

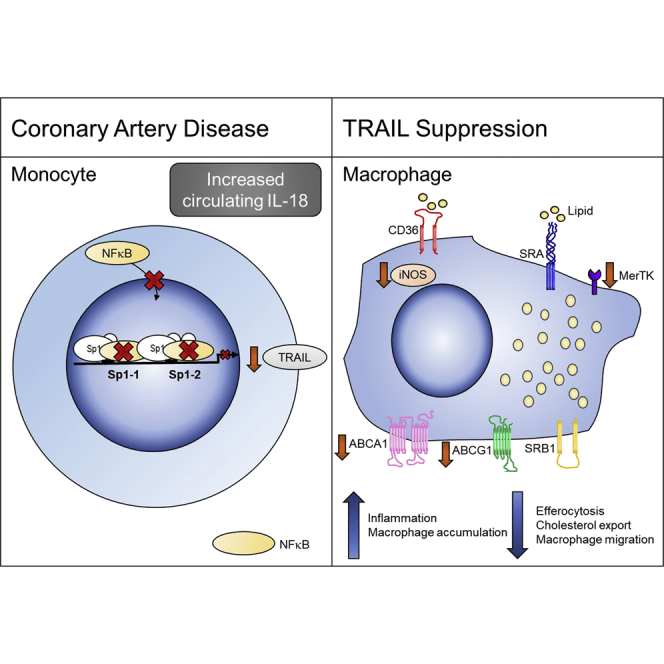
Summary
Circulating tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) levels are reduced in patients with cardiovascular disease, and TRAIL gene deletion in mice exacerbates atherosclerosis and inflammation. How TRAIL protects against atherosclerosis and why levels are reduced in disease is unknown. Here, multiple strategies were used to identify the protective source of TRAIL and its mechanism(s) of action. Samples from patients with coronary artery disease and bone-marrow transplantation experiments in mice lacking TRAIL revealed monocytes/macrophages as the main protective source. Accordingly, deletion of TRAIL caused a more inflammatory macrophage with reduced migration, displaying impaired reverse cholesterol efflux and efferocytosis. Furthermore, interleukin (IL)-18, commonly increased in plasma of patients with cardiovascular disease, negatively regulated TRAIL transcription and gene expression, revealing an IL-18-TRAIL axis. These findings demonstrate that TRAIL is protective of atherosclerosis by modulating monocyte/macrophage phenotype and function. Manipulating TRAIL levels in these cells highlights a different therapeutic avenue in the treatment of cardiovascular disease.
Subject Areas: Pathophysiology, Molecular Mechanism of Behavior, Diabetology, Immunology, Immune Response
Graphical Abstract
Highlights
-
•
Monocytes are a significant source of TRAIL in the normal circulation
-
•
Monocyte TRAIL expression is reduced, concomitant with plasma levels in CAD
-
•
TRAIL-expressing monocyte/macrophages attenuate atherosclerosis
-
•
Macrophages lacking TRAIL are dysfunctional
Pathophysiology; Molecular Mechanism of Behavior; Diabetology; Immunology; Immune Response
Introduction
Despite improvements in treatment, cardiovascular disease (CVD) remains the leading cause of death worldwide. The main cause is atherosclerosis, the pathological process underlying stroke, coronary artery disease (CAD), and peripheral vascular disease. Cells of the myeloid lineage, particularly monocyte/macrophages, play a pivotal role in atherogenesis, which is triggered by an inflammatory response to lipoproteins that are modified in the vessel wall. Here, innate responses initiate the recruitment, homing, migration, and differentiation of monocytes into macrophages, where they orchestrate a plethora of functions including phagocytosis, efferocytosis, proliferation, migration, secretion of inflammatory molecules (e.g., tumor necrosis factor alpha [TNF-α]), production of resolving molecules (e.g., interleukin [IL]-10), and cell death. In mature lesions, failure of macrophage resolution mechanisms and the subsequent increased immune response may underlie processes that exacerbate disease (Tabas, 2010).
TNF-related apoptosis-inducing ligand (TRAIL) was originally identified as a cancer-killing cytokine, but over the last 10 years it has become clear that TRAIL has pleiotropic functions beyond killing and can modulate multiple cellular processes, including proliferation, migration, and differentiation (Harith et al., 2013, Kavurma and Bennett, 2008, Kavurma et al., 2008b, Manuneedhi Cholan et al., 2017). Our particular interest is its role in CVD. In humans, low plasma TRAIL levels independently predict cardiovascular events and mortality (Volpato et al., 2011), and TRAIL is reduced in the circulation of patients with CVD (Schoppet et al., 2006), suggesting a protective role. The recent identification of 11 loss-of-function TRAIL single-nucleotide polymorphisms (SNPs) that confer increased risk of carotid artery atherosclerosis supports this view (Pott et al., 2017). We and others identified TRAIL's non-apoptotic functions in the vasculature, including in neointimal thickening, angiogenesis, and peripheral vascular disease, and in advanced atherosclerosis (Chan et al., 2010, Di Bartolo et al., 2011, Di Bartolo et al., 2013, Di Bartolo et al., 2015, Kavurma et al., 2008a, Secchiero et al., 2004a, Secchiero et al., 2006). Our atherosclerotic murine models lacking TRAIL corroborate the human association studies. For example, high-fat diet (HFD)-fed Trail−/−Apoe−/− mice exhibited 150% larger plaques than Apoe−/− mice (Di Bartolo et al., 2011). Intriguingly, these mice not only had significantly more monocyte/macrophages in their plaques but also increased monocyte/macrophage numbers in kidney (Cartland et al., 2014) and pancreata (Di Bartolo et al., 2011), suggesting that TRAIL may control macrophage accumulation in injured tissues. In contrast to our observations, TRAIL administration to diabetic Apoe−/− mice attenuated atherosclerosis, in part, by inducing macrophage death (Secchiero et al., 2006). Thus, the role of TRAIL in monocyte/macrophage function is unclear. Here we examined samples from patients with CAD and several murine models of atherosclerosis, as well as monocyte/macrophages in vitro. We identified an iNOShiTRAIL+ macrophage population that appeared critical in resolving atherosclerosis. Furthermore, we describe a unique anti-inflammatory pathway, the IL-18-TRAIL axis, in patients with CAD. These findings suggest that TRAIL is critical in modulating atherogenesis and has wider implications in mechanisms regulating inflammation.
