

Summary
Angiogenesis, the development of new blood vessels, is a key process in disease. We reported that insulin promotes translocation of transforming growth factor β (TGF-β) receptors to the plasma membrane of epithelial and fibroblast cells, thus enhancing TGF-β responsiveness. Since insulin promotes angiogenesis, we addressed whether increased autocrine TGF-β signaling participates in endothelial cell responses to insulin. We show that insulin enhances TGF-β responsiveness and autocrine TGF-β signaling in primary human endothelial cells, by inducing a rapid increase in cell surface TGF-β receptor levels. Autocrine TGF-β/Smad signaling contributed substantially to insulin-induced gene expression associated with angiogenesis, including TGF-β target genes encoding angiogenic mediators; was essential for endothelial cell migration; and participated in endothelial cell invasion and network formation. Blocking TGF-β signaling impaired insulin-induced microvessel outgrowth from neonatal aortic rings and modified insulin-stimulated blood vessel formation in zebrafish. We conclude that enhanced autocrine TGF-β signaling is integral to endothelial cell and angiogenic responses to insulin.
Subject Areas: Molecular Biology, Molecular Mechanism of Behavior, Cell Biology, Functional Aspects of Cell Biology
Graphical Abstract
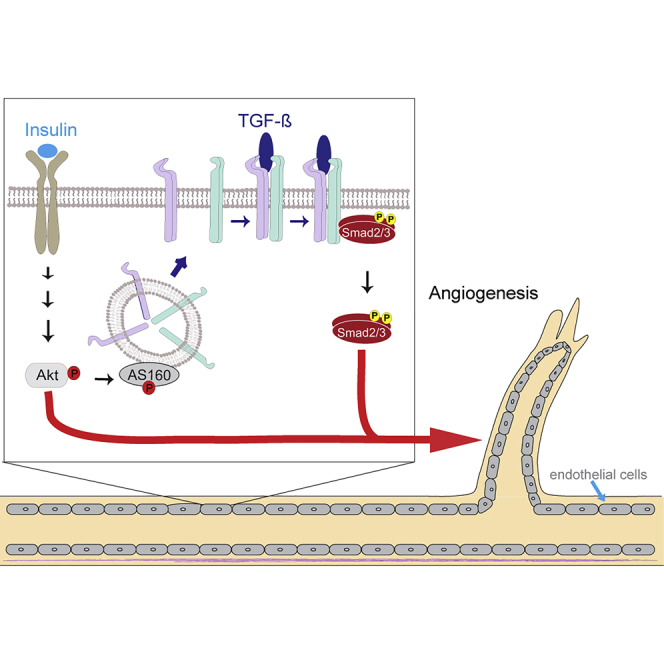
Highlights
-
•
Insulin promotes enhanced autocrine TGF-β responsiveness in endothelial cells
-
•
Autocrine TGF-β signaling contributes to insulin-induced angiogenesis gene expression
-
•
Insulin-induced endothelial migration and sprouting require autocrine TGF-β signaling
-
•
Enhanced autocrine TGF-β signaling is integral to angiogenic responses to insulin
Molecular Biology; Molecular Mechanism of Behavior; Cell Biology; Functional Aspects of Cell Biology
Introduction
Endothelial cells have indispensable roles in the control of vascular function, the formation and extension of new blood vessels during angiogenesis, and the repair of damaged vessels (Carmeliet, 2000, Kolluru et al., 2012, Michiels, 2003). In addition to their essential roles in vascular homeostasis and wound healing, they also promote the pathogenesis of various diseases (Carmeliet and Jain, 2000, Cheng and Ma, 2015, Escudero et al., 2017, Kolka and Bergman, 2013, Kolluru et al., 2012, Tahergorabi and Khazaei, 2012). Integral to physiological or pathological processes, such as development, reproduction, wound repair, and tumor growth, angiogenesis involves conversion of resting endothelial cells into activated cells that loose some endothelial characteristics, acquire the capacity to elongate and migrate, and organize themselves into endothelial tube-like structures and networks (Carmeliet, 2000). These changes are initiated and directed by extracellular growth and differentiation factors, as well as hormones that activate cell surface receptor signaling in endothelial cells that lead to changes in cell differentiation and behavior. Several of the strongly angiogenic factors, such as fibroblast growth factors and vascular endothelial growth factors (VEGFs), act through tyrosine kinase receptors (Cross and Claesson-Welsh, 2001).
Insulin, a secreted hormone, plays essential roles in regulating glucose homeostasis, cell metabolism, as well as angiogenesis and is used as standard treatment of patients with type 1 and type 2 diabetes (Escudero et al., 2017). Functional insulin receptors are expressed on endothelial cells, enabling intracellular signaling responses to hormone (Escudero et al., 2017). Insulin acts through cell surface receptor tyrosine kinases, which, following receptor autophosphorylation, induce kinase pathways, most notably the MEK1/2-Erk MAPK pathway and the PI3K-Akt-mTOR pathway (Taniguchi et al., 2006). Among many responses, insulin generally promotes cell proliferation and survival and enhances protein synthesis and cell size (Taniguchi et al., 2006). Depending on the concentration and time of exposure, insulin also induces changes in the expression of a variety of genes that are therefore seen as insulin-responsive genes (Sano et al., 2016). Consistent with the endocrine activity of insulin, endothelial cells represent a major cell population that is directly exposed to and responsive to insulin (Escudero et al., 2017, Kolka and Bergman, 2013). Insulin also induces angiogenic gene and cell responses and promotes angiogenesis in cell culture, ex vivo and in vivo (Escudero et al., 2017). Enhanced angiogenesis contributes to diabetes-associated complications, including diabetic retinopathy and nephropathy (Escudero et al., 2017), and impaired wound healing, a common problem in diabetics.
We previously documented that insulin induces a rapid increase in cell surface transforming growth factor β (TGF-β) receptors in fibroblasts and epithelial cells, through mobilization of receptors from intracellular vesicles in response to insulin-induced Akt activation (Budi et al., 2015). Increased cell surface presentation of TGF-β receptors confers increased sensitivity to TGF-β, thus enhancing autocrine TGF-β signaling responses (Budi et al., 2015), raising the possibility that the insulin-induced increase in autocrine TGF-β signaling participates in the cellular and gene expression response to insulin. Indeed, we showed that blocking TGF-β signaling attenuates or inhibits the insulin-induced expression of some genes in fibroblasts or epithelial cells (Budi et al., 2015).
TGF-β, a secreted dimeric protein, stands as the prototype of a family of cytokines and differentiation factors that act through cell surface receptors that are distinct in nature from the growth-factor-activated tyrosine kinase receptors, and, accordingly, signal differently (Hata and Chen, 2016, Robertson and Rifkin, 2016). Specifically, TGF-β binds to and activates tetrameric cell surface complexes of two pairs of structurally related dual-specificity kinases, named the type II (TβRII) and type I (TβRI) receptors. Upon ligand binding, the activated type I receptors C-terminally phosphorylate and thus activate Smad2 and Smad3 as signaling mediators that, following translocation into the nucleus, combine with DNA binding, sequence-specific transcription factors, and other coregulators to activate or repress target genes. Consequently, these Smads directly control gene expression and reprogramming in response to TGF-β, depending on the physiological context and nature of target genes (Hata and Chen, 2016, Morikawa et al., 2016). This underlying mechanism is at the basis of a plethora of biological activities of TGF-β, including growth inhibition of epithelial and endothelial cells (Goumans et al., 2002, Morikawa et al., 2016) and effects on cell differentiation of many cell types, including epithelial- and endothelial-mesenchymal transitions (Goumans et al., 2008, Lamouille et al., 2014, van Meeteren and ten Dijke, 2012). TGF-β is also essential for embryonic vascular development (Dickson et al., 1995) and induces angiogenic responses in several assays (Choi and Ballermann, 1995, Yang and Moses, 1990, Zhao et al., 2017), possibly in association with the TGF-β-induced, Smad3-mediated expression of the gene encoding VEGF-A (Goumans et al., 2002). TGF-β, however, has also been seen to inhibit angiogenesis, likely related to its growth inhibitory effects on endothelial cells (Heimark et al., 1986). Both TGF-β receptor types are required for embryonic vascular development (Larsson et al., 2001, Oshima et al., 1996).
In the present study, we examined whether insulin enhances autocrine TGF-β signaling in primary human endothelial cells and evaluated whether insulin-induced angiogenic responses incorporate increased autocrine TGF-β signaling in response to insulin. To assess the contribution of TGF-β signaling, we used primarily two inhibitors of TGF-β signaling with different mechanism of actions. The small molecule TβRI kinase inhibitor SB431542 blocks TGF-β-induced Smad2 and Smad3 activation that directs changes in gene expression, whereas a neutralizing anti-TGF-β antibody prevents all three TGF-βs from binding to their receptors, thus inhibiting all TGF-β signaling. Using primary human umbilical vein endothelial cells (HUVECs) as a model, we found that insulin treatment increases TGF-β receptor levels at the cell surface, consequently enhancing autocrine TGF-β signaling, which is required for insulin-induced endothelial cell migration and contributes to invasion and tube formation. Similar observations were also made in lung microvascular endothelial cells. Transcriptome and single gene analyses revealed that elevated autocrine TGF-β signaling also contributes to insulin-induced gene responses that have been associated with the angiogenic response. Finally, elevated autocrine TGF-β signaling in response to insulin contributed to insulin-induced angiogenic responses in cell culture, ex vivo and in vivo. These results strengthen our appreciation that one endocrine stimulus, i.e., insulin, which is commonly used to treat diabetes, engages TGF-β/Smad signaling in its outcome. Our evidence that TGF-β signaling is integrated into cell responses to insulin may have substantial implications for our understanding of and therapeutic approaches to diabetes-associated pathologies.
