on SNARE proteins and can occur independently from specialized active zones (Cirrito and others 2003; Cirrito and others 2005; Ovsepian and Dolly 2011), it is tempting to speculate that axonal swellings associated with amyloid plaques might present hotspots for the nonsynaptic release of Aβ and glutamate.
Among other histochemical changes implying a plaque-related enhancement of glutamatergic drive, the local deficit in glutamate clearance and remodeling of glutamatergic receptors have been widely discussed (Albasanz and others 2005; Paoletti and others 2013; Revett and others 2013). Downregulation of glutamate transporter around Aβ plaques as well as changes in the density and molecular composition of glutamatergic receptors have been documented in both, plaque-laden human brain and mouse models of AD, with local deficit of EAAT 1-2 and mouse GLYT-1 transporter shown explicitly (Hefendehl and others 2016; Jacob and others 2007; Revett and others 2013). Of note, while reduced glutamate transport is expected to lower its release from neurons, the build-up of glutamate in astrocytes at the same time would lead to its uncontrolled leakage from these cells into the interstitial space, promoting pathophysiology and functional changes in AD brain.
Functional Evidence for Glutamatergic Impairments at AD Amyloid Plaques
The widely held notion of gradual collapse of the brain connectome in AD has been recently confronted by data showing complex and often bilateral changes that affect synaptic and network functions at multiple levels. By combining cellular imaging and patch-clamp studies, alterations in the geometry of hippocampal pyramidal neurons has been shown in mice expressing K670N/M671L-mutated APP (APPswe) and Ml46V mutated presenilin 1 (PS1), which exhibited enhanced response to depolarizing inputs (Siskova and others 2014). As a result, hippocampal pyramidal cells in plaque-laden brain discharge bursts of action potentials, unlike wild-type and pre-plaque transgenic neurons, which exhibit sparse single spike firing. This abnormal response of pyramidal neurons translates into general hyperactivity of circuits, as detected by field potential recordings. Changes in neuronal responsiveness shown in this study were attributed to morphological abnormalities with shrinkage of the dendritic tree, lowering their electrical capacitance, and hence, making them more excitable. Similar alterations have been reported earlier in the human AD brain (Grutzendler and others 2007). Although no direct link between altered neuronal activity and amyloid plaques were shown in these studies, increased responsiveness of neurons was detectable only in animals displaying abundant plaque pathology (Siskova and others 2014).
Using neuronal Ca2+ imaging for analyzing plaque-related alterations in neuronal and glial dynamics in situ, considerable variability in spontaneous activity of these cells along with disruption of intracellular Ca2+ were documented in AD mice showing early onset plaques (Busche and others 2008; Busche and Konnerth 2015; Kuchibhotla and others 2008; Kuchibhotla and others 2009). In layer 2/3 of the frontal cortex, for instance, ~16-fold increase in the activity of selected neurons around plaques was described, while the number of quiescence neurons was reduced (Busche and others 2008) (Fig. 2A and B). The overactive neurons typically were clustered around plaques, while quiescent cells were scattered throughout the cortex. The elevated activity has been proposed to result from disruption of the balance between excitatory and inhibitory circuits, with blockade of fast glutamatergic transmission abolishing the hyperactivity. Hyperactive neurons were also reported in the hippocampus of AD mice (Busche and others 2012). Aβ is known to bind and activate a range of neuronal receptors and cause neurophysiological effects. Low-molecular-weight Aβ oligomers could also change the activity balance by disrupting the membrane integrity of neurons through the formation of cation-selective channels with Ca2+ influx (Demuro and others 2010; Lin and others 2001). Using YC3.6 FRET-based ratiometry and measurement of intracellular Ca2+ concentration, the levels of resting Ca2+ and spontaneous Ca2+ transients were determined in neurons and neurites in proximity to plaques (Kuchibhotla and others 2008) (Fig. 2C and D). Both axons and dendrites surrounding lesions showed significant Ca2+ overload, the extent of which correlated with the distance from plaques. At a cellular level, dysregulation of Ca2+ homeostasis was shown to decompartmentalize intracellular Ca2+, rendering Ca2+ transients slow and less localized.
