

Summary
The DNA damage response (DDR) protects cells against genomic instability. Surprisingly, little is known about the differences in DDR across tissues, which may affect cancer evolutionary trajectories and chemotherapy response. Using mathematical modeling and quantitative experiments, we found that the DDR is regulated differently in human breast and lung primary cells. Equal levels of cisplatin-DNA lesions caused stronger Chk1 activation in lung cells, leading to resistance. In contrast, breast cells were more resistant and showed more Chk2 activation in response to doxorubicin. Further analyses indicate that Chk1 activity played a regulatory role in p53 phosphorylation, whereas Chk2 activity was essential for p53 activation and p21 expression. We propose a novel “friction model,” in which the balance of p53 and p21 levels contributes to the apoptotic response in different tissues. Our results suggest that modulating the balance of p53 and p21 dynamics could optimize the response to chemotherapy.
Subject Areas: Bioinformatics, Mathematical Biosciences, Systems Biology, Cancer Systems Biology
Graphical Abstract
Highlights
-
•
Breast and lung cells show different sensitivities to chemotherapeutic drugs
-
•
Lung cells activate Chk1 more strongly than breast cells with chemotherapeutic drugs
-
•
Active Chk1 plays a regulatory role in p53 activation and apoptosis responses
-
•
The balance of p53 and p21 dynamics drives the apoptosis response to DNA damage
Bioinformatics; Mathematical Biosciences; Systems Biology; Cancer Systems Biology
Introduction
The DNA damage response (DDR) is an intricate signaling network that governs genomic integrity and protects against carcinogenesis. The core DDR network is formed by ATR and Chk1, ATM and Chk2, p53, and p21. ATR/Chk1 signaling is activated mostly by stresses involving single-strand DNA damage, whereas ATM and Chk2 are activated in response to double-strand breaks (DSBs) (Reinhardt and Schumacher, 2012, Smith et al., 2010, Weber and Ryan, 2015). Both ATR/Chk1 and ATM/Chk2 can activate p53, a master regulator of the DDR, which regulates cellular responses such as cell cycle arrests, repair and survival, or cell death. An important determinant of outcome is the p53 target protein p21, which can inhibit cell cycle progression and negatively regulate p53-mediated apoptosis (Abbas and Dutta, 2009).
Proper regulation of the DDR is important, because dysregulation is associated with aging and cancer (Hoeijmakers, 2009). Tumors frequently contain mutations in DDR genes and are characterized by an aberrant response to DNA damage. Strikingly, cancers originating in distinct tissues have diverse mutational signatures (Alexandrov et al., 2013, Kandoth et al., 2013, Zehir et al., 2017), suggesting that they have different DNA repair defects (Chae et al., 2016, Forestier et al., 2012, Garner and Eastman, 2011). Differences in DDR are also thought to result in specific sensitivities for DNA-damaging chemotherapy (Chae et al., 2016, Forestier et al., 2012, Liu et al., 2017, Sousa et al., 2015, Stewart-Ornstein and Lahav, 2017), which differ per cancer type. For instance, non-small-cell lung cancer responds relatively well to the DNA adduct and strand cross-linking agent cisplatin (Eastman, 1987, Gettinger and Lynch, 2011; Non-small Cell Lung Cancer Collaborative Group, 1995, Laskin and Sandler, 2005, Paoletti et al., 2011, Rosenberg et al., 1969). In contrast, the DNA-DSB-inducing drug doxorubicin is more effective in treating breast cancer (Arcamone et al., 1969, Crown, 1998, von Minckwitz, 2007).
It is unclear whether the differences in DDR in cancer types arise during tumor evolution, or whether they reflect variations present in the original tissue. It is conceivable that healthy cells have differences in the DDR network. For instance, skin cells exposed to sunlight might express a higher amount of genes involved in the repair of UV-induced DNA lesions than other tissues. In addition, mutations in DDR genes affect cancer development across tissues differently. For example, germline mutations in BRCA1/2 and Chk2 enhance cancer risk in specific tissues such as breast, whereas the added risk for tumors occurring in other tissues like lung is small (Levy-Lahad and Friedman, 2007, Michailidou et al., 2017, Nevanlinna and Bartek, 2006). Furthermore, the response to DNA damage is different across stem cell types. In particular intestinal stem cells are prone to induce apoptosis after DNA damage, whereas DNA damage in melanocytes results in terminal differentiation (Blanpain et al., 2011, Vitale et al., 2017). A systematic comparison of the DDR across tissues is lacking. Understanding what differences in DDR network arise during tumor evolution and which ones are already present will provide more insight into tissue-specific cancer risk. In addition, this knowledge could lead to more effective targeting of tumor cells for therapy.
To address how the DDR is regulated across tissues, we used non-immortalized primary epithelial cells from multiple breast and lung donors. We focused on breast and lung cells for three reasons: (1) breast and lung tissues frequently give rise to tumors; (2) certain DDR gene mutations predispose for breast cancer, but not for lung cancer; and (3) breast and lung tumors have different sensitivities for genotoxic chemotherapy. By combining modeling and experimental approaches, we show that these cell types have different apoptotic sensitivities for genotoxic chemotherapy. We demonstrate that the difference in sensitivity originates at the earliest level of DNA damage detection: in the tissue-specific regulation of the ATR-Chk1 and ATM-Chk2 network. Our modeling and experimental analyses suggest that differential activation of Chk1 and Chk2 regulates the balance of p53 and p21 dynamics and contributes to chemotherapy responses to DNA damage in breast and lung cells. This study sheds light on the tissue-specific DDR and provides clues on how to optimize chemotherapy response.