Results
Insulin Induces Increased TGF-β Receptor Levels at the Cell Surface and Enhances Autocrine TGF-β/Smad Signaling in Primary Endothelial Cells
Since insulin induces a rapid increase in cell surface presentation of both the TβRI and TβRII receptors in NMuMG epithelial cells and mouse embryonic fibroblasts (Budi et al., 2015), we evaluated the effect of this hormone on primary HUVECs. These cells express insulin receptors, as assessed by qRT-PCR and immunoblotting, and insulin treatment did not affect its expression levels (Figure S1A). Treatment of these cells with insulin, without addition of TGF-β, induced rapid increases in TβRI and TβRII levels at the plasma membrane without changes in their total abundance, as assessed by cell surface protein biotinylation (Figure 1A). Consistent with the notion that insulin-induced Akt signaling drives TGF-β receptor translocation to the cell surface, Akt VIII, a selective inhibitor of Akt1 and Akt2 activation (Calleja et al., 2009, Lindsley et al., 2005, Zhao et al., 2005), prevented the insulin-induced increase of cell surface TβRI and TβRII in endothelial cells (Figure 1B). As shown before in fibroblasts (Budi et al., 2015), insulin induced Akt-mediated phosphorylation of the Rab GTPase-activating protein AS160, which controls cell surface transport of TGF-β receptors (Figure S1B). Similar to the effects of insulin on HUVECs, insulin treatment of human microvascular lung endothelial cells (HMVEC-L), which also express the insulin receptor (Figure S1A), also resulted in increased cell surface abundance of TβRI and TβRII (Figure S1C).
Figure 1.

Insulin Enhances TGF-β Receptor Abundance at the Plasma Membrane and Induces Smad2 and Smad3 Activation through Autocrine TGF-β Signaling
(A and B) Immunoblot analyses of the TβRI and TβRII receptors in human umbilical vein endothelial cells (HUVECs), treated with 100 nM insulin, in the absence (A) or presence (B) of the Akt inhibitor AktVIII. The top two panels show the cell surface TβRI and TβRII receptors, affinity labeled by cell surface protein biotinylation, isolated by affinity adsorption to Neutravidin, and visualized by SDS-PAGE and immunoblotting. The lower panels show TβRI and TβRII in total cell lysates, with GAPDH as loading control. Inhibition of Akt activation prevents the insulin-induced increase in TβRI or TβRII abundance at the cell surface. Immunoblots are representative of three independent experiments. Control group (0) was without insulin for 540 min in starvation media.
(C) HUVECs were treated with 100 nM insulin, with or without SB431542, for the indicated times, and Smad2 and Smad3 activation was assessed by immunoblotting for C-terminally phosphorylated Smad2 (pSmad2) or Smad3 (pSmad3). Control group (0) was without insulin for 360 min in starvation media. The changes in Smad2 or Smad3 phosphorylation are densitometrically shown on the right.
(D) qRT-PCR of SMAD7 and SERPINE1 mRNA expressed by cells treated with or without insulin (Ins) in the presence or absence of SB431542 (SB) for 90 min and normalized against RPL19 mRNA.
Error bars indicate standard error of means, based on four independent experiments. *p < 0.05. In panels A, B, and C: AP, affinity purification, IB, immunoblot.
Most cells in culture express low TGF-β receptor levels at the cell surface (Massague and Kelly, 1986, Wu and Derynck, 2009), and their cell surface concentration determines the cell sensitivity to TGF-β and, thus, the level of TGF-β-induced Smad activation (Budi et al., 2015, Liu et al., 2009a, Wu and Derynck, 2009). Consequently, autocrine TGF-β signaling, commonly observed in cultured cells, is enhanced when the cell surface receptor levels are increased (Budi et al., 2015, Wu and Derynck, 2009). We therefore evaluated whether insulin induces activation of Smad2 and Smad3 without addition of exogenous TGF-β, by immunoblotting for C-terminally phosphorylated Smad2 or Smad3. This was indeed the case in both HUVECs and microvascular endothelial cells (Figures 1C and S1D). Smad2 and Smad3 activation in response to insulin was prevented by SB431542, a TβRI kinase inhibitor that blocks the activation of Smad2 and Smad3 (Callahan et al., 2002, Laping et al., 2002), as well as by LY2157299 (galunisertib), another inhibitor of the TβRI kinase, and to a lesser extent of the TβRII kinase, which also prevents TGF-β-induced Smad2 and Smad3 activation (Bueno et al., 2008) (Figure S1E). These results indicate that insulin-induced activation of Smad2 and Smad3 depend on autocrine TGF-β signaling (Figures 1C and S1D). The modest Smad activation suggested that insulin treatment might mildly activate TGF-β target genes. In agreement with this, insulin mildly but significantly induced the expression of SMAD7 and SERPINE1, direct TGF-β/Smad3 target genes (Figures 1D and S1F), and their induction was prevented by SB431542 (Figures 1D and S1F) or LY2157299 (Figure S1G).
Autocrine TGF-β Signaling Participates in the Insulin-Induced Angiogenic Gene Expression Program
To examine whether the increased autocrine TGF-β responses participate in the insulin-induced gene expression program, we performed RNA sequencing (RNA-seq) analysis of HUVECs treated with or without insulin for 90 min in the absence or presence of SB431542. Changes in gene expression with a p < 0.05 cutoff were assessed using GO-ELITE (Tables S1, S2, and S3). We focused our attention on angiogenesis- and blood-vessel-associated gene responses.
The RNA-seq data showed that insulin induced an overall increase in the expression of genes associated with angiogenesis (GO: 0001525 with an average of 1.16-fold change, adjusted p = 0.045), vasculogenesis (GO: 0001570 with an average of 1.26-fold change, adjusted p = 0.110), and tube morphogenesis (GO: 0035239 with an average of 1.7 fold change, adjusted p = 0.045) (Table S1). Enrichment analysis of the SB431542-treated group, in the absence of insulin, when compared with untreated cells, did not show enrichment for angiogenesis (GO: 0001525) but showed enrichment for genes associated with blood vessel development (GO: 0001568) and blood vessel morphogenesis (GO: 0048514) (Table S2). Inhibition of TGF-β signaling using SB431542 resulted in overall decreases in the expression of genes associated with blood vessel development and morphogenesis of 0.58-fold (adjusted p = 0.032) and 0.63-fold (adjusted p = 0.01), respectively. Enrichment analysis, comparing the gene expression in response to insulin + SB431542 versus insulin, showed that addition of SB431542 to insulin conferred an overall 0.62-fold decrease in the expression of genes associated with blood vessel development (GO: 0001568, adjusted p = 0.014) (Table S3). Furthermore, treatment with SB431542 resulted in an overall 0.54-fold decrease in the expression of the gene set associated with cell migration (GO:0016477, adjusted p = 0.014) (Table S2), a process that is integral to angiogenesis and blood vessel development. Insulin also induced an overall 1.65-fold increase in gene expression associated with actin cytoskeleton organization (GO: 0030036, adjusted p = 0.022) (Table S1), which is consistent with increased cytoskeleton remodeling during cell migration. Addition of SB431542 to insulin resulted in an overall decrease of 0.62-fold with adjusted p = 0.014, when compared with insulin treatment (Table S3). These transcriptome analyses suggest that autocrine TGF-β/Smad signaling contributes to and integrates into insulin-induced angiogenic and blood vessel development gene responses.
TGF-β Signaling Participates in the Insulin-Induced Response of Target Genes
Based on the enrichment analyses (Tables S1, S2, and S3), we next quantified the contribution of autocrine TGF-β signaling to the insulin-induced expression of several individual genes associated with angiogenesis (Figures 2A and S2A). For this purpose, we evaluated the mRNA levels by qRT-PCR after 90 min and 6 h of insulin treatment, in the absence or presence of SB431542. The selected genes comprised genes encoding transcription factors, enzymes, and transmembrane receptors and secreted growth factors with relevance to angiogenesis. We observed different temporal patterns of induction among the genes analyzed. Some showed moderate induction in response to insulin that persisted or increased by 6 h, such as SRF, RUNX1, PTGS2, HMOX1, TNFRSF12A, and GJA5 (Figure 2A). Other genes, however, showed transient induction at 90 min that diminished by 6 h. This is most apparent for the ANGPTL4 and PDGFA genes (Figure 2A). Most genes examined showed a contribution of autocrine TGF-β signaling to the insulin response. In most cases, the insulin-induced mRNA expression was inhibited or attenuated when TGF-β signaling was blocked, indicating dependence on the elevated autocrine TGF-β signaling. In contrast, SB431542 enhanced the expression of HMOX1 and ANGPT2, indicating that autocrine TGF-β signaling represses the induction of these two genes. These results are largely consistent with the RNA-seq data for both insulin-induced changes and the effect of TGF-β inhibition at 90 min and further illustrate that the induction of autocrine TGF-β signaling promotes or controls the insulin-induced gene responses.
Figure 2.