Figure 2.
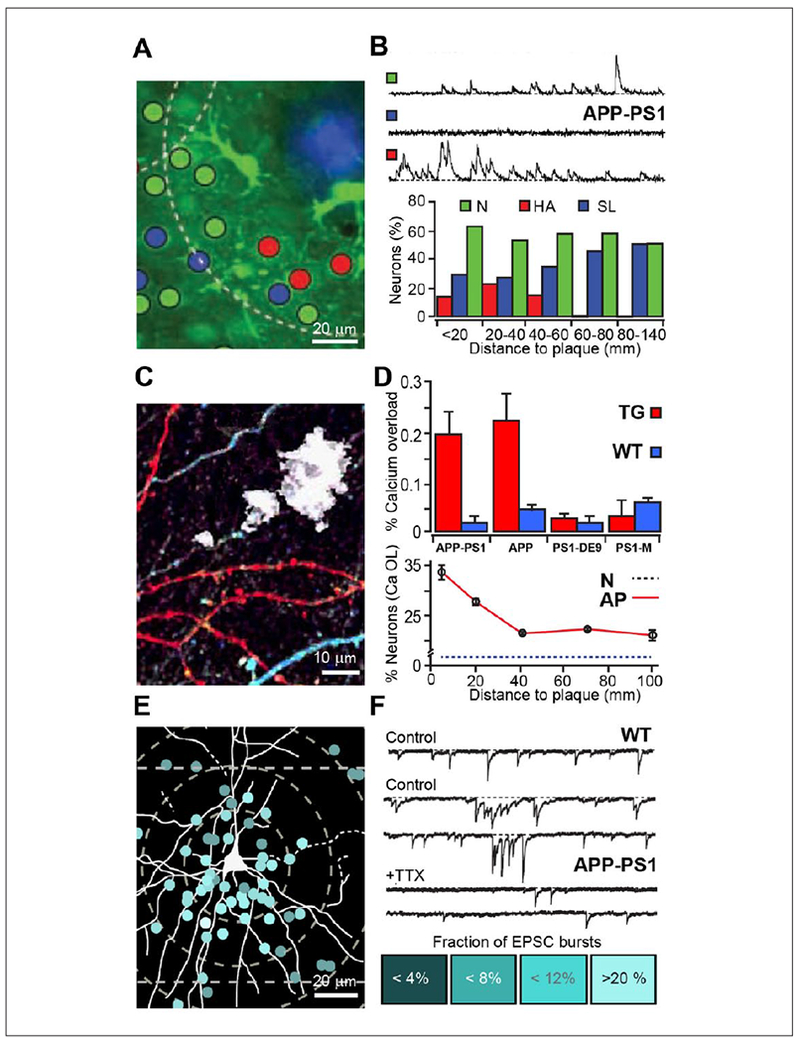
Calcium dynamics and electrophysiological activity of cortical neurons in proximity to amyloid plaques. (A, B) Spatial distribution of silent and hyperactive neurons around amyloid plaques in APP23-PS45 mouse brain. Maximal projection image (A) and recordings of spontaneous Ca2+ dynamics in normal (N), silent (SL), and hyperactive (HA) neurons with summary plot showing the relationship of the activity and the distance from the amyloid plaque. Adapted with permission from Busche and others (2008). (C, D) In vivo image of YC3.6 expressing neurites around Aβ plaques (white, C). Elevated Ca2+ colored in red. (D) Comparative summary histogram of the Ca2+ load of neurites in four Alzheimer’s disease (AD) transgenic mouse lines and corresponding wild type, demonstrating that the disruption of Ca2+ load depends on the elevated overproduction of Aβ peptide (top). In amyloid precursor protein (APP) AD model, within 25 μm from amyloid plaques, Ca2+ load was significantly higher as compared with wild-type (WT) controls (lower graph). Adapted with permission from Kuchibhotla and others (2008). (E) A representative reconstructed layer III pyramidal neuron projected over the distribution map of analyzed plaque-neuron pairs. (F) Sample current traces collected from pyramidal cells of WT, APP-PSI neurons prior and after TTX (pair top and bottom traces). The density of the RAF color below presents the percentage of sEPSC (simultaneous excitatory postsynaptic current) occurring in multi-peak bursts. Adapted with permission from Ovsepian and others (2017).