Results
An Equal Amount of Cisplatin-DNA Lesions Induces More Apoptosis in Primary Breast Than Lung Cells
The DDR pathway is a complex signaling network, which is activated upon many forms of damage and gives rise to multiple different outputs. We have focused on one DNA-damaging agent and one cellular outcome (apoptosis). To study the differences of the DDR in different cell types, we used primary breast and lung cells. We started with the platinum-containing drug cisplatin because it is a widely used chemotherapeutic that causes DNA adducts and inter- and intrastrand cross-links and is known to induce apoptosis (Eastman, 1987, Hu et al., 2016, Rosenberg et al., 1969), which can be readily quantified using MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) cell viability assay. Briefly, cells were seeded at 50% density, followed by 48 h of cisplatin exposure. This setup allowed us to measure cell death (Figure 1A). Breast cells (IC50∼ 15–20 μM) were consistently more sensitive than lung cells (IC50∼40 μM; p = 1 × 10−9; Figure 1B). A different sensitivity for apoptosis was confirmed by the appearance of apoptotic morphology as visualized by DAPI staining (Figures 1C and 1D).
Figure 1.

Primary Breast Cells Are More Sensitive to Cisplatin-Induced DNA Lesions Than Primary Lung Cells
(A) Overview of adapted MTT assay to measure cell death. Briefly, cells were seeded at 50% density and treated with high doses of cisplatin for 24 or 48 h. Treatment under these conditions results in cell death (e.g., compare the wells treated with 60 μM cisplatin at 24 and 48 h).
(B) Breast cells are more sensitive to cisplatin than lung cells. Human primary epithelial cells from six donors were treated with a range of cisplatin concentrations. After 48 h, the cell viability was determined by MTT assay. The concentration at which 50% loss of viability occurred (IC50) was calculated (n = 3–6). A t-test was used to compare IC50 values between breast and lung cells.
(C) Treatment with cisplatin results in apoptosis. Nuclear morphology of breast and lung cells was visualized by DAPI stain after 24-h exposure to 80 μM cisplatin (representative experiment, n = 4).
(D) Breast cells more frequently display apoptotic morphology (DAPI stain) after 24 h of cisplatin treatment than lung cells (representative experiment, n = 4). A two-way ANOVA with Bonferroni post-hoc test was carried out to compare differences (*p < 0.05, ***p < 0.001).
(E) Breast cells are more sensitive to equal amounts of cisplatin-induced DNA lesions at 48 h. The amount of DNA-bound platinum was determined after 48 h of cisplatin treatment (Figure S1A) and plotted against the cellular viability (MTT assay) at 48 h (representative experiment, n = 3). Regression analysis was used to test if the slope and intercept were different. Error bars represent the SD (n = 3).
To control for differential cisplatin uptake, we looked directly at the relation between platinum bound to the DNA (Pt_DNA) and cellular outcome. Breast cells contained higher levels of Pt_DNA at similar cisplatin doses (Figure S1A), and even when corrected for the amount of Pt_DNA, breast cells were consistently more sensitive for cisplatin (Figure 1E). In summary, breast cells induce apoptosis at a lower amount of DNA damage than lung cells.
Cisplatin Treatment Activates Chk1 Signaling More Strongly in Lung Cells, Resulting in Increased Resistance
To understand how an equal dose of DNA damage results in differential cell viability, we initially focused on p53 dynamics, as it is an important determinant of apoptosis. Intriguingly, p53 levels alone could not explain the differential apoptotic sensitivity. When breast and lung cells were treated with 20 and 30 μM cisplatin, DNA-bound cisplatin and total p53 levels were very similar (Figures S1B and S1C). To understand the mechanism behind their differential cisplatin sensitivity, we used protein microarrays to assess temporal changes of the proteome and phosphoproteome (covering 366 unique UniProt IDs) in breast and lung cells. We analyzed the proteome at three time points: an early stage (6 h; p53 increase becomes apparent), an intermediate stage (12 h), and a late stage (24 h), in which signs of apoptosis can be observed. We thus generated an unbiased profile of the post-DNA damage proteome in healthy breast and lung cells. Among the proteins that were differentially expressed, 17 BioCarta pathways were significantly enriched (p < 0.1; Figures 2A, 2B, and S2; Tables S1, S2, and S3). After antibody validation (Table S2), we focused on five differentially regulated signaling modules: PTEN/Akt signaling, ERK, p38, JNK signaling, and DNA damage checkpoint activation. As shown in Figures 2C and S3, differential activation (as measured by phosphorylation on key residues) was observed for proteins in the mitogen-activated protein kinase (MAPK) signaling pathway. For example, p38 MAPK was activated to a higher degree in primary breast cells. Furthermore, differences in DNA damage checkpoint activity were observed (Figures 2C and S3E). Active ATR (p-S428) was present at higher levels in lung cells than breast cells. Accordingly, its downstream target Chk1 was activated (S345) to a greater extent in lung cells.
Figure 2.

Proteomic and Functional Analysis Reveals an Increased Chk1 Activity in Lung Cells that Contributes to Cisplatin Resistance
(A) Identification of genes involved in differential cisplatin response. Breast and lung cells were treated with a cisplatin dose that caused an equal amount of DNA damage (20 μM and 30 μM, respectively). A protein array (366 unique UniProt IDs covered by both pan-specific and phospho-specific antibodies) was carried out to assess proteomic changes at 6, 12, and 24 h. Gene set enrichment was performed on differentially expressed proteins to identify differentially regulated pathways. After validation of the array results using both antibodies present on the array and antibodies from different manufacturers, inhibitors were used to screen for genes that modified the cisplatin response. For details see Transparent Methods and Figure S2.
(B) Overview of BioCarta pathways enriched for differentially expressed genes (DAVID pathway analysis). For details, see Table S2.
(C) Overview of the most differentially activated pathways after cisplatin treatment. After array antibody validation (Table S2), activity of differentially regulated pathways was determined using quantitative western blot (Figure S3). The colors indicate whether protein activity (e.g., highest phosphorylation levels) is the highest in lung (red) or breast (blue) cells.
(D) Inhibition of Chk1 activity differentially affects cisplatin sensitivity. Cells were treated for 1 h with the Chk1 inhibitor PF477736 (1 μM) or an equivalent amount of DMSO, after which cisplatin was added. Viability was determined after 48 h. Error bars represent the SD (n = 7). A two-way ANOVA with Bonferroni post-hoc test was carried out to test if differences were significant (**p < 0.01).