Relative mRNA Levels of Selected Genes, Encoding Transcription Factors, Enzymes, Membrane Proteins, and Ligands or Secreted Proteins, Known to be Involved in Angiogenesis
(A) mRNA expression of the indicated genes was measured using qRT-PCR, and values were normalized to RPL19 mRNA. Error bars indicate standard error of the means, based on three independent experiments. Inhibition of TGF-β signaling using SB431542 (SB) repressed the insulin-induced changes of the mRNA expression of many but not all genes. The statistical significance was determined by Wilcoxon test; *p < 0.05, **p < 0.0083.
(B) Selected disease and Bio function analysis using IPA comparing different treatment groups. Values are activation Z scores. Z score represents the IPA regulation trend (Z score > 0: up-regulation [blue]; Z score < 0: down-regulation [red]).
The identities of insulin-responsive genes regulated by autocrine TGF-β signaling are informative in assessing the role of TGF-β in insulin-induced angiogenic gene responses. Among the intracellular signaling effectors, the induction of cyclooxygenase-2 expression (Howe, 2007, Tsujii et al., 1998), encoded by PTGS2, fully depended on TGF-β signaling, whereas the expression of heme-oxygenase 1 from the HMOX1 gene (Loboda et al., 2008) was repressed by autocrine TGF-β signaling. Remarkably, the insulin-induced expression of transcription factors known to contribute to angiogenic responses, namely, SRF and RUNX1 (Franco et al., 2008, Franco et al., 2013, Franco and Li, 2009, Iwatsuki et al., 2005, Lam et al., 2017, Sangpairoj et al., 2017), depended on autocrine TGF-β signaling (Figure 2A). Equally if not more striking is the substantial role of the increased autocrine TGF-β signaling in the induction by insulin of genes encoding secreted cytokines that promote angiogenesis and vasculogenesis (Babapoor-Farrokhran et al., 2015, Carmeliet et al., 1996, Leung et al., 1989, Okochi-Takada et al., 2014, Perdiguero et al., 2011; Risau et al., 1992). Specifically, the insulin-induced expression of angiopoietin-like 4 (ANGPTL4) and PDGFA depended on TGF-β signaling, whereas autocrine TGF-β signaling repressed the expression of the secreted angiogenic factor angiopoietin-2 (ANGPT2) and the VEGF-related placental growth factor (Figure 2A). Finally, insulin-induced expression of the “TNF receptor-related weak inducer of apoptosis” (TWEAKR, encoded by TNFRSF12A), which also promotes angiogenesis (Wiley et al., 2001), also depended on TGF-β signaling. We also observed transcripts with little to no change in response to insulin or SB431542. FLT1, which encodes a VEGF receptor, and MMP2 encoding matrix metalloprotease 2 and ADAM15, which associate with extracellular matrix remodeling, were some genes that showed little to no changes (Figure S2A). Although our data of differential VEGFA expression did not pass our p < 0.05 cutoff in the enrichment analysis, insulin has been reported to increase VEGFA expression in certain cells (Hale et al., 2013, Jiang et al., 2003, Lu et al., 1999, Miele et al., 2000). Quantified using qRT-PCR, VEGFA mRNA was not increased by 90 min and was modestly increased by 6 h in response to 100 nM insulin (Figure S2A). Increasing the insulin concentration, however, revealed a concentration-dependent increase of VEGFA mRNA expression by 6 h, which fully depended on autocrine TGF-β/Smad signaling, as it was prevented by SB431542 (Figure S2A and S2B). Addition of the pan-TGF-β neutralizing 1D11 antibody, which prevents ligand binding to its receptors, confirmed the participation of autocrine TGF-β signaling in insulin-induced expression changes of select target genes, similar to the addition of SB431542 but often less pronounced (Figure S2C). Together, these data further support the contribution and roles of TGF-β signaling in insulin-induced angiogenesis gene responses.
Ingenuity pathway analysis (IPA) of the RNA-seq data revealed activation as well as inhibition of states of “associated diseases and biological functions” (Table S4). We focused our attention on five IPA-based biological functions that are altered in response to insulin and relevant to angiogenesis (Figure 2B). Insulin-activated transcripts included those associated with “cell migration,” “cell movement,” “invasion,” as well as “angiogenesis” and “vasculogenesis.” SB431542 in the absence of insulin inhibited gene responses associated with cell migration, movement, and invasion but activated those associated with angiogenesis and vasculogenesis, strongly suggesting that autocrine TGF-β signaling promotes cell migration and invasion and has an inhibitory effect on some genes associated with angiogenesis and vasculogenesis in HUVECs (Table S4). Combining SB431542 with insulin resulted in repression of all five insulin-induced response groups, thus reflecting contributions of autocrine TGF-β signaling to the insulin responses of these functional groups of genes (Figure 2B).
Autocrine TGF-β Signaling Promotes Insulin-Induced Cell Migration
Insulin has been shown to stimulate cell migration (Budi et al., 2015, Jin et al., 2014, Liu et al., 2009c, Shanley et al., 2004), which is now also supported by our transcriptome analyses of insulin-treated endothelial cells (Figure 2B). Since endothelial cell migration is integral to the angiogenic response, we asked whether insulin promotes migration of primary HUVECs and whether the increased autocrine TGF-β signaling contributes to the insulin-induced migration response. Serum-starved cell monolayers were “wounded” by scratching and stimulated for 8 h with insulin in the presence or absence of SB431542 or the TGF-β neutralizing antibody 1D11. Insulin promoted endothelial cell migration, scored by measuring the width of the cell-free wound before and after treatment (Figure 3A). The TβRI kinase inhibitors SB431542 and LY2157299 and the 1D11 anti-TGF-β antibody prevented the insulin-induced cell migration (Figures 3A, 3B, and S3A). No differences in cell number were apparent after the 8 h time interval used to score cell migration (Figure S3B). As in HUVECs, LY2157299, SB431542, and the 1D11 antibody also blocked the insulin-induced migratory response of microvascular endothelial cells (Figures S3A and S3C).
Figure 3.

Contribution of TGF-β Signaling to Insulin-Induced Cell Migration and Invasion
(A–C) Migration of HUVECs measured in a monolayer wounding assay. Confluent monolayers were scratched with a pipette tip at time 0 and cells were allowed to migrate into the wounded area for 8 h, in the presence or absence of insulin with or without SB431542 (A and B) or the neutralizing anti-TGF-β antibody 1D11 (C). The migration distance is graphically presented. Error bars indicate standard error of the means of three experiments. The statistical significance was determined by Wilcoxon test; *p < 0.05, **p < 0.0083.
(D and E) mRNA expression of RHOB, encoding a Rho-related GTP-binding protein, SNAI1 and SNAI2 (D), as well as CDH5, encoding VE-cadherin and FN, encoding fibronectin (E), after 90 min or 6 h insulin treatment, in the presence or absence of SB431542, determined by qRT-PCR and normalized against RPL19 mRNA. Statistical significance was determined by Wilcoxon test; *p < 0.05, **p < 0.0083. Ab, antibody.
(F) Immunoblots of extracted HUVEC lysates treated for 48 h in the presence or absence of insulin with or without SB431542. Protein expression changes were assessed by immunoblotting for Snail2, N-cadherin, VE-cadherin, and fibronectin, with GAPDH as loading control. IB, immunoblot.
(G) Invasion of HUVECs in Transwell assays in the presence or absence of insulin, with or without SB431542 or 1D11 antibody. HUVECs were treated with insulin with or without SB431542 or the anti-TGF-β neutralizing antibody 1D11 for 8 h, and the invaded cells at the bottom filter surface were DAPI-stained and counted. The microscopic fields show cells that invaded through the filter as black dots against a white background. The results, shown graphically, are averages of five random microscopic fields of three separate experiments, each conducted in duplicate.
Scale bar, 100 μM. Statistical significance was determined by Wilcoxon test; *p < 0.05, **p < 0.0083. ns, non-significant. SB, SB431542. Ins, insulin.
We also observed concomitant increases in the expression of several genes that might contribute to the increased cell migration. qRT-PCR analyses showed that insulin enhanced the expression of RHOB, a gene implicated in activation of cell migration (Howe and Addison, 2012) after 6 h, albeit not yet at 90 min, and this increase was prevented when TGF-β signaling was blocked using SB431542 (Figure 3D). Insulin also induced the expression of SNAI1 and SNAI2 (Figures 3D and 3F), which are known to drive the process of endothelial-mesenchymal transition that enables the cells to acquire a migratory and invasive phenotype (Lamouille et al., 2014). We also observed an increased fibronectin mRNA expression, consistent with initiation of endothelial-mesenchymal transition, although decreased VE-cadherin mRNA expression was not yet apparent after 6 h of insulin (Figure 3E). That the cells undergo at least a partial endothelial-mesenchymal transition is more apparent after 48 h of insulin treatment, which resulted in increased fibronectin and N-cadherin expression and decreased VE-cadherin expression (Figure 3F). The insulin-induced increase in RhoB expression and changes in the expression of genes that characterize endothelial-mesenchymal transition were prevented in the presence of SB431542 (Figures 3D–3F), indicating contributions of autocrine TGF-β signaling. These data strongly suggest that insulin-stimulated cell migration requires autocrine TGF-β signaling and is facilitated by changes in gene expression that mark endothelial-mesenchymal transition and are induced by the insulin-induced increase in autocrine TGF-β signaling.