While it was generally assumed that Ca2+ dysregulation and altered activity of neurons at plaques is caused by Aβ oligomers floating in the interstitial fluid, disruptions of glutamatergic signaling also could play a role. In fact, focal increase in glutamatergic drive around plaques is expected not only to drive synaptic hyperactivity and rise of intracellular Ca2+ but also to initiate degenerative axonal swelling and synaptic loss (Hiruma and others 1999; Hiruma and others 2003). Using a genetically encoded glutamate sensor iGluSnFR, a significant rise of extracellular glutamate level at plaques with reduced glutamate clearance was shown, with disruptive effects on processing of sensory inputs by local circuits (Hefendehl and others 2016). These changes were attributed to downregulation of the GLT-1 at plaques with slower glutamate clearance. Analysis of electrophysiological changes in proximity of amyloid lesions in the frontal cortex showed major disruption in spontaneous and evoked synaptic activity in layer 2/3 pyramidal cells of APP-PS1 and APP23 AD mice (expressing K670N/M671L-mutated APP [APPswe]), which caused significant changes in local field potentials and excitatory synaptic currents (Ovsepian and others 2017) (Fig. 2E and F). Plaque related alterations were detectable from the early stages of the pathology, without changes in the passive properties of neurons, attributing changes to disruptions of synaptic inputs. Using pharmacological tools, it was shown that abnormal glutamatergic inputs to pyramidal neurons depend on loaded presynaptic Ca2+ stores and could be greatly attenuated by depletion of IP3-sensitive stores by SERCA ATPase inhibitors. Because anomalous excitatory activity could be also observed in response to unitary synaptic inputs, their generation was attributed to the presynaptic terminals, whereas inhibition of the hyperactivity by selective antagonists of type 1 metabotropic glutamatergic receptors (mGluRl) implied underlying metabotropic mechanisms. The strong enrichment of presynaptic swellings and dystrophies around plaques with SNARE proteins SNAP-25 and syntaxin, as well as, vGluTl and accumulation of putative small synaptic vesicles inside dystrophies suggests that elevated local glutamatergic drive and hyperactivity could result from the ectopic release of glutamate with paracrine effects (Ovsepian and others 2017). Multiple small vesicles juxtaposed at the surface membrane of dystrophies, in the absence of post-synaptic elements, are also consistent with possible ectopic release of glutamate from axonal swellings. Together with reduced clearance, the ectopic release of glutamate from axonal swellings at plaques could cause not only local disruption in neuronal activity but also induce waves of hyperactivity spreading over extended cortical circuits to contribute toward seizures and cognitive decline of AD and overall degenerative process.
Plaque-Related Local Functional Changes in the Context of Global Brain Activity of AD
In the nervous system, glutamate is not only the principal mediator of synaptic transmission at excitatory synapses but also a powerful regulator of the state and general activity of neural circuits. It is thought that extracellular glutamate plays an important regulatory role in the healthy brain, with its concentrations finely adjusted during different behavioral states by transport and clearance mechanisms (Featherstone and Shippy 2008). The physiological levels of ambient glutamate greatly vary throughout the brain, with fluctuations of tonic glutamatergic drive reported in association with circadian changes as well as alterations of cognitive states. Changes in the level of ambient glutamate are also thought to underlie mood disruptions, with strong irregularities in its amounts reported during seizures, ischemia and fever (Nyitrai and others 2006; Wahl and others 1994) as well as in some neurodegenerative disease (Lewerenz and Maher 2015). Assuming that the physiological levels of nonsynaptic glutamate stay below the level known to trigger cytotoxicity (<5 μM), its concentrations during normal processes must not exceed low micromolar amounts. Of note, a moderate increase of extracellular glutamate is capable of causing axonal swellings and dystrophies, with fast necrotic changes also induced by concentrations greater than 100 μM (Cheung and others 1998; Hiruma and others 2003). With the EC50 (half maximal effective concentration) of glutamate sensor iGluSnFR ~5 μM, it is conceivable that during bursts of neuronal activity, the levels of glutamate around amyloid lesions may reach amounts sufficient for activating nonsynaptic NMDA and mGluR receptors (EC50 ranging between ~0.5 and ~20 μM) (Fig. 3).