To investigate if the differentially regulated pathways affect cisplatin-induced apoptosis, we negatively regulated essential proteins in each pathway using specific inhibitors that produce fewer side effects than small interfering RNAs. Inhibitors for phosphatidylinositol 3-kinase, JNK, p38 kinase, and MEK did not significantly alter the cisplatin response (Figure S4). We next focused on ATR, Chk1, and Chk2 using specific inhibitors (VE822 for ATR, PF477736 for Chk1, Chk2 inhibitor II for Chk2) (Figure S5A). Strikingly, Chk1 inhibition sensitized primary lung cells to cisplatin (p < 0.001), but it had no effect on breast cells (Figure 2D). Similar effects of Chk1 inhibition were observed in primary cells obtained from different donors (Table S4), and ATR inhibition sensitized lung cells too (Figures S5B and S5C). We also found that cell viability responses in lung cells were affected to a lesser extent by Chk2 inhibition than by Chk1 inhibition (Figure S5D). Taken together, these results indicate that the higher activity of Chk1 in lung cells contributes to cisplatin resistance of lung cells.
Cell-Type-Specific DNA Damage Signaling Dynamics after Cisplatin Treatment
The identification of Chk1 as a modifier of the apoptotic sensitivity for cisplatin was unexpected because its downstream target p53 accumulates to similar levels in breast and lung cells (Figure S1C). We therefore decided to compare the activity of the Chk1-Chk2-p53 signaling network in breast and lung cells more closely. We quantitatively measured the total levels and activities of Chk1, Chk2, p53, and p21 (Figure 3A). After an equal amount of cisplatin-induced DNA damage, Chk1 activity, as measured by phosphorylation of sites S345, S317, and S296, increased to a much higher level in the lung cells (Figure 3A, p < 0.01, Figure S6). In contrast, Chk2 activation (T68, S516) was only moderately increased in both cell types. Although total p53 levels were similar in breast and lung cells, p53 was phosphorylated (S15/S20) to a larger extent in lung cells (Figure 3A, p < 0.01, Figure S6), correlating with higher Chk1 activation. In addition, p21 levels increased in lung cells but not in breast cells. Together, these results show that primary breast and lung cells display different dynamics of Chk1 activation, p53 phosphorylation, and p21 protein expression after cisplatin treatment.
Figure 3.

Cisplatin Treatment Results in Different DNA Damage Signaling Dynamics in Breast and Lung Cells
(A) Relative change of Chk1, Chk2, p53, and p21 dynamics after cisplatin treatment in breast and lung cells. Protein levels were analyzed using quantitative western blot and normalized by the number cells loaded in each sample. For every blot the highest expression value was set to 1, after which the average value was calculated for five to eight blots. Depicted is the average value normalized to the maximum value per graph. Error bars represent the SEM. Two-way ANOVAs were used to test if a protein was differentially expressed in breast versus lung cells. See also Figure S6.
(B) Comparison of model simulations to the experimental data of DNA damage signaling dynamics. To compare with the corresponding experimental data, the model simulation data were scaled as relative values (normalized to the maximal values in breast and lung cells) and depicted as blue lines. Data from individual blots are depicted as “+.” The average values are shown as red circles. Note that some data points with the same values appear overlapped in this scatterplot.
A Quantitative Model Predicts Differential Roles of Chk1 and Chk2 in the Regulation of p53 Phosphorylation
To better understand the relation between the dynamics of the DDR proteins and the chemotherapy response (apoptosis), we developed an integrated mathematical model based on our current data and previous studies (Barr et al., 2017, Batchelor et al., 2008, Batchelor et al., 2011, Ma et al., 2005, Stewart-Ornstein and Lahav, 2017, Zhang et al., 2011). This new model consists of coupled ordinary differential equations, and it focuses on the key interactions between Chk1, Chk2, p53, and p21 (Figure S7). In the model, we used the same model structure and reaction kinetics for breast and lung cells. Most parameters have the same values, except that some parameters are set with cell-type-specific values to distinguish different cisplatin-DNA binding dynamics, differential activation, and degradation of proteins observed in breast and lung cells (list of parameter values is given in Tables S5–S7). A detailed description of the model development, model simulation, and parameter estimation is provided in the Supplemental Information.
The mathematical model was fitted to the experimental datasets that captured the temporal profiles of cisplatin-DNA binding dynamics; total levels of Chk1, Chk2, p53, and p21; the activation of Chk1, Chk2, and p53; as well as cell viability responses in breast and lung cells. In total, 98 average values (from 564 measured data points) were used to estimate the kinetic parameters of the model for cisplatin treatment. As shown in Figures 3B and S8, the model simulations quantitatively fit the signaling dynamics of Chk1, Chk2, p53, and p21 proteins and DNA-bound cisplatin.
Intriguingly, the model suggested that the contribution of Chk1 and Chk2 to the regulation of p53 and p21 dynamics is different. As shown in Figure S9, the modeling analyses indicate that Chk2 activities in breast and lung cells are relatively high and give rise to a saturated p53 phosphorylation rate. On the other hand, in breast cells Chk1 activity is too low to induce p53 phosphorylation, whereas its level in lung cells is relatively high and induces additional p53 phosphorylation on top of Chk2's activity. We next studied what happens to downstream p53 and p21 responses after in silico inhibition of Chk1 and Chk2 activities (details are in Supplemental Information). The model predicted that Chk1 inhibition has almost no effect on p53 and p21 dynamics in breast cells, where Chk1 phosphorylation is low (Figure 4A). In contrast, in lung cells a strong effect of Chk1 inhibition on p53 and p21 dynamics is expected (Figure 4B). Interestingly, it is also predicted that Chk2 inhibition will strongly decrease the levels of total p53, p53 phosphorylation, and p21 in both breast and lung cells (Figures 4A and 4B). These results thus indicate that the contributions of Chk1 and Chk2 in regulating p53 and p21 dynamics are different.
Figure 4.