We also evaluated whether TGF-β signaling plays a role in insulin-induced invasion of HUVECs using Transwell assays that quantify the number of cells that invade through extracellular matrix (™Matrigel) (Figure 3G). Cells were treated for 8 h with or without insulin in the absence or presence of SB431542 or the 1D11 antibody, and in the presence of mitomycin C, which was added to inhibit cell proliferation (Coomber, 1992, Wu et al., 2015). Addition of mitomycin C did not affect TGF-β-induced Smad activation and gene expression (Figure S3D). In the presence of insulin, the number of invading cells increased about 2-fold compared with control treatment (Figure 3G). Both the 1D11 antibody and SB431542 prevented insulin to enhance cell invasion, although SB431542 treatment resulted in an increased number of invading cells (Figure 3G). As with SB431542, inhibiting TGF-β signaling using LY2157299 also prevented an insulin-induced increase in cell invasion and induced a higher basal cell invasion (Figure S3E). These data suggest that insulin-induced autocrine TGF-β signaling participates in insulin-induced endothelial cell invasion, consistent with its role in cell migration and the insulin-induced cell plasticity response.
In endothelial cells, the type I receptor ALK1, also known as ACVRL1, has been shown to contribute to cell migration (David et al., 2007, Lamouille et al., 2002) and to also be activated by TGF-β (Goumans et al., 2003). We therefore evaluated the contribution of ALK1 to the insulin response. Insulin induces a mild increase of ALK1 at the cell surface with no change in mRNA expression (Figures S3F and S3G). However, this increase did not result in increased expression of ID1, a TGF-β-inducible ALK1 target gene (Figure S3G). Inhibition of ALK1 and related BMP-responsive type I receptors using LDN-193189 enhanced the basal level of cell migration and invasion and allowed insulin to decrease endothelial cell migration, without a significant effect on cell invasion (Figure S3H). These data suggest that ALK1 might participate in the insulin-induced cell migration response.
TGF-β Signaling Participates in Insulin-Induced Endothelial Network Formation in Culture
The ability of extracellular factors to control angiogenesis is often scored using a cell culture assay for endothelial network formation. In this assay, HUVECs, cultured on Matrigel, differentiate gradually with formation of cell adhesion junction complexes and form tube-like structures that connect to each other through node- and mesh-like structures (Arnaoutova and Kleinman, 2010, DeCicco-Skinner et al., 2014). Insulin has been shown to promote the formation of such an endothelial network when the endothelial cells are cultured in Matrigel. To examine whether autocrine TGF-β signaling contributes to the insulin-induced network formation, we performed these assays using HUVECs in the presence or absence of the TβRI kinase inhibitor SB431542 or the 1D11 antibody (Figure 4).
Figure 4.

Effects of Inhibiting TGF-β Signaling in Insulin-Induced Endothelial Network Formation
(A) HUVECs were plated onto Matrigel with or without insulin in the absence or presence of SB431542. Representative images are shown after 6 h of treatment.
(B and C) Quantitative analyses of numbers of nodes (red) and meshes (blue), relative to untreated controls, in experiments with or without SB431542 (B) or 1D11 antibody (C).
Error bars represent mean ± SEM of three independent experiments. Statistical significance was determined by Wilcoxon test; *p < 0.05, **p < 0.0083, ***p < 0.0001. Scale bar, 500 μM.
Besides visible changes in appearance, the formation of the network can be quantified using several parameters, most notably the number of nodes and meshes formed (Figure 4A). Insulin significantly enhanced the total number of nodes and meshes (Figures 4B and 4C), thus promoting network formation. Addition of SB431542 or the 1D11 antibody augmented these parameters of network formation, both in the absence or presence of insulin, although the antibody was less effective (Figures 4B and 4C). These results strongly suggest that autocrine TGF-β signaling dampens the basal and insulin-induced network formation and further illustrate the functional cross talk between the insulin-induced increase in autocrine TGF-β signaling and a response to insulin that is not secondary to TGF-β signaling.
Insulin and SB431542 Control Intersomitic Vessel Development in Zebrafish Embryos
Since insulin and inhibition of TGF-β signaling both enhanced the formation of endothelial tube-like structures in culture, we evaluated the effects of insulin and SB431542 on the development of the vasculature in transgenic fli:gfp zebrafish embryos (Lawson and Weinstein, 2002), in which endothelial cells express GFP and therefore are readily visible in the transparent embryos (Figure 5A). This in vivo model allows easy visualization of vascular patterning defects, especially since the vasculature of the trunk has an extremely regular pattern. Incubation of zebrafish embryos with high glucose results in the development of an additional vessel arising from the intersegmental vessel (ISV), and inhibiting Akt activation prevents this hyperbranching effect (Jorgens et al., 2015). To follow changes in glucose levels and effects of insulin, we analyzed the expression of phosphoenol-pyruvate carboxykinase 1 (PCK1), which is transcriptionally repressed in response to insulin and therefore a sensitive marker of blood glucose levels (Elo et al., 2007). This surrogate marker approach was necessary since the small amount of larval blood available precludes accurate analysis of insulin-induced changes in blood glucose levels. Adding insulin inhibited PCK1 expression (Figure 5B), illustrating the response to insulin.
Figure 5.

Effects of Insulin and SB431542 on Intersomitic Vessels in Zebrafish Embryos
(A) Scheme representing the treatment protocol. Tg(fli:gfp) zebrafish embryos were treated for 72 h.
(B) Expression of pck1 mRNA was quantified by qRT-PCR. RNA was extracted from pool of larvae that had received insulin with DMSO or DMSO control for 48 h.
(C) Altered blood vessels in insulin- and SB431542-treated embryos. Representative images show the trunk vasculature of 96 hpf tg(fli:EGFP) zebrafish larvae. The boxes mark the region shown at higher magnification in the lower right corner of the panel.
(D) Branching quantification of zebrafish blood vessels at 96 hpf, as seen in C in control solution DMSO, SB431542, insulin, and insulin + SB431542. Bars represent mean ± SD. Zf, zebrafish. Ctrl, control.
In this system, insulin treatment for 96 h induced hyper-branching of blood vessels, which appear as small vessel structures that sprout from the intersomitic vessels and grow horizontally toward neighboring intersomitic vessels (Figure 5C). This effect was similar to that of high glucose treatment (Jorgens et al., 2015). Inhibiting TGF-β signaling for 96 h using SB431542 induced the formation of misdirected small vessels that emerged from the upper part of the intersomitic vessels, whereas combining insulin with SB431542 resulted in both extra branching and misdirected vessels (Figures 5C and 5D). Even though our data support an angiogenic response to insulin, we could not dissect the direct contribution of TGF-β signaling to the insulin-induced intersomitic vessel sprouting. The changes in the vascular pattern when TGF-β signaling is inhibited suggest a role of TGF-β signaling during early embryo vessel formation that is independent of insulin-induced blood vessel hyper-branching.
Blocking TGF-β Signaling Inhibits Insulin-Induced Microvessel Outgrowth from Neonatal Aortic Rings
In addition to analysis in the zebrafish model, we evaluated the contribution of TGF-β signaling to insulin-induced microvessel outgrowth ex vivo, following tissue explant of neonatal mouse aortae, using an aortic ring assay (Bellacen and Lewis, 2009) (Figure 6A). This assay relies on the capacity of the aortic explants to form new endothelial microvessels, whose outgrowth involves interactions of different cell types, including endothelial cells, pericytes, fibroblasts, macrophages, and dendritic cells (Nicosia, 2009). In this assay, 1 mm transverse sections of aortae of 6-day-old mice are incubated in Matrigel for 96 h, allowing outgrowth of endothelial cells from the intimal layer. Consistent with its angiogenic effects, insulin stimulated microvessel sprouting from aortic rings that was more extensive than sprouting seen from control-treated aortae (Figures 6B and 6C). The insulin-promoted new microvessels were endothelial in nature, apparent by mTomato expression from the Cdh5 promoter (Figure S4), and their outgrowth was strongly attenuated in the presence of SB431542. Basal microvessel sprouting was minimal without addition of insulin, and SB431542 had no significant effect in reducing it (Figures 6B and 6C). A pan-neutralizing anti-TGF-β antibody that prevents receptor binding of all three TGF-β ligands also decreased insulin-induced microvessel outgrowth from aortic rings, whereas non-immune IgG had no effect (Figures 6B and 6C). These findings indicate that autocrine TGF-β/Smad signaling participates in and contributes to the angiogenic activity of insulin in the microvessel outgrowth assay.
Figure 6.

Contribution of TGF-β Signaling in Insulin-Induced Mouse Aortic Ring Microvessel Sprouting
(A) Scheme of the mouse aorta ring experiments. Segments of aorta ring from 6-day-old mice were embedded in Matrigel and incubated with insulin and/or SB431542 or a neutralizing anti-TGF-β antibody or control IgG for 96 h.
(B) Microvessel sprouting from aortic ring segments cultured ex vivo for 96 h with or without insulin in the presence or absence of SB431542 or anti-TGF-β antibody or control IgG. The microvessel growth in response to insulin is blocked by SB431542 or anti-TGF-β antibody in a mouse aorta ring assay. Representative images are shown.
(C) Quantitative analysis of the ex vivo microvessel growth, measured as surface area to which vessels are extended. Error bars show ±SD. The experiments were repeated twice. Ctrl, control.