Results
Circulating TRAIL Is Reduced in Patients with CAD and Associates with Reduced Monocyte TRAIL mRNA
To assess local coronary production of TRAIL, we measured plasma concentrations in blood sampled from three sites around the heart, collected from control subjects and stable and unstable patients with CAD. Regardless of sampling site, TRAIL was significantly lower in plasma from patients with CAD, ∼45% less than control levels (Figure 1A). In contrast, TNF-α (Figure 1B), a cytokine with high sequence homology to TRAIL, was similar in all samples. No differences were found in the TRAIL arterial-coronary sinus gradient (Figure 1C), indicating that the source of TRAIL was systemic rather than directly from the heart. Plasma from a second cohort of patients with unstable CAD also contained markedly less TRAIL at ∼70% less than control levels (Figure 1D). TRAIL mRNA in monocytes from the same individuals was also strikingly reduced (∼70%; Figure 1E), implicating these cells as a major source of TRAIL in healthy circulation, which may be compromised in patients with CAD.
Figure 1.

Circulating TRAIL Is Reduced in Patients with CAD, Associating with Reduced Monocyte TRAIL mRNA
(A–C) Blood from the coronary sinus, aortic root (arterial), and lower right atrium (venous) were collected and assayed for (A) TRAIL and (B) TNF-α by ELISA. (C) The coronary sinus-arterial TRAIL gradient. Patient samples are n = 8–15/group.
(D and E) Venous blood was collected from healthy controls (n = 7) and a second cohort of patients with CAD (n = 11) and assayed for (D) plasma TRAIL and (E) monocyte TRAIL gene expression by qPCR. mRNA expression was normalized to GAPDH.
Results are mean ± SEM. One-way ANOVA or t-test; *p < 0.05, **p < 0.01, ***p < 0.001.
TRAIL Expressing Monocyte/Macrophages Protect against Atherosclerosis
Wildtype mice do not develop significant atherosclerosis without the disruption of genes critical in cholesterol metabolism. Apolipoprotein E (ApoE) is key in maintaining plasma cholesterol concentrations. Deletion in mice causes hypercholesterolemia and spontaneous development of atherosclerotic lesions that are accelerated by HFD feeding (Meir and Leitersdorf, 2004). We showed that TRAIL deletion in Apoe−/− mice (Trail−/−Apoe−/−) exacerbated plasma cholesterol levels, and macrophage-rich plaques were larger and more frequent than in Apoe−/− mice fed a HFD (Di Bartolo et al., 2011). These findings indicated a protective role for TRAIL.
Monocytes originate in bone marrow, and because they were a significant source of TRAIL in people (Figure 1), we predicted that TRAIL-expressing (TRAIL+) bone marrow afforded protection in atherosclerosis. To investigate this, we examined two sources using bone marrow chimeras: TRAIL expressed only in the bone marrow (BM-TRAIL) and TRAIL expressed everywhere except the bone marrow (parenchymal-TRAIL). Results were compared with mice with no TRAIL in bone marrow or parenchyma (null-TRAIL). The bone marrow chimera study design is illustrated in Figure 2A. Brachiocephalic arteries of BM-TRAIL mice had significantly reduced atherosclerotic lesion size compared with the null- and parenchymal-TRAIL lesions, which had ∼50% more atherosclerosis (Figure 2B). Body weight and plasma chemistries, including total cholesterol, triglycerides, glucose and insulin were similar between groups (Table 1). Interestingly, atherosclerotic lesions from BM-TRAIL mice were of similar size to those in Trail−/−Apoe−/− samples harvested after only 5–8 weeks HFD (Figure 2C), suggesting that TRAIL expressed in the bone marrow slows atherogenesis. Depletion of macrophages in HFD-fed Trail−/−Apoe−/− using clodronate liposomes dramatically reduced atherosclerotic plaque size (Figure 2D), confirming that a significant number of cells in the plaque are of monocyte/macrophage origin. Immunohistochemical assessment showed significantly lower numbers of Mac3+ macrophages in BM-TRAIL plaque compared with those in lesions from null- or parenchymal-TRAIL mice (Figure 2E). Furthermore, TRAIL mRNA expression from BM-TRAIL aortae was ∼50-fold less than parenchymal-TRAIL samples (Figure 2F) reinforcing the profound effect of TRAIL+ monocyte/macrophages in reducing lesion size (Figure 2B); TRAIL was undetectable in aortae from null-TRAIL mice (Figure 2F). In agreement with this, plasma from BM-TRAIL mice had significantly reduced TRAIL concentrations, ∼2.5-fold less than in plasma from parenchymal-TRAIL mice (Table 1). By combining plasma TRAIL concentrations from BM-TRAIL and parenchymal-TRAIL mice (Table 1), we estimated that ∼30% of plasma TRAIL was derived from cells originating in the bone marrow. Collectively, these findings indicate that BM cells are a protective source of TRAIL and that TRAIL+ monocyte/macrophages markedly reduce atherosclerosis.
Figure 2.

BM-TRAIL Attenuates Atherosclerosis
(A) BMT study design.
(B) Atherosclerosis is reduced in BM-TRAIL vs. null- or parenchymal-TRAIL treatment groups. Left, representative H&E brachiocephalic arteries. Right, quantification of plaque area (n = 5–8/group; scale bar, 20 μm).
(C) Progression of atherosclerosis in Trail−/−Apoe−/− mice in response to an HFD over 12 weeks. Left, representative H&E brachiocephalic arteries. Right, quantification of plaque area (n = 3–4/group).
(D) Atherosclerosis was reduced in HFD Trail−/−Apoe−/− mice treated with clodronate liposomes. Left, representative H&E brachiocephalic arteries. Right, quantification of plaque area (n = 5–7/group; scale bar, 20 μm).
(E) BM-TRAIL reduces Mac3+ staining in plaque. Left, Mac3+ staining. Right, quantification of staining (n = 5–8/group; scale bar, 20 μm).
(F) Aortic TRAIL mRNA expression in BMT mice by qPCR (n = 5–8/group).
Results are mean ± SEM. One-way ANOVA or Mann-Whitney U-test; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Table 1.