Inhibitor Studies Reveal Different Roles for Chk1 and Chk2 in Breast and Lung Cells
(A and B) Model predictions for the inhibition of Chk1 and Chk2 in breast (A) and lung cells (B).
(C and D) Inhibition of Chk1 and Chk2 alter p53 and p21 dynamics. Primary breast (C) and lung cells (D) were pretreated for 1 h with inhibitors against Chk1 (PF477736; 1 μM) and Chk2 (Inhibitor II, 10 μM) or an equivalent amount of DMSO, followed by cisplatin treatment. Depicted is the average relative expression (n = 3–7) for each condition normalized to the maximum value. Error bars represent the SEM. Two-way ANOVAs were used to test if a protein was differentially expressed between treatments.
See also Figures S10 and S11.
To experimentally validate the model predictions, we treated breast and lung cells with inhibitors for Chk1 and Chk2, or an equivalent amount of DMSO, followed by cisplatin treatment. As predicted by the model, Chk1 inhibition did not have a significant effect on downstream targets in breast cells (Figures 4C and S10). In lung cells, however, Chk1 inhibition significantly altered p53 and p21 dynamics. Total p53 levels were unaffected, whereas p53 phosphorylation was diminished and p21 was not upregulated after cisplatin treatment (Figures 4D and S11). In line with model predictions, Chk2 inhibition resulted in the loss of p21 expression in both breast and lung cells. In addition, total and phospho-p53 levels remained low when Chk2 activity was inhibited (Figures 4C and 4D). Together, our model analyses and experimental data suggest that Chk1 activity plays a regulatory role in p53 activation, whereas Chk2 is essential for p53 activation and expression.
Breast Cells Are More Resistant to DNA Double-Strand Breaks Than Lung Cells
Chk1 is strongly activated by single-strand DNA damage, whereas Chk2 is mostly activated by DNA DSBs (Bartek and Lukas, 2003, Smith et al., 2010). We therefore wondered what happens if we treat primary breast and lung cells with the DSB-inducing drug doxorubicin. Doxorubicin induces DNA adducts and also causes DNA DSBs by inhibiting topoisomerase II activity (Arcamone et al., 1969, Yang et al., 2014). When cell viability response data were plotted against the level of intracellular doxorubicin at 48 h (Figure S12), breast cells were more resistant than lung cells (Figure 5A). Surprisingly, this is opposite to the effect of cisplatin, which induces more apoptosis in breast cells, and suggests that the sensitivities of breast and lung cells to genotoxic stress depend on the nature of DNA damage.
Figure 5.

Breast Cells Are More Resistant to Doxorubicin Than Lung Cells
(A) Cell viability response to equal intracellular amounts of doxorubicin. After 48 h of doxorubicin treatment, intracellular doxorubicin fluorescence was determined by fluorescence-activated cell sorting (Figure S12). The intracellular doxorubicin levels (arbitrary units) were plotted against the cellular viability (as determined by MTT assay) (n = 3). Error bars represent the SD. Regression analysis was used to test if the slope and intercept were different.
(B) Relative change of Chk1, Chk2, p53, and p21 dynamics after doxorubicin treatment in breast and lung cells. Depicted is the average relative expression per treatment (n = 4–6) normalized to the maximum value per graph. Error bars represent the SEM. Two-way ANOVAs were used to test if a protein was differentially expressed in breast versus lung cells. See also Figure S13.
(C) Comparison of model simulations to the experimental data in response to doxorubicin treatment. To compare with the corresponding experimental data, the model simulation data were scaled as relative values (normalized to the maximal values in breast and lung cells) and depicted as blue lines. Data from individual blots are depicted as “+.” The average values are shown as red circles. Note that some data points with the same values appear overlapped in this scatterplot.
To compare how DNA damage signaling is differently regulated after doxorubicin treatment, we quantitatively measured the dynamics of DNA-damaging signaling proteins. Interestingly, doxorubicin treatment led to strong activation of both ATM (S1981) and Chk2 (T68 and S516) in both cell types (Figures 5B and S13), although breast cells showed higher levels of active Chk2. In addition, lung cells showed a significant higher level of ATR and Chk1 activation (S345, S317, S296) than breast cells. The induction of total and phospho-p53 was higher in lung cells, whereas p21 levels showed a weak increase in both breast and lung cells.
To understand the DDR to doxorubicin treatment, we slightly modified the values of the model parameters that are specific for cisplatin treatment. These parameters were estimated with 94 average values from 412 measured data points for doxorubicin treatment (parameters values are listed in Table S7). As shown in Figures 5C and S14, the models for doxorubicin treatment fitted well to the temporal profiles of different DNA-damage-signaling proteins in both breast and lung cells. Similar to the cisplatin model, the doxorubicin model predicted that Chk2 activity is required for p53 phosphorylation and accumulation, as well as p21 induction in both breast and lung cells (Figures S15A and S15B). Chk1 activity contributed to p53 phosphorylation, p53 accumulation, and p21 induction in lung cells. However, it has only a minor contribution in breast cells. The model predictions were confirmed by experiments with Chk1 and Chk2 inhibitors (Figures S15C, S15D, S16, and S17).
A “Friction Model” Suggests that the Balance of p53 and p21 Dynamics Contributes to Chemotherapy Responses
The different apoptotic sensitivities to cisplatin and doxorubicin are counterintuitive. Therefore, it is interesting to investigate how the same DNA-damaging core network can lead to a different apoptotic sensitivity depending on the cell type and the nature of chemotherapeutic agents. Previous studies have indicated that a threshold mechanism mediates p53 cell fate decision to induce apoptosis (Kracikova et al., 2013, Paek et al., 2016). In this work, we proposed a novel “friction model” to describe the cell apoptosis response using a coarse-grained approach, in which a hypothetical threshold mechanism is used to trigger apoptosis. The model assumes that cells undergo apoptosis when the total p53 level is higher than a certain threshold, θ. Instead of using a fixed threshold, we assume that the threshold θ is proportional to the p21 expression level as p21 can act as a negative regulator for apoptosis (Abbas and Dutta, 2009, Roninson, 2002). The dynamic threshold is analogous to the friction force exerted by a surface that resists the motion of an object, which is proportional to its mass. To move cells to apoptosis, the driving force (caused by p53) should be larger than the friction force (induced by p21). As shown in Figure S18, this simplified friction model was able to reproduce the different cell viability responses in breast and lung cells after cisplatin or doxorubicin treatment.