Figure 3.
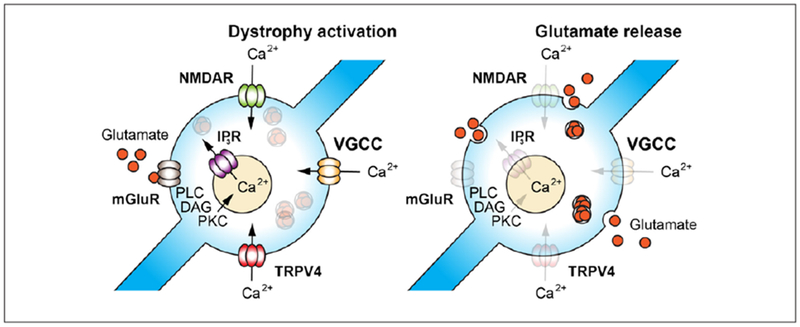
Schematic diagrams illustrating the hypothetical mechanisms of the activation of nonsynaptic glutamate release from dystrophies surrounding amyloid plaques. Ca2+ influx activated by nonsynaptic glutamate through stimulation of presynaptic NMDA receptors and mobilization of intracellular Ca2+ via activation of metabotropic glutamatergic receptors, as well as depolarization induced Ca2+ influx through voltage gated Ca2+ channels or activation of mechanosensing TRPV4 receptors (left), which can stimulate glutamate release from dystrophic neurites (right) with diffuse paracrine effects on surrounding neurons and synaptic terminals.
As mentioned earlier, poor clearance and the plausible ectopic release of glutamate from dystrophies are two key drivers of high levels of extracellular glutamate at plaques. While typically glutamate is released from specialized sites, numerous examples of its nonsynaptic secretion in the absence of active zones have also been shown (Matsui and Jahr 2004; Ovsepian and Dolly 2011). The histochemical and functional data discussed above suggest that nonsynaptic release of glutamate with paracrine effects might underlie the pathological hyperactivity of cortical circuits associated with amyloid plaques. Another intriguing posibility related to amyloid plaques and dystrophies is the likely activation of mechanosensitive TRPV4 and Cav1.2 channels in dystrophic axons (Daschil and others 2013). The enrichment of Cav1.2 channel subunit at dystrophies surrounding plaques and the ubiquitous expression of the TRPV4 channel in axons throughout the brain are expected to render swellings hyperactive, given that mechanosensitive TRPV4 channels enable nonselective cation influx and lead to membrane depolarization (Gu and Gu 2014; Kanju and Liedtke 2016). Functional abnormalities in Cav1.2 and TRPV4 channels are known not only to disrupt the synaptic activity and transmitter release but are also suspected in hyperactivity and generation of seizures. TRPV4 is, for instance, known to cause seizures under various developmental aberrations, through activation of Ca2+ influx, while certain mutations of Cavl .2 are known to cause episodic ataxia and epilepsy (Kanju and Liedtke 2016; Ovsepian and Friel 2008; Zamponi and others 2010). Age-related changes in TRPV4 functions have been also implicated in pathogenesis of neurodegenerative disease. In light of the enhanced activity of neurons around plaques and emerging evidence for recurrent seizures in AD patients, there is pressing need for future research and elucidation of the role of Cavl .2 and TRPV4 channels as well as other key regulators of neurotransmitter release in augmented glutamatergic drive at dystrophies.