Body Weight and Plasma Chemistries from Bone Marrow Chimera Mice
| Null | BM | Parenchymal | |
|---|---|---|---|
| Body weight (g) | 28.38 ± 1.9 | 27.20 ± 0.8 | 28.17 ± 0.7 |
| Glucose (mmol/L) | 7.58 ± 0.8 | 7.16 ± 0.6 | 8.98 ± 0.3 |
| Insulin (pmol/L) | 44.87 ± 3.2 | 37.73 ± 2.1 | 40.75 ± 2.8 |
| Cholesterol (mmol/L) | 18.04 ± 3.3 | 19.28 ± 2.4 | 21.95 ± 5.9 |
| Triglycerides (mmol/L) | 2.29 ± 0.3 | 1.99 ± 0.3 | 1.96 ± 0.5 |
| TRAIL (pg/mL) | 0 | 810.7 ± 108.2 | 1966 ± 503.5* |
Body weight and plasma chemistries from null-, BM, and parenchymal-TRAIL mice at euthanasia (n = 5–8/group). Results are mean ± SEM. One-way ANOVA; *p < 0.05 vs. BM.
To confirm that TRAIL+ monocyte/macrophages could also reduce inflammation and macrophage numbers in other tissues, we examined pancreata from bone marrow chimera mice. There was ∼50% less Mac3+ immunoreactivity in pancreata from BM-TRAIL mice compared with that in null-TRAIL pancreatic islets (Figure S1A). Macrophage accumulation was similarly reduced in parenchymal-TRAIL islets (Figure S1A). These indicate that TRAIL expression reduced monocyte/macrophage content regardless of its source. However, although reconstitution with TRAIL+ monocyte/macrophages reduced atherosclerosis in BM-TRAIL mice, this did not affect glucose handling. Glucose tolerance testing indicated that parenchymal-TRAIL mice had improved glucose clearance compared with null- or BM-TRAIL mice (Figure S1B) associating with significantly reduced active caspase-3 staining (Figure S1C). Active caspase-3 immunoreactivity was also reduced in BM-TRAIL islets, although this did not reach significance (Figure S1C). No differences in insulin tolerance, insulin expression, islet area, or islet number between groups were observed (Figures S1D–S1G). These findings suggest that TRAIL+ monocyte/macrophages appear to have functions distinct from TRAIL+ parenchymal cells, highlighting tissue- and cell-specific responses.
Macrophages Lacking TRAIL Have Impaired Cholesterol Metabolism
Atherosclerosis associates with expanded hematopoietic stem cell numbers (Murphy et al., 2011). Because HFD Trail−/−Apoe−/− mice develop larger plaques than Apoe−/− (Di Bartolo et al., 2011), and TRAIL+ monocyte/macrophage reconstitution attenuated atherosclerosis with concomitantly less macrophage accumulation in vascular tissues (Figure 2), we assessed whether inherent differences in hematopoietic stem cell numbers were associated with TRAIL deletion. No differences in hematopoietic stem cells, common myeloid, lymphoid, erythroid or granulocyte progenitors, or monocyte precursors in bone marrow from Apoe−/− (TRAIL+ve) vs. Trail−/−Apoe−/− (TRAIL−ve) mice were apparent (Figure S2), suggesting that the changes we observed in Figure 2 occurred once the cells had left the bone marrow and differentiated.
Cholesterol accumulation in macrophages is a defining feature of atherosclerosis (Jessup and Kritharides, 2008). Interestingly, TRAIL+ve macrophages loaded with acetlyated LDL (acLDL) had significantly reduced TRAIL mRNA expression (Figure 3A). Macrophage cholesterol homeostasis is maintained by uptake and export; cholesterol uptake can occur via scavenger receptors CD36 and SRA, and by phagocytosis of aggregated LDL, whereas cholesterol export occurs primarily via reverse cholesterol transporters ABCA1 and ABCG1 (Du et al., 2015, Out et al., 2008). We found that TRAIL−ve (Trail−/−Apoe−/−) macrophages had no change in CD36 or SRA mRNA (Figure 3B) or phagocytic ability (Figure 3C). On the other hand, ABCA1 and ABCG1 mRNA expression was significantly reduced (Figure 3D), and TRAIL−ve macrophages had a diminished capacity to export cholesterol compared to TRAIL+ve macrophages, as evidenced by reduced cholesterol efflux to Apo-A1 (Figure 3E). These findings indicate that TRAIL deletion in macrophages impairs reverse cholesterol transport, making macrophages more lipid laden.
Figure 3.

TRAIL Deletion Alters Cholesterol Metabolism in Macrophages
(A) TRAIL expression in TRAIL+ve macrophages in response to acLDL for 48 h followed by 16 h equilibration.
(B–D) (B) Scavenger receptor mRNA expression, (C) phagocytosis of beads, and (D) mRNA expression of cholesterol transporters ABCA1, ABCG1, and SRB1 in basal macrophages.
(E) Cholesterol efflux is impaired with TRAIL deletion, where cells were 3H-acLDL loaded for 48 h, followed by 16 h equilibration and efflux to ApoA1 for 4 h (n = 3/group).
Results are mean ± SEM. t-test; **p < 0.01 and ***p < 0.001.
Macrophages Lacking TRAIL Exacerbate Inflammation and Impair Efferocytosis
Activated monocyte/macrophages express and secrete inflammatory cytokines. The mechanisms by which TRAIL may modulate inflammation in the vessel wall were analyzed, and the expression of inflammatory cytokines involved in the progression of atherosclerosis was measured. Consistent with accelerated atherosclerosis in mice with TRAIL deletion (Figures 2B and 2C), TRAIL−ve macrophages had >2-fold higher CCL-2 mRNA (Figure 4A), a potent monocyte chemoattractant implicated in atherosclerosis progression (Aiello et al., 1999, Gu et al., 1998). TRAIL−ve macrophages also had a more reactive, inflammatory phenotype following lipopolysaccharide (LPS) challenge, with significantly higher TNF-α, IL-1β, and IL-6 mRNA expression than TRAIL+ve macrophages (Figure 4B). Correspondingly, anti-inflammatory IL-10 mRNA was significantly reduced with TRAIL deletion (Figure 4B). Similarly, acLDL-loaded TRAIL−ve macrophages expressed more IL-1β and IL-6 but not TNFα (Figure 4C). CD11b expression is reduced on macrophages involved in the resolution of inflammation (Ortega-Gomez et al., 2013). We showed that CD11b expression on TRAIL−ve macrophages was elevated, both at the level of mRNA and protein (Figures 4C and 4D). Collectively, these findings imply that TRAIL deletion promotes an inflammatory macrophage.
Figure 4.

TRAIL Deletion Alters Macrophage Phenotype and Function
(A–C) (A) CCL-2 expression in basal macrophages. Inflammatory cytokine expression in macrophages in response to (B) LPS (20 ng/mL) challenge for 24 h or (C) acLDL loading for 48 h followed by 16 h equilibration.