Additional modeling analyses indicated that the balance between p53 and p21 dynamics could explain the differential sensitivities to diverse chemotherapeutic agents. In case of cisplatin treatment, both breast and lung cells have a similar accumulation of p53 protein (similar driving forces), whereas p21 expression is induced more in lung cells. Therefore the overall friction force is larger in lung cells, which makes lung cells more resistant to apoptosis after cisplatin treatment (Figures 6A and 6B). Indeed, an inhibitor of p21 transcription could sensitize lung cells to cisplatin treatment (p < 0.001; Figure S19). On the other hand, after doxorubicin treatment the p53 level in lung cells is higher, whereas p21 dynamics is similar. Hence, the driving force is lower in breast cells, explaining why breast cells are more resistant (Figures 6C and 6D). In summary, the model suggests that the balance of p53 and p21 dynamics contributes to the decision to undergo apoptosis after DNA damage.
Figure 6.

Mathematical Modeling Shows that the Balance of p53 and p21 Levels Determines Cell Viability after DNA Damage
(A–D) Model simulations for the dynamics of cell viability, apoptosis rate, p53, p21, and threshold for apoptosis in response to cisplatin (A and B) or doxorubicin (C and D) in breast and lung cells.
Discussion
We report here that primary non-transformed breast and lung cells have different sensitivities for DNA-damaging agents. Breast cells are more sensitive to the DNA adduct and cross-linking agent cisplatin, whereas lung cells are more sensitive to the DNA adduct and DSB-inducing agent doxorubicin. These differences in sensitivity can be explained by the differential activation of Chk1, which is phosphorylated to a greater extent in lung cells. In contrast, in breast cells, Chk2 is activated to a higher extent after doxorubicin treatment. The differential activities of Chk1 and Chk2 affect p53 and p21 dynamics and hence contribute to drug sensitivity. Although differences in p53 and p21 dynamics and downstream pathways have been described in tissue-specific stem cells before (Blanpain et al., 2011, Insinga et al., 2013, Vitale et al., 2017), they were considered to be stem cell properties that change after differentiation (Insinga et al., 2013). Our study shows for the first time that healthy epithelial cell types have a different DDR. Moreover, our observation that differences in DDR can already be observed at the level of DNA damage detection is novel.
An intriguing question is why breast and lung cells show different Chk1 and Chk2 activities. The ATR-Chk1 pathway is most strongly activated by single-strand DNA damage, whereas the ATM-Chk2 pathway plays a role in DSB repair. As cell types experience different kinds of DNA damage, it may be advantageous to tailor DDR protein dynamics to the form of DNA damage that occurs most frequently. For instance, the high oxygen levels in lung likely result in increased numbers of free radicals, which can damage the DNA by causing DNA adducts and other forms of single-strand damage (Cadet and Wagner, 2013, Pham-Huy et al., 2008). This may explain why Chk1 is activated to a higher extent in lung cells than breast cells. In contrast, in breast, metabolic by-products of estrogen signaling (Yasuda et al., 2017) are known to give rise to DSBs (Savage et al., 2014), indicating that breast cells may need enhanced ATM-Chk2 signaling. The causes of the differences in Chk1 and Chk2 activities in breast and lung cells are unknown. This will be the subject of future study.
We observed that primary breast cells are more sensitive to cisplatin, and lung cells, to doxorubicin treatment. Paradoxically, first-line chemotherapeutic regimens for breast and lung cancer are mostly based on doxorubicin or cisplatin, respectively (Crown, 1998, Laskin and Sandler, 2005, Paoletti et al., 2011, von Minckwitz, 2007). It is likely that during tumorigenesis breast and lung cancer cells become more sensitive to doxorubicin and cisplatin, respectively, as breast cancer cell lines respond well to doxorubicin and non-small-cell lung cancer cell lines are generally sensitive to cisplatin (Alley et al., 1988, Shoemaker, 2006). The goal of cancer therapy is to destroy cancer cells while sparing healthy cells. The difference in sensitivity between normal and transformed cells would provide a window to selectively kill cancer cells. It would be interesting to investigate if and how the sensitivity for DNA damage changes during tumorigenesis.
Chk1 inhibitors have recently attracted attention as sensitizers for chemotherapy as Chk1 is frequently overexpressed in tumors (Zhang and Hunter, 2014). Our model confirms that Chk1 overexpression could lead to increased cisplatin resistance and hence provides a rationale for co-treatment of Chk1 inhibitors with cisplatin. Indeed, Chk1 inhibition has been found to decrease cisplatin resistance in cancer cell lines (Gadhikar et al., 2013, Li et al., 2016, Thompson et al., 2012). However, the observation that Chk1 inhibition did not sensitize breast cells to cisplatin may indicate that only tumors with overactive Chk1 may respond to Chk1 inhibition, and hence calls for careful patient stratification in clinical trials.
Both ATR-Chk1 and ATM-Chk2 signaling are known to regulate p53 stabilization and phosphorylation. Our model predicts that Chk1 and Chk2 affect p53 dynamics differently. Chk2 activity is required for p53 phosphorylation, whereas Chk1 activity is not essential, but contributes to p53 phosphorylation (Figure 7A). As a consequence of its higher activity in lung cells, Chk1 contributes to p53 phosphorylation in lung cells but not in breast cells. As expected, a Chk1 inhibitor reduced p53 phosphorylation levels only in lung cells. Although Chk1 has been demonstrated to be able to phosphorylate p53 on S15 and S20 (Shieh et al., 2000), some studies reported that Chk1 inhibition did not diminish p53 phosphorylation (Tian et al., 2002, Wu et al., 2013). Our data indicate that this may be a cell-type or DNA-damage-specific effect.