The greater propensity of the amyloid plaque load in the limbic and associative cortices comprising dense local and long-range connections is expected to favor generation of seizures and their spread over wider brain areas (Ovsepian and O’Leary 2016). With growing evidence for early onset functional abnormalities in cortical circuits of AD animal models and humans, described herein impairments of glutamatergic drive are of major interest and warrant careful future research. Along with settling the controversy over the role of plaques in pathobiology of AD, the outcome of upcoming studies could afford major advances towards developing new concepts of brain functions and disease models, an investment holding major scientific rewards and potential clues for therapy development.
Acknowledgments
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Programme for Research in Third Level Institutions (PRTLI) Cycle 4 from the Higher Education Authority of Ireland and the Neuroscience Section Grant for Target-Driven Therapeutics and Theranostics Research (S.V.O. and J.O.D.).
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Adams LA, Munoz DG. 1993. Differential incorporation of processes derived from different classes of neurons into senile plaques in Alzheimer’s disease. Acta Neuropathol 86(4):365–70. [DOI] [PubMed] [Google Scholar]
- Albasanz JL, Dalfo E, Ferrer I, Martin M. 2005. Impaired metabotropic glutamate receptor/phospholipase C signaling pathway in the cerebral cortex in Alzheimer’s disease and dementia with Lewy bodies correlates with stage of Alzheimer’s disease-related changes. Neurobiol Dis 20(3):685–93. [DOI] [PubMed] [Google Scholar]
- Armstrong RA. 1995. Is the clustering of β-amyloid (Aβ) deposits in the frontal cortex of Alzheimer patients determined by bloodvessels? Neurosci Lett 195(2): 121–4. [DOI] [PubMed] [Google Scholar]
- Burgold S, Bittner T, Dorostkar MM, Kieser D, Fuhrmann M, Mitteregger G, and others 2011. In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol 121 (3):327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, and others 2012. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 109(22): 8740–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, and others 2008. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321(5896): 1686–9. [DOI] [PubMed] [Google Scholar]
- Busche MA, Konnerth A. 2015. Neuronal hyperactivity-a key defect in Alzheimer’s disease? Bioessays 37(6):624–32. [DOI] [PubMed] [Google Scholar]
- Caughey B, Lansbury PT. 2003. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26:267–98. [DOI] [PubMed] [Google Scholar]
- Cheung NS, Pascoe CJ, Giardina SF, John CA, Beart PM. 1998. Micromolar L-glutamate induces extensive apoptosis in an apoptotic-necrotic continuum of insult-dependent, excitotoxic injury in cultured cortical neurones. Neuropharmacology 37(10-11): 1419–29. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, May PC, O’Dell MA, Taylor JW, Parsadanian M, Cramer JW, and others 2003. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-β metabolism and half-life. J Neurosci 23(26):8844–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, and others 2005. Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron 48(6): 913–22. [DOI] [PubMed] [Google Scholar]
- Daschil N, Obermair GJ, Flucher BE, Stefanova N, Hutter-Paier B, Windisch M, and others 2013. CaV1.2 calcium channel expression in reactive astrocytes is associated with the formation of amyloid-β plaques in an Alzheimer’s disease mouse model. J Alzheimers Dis 37(2):439–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies L, Wolska B, Hilbich C, Multhaup G, Martins R, Simms G, and others 1988. A4 amyloid protein deposition and the diagnosis of Alzheimer’s disease: prevalence in aged brains determined by immunocytochemistry compared with conventional neuropathologic techniques. Neurology 38(11): 1688–93. [DOI] [PubMed] [Google Scholar]
- Demuro A, Parker I, Stutzmann GE. 2010. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem 285(17):12463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW.1997. The pathogenesis of senile plaques. J Neuropathol Exp Neurol 56(4):321–39. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH. 1988. Alzheimer’s disease. A double-labeling immunohistochemical study of senile plaques. Am J Pathol 132(1 ):86–101. [PMC free article] [PubMed] [Google Scholar]