Discussion
We previously reported that, in fibroblasts and epithelial cells, insulin induces, through Akt activation, the rapid mobilization of the TβRI and TβRII receptors from intracellular stores to the plasma membrane, thus enhancing TGF-β responsiveness and autocrine TGF-β signaling (Budi et al., 2015). This raised the possibility that insulin-induced autocrine TGF-β signaling through Smad2 or Smad3 participates in insulin-induced gene responses, which was confirmed for a few selected genes in immortalized epithelial and fibroblast cell lines (Budi et al., 2015). To evaluate the extent to which autocrine TGF-β signaling is integrated in the cellular response to insulin, we now studied the response of primary endothelial cells, a cell population that is directly exposed to insulin in circulation and whose angiogenic activation promotes the progression of a variety of diseases, including heart disease, diabetes-associated complications, fibroses, and cancer (Aoki et al., 2006, Escudero et al., 2017, Kizu et al., 2009, Pardali et al., 2017, Rajendran et al., 2013). Our results allowed us to make conclusions of physiological relevance. (1) Insulin increases the cell surface levels of the TGF-β receptors, TβRI and TβRII, through Akt activation in human primary endothelial cells, thus enhancing autocrine TGF-β signaling. (2) Insulin-induced autocrine TGF-β signaling participates in the insulin-induced transcriptional response of human endothelial cells, including gene responses associated with angiogenesis. (3) Insulin-induced autocrine TGF-β signaling participates in insulin-induced angiogenic responses, including cell migration and endothelial network formation in culture and angiogenesis ex vivo. In summary, we report that enhanced autocrine TGF-β signaling in response to insulin is integral to insulin-induced angiogenesis.
Our analyses of the transcriptome and mRNA expression of individual genes in response to insulin supports the notion that autocrine TGF-β signaling through TβRI and Smads integrates into the overall endothelial gene expression response to insulin and the expression of some genes that are known to function in angiogenesis. Indeed, many insulin-induced gene responses were inhibited or attenuated when TGF-β-induced Smad activation was blocked. Among these genes, several encode transcription factors that contribute to the angiogenic response, e.g., SRF and RUNX1 (Franco et al., 2008, Franco et al., 2013, Franco and Li, 2009, Iwatsuki et al., 2005, Lam et al., 2017), and their induction in response to insulin depended on autocrine TGF-β signaling. Even more striking is the contribution of autocrine TGF-β signaling to the activation of insulin-induced genes encoding secreted, pro-angiogenic cytokines. Specifically, the insulin-induced expression of angiopoietin-like 4, PDGF-A, and VEGF-A, all of which have been individually linked to induction of angiogenesis (Andrae et al., 2008, Babapoor-Farrokhran et al., 2015, Carmeliet et al., 1996, Heldin, 2013, Hu et al., 2016, Leung et al., 1989, Okochi-Takada et al., 2014, Perdiguero et al., 2011, Risau et al., 1992), depended on TGF-β signaling. The induction of an angiogenic response to insulin has been functionally linked to activation of VEGF-A expression (Liu et al., 2009b, Lu et al., 1999, Yamagishi et al., 1999), whereas the VEGFA gene is known to be directly induced by Smad3 in response to TGF-β (Kobayashi et al., 2005, Shi et al., 2014). Moreover, increased ANGPTL4 expression is commonly seen in diabetic retinopathy (Babapoor-Farrokhran et al., 2015). Additionally, consistent with the notion that developing nerves and blood vessels use similar guidance mechanisms (Arese et al., 2011, Carmeliet, 2003), insulin induced changes in the expression of genes that participate in axon development and guidance (Inoue et al., 2008, Wickramasinghe et al., 2008, Yoshikawa et al., 2007), such as RHOB, SRF, RUNX1, and also their induction was inhibited by blocking TGF-β signaling. This suggests that TGF-β signaling may also participate in insulin-induced changes in neurogenesis and neuronal guidance.
Insulin has been shown to stimulate angiogenesis, as assessed by cell culture and ex vivo and in vivo assays (Escudero et al., 2017). Subcutaneous injection of insulin in mice induces the development of new and extensively branched microvessels (Dhall et al., 2016, Liu et al., 2009b), and topic insulin injection into foot ulcer wounds of diabetic patients promotes increased microvessel density that helps improve wound healing (Zhang and Lv, 2016). To define the contribution of autocrine TGF-β signaling in insulin-induced angiogenic responses, we used the pharmacological inhibitor SB431542 to block the TβRI kinase activity, and, consequently, TGF-β-induced Smad activation, and verified key results using another TβRI kinase inhibitor, LY2157299 (galunisertib), and/or specific pan-neutralizing anti-TGF-β monoclonal antibodies that prevent all TGF-β signaling. Generating primary HUVECs that stably and quantitatively lack TβRII or TβRI expression by small interfering RNA-mediated silencing was not feasible, and their silencing appears to be detrimental to these endothelial cells in culture. We used several assays that mimic major aspects of angiogenesis to assess the contribution of TGF-β signaling. Endothelial cell migration is essential to angiogenesis (Lamalice et al., 2007), and migration in many different cell lines is known to be induced by insulin (Budi et al., 2015, Jin et al., 2014, Liu et al., 2009c, Shanley et al., 2004). Our results indicate that insulin does not induce endothelial cell motility when TGF-β signaling is blocked and therefore that insulin-induced cell migration depends on autocrine TGF-β signaling. Furthermore, neutralizing anti-TGF-β antibody, SB431542, and LY2157299 prevented the insulin-induced increase of invasiveness, assessed in Transwell assays, although blocking TGF-β signaling enhanced the basal level of cell invasion. These results strongly suggest that TGF-β signaling participates in insulin-induced invasion. Since both kinase inhibitors block the TβRI kinase and thus prevent Smad2 and Smad3 activation, and the neutralizing 1D11 antibody prevents ligand binding to the receptors, and thus all ligand-induced TGF-β signaling pathways, differences in their effects may be anticipated and may highlight contributions of non-Smad signaling, e.g., of TGF-β-induced Akt-mTOR activation, which does not depend on the TβRI kinase activity (Hamidi et al., 2017). Additionally, TGF-β1-induced ALK1 activation, which is not prevented by SB431542, may contribute to cell invasion, although insulin did not induce ID1 expression in HUVECs (Figures S3G and S3H), and inhibition of other kinases by SB431542, such as the ALK4 or ALK7 receptors, may also affect the outcome.
Based on our data, we surmise that TGF-β-induced Smad3 activation may participate in insulin-induced endothelial cell migration and invasion, in part through the induction of a mild or partial endothelial-mesenchymal transition (EndoMT) response. Indeed, insulin mildly induced the expression of SNAI1 and SNAI2, which are known to drive EndoMT, are activated by Smad3 in response to TGF-β, and are required for endothelial cell invasion (Lamouille et al., 2014). Insulin also induced increased fibronectin and N-cadherin expression and decreased VE-cadherin expression, which also mark EndoMT (Goumans et al., 2008, Piera-Velazquez et al., 2011). These EndoMT-defining responses were blocked when TGF-β signaling was blocked, highlighting the contribution of TGF-β signaling.
The endothelial network formation that we observed in vitro and in the mouse aortic ring assay provides strong evidence for participation of TGF-β signaling in the angiogenic responses to insulin. However, we did not see a repression of insulin-induced endothelial network formation when TGF-β signaling was inhibited using either SB431542 or the 1D11 antibody but instead observed an increase in basal and insulin-induced mesh and node formation. The enhanced formation of nodes and mesh-like structures, when TGF-β signaling is inhibited, is reminiscent of the SB431542-induced increase in VEGF-induced capillary sprouting by HUVECs in a spheroid angiogenesis assay (Liu et al., 2009d). Inhibition of TGF-β signaling strongly attenuated the ability of insulin to induce microvessel outgrowth from aortic ring segments ex vivo. This effect, similar to the requirement of TGF-β signaling for insulin-induced endothelial cell migration, highlights the contribution of autocrine TGF-β signaling to the insulin-induced angiogenic response. These observations are in line with the pro-angiogenic effects of TGF-β in the same microvessel outgrowth assay (Zhao et al., 2017) as well as in corneal and chicken chorioallantoic membrane angiogenesis assays (Phillips et al., 1994, Yang and Moses, 1990) and the requirement for TGF-β in murine vascular development (Dickson et al., 1995). Altogether, our data provide strong support for the participation of the insulin-induced increase in TGF-β responsiveness and autocrine TGF-β signaling in insulin-induced angiogenic responses.
We recognize that both ALK1 and TβRI/ALK5 signaling mediate TGF-β responses in endothelial cells and angiogenesis (Goumans et al., 2002, Lamouille et al., 2002, Liu et al., 2009d, Mallet et al., 2006, Oh et al., 2000, Rochon et al., 2016), and it has been proposed that the activities from both type I receptors may balance each other in the control of angiogenesis (Goumans et al., 2002, Oh et al., 2000). Our results using the kinase inhibitors SB431542 and LY2157299 (galunisertib) and neutralizing anti-TGF-β antibodies revealed that TGF-β signaling, and specifically TGF-β signaling through TβRI/ALK5, contributes to insulin-induced angiogenic response and target gene expression. They are, however, not informative about a possible role of ALK1 in insulin-induced angiogenic responses. Such role may be suggested by the effect of the small molecule inhibitor LDN-193189, which targets ALK1 as well as related BMP type I receptors, on insulin-induced cell migration and invasion, although LDN-193189 did not affect insulin-induced ID1 expression, a Smad1/5-directed target of activated ALK1 and other BMP type 1 receptors. Further research is required to address possible contributions of ALK1 or related BMP type I receptors in insulin-induced angiogenesis.