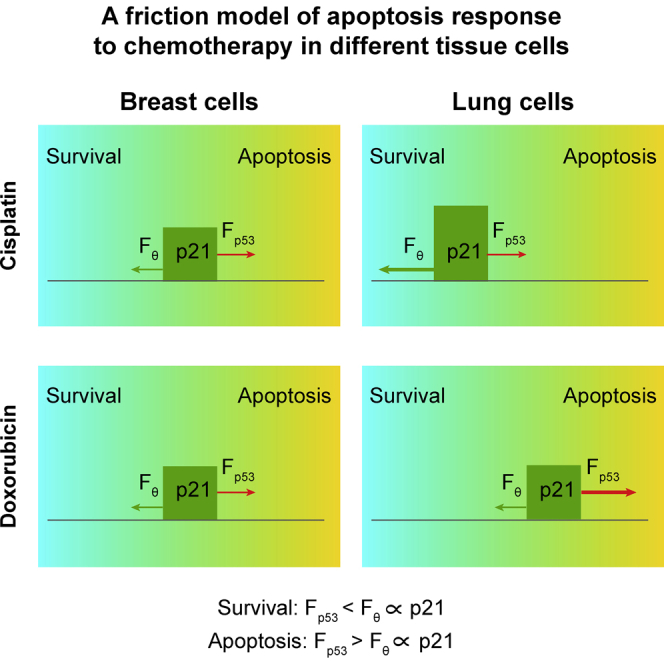
Figure 7.

A Friction Model of Cell Apoptosis Response to DNA Damage Signaling
(A) A simple scheme for Chk1, Chk2, p53, and p21 protein interactions.
(B) A cartoon of the “friction model.” A cyan-to-orange gradient indicates the balance between survival (cyan region) and apoptosis (orange region). Cellular outcome to DNA damage is determined by the balance of the driving force (caused by p53) and the friction force (induced by p21). To move cells to the apoptotic state, the driving force should be larger than the friction force. The size of arrows depicts the relative strength of the driving and friction forces in breast and lung cells in response to cisplatin or doxorubicin treatment. p-Chk1 and p-Chk2 contribute to p53 phosphorylation, and both p-p53 and Chk2 can induce p21 transcription. However, whether p21 expression increases also depends on the nature of the stress.
We propose a “friction model” to explain the differences in apoptotic sensitivity for cisplatin and doxorubicin in breast and lung cells. The induction of apoptosis is a complex process that may involve caspase-dependent or caspase-independent pathways and is regulated by pro- and anti-survival proteins. We did not incorporate these effects in our model because we found that a simplified version of apoptosis induction, based on the balance between p53 and p21 dynamics, was sufficient to explain the apoptotic response. Moreover, additional processes will increase the complexity of the model, and studying these detailed regulations is not the focus of this work. Our new friction model incorporates the idea of a dynamic p53 threshold for apoptosis induction (Paek et al., 2016), but defines it as a function of p21 levels. After cisplatin treatment, p53 dynamics are similar in breast and lung cells causing a similar apoptotic stimulus (Figure 7B). The difference in cisplatin resistance can be explained because in lung cells increased amounts of p53 phosphorylation (as result of increased Chk1 activity) give rise to p21 induction, resulting in an increased apoptotic threshold. In contrast, after doxorubicin treatment p21 dynamics are similar in breast and lung cells. In this case, increased p53 phosphorylation in lung cells leads to stabilization of p53, which translates into a higher apoptotic stimulus and enhanced sensitivity to doxorubicin (Figure 7B). A possible role for p21 as a friction force is supported by data for the p21 inhibitor UC2288. Although the mechanism of action of this inhibitor is incompletely understood, we observed down-regulation of p21 after treatment. We cannot exclude, however, that the role of p21 is cellular context dependent. In addition, it is likely that other proteins could contribute to the friction force as well, such as the inhibitors of apoptosis (IAP) family proteins (Deveraux and Reed, 1999, Paek et al., 2016).
In summary, our study provides new insights into the apoptotic sensitivity for DNA-damaging agents. It will be interesting to investigate how modulating Chk1 levels and the balance between p53 and p21 may improve the effectiveness of chemotherapy.
Limitations of the Study
In this study we made use of primary epithelial cells to investigate the DDR in healthy cells. Although this study provided novel insights into how Chk1, Chk2, p53, and p21 collectively affect the apoptotic response to DNA-damaging chemotherapy in different tissues, subsequent studies are necessary to investigate the implication for cancer cells and cancer treatment. In addition, in this study, we described a friction model to explain the difference in apoptotic sensitivity between breast and lung cells for two different anti-cancer drugs. This friction model is based on the balance of p53 and p21. It is likely, however, that other anti-apoptotic proteins (such as the IAP family of proteins) could contribute to the friction force as well. Furthermore, in our mathematical model, different p53 negative regulators are lumped as one abstract variable “negative feedback regulator”, which makes it difficult to study the contributions of specific negative regulators. As different p53 negative regulators may affect p53 expression and activity in different ways, their exact roles and contributions need to be further studied.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to thank Joris Pothof, Maikel Wouters, and Serena Bruens for providing cell line samples; Peter de Bruijn for measuring platinum concentrations in DNA samples; Uta Marchfelder for setting up the FACS; and Susanne Freier for help in experiments. We are also grateful to Alexander Meissner for his critical reading of this manuscript. This work was supported by a grant to Z.Z. from the Federal Ministry of Education and Research (BMBF, Germany) within the e:Bio project (031A309).
Author Contributions
M.T.v.J. designed and performed the experiments; Z.Z. developed the mathematical model and performed modeling analyses; D.D. assisted at the western blot experiments; and E.A.C.W. provided insights and commented on the results. M.T.v.J. and Z.Z. analyzed the data and wrote the manuscript. Z.Z. supervised the project.
Declaration of Interests
The authors declare no competing interests.