- Featherstone DE, Shippy SA. 2008. Regulation of synaptic transmission by ambient extracellular glutamate. Neuroscientist 14(2): 171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC.2007. Mechanisms of amyloid plaque pathogenesis. Acta Neuropathol 114(6): 551–71. [DOI] [PubMed] [Google Scholar]
- Geddes JW, Cotman CW. 1989. Plasticity, pathology, and Alzheimer’s disease. Neurobiol Aging 10(5):571–3. [DOI] [PubMed] [Google Scholar]
- Grutzendler J, Helmin K, Tsai J, Gan WB. 2007. Various dendritic abnormalities are associated with fibrillar amyloid deposits in Alzheimer’s disease. Ann N Y Acad Sci 1097:30–9. [DOI] [PubMed] [Google Scholar]
- Gu Y, Gu C. 2014. Physiological and pathological functions of mechanosensitive ion channels. Mol Neurobiol 50(2): 339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefendehl JK, LeDue J, Ko RW, Mahler J, Murphy TH, MacVicar BA. 2016. Mapping synaptic glutamate transporter dysfunction in vivo to regions surrounding Aβ plaques by iGluSnFR two-photon imaging. Nat Commun 7:13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiruma H, Katakura T, Takahashi S, Ichikawa T, Kawakami T. 2003. Glutamate and amyloid β-protein rapidly inhibit fast axonal transport in cultured rat hippocampal neurons by different mechanisms. J Neurosci 23(26):8967–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiruma H, Nishida S, Katakura T, Kusakabe T, Takenaka T, Kawakami T. 1999. Extracellular potassium rapidly inhibits axonal transport of particles in cultured mouse dorsal root ganglion neurites. J Neurobiol 38(2):225–33. [DOI] [PubMed] [Google Scholar]
- Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, and others 2007. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J Alzheimers Dis 11 (1):97–116. [DOI] [PubMed] [Google Scholar]
- Kandalepas PC, Sadleir KR, Eimer WA, Zhao J, Nicholson DA, Vassar R. 2013. The Alzheimer’s β-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol 126(3):329–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanju P, Liedtke W. 2016. Pleiotropic function of TRPV4 ion channels in the central nervous system. Exp Physiol 101(12): 1472–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai M, Cras P, Richey P, Tabaton M, Lowery DE, Gonzalez-DeWhitt PA, and others 1992. Subcellular localization of amyloid precursor protein in senile plaques of Alzheimer’s disease. Am J Pathol 140(4):947–58. [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. 2008. Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59(2):214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. 2009. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323(5918): 1211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Maher P. 2015. Chronic glutamate toxicity in neurodegenerative diseases-what is the evidence? Front Neurosci 9:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Bhatia R, Lal R. 2001. Amyloid β protein forms ion channels: implications for Alzheimer’s disease pathophysiology. FASEB J 15(13):2433–44. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, Alford M, Albright T, DeTeresa R, and others 1991. Patterns of aberrant sprouting in Alzheimer’s disease. Neuron 6(5):729–39. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, Alford M, DeTeresa R, Terry R. 1993. An antibody against phosphorylated neurofilaments identifies a subset of damaged association axons in Alzheimer’s disease. Am J Pathol 142(3):871–82. [PMC free article] [PubMed] [Google Scholar]
- Matsui K, Jahr CE. 2004. Differential control of synaptic and ectopic vesicular release of glutamate. J Neurosci 24(41):8932–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, and others 2008. Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 451(7179): 720–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitew S, Kirkcaldie MT, Dickson TC, Vickers JC. 2013. Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol Aging 34(10):2341–51. [DOI] [PubMed] [Google Scholar]
- Miyawaki K, Nakayama H, Nakamura S, Uchida K, Doi K. 2001. Three-dimensional structures of canine senile plaques. Acta Neuropathol 102(4): 321–8. [DOI] [PubMed] [Google Scholar]