(D–G) (D) CD11b mRNA expression in basal macrophages and (E) flow cytometry analysis of CD11b+F4/80+ macrophages. Macrophage (F) cell counts after 7 days differentiation and (G) apoptosis determined by Annexin V staining after 7, 14, and 21 days differentiation.
(H and I) (H) MerTK mRNA expression and (I) efferocytosis in macrophages. Left, quantification. Right, representative images where macrophages are stained blue, and apoptotic thymocytes are stained in green.
(J) Migration of macrophages to CCL-19, in a Transwell assay. n = 3/group. Results are mean ± SEM. t-test; *p < 0.05.
Apoptosis is triggered by inflammation in atherosclerotic lesions, and macrophage apoptosis under these circumstances can be detrimental if not coupled with effective efferocytosis (Kavurma et al., 2017). We next examined cell viability, apoptosis, and expression of genes important in efferocytosis. Viability of TRAIL+ve and TRAIL−ve macrophages was similar (Figure 4F), and levels of apoptosis were low, with no difference over 21 days in culture (Figure 4G). In contrast, MerTK mRNA was ∼50% less in TRAIL−ve macrophages (Figure 4H), associating with a significant reduction in efferocytosis (Figure 4I). Of note, MerTK is a transmembrane protein, mutation of which disrupts signaling, resulting in dysfunctional efferocytosis in Apoe−/− mice (Thorp et al., 2008). TRAIL−ve macrophages also had significantly reduced migration to CCL-19 (Figure 4J), a chemokine implicated in macrophage egress and plaque regression (Feig, 2014). These findings suggest that macrophages lacking TRAIL may be defective in removing dead/dying cells and modulating tissue repair.
TRAIL Deletion Effects the Nitric Oxide Synthase Pathway
The nitric oxide (NO) pathway is critical for maintenance of cardiovascular homeostasis and contributes to maintaining an anti-thrombotic, anti-inflammatory, and anti-atherogenic state within the vessel wall. Macrophages produce and respond to NO, and the inducible nitric oxide synthase (iNOS) pathway catalyzes production of NO and citrulline from L-arginine. We focused on this because we reported that physiologically relevant concentrations of TRAIL stimulate phosphorylation of endothelial-derived NOS (eNOS) and increased intracellular NO concentrations in human endothelial cells, promoting their proliferation, migration, and differentiation (Cartland et al., 2016, Di Bartolo et al., 2015). Plasma nitrate/nitrite levels are an indicator of NO production in vivo. These were reduced in samples from HFD Trail−/−Apoe−/− mice (Figure 5A). Furthermore, arginine and citrulline levels were significantly less in Trail−/− plasma, although levels of the endogenous NOS inhibitor, asymmetric dimethylarginine, remained unchanged (Table 2). Plasma from BM-TRAIL mice also had significantly less citrulline compared with null- or parenchymal-TRAIL (Figure 5B), whereas plasma arginine levels were similar between groups (data not shown).
Figure 5.

TRAIL Deletion Impairs NO Signaling
(A) Nitrate/nitrite in plasma from 12-week HFD Apoe−/− and Trail−/−Apoe−/− mice (n = 7–8/group).
(B) Citrulline levels in plasma from BMT mice. Metabolite peaks were quantified as area under the curve (n = 5–8/group).
(C and D) (C) Hierarchical clustering of 845 differentially expressed genes showing opposing gene expression patterns and (D) gene ontology (GO) enrichment analysis. Analyses were performed as described in the Methods.
(E–J) (E) iNOS mRNA expression in TRAIL+ve vs. TRAIL−ve macrophages, (F) after 20 ng/mL LPS stimulation for 24 h or (G) after acLDL loading for 48 h followed by 16 h equilibration (n = 3). iNOS mRNA expression in aortic tissues from (H) 6-week wildtype vs. Trail−/− mice (6–7/group), (I) 12-week HFD Apoe−/− vs. Trail−/−Apoe−/−, and (J) from null-, BM- and parenchymal-TRAIL mice (n = 5–6/group). mRNA expression was normalized to β-actin.
Results are mean ± SEM. One-way ANOVA, t-test or Mann-Whitney U-test; *p < 0.05, **p < 0.01, ***p < 0.001, and p < 0.0001.
Table 2.
Metabolomic Changes in 6-Week-Old Trail−/− vs. Wildtype Plasma
| Metabolite | Wildtype | Trail−/− |
|---|---|---|
| ADMA | 0.4932 ± 0.1219 | 0.3556 ± 0.08856 |
| Arginine | 0.7568 ± .09599 | 0.5087 ± 0.04466** |
| Citrulline | 0.1864 ± 0.0275 | 0.1483 ± 0.006803* |
| Cysteine | 2.097 ± 0.1294 | 1.164 ± 0.1087** |
| Methionine | 0.8381 ± .04502 | 0.7719 ± 0.1206 |
ADMA, asymmetric dimethylarginine.
Metabolite peaks quantified as area under the curve. Results are mean ± SD (n = 6); *p < 0.05, **p < 0.01.
We also performed transcriptome analysis of aortic tissues from Trail−/− and wildtype mice and found 845 genes were differentially expressed. Hierarchical clustering of these demonstrated opposing gene expression patterns, and gene ontology analysis indicated enrichment for biological processes (Figures 5C and 5D). Interestingly, cysteine/methionine metabolism was identified as a top pathway altered by TRAIL deletion (enrichment score = 3.57; p = 0.028), and cysteine levels in plasma from these mice were ∼45% less than in wildtype mice, whereas methionine concentrations did not change (Table 2). These findings are significant because cysteine is important for NOS function (Forstermann and Sessa, 2012).
iNOS is expressed by macrophages, and TRAIL−ve macrophages had significantly less iNOS expression constitutively, following LPS or acLDL stimulation (Figures 5E–5G). iNOS mRNA was ∼40% less in aortic tissue from Trail−/− compared with wildtype mice (Figure 5H), and was reduced by ∼90% in samples from HFD Trail−/−Apoe−/− mice (Figure 5I). Importantly, iNOS mRNA in aortae from reconstituted BM-TRAIL mice was restored when compared with null- and parenchymal-TRAIL samples (Figure 5J). Thus, TRAIL appears integral to iNOS expression in macrophages and iNOShiTRAIL+ macrophages may help to maintain levels of arginine and cysteine necessary to attenuate atherosclerosis.