Published: February 22, 2019
Footnotes
Supplemental Information includes Transparent Methods, 19 figures, and 7 tables and can be found with this article online at https://doi.org/10.1016/j.isci.2019.01.001.
Supplemental Information
Kinexus KAM-900P arrays were hybridized with control treatment (0 h) and cisplatin-treated (6, 12, or 24 h) breast or lung cells. Raw data show the expression value for each antibody (present in duplicate) after scanning and Imagene spot quantification for all samples. See the corresponding Excel file.
After pathway enrichment analysis of differentially expressed genes (Figure S2), antibodies recognizing key proteins from the BioCarta pathways were validated on western blot. The results from the western blot are included in the table. See the corresponding Excel file.
The table indicates the experimental conditions for the antibodies used for western blot. After blotting, proteins were blocked for 1 h, followed by overnight first antibody incubation. See the corresponding Excel file.
References
- Abbas T., Dutta A. p21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alley M.C., Scudiero D.A., Monks A., Hursey M.L., Czerwinski M.J., Fine D.L., Abbott B.J., Mayo J.G., Shoemaker R.H., Boyd M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988;48:589–601. [PubMed] [Google Scholar]
- Arcamone F., Cassinelli G., Fantini G., Grein A., Orezzi P., Pol C., Spalla C. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnol. Bioeng. 1969;11:1101–1110. doi: 10.1002/bit.260110607. [DOI] [PubMed] [Google Scholar]
- Barr A.R., Cooper S., Heldt F.S., Butera F., Stoy H., Mansfeld J., Novak B., Bakal C. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat. Commun. 2017;8:14728. doi: 10.1038/ncomms14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J., Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- Batchelor E., Mock C.S., Bhan I., Loewer A., Lahav G. Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol. Cell. 2008;30:277–289. doi: 10.1016/j.molcel.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor E., Loewer A., Mock C., Lahav G. Stimulus-dependent dynamics of p53 in single cells. Mol. Syst. Biol. 2011;7:488. doi: 10.1038/msb.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C., Mohrin M., Sotiropoulou P.A., Passegue E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell. 2011;8:16–29. doi: 10.1016/j.stem.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Cadet J., Wagner J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013;5 doi: 10.1101/cshperspect.a012559. a012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae Y.K., Anker J.F., Carneiro B.A., Chandra S., Kaplan J., Kalyan A., Santa-Maria C.A., Platanias L.C., Giles F.J. Genomic landscape of DNA repair genes in cancer. Oncotarget. 2016;7:23312–23321. doi: 10.18632/oncotarget.8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crown J. Evolution in the treatment of advanced breast cancer. Semin. Oncol. 1998;25:12–17. [PubMed] [Google Scholar]
- Deveraux Q.L., Reed J.C. IAP family proteins–suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- Eastman A. The formation, isolation and characterization of DNA adducts produced by anticancer platinum complexes. Pharmacol. Ther. 1987;34:155–166. doi: 10.1016/0163-7258(87)90009-x. [DOI] [PubMed] [Google Scholar]
- Forestier A., Sarrazy F., Caillat S., Vandenbrouck Y., Sauvaigo S. Functional DNA repair signature of cancer cell lines exposed to a set of cytotoxic anticancer drugs using a multiplexed enzymatic repair assay on biochip. PLoS One. 2012;7:e51754. doi: 10.1371/journal.pone.0051754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadhikar M.A., Sciuto M.R., Alves M.V., Pickering C.R., Osman A.A., Neskey D.M., Zhao M., Fitzgerald A.L., Myers J.N., Frederick M.J. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol. Cancer Ther. 2013;12:1860–1873. doi: 10.1158/1535-7163.MCT-13-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner K.M., Eastman A. Variations in Mre11/Rad50/Nbs1 status and DNA damage-induced S-phase arrest in the cell lines of the NCI60 panel. BMC Cancer. 2011;11:201–213. doi: 10.1186/1471-2407-11-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gettinger S., Lynch T. A decade of advances in treatment for advanced non-small cell lung cancer. Clin. Chest Med. 2011;32:839–851. doi: 10.1016/j.ccm.2011.08.017. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- Hu J., Lieb J.D., Sancar A., Adar S. Cisplatin DNA damage and repair maps of the human genome at single-nucleotide resolution. Proc. Natl. Acad. Sci. U S A. 2016;113:11507–11512. doi: 10.1073/pnas.1614430113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insinga A., Cicalese A., Faretta M., Gallo B., Albano L., Ronzoni S., Furia L., Viale A., Pelicci P.G. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proc. Natl. Acad. Sci. U S A. 2013;110:3931–3936. doi: 10.1073/pnas.1213394110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kracikova M., Akiri G., George A., Sachidanandam R., Aaronson S.A. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013;20:576–588. doi: 10.1038/cdd.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin J.J., Sandler A.B. First-line treatment for advanced non-small-cell lung cancer. Oncology (Williston Park) 2005;19:1671–1676. discussion 1678–1680. [PubMed] [Google Scholar]
- Levy-Lahad E., Friedman E. Cancer risks among BRCA1 and BRCA2 mutation carriers. Br. J. Cancer. 2007;96:11–15. doi: 10.1038/sj.bjc.6603535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.C., Yang J.C., Lu M.C., Lee C.L., Peng C.Y., Hsu W.Y., Dai Y.H., Chang F.R., Zhang D.Y., Wu W.J. ATR-Chk1 signaling inhibition as a therapeutic strategy to enhance cisplatin chemosensitivity in urothelial bladder cancer. Oncotarget. 2016;7:1947–1959. doi: 10.18632/oncotarget.6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Chang H., Li X.H., Qi Y.F., Wang J.O., Zhang Y., Yang X.H. Network meta-analysis on the effects of DNA damage response-related gene mutations on overall survival of breast cancer based on TCGA database. J. Cell Biochem. 2017;118:4728–4734. doi: 10.1002/jcb.26140. [DOI] [PubMed] [Google Scholar]