Although the insulin levels used in this study were higher than the normal insulin levels in blood, our findings may have substantial implications for the understanding of diabetes-associated complications in patients treated with insulin. Excessive formation of immature blood vessels is seen in association with tissue fibrosis, e.g., in the retina or kidney, in diabetic patients (Forbes and Cooper, 2013, Martin et al., 2003, Nakagawa et al., 2009), and, based on our findings, may result in part from the increased TGF-β responsiveness and the integration of enhanced autocrine TGF-β signaling in response to insulin. Furthermore, many diabetes-associated complications are associated with fibrosis (Ban and Twigg, 2008), which is driven by increased TGF-β signaling (Hills and Squires, 2011, Lamouille et al., 2014, Van Geest et al., 2010). Accordingly, these fibrotic lesions show increased Smad3 activation (Inazaki et al., 2004, Kim et al., 2003). As we reported before, the increased TGF-β signaling may be initiated by high glucose- or insulin-induced increases in TGF-β responsiveness (Budi et al., 2015, Wu and Derynck, 2009). Our findings may also be relevant in the context of cancer progression, since carcinomas depend on tumor angiogenesis. Higher rates of certain cancer types have been associated with type 2 and type 1 diabetes and insulin plasma levels (Belfiore, 2007, Frasca et al., 2008, Giovannucci et al., 2010, Huang et al., 2011, Kalla Singh et al., 2011). Additionally, tumor cells themselves often show an increased expression of insulin-related growth factor (IGF) 1 (Pollak, 2008), which acts through insulin and IGF-1 receptors, similar to insulin (Varewijck and Janssen, 2012). The extent to which the insulin-induced upregulation of TGF-β responsiveness contributes to diabetes- or cancer-associated angiogenesis is worth consideration.
Limitations of the Study
We showed that insulin-induced autocrine TGF-β signaling contributes to many aspects of angiogenesis, in cell culture, ex vivo, and in zebrafish. Owing to technical limitations and, in the case of humans, to ethical considerations, the importance of insulin-induced autocrine TGF-β signaling in these same aspects of angiogenesis may not be defined in the whole organism in vivo, i.e., in mice or humans. Our RNA-seq study showed integration of TGF-β signaling in the insulin-regulated expression of many genes that are associated with angiogenesis. Levels of RNA may serve as useful indication of their regulation but are not necessarily predictive of the protein levels. Further study is needed to examine the abundance of the proteins expressed by genes associated with angiogenesis in endothelial cells. In vertebrates, the integrity and functions of endothelial cells that line the vessel wall are defined through functional contacts with many cells, including pericytes, smooth muscle cells, and blood cells. Our study focuses on the endothelial cells, with less focus on these other cells that provide additional signals to the endothelial cells. Lastly, consistent with most if not all studies in cell culture, insulin used in this study was used at higher levels than those that are seen as normal physiological levels in circulation.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank the UCSF zebrafish core facility for zebrafish care, Daniel Hart for the advice and suggestions on zebrafish work, Alex Williams from the Gladstone Bioinformatics core for RNA-seq analyses, Carlos O. Valenzuela at UCSF's Cardiovascular Research Institute for providing Tg(Cdh5-cre)1Spe x Gt(ROSA)26Sortm14(CAG-tdTomato)Hze mice, and K. Brückner and the Derynck laboratory members for valuable suggestions. This research was sponsored by NIH grant RO1-CA136690 to R.D. and RO1-HL122869 to R.J.A. and a Juvenile Diabetes Research Fund (JDRF) advanced postdoctoral fellowship 3-APF-2017-392-A-N support to E.H.B.
Author Contributions
Conceptualization, E.H.B. and R.D.; Methodology, E.H.B., R.D., and R.J.A.; Investigation, E.H.B., S.H., and O.M.; Writing – Original Draft, E.H.B. and R.D.; Writing – Review and & Editing, E.H.B., R.D., O.M, and R.J.A.; Funding Acquisition, E.H.B., R.D., and R.J.A; Resources, E.H.B., R.D., O.M, and R.J.A.; Supervision, R.D. and R.J.A.
Declaration of Interests
The authors have no competing interests. R.D. is a Founder of Pliant Therapeutics and a consultant to Genentech, Inc. and NGM Biopharmaceuticals, Inc. R.J.A. has sponsored research agreements with Pfizer Corp. and Plexxikon and is a co-inventor of a patent on the use of anti-TGF-β antibodies that is co-owned by UCSF and Xoma.
Published: January 25, 2019
Footnotes
Supplemental Information includes Transparent Methods, four figures, and five tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.12.038.
Supplemental Information
Data comparing insulin treatment and control treatment are organized by lowest adjusted p-value and p < 0.05 was considered as a statistically significant difference.
Data comparing SB431542 treatment and control treatment are organized by lowest adjusted p-value, and p < 0.05 was considered as a statistically significant difference.
Data comparing insulin + SB431542 treatment and insulin treatment are organized by lowest adjusted p-value, and p < 0.05 was considered to indicate a statistically significant difference.
A positive or negative z-score value indicates that a function is predicted to be increased or decreased when compared to the indicated group.
Primer sequences are given in 5′ to 3′ direction.
References
- Andrae J., Gallini R., Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki R., Ikarugi H., Naemura A., Ijiri Y., Yamashita T., Yamamoto J. Endothelial dysfunction precedes atherosclerotic lesions and platelet activation in high fat diet-induced prothrombotic state. Thromb. Res. 2006;117:529–535. doi: 10.1016/j.thromres.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Arese M., Serini G., Bussolino F. Nervous vascular parallels: axon guidance and beyond. Int. J. Dev. Biol. 2011;55:439–445. doi: 10.1387/ijdb.103242ma. [DOI] [PubMed] [Google Scholar]
- Arnaoutova I., Kleinman H.K. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat. Protoc. 2010;5:628–635. doi: 10.1038/nprot.2010.6. [DOI] [PubMed] [Google Scholar]
- Babapoor-Farrokhran S., Jee K., Puchner B., Hassan S.J., Xin X., Rodrigues M., Kashiwabuchi F., Ma T., Hu K., Deshpande M. Angiopoietin-like 4 is a potent angiogenic factor and a novel therapeutic target for patients with proliferative diabetic retinopathy. Proc. Natl. Acad. Sci. U S A. 2015;112:E3030–E3039. doi: 10.1073/pnas.1423765112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban C.R., Twigg S.M. Fibrosis in diabetes complications: pathogenic mechanisms and circulating and urinary markers. Vasc. Health Risk Manag. 2008;4:575–596. doi: 10.2147/vhrm.s1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belfiore A. The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr. Pharm. Des. 2007;13:671–686. doi: 10.2174/138161207780249173. [DOI] [PubMed] [Google Scholar]
- Bellacen K., Lewis E.C. Aortic ring assay. J. Vis. Exp. 2009;33:1564. doi: 10.3791/1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budi E.H., Muthusamy B.P., Derynck R. The insulin response integrates increased TGF-β signaling through Akt-induced enhancement of cell surface delivery of TGF-β receptors. Sci. Signal. 2015;8:ra96. doi: 10.1126/scisignal.aaa9432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno L., de Alwis D.P., Pitou C., Yingling J., Lahn M., Glatt S., Troconiz I.F. Semi-mechanistic modelling of the tumour growth inhibitory effects of LY2157299, a new type I receptor TGF-β kinase antagonist, in mice. Eur. J. Cancer. 2008;44:142–150. doi: 10.1016/j.ejca.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Callahan J.F., Burgess J.L., Fornwald J.A., Gaster L.M., Harling J.D., Harrington F.P., Heer J., Kwon C., Lehr R., Mathur A. Identification of novel inhibitors of the transforming growth factor beta1 (TGF- β1) type 1 receptor (ALK5) J. Med. Chem. 2002;45:999–1001. doi: 10.1021/jm010493y. [DOI] [PubMed] [Google Scholar]
- Calleja V., Laguerre M., Parker P.J., Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 2009;7:e17. [Google Scholar]
- Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Blood vessels and nerves: common signals, pathways and diseases. Nat. Rev. Genet. 2003;4:710–720. doi: 10.1038/nrg1158. [DOI] [PubMed] [Google Scholar]