IL-18 Negatively Regulates TRAIL Expression in Monocytes
We have identified an iNOShiTRAIL+ macrophage population that appears critical in moderating atherosclerosis. Mechanisms whereby TRAIL is reduced in monocytes from people with CAD are unknown. We previously showed that patients with CAD had elevated plasma IL-18, IL-1β, and IL-6 concentrations (Martinez et al., 2015). Here, a significant inverse correlation between plasma TRAIL and IL-18 was observed in these patient samples (R2 = 0.2344, p = 0.0105), but not between TRAIL and IL-1β, or IL-6 (Figure 6A), suggesting an IL-18-TRAIL axis. In fact, IL-18 mRNA was significantly increased in aortic tissues from atherosclerotic Trail−/−Apoe−/− mice (Figure 6B). To confirm a direct relationship, human monocytes were incubated with recombinant IL-18 and TRAIL mRNA was measured. IL-18 significantly repressed TRAIL mRNA expression by ∼60%, without affecting TNF-α (Figures 6C and 6D). We examined the mechanism of this repression further and measured the direct effect of IL-18 on NF-κB p65 and Sp1 expression, two transcription factors that we showed control TRAIL transcriptional activity in vascular cells by combinatorial interaction (Chan et al., 2010). IL-18 reduced NF-κB p65 mRNA expression in human monocytes by ∼50%, without affecting Sp1 levels (Figures 6E and 6F). IL-18 also reduced NF-κB p65 activity, because the phosphorylated form (p-NFκB p65) was significantly reduced, ∼50% (Figure 6G). NF-κB enrichment on the endogenous TRAIL promoter was significantly reduced in healthy monocytes exposed to IL-18, whereas Sp1 enrichment remained unaltered (Figure 6H). These findings indicate that IL-18 moderates TRAIL transcription by altering NF-κB-binding activity. This mechanism may account for the reduced systemic TRAIL levels observed in CAD.
Figure 6.

IL-18 Represses TRAIL Expression in Monocytes
(A) Spearman's correlation between plasma TRAIL and IL-18, IL-1β, and IL-6.
(B) Aortic IL-18 mRNA expression in 12-week HFD Apoe−/− and Trail−/−Apoe−/− (n = 5–7). (C–F) Monocytes were isolated from healthy human donors and treated with recombinant human IL-18 (30 ng/mL) for 24 h followed by assessment of (C) TRAIL, (D) TNF-α, (E) NF-κB p65, and (F) Sp1 mRNA expression measured by qPCR (n = 3).
(G) IL-18 (30 ng/mL) represses NF-κB p-p65 protein expression in human monocytes as assessed by western blotting, quantification on right (n = 3). (H) IL-18 (30 ng/mL) reduces NF-κB enrichment on the endogenous TRAIL promoter, but Sp1 enrichment remained unaltered.
Results are mean ± SEM. One-way ANOVA or t-test; *p < 0.05 and **p < 0.01.
Discussion
Despite aggressive risk factor management, many patients with CVD suffer heart attacks, highlighting the need for new biomarkers, prognostic factors, and therapies. Here, we have identified that monocytes from healthy human donors are a significant source of TRAIL, whereas monocytes from patients with CAD express significantly less TRAIL mRNA, concomitant with reduced plasma levels. In mice with severe disease, reconstitution of TRAIL+ monocyte/macrophages markedly attenuated atherosclerosis by reducing macrophage accumulation and increasing iNOS levels. Furthermore, we identified IL-18 as a negative regulator of TRAIL transcription in human monocytes. The recent identification of TRAIL SNPs associating with atherosclerosis (Pott et al., 2017) further underpins the biological significance of our findings. Circulating TRAIL levels, and/or TRAIL/IL-18 ratios, may predict an individual's risk of developing CVD. Our work also highlights the potential for targeting monocyte/macrophage-derived TRAIL in atherogenesis for the treatment of CVD.
Much of TRAIL biology has focused on its ability to kill cancer cells. However, low TRAIL concentrations positively associate with CVD in people (Schoppet et al., 2006), suggesting a role that goes beyond that of killing. Indeed, non-apoptotic functions, including stimulation of cell survival, proliferation, and migration, particularly in vascular cells (Azahri et al., 2012, Cartland et al., 2014, Cartland et al., 2017, Chan et al., 2010, Di Bartolo et al., 2015, Harith et al., 2015, Kavurma et al., 2008a, Secchiero et al., 2004a, Secchiero et al., 2004b), and even cancer cells, (Lancaster et al., 2003, Secchiero et al., 2005) have been described. However, in CVD, because TRAIL's cellular source is unknown, the reason why concentrations were reduced and the direct impact this could have on pathogenesis were unclear. Here we show that monocyte/macrophages represent a significant source of TRAIL in healthy individuals, which was suppressed in monocytes from patients with CAD. We identified IL-18 as a negative regulator of TRAIL expression in these cells. IL-18 is synthesized as an inactive precursor, expressed constitutively by macrophages and other cells within the vessel wall (Gerdes et al., 2002, Gracie et al., 2003). This has significant implications because circulating IL-18 levels are increased in patients with CAD (Martinez et al., 2015, Robertson et al., 2016) and IL-18 is linked to atherosclerosis in mice (Elhage et al., 2003, Whitman et al., 2002), with mature IL-18 secreted primarily from monocyte/macrophages following caspase-1 cleavage (Gerdes et al., 2002, Gracie et al., 2003). IL-18 can also modulate activity of multiple transcription factors, including NF-κB and Sp1 (Reddy et al., 2010), and critically, we showed that IL-18 influenced NF-κB binding ability on the endogenous TRAIL promoter, contributing to suppressed TRAIL transcription. These suggest a mechanism for reduced TRAIL levels observed in patients with CAD.