- Ma L., Wagner J., Rice J.J., Hu W., Levine A.J., Stolovitzky G.A. A plausible model for the digital response of p53 to DNA damage. Proc. Natl. Acad. Sci. U S A. 2005;102:14266–14271. doi: 10.1073/pnas.0501352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidou K., Lindstrom S., Dennis J., Beesley J., Hui S., Kar S., Lemacon A., Soucy P., Glubb D., Rostamianfar A. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551:92–94. doi: 10.1038/nature24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Minckwitz G. Docetaxel/anthracycline combinations for breast cancer treatment. Expert Opin. Pharmacother. 2007;8:485–495. doi: 10.1517/14656566.8.4.485. [DOI] [PubMed] [Google Scholar]
- Nevanlinna H., Bartek J. The CHEK2 gene and inherited breast cancer susceptibility. Oncogene. 2006;25:5912–5919. doi: 10.1038/sj.onc.1209877. [DOI] [PubMed] [Google Scholar]
- Non-small Cell Lung Cancer Collaborative Group Chemotherapy in non-small cell lung cancer: a meta-analysis using updated data on individual patients from 52 randomised clinical trials. BMJ. 1995;311:899–909. [PMC free article] [PubMed] [Google Scholar]
- Paek A.L., Liu J.C., Loewer A., Forrester W.C., Lahav G. Cell-to-cell variation in p53 dynamics leads to fractional killing. Cell. 2016;165:631–642. doi: 10.1016/j.cell.2016.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti L., Pastis N.J., Denlinger C.E., Silvestri G.A. A decade of advances in treatment of early-stage lung cancer. Clin. Chest Med. 2011;32:827–838. doi: 10.1016/j.ccm.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham-Huy L.A., He H., Pham-Huy C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008;4:89–96. [PMC free article] [PubMed] [Google Scholar]
- Reinhardt H.C., Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012;28:128–136. doi: 10.1016/j.tig.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roninson I.B. Oncogenic functions of tumour suppressor p21(Waf1/Cip1/Sdi1): association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett. 2002;179:1–14. doi: 10.1016/s0304-3835(01)00847-3. [DOI] [PubMed] [Google Scholar]
- Rosenberg B., VanCamp L., Trosko J.E., Mansour V.H. Platinum compounds: a new class of potent antitumour agents. Nature. 1969;222:385–386. doi: 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- Savage K.I., Matchett K.B., Barros E.M., Cooper K.M., Irwin G.W., Gorski J.J., Orr K.S., Vohhodina J., Kavanagh J.N., Madden A.F. BRCA1 deficiency exacerbates estrogen-induced DNA damage and genomic instability. Cancer Res. 2014;74:2773–2784. doi: 10.1158/0008-5472.CAN-13-2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh S.Y., Ahn J., Tamai K., Taya Y., Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- Shoemaker R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Smith J., Tho L.M., Xu N., Gillespie D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- Sousa F.G., Matuo R., Tang S.W., Rajapakse V.N., Luna A., Sander C., Varma S., Simon P.H., Doroshow J.H., Reinhold W.C. Alterations of DNA repair genes in the NCI-60 cell lines and their predictive value for anticancer drug activity. DNA Repair (Amst) 2015;28:107–115. doi: 10.1016/j.dnarep.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart-Ornstein J., Lahav G. p53 dynamics in response to DNA damage vary across cell lines and are shaped by efficiency of DNA repair and activity of the kinase ATM. Sci. Signal. 2017;10 doi: 10.1126/scisignal.aah6671. eaah6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R., Meuth M., Woll P., Zhu Y., Danson S. Treatment with the Chk1 inhibitor Go6976 enhances cisplatin cytotoxicity in SCLC cells. Int. J. Oncol. 2012;40:194–202. doi: 10.3892/ijo.2011.1187. [DOI] [PubMed] [Google Scholar]
- Tian H., Faje A.T., Lee S.L., Jorgensen T.J. Radiation-induced phosphorylation of Chk1 at S345 is associated with p53-dependent cell cycle arrest pathways. Neoplasia. 2002;4:171–180. doi: 10.1038/sj.neo.7900219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale I., Manic G., De Maria R., Kroemer G., Galluzzi L. DNA damage in stem cells. Mol. Cell. 2017;66:306–319. doi: 10.1016/j.molcel.2017.04.006. [DOI] [PubMed] [Google Scholar]
- Weber A.M., Ryan A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015;149:124–138. doi: 10.1016/j.pharmthera.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Wu G., Lin N., Xu L., Liu B., Feitelson M.A. UCN-01 induces S and G2/M cell cycle arrest through the p53/p21(waf1) or CHK2/CDC25C pathways and can suppress invasion in human hepatoma cell lines. BMC Cancer. 2013;13:167. doi: 10.1186/1471-2407-13-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F., Teves S.S., Kemp C.J., Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta. 2014;1845:84–89. doi: 10.1016/j.bbcan.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M.T., Sakakibara H., Shimoi K. Estrogen- and stress-induced DNA damage in breast cancer and chemoprevention with dietary flavonoid. Genes Environ. 2017;39:10. doi: 10.1186/s41021-016-0071-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehir A., Benayed R., Shah R.H., Syed A., Middha S., Kim H.R., Srinivasan P., Gao J., Chakravarty D., Devlin S.M. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer. 2014;134:1013–1023. doi: 10.1002/ijc.28226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.P., Liu F., Wang W. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. U S A. 2011;108:8990–8995. doi: 10.1073/pnas.1100600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Kinexus KAM-900P arrays were hybridized with control treatment (0 h) and cisplatin-treated (6, 12, or 24 h) breast or lung cells. Raw data show the expression value for each antibody (present in duplicate) after scanning and Imagene spot quantification for all samples. See the corresponding Excel file.
After pathway enrichment analysis of differentially expressed genes (Figure S2), antibodies recognizing key proteins from the BioCarta pathways were validated on western blot. The results from the western blot are included in the table. See the corresponding Excel file.
The table indicates the experimental conditions for the antibodies used for western blot. After blotting, proteins were blocked for 1 h, followed by overnight first antibody incubation. See the corresponding Excel file.