- Carmeliet P., Ferreira V., Breier G., Pollefeyt S., Kieckens L., Gertsenstein M., Fahrig M., Vandenhoeck A., Harpal K., Eberhardt C. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- Carmeliet P., Jain R.K. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- Cheng R., Ma J.X. Angiogenesis in diabetes and obesity. Rev. Endocr. Metab. Disord. 2015;16:67–75. doi: 10.1007/s11154-015-9310-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M.E., Ballermann B.J. Inhibition of capillary morphogenesis and associated apoptosis by dominant negative mutant transforming growth factor-beta receptors. J. Biol. Chem. 1995;270:21144–21150. doi: 10.1074/jbc.270.36.21144. [DOI] [PubMed] [Google Scholar]
- Coomber B.L. Influence of mitomycin C on endothelial monolayer regeneration in vitro. J. Cell Biochem. 1992;50:293–300. doi: 10.1002/jcb.240500310. [DOI] [PubMed] [Google Scholar]
- Cross M.J., Claesson-Welsh L. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 2001;22:201–207. doi: 10.1016/s0165-6147(00)01676-x. [DOI] [PubMed] [Google Scholar]
- David L., Mallet C., Vailhe B., Lamouille S., Feige J.J., Bailly S. Activin receptor-like kinase 1 inhibits human microvascular endothelial cell migration: potential roles for JNK and ERK. J. Cell Physiol. 2007;213:484–489. doi: 10.1002/jcp.21126. [DOI] [PubMed] [Google Scholar]
- DeCicco-Skinner K.L., Henry G.H., Cataisson C., Tabib T., Gwilliam J.C., Watson N.J., Bullwinkle E.M., Falkenburg L., O'Neill R.C., Morin A., Wiest J.S. Endothelial cell tube formation assay for the in vitro study of angiogenesis. J. Vis. Exp. 2014;91:e51312. doi: 10.3791/51312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhall S., Alamat R., Castro A., Sarker A.H., Mao J.H., Chan A., Hang B., Martins-Green M. Tobacco toxins deposited on surfaces (third hand smoke) impair wound healing. Clin. Sci. (Lond). 2016;130:1269–1284. doi: 10.1042/CS20160236. [DOI] [PubMed] [Google Scholar]
- Dickson M.C., Martin J.S., Cousins F.M., Kulkarni A.B., Karlsson S., Akhurst R.J. Defective haematopoiesis and vasculogenesis in transforming growth factor-β1 knock out mice. Development. 1995;121:1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- Elo B., Villano C.M., Govorko D., White L.A. Larval zebrafish as a model for glucose metabolism: expression of phosphoenolpyruvate carboxykinase as a marker for exposure to anti-diabetic compounds. J. Mol. Endocrinol. 2007;38:433–440. doi: 10.1677/JME-06-0037. [DOI] [PubMed] [Google Scholar]
- Escudero C.A., Herlitz K., Troncoso F., Guevara K., Acurio J., Aguayo C., Godoy A.S., Gonzalez M. Pro-angiogenic role of insulin: from physiology to pathology. Front. Physiol. 2017;8:204. doi: 10.3389/fphys.2017.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes J.M., Cooper M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013;93:137–188. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- Franco C.A., Blanc J., Parlakian A., Blanco R., Aspalter I.M., Kazakova N., Diguet N., Mylonas E., Gao-Li J., Vaahtokari A. SRF selectively controls tip cell invasive behavior in angiogenesis. Development. 2013;140:2321–2333. doi: 10.1242/dev.091074. [DOI] [PubMed] [Google Scholar]
- Franco C.A., Li Z. SRF in angiogenesis: branching the vascular system. Cell Adh. Migr. 2009;3:264–267. doi: 10.4161/cam.3.3.8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco C.A., Mericskay M., Parlakian A., Gary-Bobo G., Gao-Li J., Paulin D., Gustafsson E., Li Z. Serum response factor is required for sprouting angiogenesis and vascular integrity. Dev. Cell. 2008;15:448–461. doi: 10.1016/j.devcel.2008.07.019. [DOI] [PubMed] [Google Scholar]
- Frasca F., Pandini G., Sciacca L., Pezzino V., Squatrito S., Belfiore A., Vigneri R. The role of insulin receptors and IGF-I receptors in cancer and other diseases. Arch. Physiol. Biochem. 2008;114:23–37. doi: 10.1080/13813450801969715. [DOI] [PubMed] [Google Scholar]
- Giovannucci E., Harlan D.M., Archer M.C., Bergenstal R.M., Gapstur S.M., Habel L.A., Pollak M., Regensteiner J.G., Yee D. Diabetes and cancer: a consensus report. Diabetes Care. 2010;33:1674–1685. doi: 10.2337/dc10-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans M.J., Valdimarsdottir G., Itoh S., Lebrin F., Larsson J., Mummery C., Karlsson S., ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol. Cell. 2003;12:817–828. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- Goumans M.J., Valdimarsdottir G., Itoh S., Rosendahl A., Sideras P., ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002;21:1743–1753. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans M.J., van Zonneveld A.J., ten Dijke P. Transforming growth factor β-induced endothelial-to-mesenchymal transition: a switch to cardiac fibrosis? Trends Cardiovasc. Med. 2008;18:293–298. doi: 10.1016/j.tcm.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Hale L.J., Hurcombe J., Lay A., Santamaria B., Valverde A.M., Saleem M.A., Mathieson P.W., Welsh G.I., Coward R.J. Insulin directly stimulates VEGF-A production in the glomerular podocyte. Am. J. Physiol. Renal. Physiol. 2013;305:F182–F188. doi: 10.1152/ajprenal.00548.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi A., Song J., Thakur N., Itoh S., Marcusson A., Bergh A., Heldin C.H., Landström M. TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Sci. Signal. 2017;10:eaal4186. doi: 10.1126/scisignal.aal4186. [DOI] [PubMed] [Google Scholar]
- Hata A., Chen Y.G. TGF-β signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016;8:a022061. doi: 10.1101/cshperspect.a022061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimark R.L., Twardzik D.R., Schwartz S.M. Inhibition of endothelial regeneration by type-β transforming growth factor from platelets. Science. 1986;233:1078–1080. doi: 10.1126/science.3461562. [DOI] [PubMed] [Google Scholar]
- Heldin C.H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013;11:97. doi: 10.1186/1478-811X-11-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hills C.E., Squires P.E. The role of TGF-β and epithelial-to mesenchymal transition in diabetic nephropathy. Cytokine Growth Factor Rev. 2011;22:131–139. doi: 10.1016/j.cytogfr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- Howe G.A., Addison C.L. RhoB controls endothelial cell morphogenesis in part via negative regulation of RhoA. Vasc. Cell. 2012;4:1. doi: 10.1186/2045-824X-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe L.R. Inflammation and breast cancer. Cyclooxygenase/prostaglandin signaling and breast cancer. Breast Cancer Res. 2007;9:210. doi: 10.1186/bcr1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K., Babapoor-Farrokhran S., Rodrigues M., Deshpande M., Puchner B., Kashiwabuchi F., Hassan S.J., Asnaghi L., Handa J.T., Merbs S. Hypoxia-inducible factor 1 upregulation of both VEGF and ANGPTL4 is required to promote the angiogenic phenotype in uveal melanoma. Oncotarget. 2016;7:7816–7828. doi: 10.18632/oncotarget.6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Morehouse C., Streicher K., Higgs B.W., Gao J., Czapiga M., Boutrin A., Zhu W., Brohawn P., Chang Y. Altered expression of insulin receptor isoforms in breast cancer. PLoS One. 2011;6:e26177. doi: 10.1371/journal.pone.0026177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inazaki K., Kanamaru Y., Kojima Y., Sueyoshi N., Okumura K., Kaneko K., Yamashiro Y., Ogawa H., Nakao A. Smad3 deficiency attenuates renal fibrosis, inflammation, and apoptosis after unilateral ureteral obstruction. Kidney Int. 2004;66:597–604. doi: 10.1111/j.1523-1755.2004.00779.x. [DOI] [PubMed] [Google Scholar]
- Inoue K., Shiga T., Ito Y. Runx transcription factors in neuronal development. Neural Dev. 2008;3:20. doi: 10.1186/1749-8104-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatsuki K., Tanaka K., Kaneko T., Kazama R., Okamoto S., Nakayama Y., Ito Y., Satake M., Takahashi S., Miyajima A. Runx1 promotes angiogenesis by downregulation of insulin-like growth factor-binding protein-3. Oncogene. 2005;24:1129–1137. doi: 10.1038/sj.onc.1208287. [DOI] [PubMed] [Google Scholar]
- Jiang Z.Y., He Z., King B.L., Kuroki T., Opland D.M., Suzuma K., Suzuma I., Ueki K., Kulkarni R.N., Kahn C.R., King G.L. Characterization of multiple signaling pathways of insulin in the regulation of vascular endothelial growth factor expression in vascular cells and angiogenesis. J. Biol. Chem. 2003;278:31964–31971. doi: 10.1074/jbc.M303314200. [DOI] [PubMed] [Google Scholar]
- Jin S.Y., Kim E.K., Ha J.M., Lee D.H., Kim J.S., Kim I.Y., Song S.H., Shin H.K., Kim C.D., Bae S.S. Insulin regulates monocyte trans-endothelial migration through surface expression of macrophage-1 antigen. Biochim. Biophys. Acta. 2014;1842:1539–1548. doi: 10.1016/j.bbadis.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Jorgens K., Stoll S.J., Pohl J., Fleming T.H., Sticht C., Nawroth P.P., Hammes H.P., Kroll J. High tissue glucose alters intersomitic blood vessels in zebrafish via methylglyoxal targeting the VEGF receptor signaling cascade. Diabetes. 2015;64:213–225. doi: 10.2337/db14-0352. [DOI] [PubMed] [Google Scholar]