NO is a powerful vasodilator and plays important roles in inflammation and maintaining vessel tone (MacMicking et al., 1997). iNOS is generally expressed by proinflammatory macrophages in atherosclerosis (Moore et al., 2013). We found markedly less iNOS mRNA in Trail−/−Apoe−/− macrophages and in vascular tissue from Trail−/− and Trail−/−Apoe−/− mice, particularly from the latter that were fed a HFD and manifested a strong atherosclerotic phenotype. Reconstitution of Trail−/−Apoe−/− mice with bone marrow expressing TRAIL markedly increased aortic iNOS mRNA, and concentrations of its metabolite citrulline were significantly less than in null- or parenchymal-TRAIL expressing samples. Thus, TRAIL+ monocyte/macrophages may contribute to vascular protection by increasing iNOS gene expression, thereby facilitating NO production. Our findings, and those of Lu et al. (2015), challenge the concept of iNOS as a marker of inflammatory macrophages. For example, macrophages from iNOS−/− mice showed increased polarization to an inflammatory macrophage (Lu et al., 2015), and NO generated by iNOS reduced leukocyte-endothelium interactions (Hickey et al., 1997). IL-18 also modulates iNOS via NF-κB-dependent mechanisms (Jablonska et al., 2006); however, an interesting control mechanism worthy of consideration in this context is the prevention of IL-18 release from macrophages by NO-mediated suppression of caspase-1 (Kim et al., 2002). Taken together, our findings suggest that TRAIL regulates the iNOS pathway in macrophages, and it is tantalizing to speculate that TRAIL-induced iNOS attenuates their infiltration via NO to reduce atherogenesis. Notably, these may also be influenced by IL-18.
In chronic CVD, macrophages change from a resolving to a pro-inflammatory phenotype, propagating the inflammatory milieu of the plaque (reviewed in Kavurma et al., 2017). Here we propose TRAIL as a player in macrophage biology, protecting against atherosclerosis and macrophage accumulation, and provide mechanistic insights into monocyte/macrophage function(s) in CAD. We propose that the reduced TRAIL expression in monocyte/macrophages from patients with CAD may impair these processes, i.e., by promoting a more inflammatory monocyte/macrophage, increasing cellular cholesterol accumulation in lesions, aggravated by the inability of these cells to egress, or efferocytose, which collectively result in severe atherosclerosis. Interestingly, TRAIL+ monocyte/macrophage reconstitution also reduced inflammation in pancreata. Thus, impairment of macrophage processes as observed in atheroma may also be evident in pancreatic islets. Indeed, during islet inflammation, proinflammatory macrophages contribute to β-cell dysfunction in diabetes, yet resolving macrophages are essential for physiological β-cell proliferation and survival (Eguchi and Nagai, 2017). Whether TRAIL's pro-resolving function(s) in macrophages affect β-cell function is unclear. Interestingly, however, TRAIL+ parenchymal cells also inhibited MAC3+ cell accumulation to the same degree as reconstitution of TRAIL+ monocyte/macrophages. TRAIL-expressing parenchymal cells also improved whole-body glucose clearance in response to a glucose tolerance test. These findings suggest two things: that when monocyte/macrophage accumulation is the driver of disease, as in atherosclerosis, TRAIL+ monocyte macrophages are critical in reducing inflammation and disease. However, when monocyte/macrophage accumulation may not be the primary driver of disease, such as in diabetes, TRAIL-expressing parenchymal cells play a more prominent role in protection, suggesting a tissue/cell-specific role. These findings are significant as they provide important information on which tissue/cells to target in the treatment of atherosclerosis vs. metabolic diseases dependent on TRAIL signaling.
In summary, we have identified a unique population of iNOShiTRAIL+ monocyte/macrophages, and these appear to be potent suppressors of inflammation and atherosclerosis. Furthermore, we suggest TRAIL is a key modulator of macrophage phenotype and function. Our findings are important because TRAIL expression is low in monocytes from people with CAD, concomitant with high IL-18 and low TRAIL concentrations in plasma. These studies highlight an exciting immunotherapeutic potential in the treatment of atherosclerosis. Results may spawn wider interest in the role of this macrophage phenotype in chronic inflammatory conditions.
Limitations of the Study
Atherosclerosis is a multifactorial disease involving other immune cells or their interactions. The study design did not investigate the effect of TRAIL on other bone-marrow-originating cells, including neutrophils, T cells, and B cells, implicated in plaque progression or their interaction with monocyte/macrophages. Furthermore, our investigations into the cause of TRAIL suppression focused only on IL-18, yet owing to the complex nature of atherosclerosis, other factors are also likely to contribute.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to thank Professor Paul Pilowsky for reading the manuscript. This work was supported by a University of Sydney Early Career Research Grant (S.P.C.), a University of Sydney Sydney Medical School Foundation Fellowship (J.F.O.) a Heart Foundation of Australia Career Development Award (CR 12S 6833; M.M.K.) and a Perpetual IMPACT Philanthropy Grant (IPAP2018/1674; M.M.K. and S.P.C.).
Author Contributions
S.P.C., S.P., and M.M.K. were responsible for experimentation, conception, design, analysis and interpretation of data, manuscript preparation, and intellectual input. S.W.G., R.C.Y.L., and A.J.M. were responsible for experimentation, analysis and interpretation of data, manuscript preparation, and intellectual input. G.J.M., S.R., Y.C.K., and P.M.C. were responsible for experimentation, data analysis and interpretation, and manuscript preparation. M.K., J.F.O., M.A.K., M.R.B., W.J., L.K., S.M., and C.G. were responsible for reagents, interpretation of data, intellectual input, and manuscript preparation. All authors gave final approval for publication.
Declaration of Interests
Trail−/− mice were originally sourced from Amgen.
Published: February 22, 2019
Footnotes
Supplemental Information includes Transparent Methods, two figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.12.037.