- Morris GP, Clark IA, Vissel B. 2014. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Conunun 2:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA.2014. Alzheimer neurodegeneration, autophagy, and Aβ secretion: the ins and outs (comment on DOI 10.1002/bies.201400002). Bioessays 36(6):547. [DOI] [PubMed] [Google Scholar]
- Nyitrai G, Kekesi KA, Juhasz G. 2006. Extracellular level of GABA and Glu: in vivo microdialysis-HPLC measurements. Curr Top Med Chem 6(10):935–40. [DOI] [PubMed] [Google Scholar]
- Ovsepian SV, Blazquez-Llorca L, Freitag SV, Rodrigues EF, Herms J. 2017. Ambient glutamate promotes paroxysmal hyperactivity in cortical pyramidal neurons at amyloid plaques via presynaptic mGluRl receptors. Cereb Cortex 27(10):4733–49. [DOI] [PubMed] [Google Scholar]
- Ovsepian SV, Dolly JO. 2011. Dendritic SNAREs add a new twist to the old neuron theory. Proc Natl Acad Sci U S A 108(48): 19113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovsepian SV, Friel DD. 2008. The leaner P/Q-type calcium channel mutation renders cerebellar Purkinje neurons hyperexcitable and eliminates Ca2+-Na+ spike bursts. Eur J Neurosci 27(1 ):93–103. [DOI] [PubMed] [Google Scholar]
- Ovsepian SV, O’Leary VB. 2016. Neuronal activity and amyloid plaque pathology: an update. J Alzheimers Dis 49( 1): 13–9. [DOI] [PubMed] [Google Scholar]
- Ovsepian SV, O’Leary VB, Zaborszky L, Ntziachristos V, Dolly JO. 2018. Synaptic vesicle cycle and amyloid β: biting the hand that feeds. Alzheimers Dement 14(4):502–13. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. 2013. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14(6):383–400. [DOI] [PubMed] [Google Scholar]
- Revett TJ, Baker GB, Jhamandas J, Kar S. 2013. Glutamate system, amyloid ss peptides and tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatry Neurosci 38(1):6–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifenburg RP, Perry G. 1995. Dystrophic neurites define diffuse as well as core-containing senile plaques in Alzheimer’s disease. Neurodegeneration 4(2):235–7. [PubMed] [Google Scholar]
- Sadleir KR, Kandalepas PC, Buggia-Prevot V, Nicholson DA, Thinakaran G, Vassar R. 2016. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer’s disease. Acta Neuropathol 132(2):235–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimemi A, Meabon JS, Woltjer RL, Sullivan JM, Diamond JS, Cook DG. 2013. Amyloid-β1-42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J Neurosci 33(12):5312–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, Qian J, Monsell SE, Blacker D, Gomez-Isla T, Betensky RA, and others 2014. Mild to moderate Alzheimer dementia with insufficient neuropathological changes. Ann Neurol 75(4):597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siskova Z, Justus D, Kaneko H, Friedrichs D, Henneberg N, Beutel T, and others 2014. Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron 84(5): 1023–33. [DOI] [PubMed] [Google Scholar]
- Su JH, Cummings BJ, Cotman CW. 1998. Plaque biogenesis in brain aging and Alzheimer’s disease. II. Progressive transformation and developmental sequence of dystrophic neurites. Acta Neuropathol 96(5):463–71. [DOI] [PubMed] [Google Scholar]
- Vickers JC, Kirkcaldie MT, Phipps A, King AE. 2016a. Alterations in neurofilaments and the transformation of the cytoskeleton in axons may provide insight into the aberrant neuronal changes of Alzheimer’s disease. Brain Res Bull 126(Pt 3): 324–33. [DOI] [PubMed] [Google Scholar]
- Vickers JC, Mitew S, Woodhouse A, Fernandez-Martos CM, Kirkcaldie MT, Canty AJ, and others 2016b. Defining the earliest pathological changes of Alzheimer’s disease. Curr Alzheimer Res 13(3):281–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl F, Obrenovitch TP, Hardy AM, Plotkine M, Boulu R, Symon L. 1994. Extracellular glutamate during focal cerebral ischaemia in rats: time course and calcium dependency. J Neurochem 63(3): 1003–11. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Lory P, Perez-Reyes E. 2010. Role of voltage-gated calcium channels in epilepsy. Pflugers Arch 460(2):395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]