Supplemental Information
References
- Aiello R.J., Bourassa P.A., Lindsey S., Weng W., Natoli E., Rollins B.J., Milos P.M. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 1999;19:1518–1525. doi: 10.1161/01.atv.19.6.1518. [DOI] [PubMed] [Google Scholar]
- Azahri N.S., Di Bartolo B.A., Khachigian L.M., Kavurma M.M. Sp1, acetylated histone-3 and p300 regulate TRAIL transcription: mechanisms for PDGF-BB-mediated VSMC proliferation and migration. J. Cell. Biochem. 2012;13:2597–2606. doi: 10.1002/jcb.24135. [DOI] [PubMed] [Google Scholar]
- Cartland S.P., Erlich J.H., Kavurma M.M. TRAIL deficiency contributes to diabetic nephropathy in fat-fed ApoE-/- mice. PLoS One. 2014;9:e92952. doi: 10.1371/journal.pone.0092952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartland S.P., Genner S.W., Zahoor A., Kavurma M.M. Comparative evaluation of TRAIL, FGF-2 and VEGF-A-induced angiogenesis in vitro and in vivo. Int. J. Mol. Sci. 2016;17 doi: 10.3390/ijms17122025. e2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartland S.P., Harith H.H., Genner S.W., Dang L., Cogger V.C., Vellozzi M., Di Bartolo B.A., Thomas S.R., Adams L.A., Kavurma M.M. Non-alcoholic fatty liver disease, vascular inflammation and insulin resistance are exacerbated by TRAIL deletion in mice. Sci. Rep. 2017;7:1898. doi: 10.1038/s41598-017-01721-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J., Prado-Lourenco L., Khachigian L.M., Bennett M.R., Di Bartolo B.A., Kavurma M.M. TRAIL promotes VSMC proliferation and neointima formation in a FGF-2-, Sp1 phosphorylation-, and NFkappaB-dependent manner. Circ. Res. 2010;106:1061–1071. doi: 10.1161/CIRCRESAHA.109.206029. [DOI] [PubMed] [Google Scholar]
- Di Bartolo B.A., Cartland S.P., Harith H.H., Bobryshev Y.V., Schoppet M., Kavurma M.M. TRAIL-deficiency accelerates vascular calcification in atherosclerosis via modulation of RANKL. PLoS One. 2013;8:e74211. doi: 10.1371/journal.pone.0074211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bartolo B.A., Cartland S.P., Prado-Lourenco L., Griffith T.S., Gentile C., Ravindran J., Azahri N.S., Thai T., Yeung A.W., Thomas S.R. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) promotes angiogenesis and ischemia-induced neovascularization via NADPH oxidase 4 (NOX4) and nitric oxide-dependent mechanisms. J. Am. Heart Assoc. 2015;4 doi: 10.1161/JAHA.115.002527. e002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bartolo B.A., Chan J., Bennett M.R., Cartland S., Bao S., Tuch B.E., Kavurma M.M. TNF-related apoptosis-inducing ligand (TRAIL) protects against diabetes and atherosclerosis in Apoe ( -/- ) mice. Diabetologia. 2011;54:3157–3167. doi: 10.1007/s00125-011-2308-0. [DOI] [PubMed] [Google Scholar]
- Du X.M., Kim M.J., Hou L., Le Goff W., Chapman M.J., Van Eck M., Curtiss L.K., Burnett J.R., Cartland S.P., Quinn C.M. HDL particle size is a critical determinant of ABCA1-mediated macrophage cellular cholesterol export. Circ. Res. 2015;116:1133–1142. doi: 10.1161/CIRCRESAHA.116.305485. [DOI] [PubMed] [Google Scholar]
- Eguchi K., Nagai R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Invest. 2017;127:14–23. doi: 10.1172/JCI88877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhage R., Jawien J., Rudling M., Ljunggren H.G., Takeda K., Akira S., Bayard F., Hansson G.K. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc. Res. 2003;59:234–240. doi: 10.1016/s0008-6363(03)00343-2. [DOI] [PubMed] [Google Scholar]
- Feig J.E. Regression of atherosclerosis: insights from animal and clinical studies. Ann. Glob. Health. 2014;80:13–23. doi: 10.1016/j.aogh.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstermann U., Sessa W.C. Nitric oxide synthases: regulation and function. Eur. Heart J. 2012;33:829–837. doi: 10.1093/eurheartj/ehr304. 837a-837d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes N., Sukhova G.K., Libby P., Reynolds R.S., Young J.L., Schonbeck U. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J. Exp. Med. 2002;195:245–257. doi: 10.1084/jem.20011022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracie J.A., Robertson S.E., McInnes I.B. Interleukin-18. J. Leukoc. Biol. 2003;73:213–224. doi: 10.1189/jlb.0602313. [DOI] [PubMed] [Google Scholar]
- Gu L., Okada Y., Clinton S.K., Gerard C., Sukhova G.K., Libby P., Rollins B.J. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell. 1998;2:275–281. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- Harith H.H., Di Bartolo B.A., Cartland S.P., Genner S., Kavurma M.M. Insulin promotes VSMC proliferation and apoptosis via differential regulation of TNF-related apoptosis inducing ligand. J. Diabetes. 2015;8:568–578. doi: 10.1111/1753-0407.12339. [DOI] [PubMed] [Google Scholar]
- Harith H.H., Morris M.J., Kavurma M.M. On the TRAIL of obesity and diabetes. Trends Endocrinol. Metab. 2013;24:578–587. doi: 10.1016/j.tem.2013.07.001. [DOI] [PubMed] [Google Scholar]
- Hickey M.J., Sharkey K.A., Sihota E.G., Reinhardt P.H., Macmicking J.D., Nathan C., Kubes P. Inducible nitric oxide synthase-deficient mice have enhanced leukocyte-endothelium interactions in endotoxemia. FASEB J. 1997;11:955–964. doi: 10.1096/fasebj.11.12.9337148. [DOI] [PubMed] [Google Scholar]
- Jablonska E., Puzewska W., Charkiewicz M. Effect of IL-18 on leukocyte expression of iNOS and phospho-IkB in patients with squamous cell carcinoma of the oral cavity. Neoplasma. 2006;53:200–205. [PubMed] [Google Scholar]
- Jessup W., Kritharides L. Lipid metabolism: recent progress in defining the contributions of cholesterol transporters to cholesterol efflux in vitro and in vivo. Curr. Opin. Lipidol. 2008;19:212–214. doi: 10.1097/MOL.0b013e3282f6a476. [DOI] [PubMed] [Google Scholar]
- Kavurma M.M., Bennett M.R. Expression, regulation and function of trail in atherosclerosis. Biochem. Pharmacol. 2008;75:1441–1450. doi: 10.1016/j.bcp.2007.10.020. [DOI] [PubMed] [Google Scholar]
- Kavurma M.M., Rayner K.J., Karunakaran D. The walking dead: macrophage inflammation and death in atherosclerosis. Curr. Opin. Lipidol. 2017;28:91–98. doi: 10.1097/MOL.0000000000000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavurma M.M., Schoppet M., Bobryshev Y.V., Khachigian L.M., Bennett M.R. Trail stimulates proliferation of vascular smooth muscle cells via activation of NF-kappa B and induction of insulin-like growth factor-1 receptor. J. Biol. Chem. 2008;283:7754–7762. doi: 10.1074/jbc.M706927200. [DOI] [PubMed] [Google Scholar]
- Kavurma M.M., Tan N.Y., Bennett M.R. Death receptors and their ligands in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008;28:1694–1702. doi: 10.1161/ATVBAHA.107.155143. [DOI] [PubMed] [Google Scholar]
- Kim P.K., Kwon Y.G., Chung H.T., Kim Y.M. Regulation of caspases by nitric oxide. Ann. N. Y. Acad. Sci. 2002;962:42–52. doi: 10.1111/j.1749-6632.2002.tb04054.x. [DOI] [PubMed] [Google Scholar]
- Lancaster J.M., Sayer R., Blanchette C., Calingaert B., Whitaker R., Schildkraut J., Marks J., Berchuck A. High expression of tumor necrosis factor-related apoptosis-inducing ligand is associated with favorable ovarian cancer survival. Clin. Cancer Res. 2003;9:762–766. [PubMed] [Google Scholar]
- Lu G., Zhang R., Geng S., Peng L., Jayaraman P., Chen C., Xu F., Yang J., Li Q., Zheng H. Myeloid cell-derived inducible nitric oxide synthase suppresses M1 macrophage polarization. Nat. Commun. 2015;6:6676. doi: 10.1038/ncomms7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking J., Xie Q.W., Nathan C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- Manuneedhi Cholan P., Cartland S.P., Kavurma M.M. NADPH oxidases, angiogenesis, and peripheral artery disease. Antioxidants (Basel) 2017;6 doi: 10.3390/antiox6030056. e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez G.J., Robertson S., Barraclough J., Xia Q., Mallat Z., Bursill C., Celermajer D.S., Patel S. Colchicine acutely suppresses local cardiac production of inflammatory cytokines in patients with an acute coronary syndrome. J. Am. Heart Assoc. 2015;4:e002128. doi: 10.1161/JAHA.115.002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meir K.S., Leitersdorf E. Atherosclerosis in the apolipoprotein-E-deficient mouse: a decade of progress. Arterioscler. Thromb. Vasc. Biol. 2004;24:1006–1014. doi: 10.1161/01.ATV.0000128849.12617.f4. [DOI] [PubMed] [Google Scholar]
- Moore K.J., Sheedy F.J., Fisher E.A. Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy A.J., Akhtari M., Tolani S., Pagler T., Bijl N., Kuo C.L., Wang M., Sanson M., Abramowicz S., Welch C. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Invest. 2011;121:4138–4149. doi: 10.1172/JCI57559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Gomez A., Perretti M., Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol. Med. 2013;5:661–674. doi: 10.1002/emmm.201202382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Out R., Jessup W., Le Goff W., Hoekstra M., Gelissen I.C., Zhao Y., Kritharides L., Chimini G., Kuiper J., Chapman M.J. Coexistence of foam cells and hypocholesterolemia in mice lacking the ABC transporters A1 and G1. Circ. Res. 2008;102:113–120. doi: 10.1161/CIRCRESAHA.107.161711. [DOI] [PubMed] [Google Scholar]
- Pott J., Burkhardt R., Beutner F., Horn K., Teren A., Kirsten H., Holdt L.M., Schuler G., Teupser D., Loeffler M. Genome-wide meta-analysis identifies novel loci of plaque burden in carotid artery. Atherosclerosis. 2017;259:32–40. doi: 10.1016/j.atherosclerosis.2017.02.018. [DOI] [PubMed] [Google Scholar]
- Reddy V.S., Prabhu S.D., Mummidi S., Valente A.J., Venkatesan B., Shanmugam P., Delafontaine P., Chandrasekar B. Interleukin-18 induces EMMPRIN expression in primary cardiomyocytes via JNK/Sp1 signaling and MMP-9 in part via EMMPRIN and through AP-1 and NF-kappaB activation. Am. J. Physiol. Heart Circ. Physiol. 2010;299:H1242–H1254. doi: 10.1152/ajpheart.00451.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson S., Martinez G.J., Payet C.A., Barraclough J.Y., Celermajer D.S., Bursill C., Patel S. Colchicine therapy in acute coronary syndrome patients acts on caspase-1 to suppress NLRP3 inflammasome monocyte activation. Clin. Sci. (Lond.) 2016;130:1237–1246. doi: 10.1042/CS20160090. [DOI] [PubMed] [Google Scholar]
- Schoppet M., Sattler A.M., Schaefer J.R., Hofbauer L.C. Osteoprotegerin (OPG) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) levels in atherosclerosis. Atherosclerosis. 2006;184:446–447. doi: 10.1016/j.atherosclerosis.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Secchiero P., Candido R., Corallini F., Zacchigna S., Toffoli B., Rimondi E., Fabris B., Giacca M., Zauli G. Systemic tumor necrosis factor-related apoptosis-inducing ligand delivery shows antiatherosclerotic activity in apolipoprotein E-null diabetic mice. Circulation. 2006;114:1522–1530. doi: 10.1161/CIRCULATIONAHA.106.643841. [DOI] [PubMed] [Google Scholar]
- Secchiero P., Gonelli A., Carnevale E., Corallini F., Rizzardi C., Zacchigna S., Melato M., Zauli G. Evidence for a proangiogenic activity of TNF-related apoptosis-inducing ligand. Neoplasia. 2004;6:364–373. doi: 10.1593/neo.03421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secchiero P., Tiribelli M., Barbarotto E., Celeghini C., Michelutti A., Masolini P., Fanin R., Zauli G. Aberrant expression of TRAIL in B chronic lymphocytic leukemia (B-CLL) cells. J. Cell Physiol. 2005;205:246–252. doi: 10.1002/jcp.20392. [DOI] [PubMed] [Google Scholar]
- Secchiero P., Zerbinati C., Rimondi E., Corallini F., Milani D., Grill V., Forti G., Capitani S., Zauli G. TRAIL promotes the survival, migration and proliferation of vascular smooth muscle cells. Cell Mol. Life Sci. 2004;61:1965–1974. doi: 10.1007/s00018-004-4197-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorp E., Cui D., Schrijvers D.M., Kuriakose G., Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe-/- mice. Arterioscler. Thromb. Vasc. Biol. 2008;28:1421–1428. doi: 10.1161/ATVBAHA.108.167197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpato S., Ferrucci L., Secchiero P., Corallini F., Zuliani G., Fellin R., Guralnik J.M., Bandinelli S., Zauli G. Association of tumor necrosis factor-related apoptosis-inducing ligand with total and cardiovascular mortality in older adults. Atherosclerosis. 2011;215:452–458. doi: 10.1016/j.atherosclerosis.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman S.C., Ravisankar P., Daugherty A. Interleukin-18 enhances atherosclerosis in apolipoprotein E(-/-) mice through release of interferon-gamma. Circ. Res. 2002;90:E34–E38. doi: 10.1161/hh0202.105292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.