- Kalla Singh S., Brito C., Tan Q.W., De Leon M., De Leon D. Differential expression and signaling activation of insulin receptor isoforms A and B: a link between breast cancer and diabetes. Growth Factors. 2011;29:278–289. doi: 10.3109/08977194.2011.616200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.H., Kim B.K., Moon K.C., Hong H.K., Lee H.S. Activation of the TGF-β/Smad signaling pathway in focal segmental glomerulosclerosis. Kidney Int. 2003;64:1715–1721. doi: 10.1046/j.1523-1755.2003.00288.x. [DOI] [PubMed] [Google Scholar]
- Kizu A., Medici D., Kalluri R. Endothelial-mesenchymal transition as a novel mechanism for generating myofibroblasts during diabetic nephropathy. Am. J. Pathol. 2009;175:1371–1373. doi: 10.2353/ajpath.2009.090698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Liu X., Wen F.Q., Fang Q., Abe S., Wang X.Q., Hashimoto M., Shen L., Kawasaki S., Kim H.J. Smad3 mediates TGF-β1 induction of VEGF production in lung fibroblasts. Biochem. Biophys. Res. Commun. 2005;327:393–398. doi: 10.1016/j.bbrc.2004.12.032. [DOI] [PubMed] [Google Scholar]
- Kolka C.M., Bergman R.N. The endothelium in diabetes: its role in insulin access and diabetic complications. Rev. Endocr. Metab. Disord. 2013;14:13–19. doi: 10.1007/s11154-012-9233-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolluru G.K., Bir S.C., Kevil C.G. Endothelial dysfunction and diabetes: effects on angiogenesis, vascular remodeling, and wound healing. Int. J. Vasc. Med. 2012;2012:918267. doi: 10.1155/2012/918267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam J.D., Oh D.J., Wong L.L., Amarnani D., Park-Windhol C., Sanchez A.V., Cardona-Velez J., McGuone D., Stemmer-Rachamimov A.O., Eliott D. Identification of RUNX1 as a mediator of aberrant retinal angiogenesis. Diabetes. 2017;66:1950–1956. doi: 10.2337/db16-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamalice L., Le Boeuf F., Huot J. Endothelial cell migration during angiogenesis. Circ. Res. 2007;100:782–794. doi: 10.1161/01.RES.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- Lamouille S., Mallet C., Feige J.J., Bailly S. Activin receptor-like kinase 1 is implicated in the maturation phase of angiogenesis. Blood. 2002;100:4495–4501. doi: 10.1182/blood.V100.13.4495. [DOI] [PubMed] [Google Scholar]
- Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laping N.J., Grygielko E., Mathur A., Butter S., Bomberger J., Tweed C., Martin W., Fornwald J., Lehr R., Harling J. Inhibition of transforming growth factor (TGF)-β1-induced extracellular matrix with a novel inhibitor of the TGF-β type I receptor kinase activity: SB-431542. Mol. Pharmacol. 2002;62:58–64. doi: 10.1124/mol.62.1.58. [DOI] [PubMed] [Google Scholar]
- Larsson J., Goumans M.J., Sjostrand L.J., van Rooijen M.A., Ward D., Leveen P., Xu X., ten Dijke P., Mummery C.L., Karlsson S. Abnormal angiogenesis but intact hematopoietic potential in TGF-β type I receptor-deficient mice. EMBO J. 2001;20:1663–1673. doi: 10.1093/emboj/20.7.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson N.D., Weinstein B.M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 2002;248:307–318. doi: 10.1006/dbio.2002.0711. [DOI] [PubMed] [Google Scholar]
- Leung D.W., Cachianes G., Kuang W.J., Goeddel D.V., Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- Lindsley C.W., Zhao Z., Leister W.H., Robinson R.G., Barnett S.F., Defeo-Jones D., Jones R.E., Hartman G.D., Huff J.R., Huber H.E., Duggan M.E. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg. Med. Chem. Lett. 2005;15:761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Liu C., Xu P., Lamouille S., Xu J., Derynck R. TACE-mediated ectodomain shedding of the type I TGF-β receptor downregulates TGF-β signaling. Mol. Cell. 2009;35:26–36. doi: 10.1016/j.molcel.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Petreaca M., Martins-Green M. Cell and molecular mechanisms of insulin-induced angiogenesis. J. Cell Mol. Med. 2009;13:4492–4504. doi: 10.1111/j.1582-4934.2008.00555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Petreaca M., Yao M., Martins-Green M. Cell and molecular mechanisms of keratinocyte function stimulated by insulin during wound healing. BMC Cell Biol. 2009;10:1. doi: 10.1186/1471-2121-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Kobayashi K., van Dinther M., van Heiningen S.H., Valdimarsdottir G., van Laar T., Scharpfenecker M., Lowik C.W., Goumans M.J., ten Dijke P., Pardali E. VEGF and inhibitors of TGFβ type-I receptor kinase synergistically promote blood-vessel formation by inducing α5-integrin expression. J. Cell Sci. 2009;122:3294–3302. doi: 10.1242/jcs.048942. [DOI] [PubMed] [Google Scholar]
- Loboda A., Jazwa A., Grochot-Przeczek A., Rutkowski A.J., Cisowski J., Agarwal A., Jozkowicz A., Dulak J. Heme oxygenase-1 and the vascular bed: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2008;10:1767–1812. doi: 10.1089/ars.2008.2043. [DOI] [PubMed] [Google Scholar]
- Lu M., Amano S., Miyamoto K., Garland R., Keough K., Qin W., Adamis A.P. Insulin-induced vascular endothelial growth factor expression in retina. Invest. Ophthalmol. Vis. Sci. 1999;40:3281–3286. [PubMed] [Google Scholar]
- Mallet C., Vittet D., Feige J.J., Bailly S. TGFβ1 induces vasculogenesis and inhibits angiogenic sprouting in an embryonic stem cell differentiation model: respective contribution of ALK1 and ALK5. Stem Cells. 2006;24:2420–2427. doi: 10.1634/stemcells.2005-0494. [DOI] [PubMed] [Google Scholar]
- Martin A., Komada M.R., Sane D.C. Abnormal angiogenesis in diabetes mellitus. Med. Res. Rev. 2003;23:117–145. doi: 10.1002/med.10024. [DOI] [PubMed] [Google Scholar]
- Massague J., Kelly B. Internalization of transforming growth factor-β and its receptor in BALB/c 3T3 fibroblasts. J. Cell Physiol. 1986;128:216–222. doi: 10.1002/jcp.1041280212. [DOI] [PubMed] [Google Scholar]
- Michiels C. Endothelial cell functions. J. Cell Physiol. 2003;196:430–443. doi: 10.1002/jcp.10333. [DOI] [PubMed] [Google Scholar]
- Miele C., Rochford J.J., Filippa N., Giorgetti-Peraldi S., Van Obberghen E. Insulin and insulin-like growth factor-I induce vascular endothelial growth factor mRNA expression via different signaling pathways. J. Biol. Chem. 2000;275:21695–21702. doi: 10.1074/jbc.M000805200. [DOI] [PubMed] [Google Scholar]
- Morikawa M., Derynck R., Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016;8:a021873. doi: 10.1101/cshperspect.a021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T., Kosugi T., Haneda M., Rivard C.J., Long D.A. Abnormal angiogenesis in diabetic nephropathy. Diabetes. 2009;58:1471–1478. doi: 10.2337/db09-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicosia R.F. The aortic ring model of angiogenesis: a quarter century of search and discovery. J. Cell Mol. Med. 2009;13:4113–4136. doi: 10.1111/j.1582-4934.2009.00891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S.P., Seki T., Goss K.A., Imamura T., Yi Y., Donahoe P.K., Li L., Miyazono K., ten Dijke P., Kim S., Li E. Activin receptor-like kinase 1 modulates transforming growth factor-β1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. U S A. 2000;97:2626–2631. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okochi-Takada E., Hattori N., Tsukamoto T., Miyamoto K., Ando T., Ito S., Yamamura Y., Wakabayashi M., Nobeyama Y., Ushijima T. ANGPTL4 is a secreted tumor suppressor that inhibits angiogenesis. Oncogene. 2014;33:2273–2278. doi: 10.1038/onc.2013.174. [DOI] [PubMed] [Google Scholar]
- Oshima M., Oshima H., Taketo M.M. TGF-β receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 1996;179:297–302. doi: 10.1006/dbio.1996.0259. [DOI] [PubMed] [Google Scholar]
- Pardali E., Sanchez-Duffhues G., Gomez-Puerto M.C., ten Dijke P. TGF-β-induced endothelial-mesenchymal transition in fibrotic diseases. Int. J. Mol. Sci. 2017;18:E2157. doi: 10.3390/ijms18102157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdiguero E.G., Galaup A., Durand M., Teillon J., Philippe J., Valenzuela D.M., Murphy A.J., Yancopoulos G.D., Thurston G., Germain S. Alteration of developmental and pathological retinal angiogenesis in angptl4-deficient mice. J. Biol. Chem. 2011;286:36841–36851. doi: 10.1074/jbc.M111.220061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips G.D., Stone A.M., Schultz J.C., Jones B.D., Whitehead R.A., Knighton D.A. Whitehead R.A. and Knighton D.A., Transforming growth factor alpha (tgf-a) induced angiogenesis: direct versus indirect. Endothelium. 1994;2:297–303. [Google Scholar]
- Piera-Velazquez S., Li Z., Jimenez S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011;179:1074–1080. doi: 10.1016/j.ajpath.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- Rajendran P., Rengarajan T., Thangavel J., Nishigaki Y., Sakthisekaran D., Sethi G., Nishigaki I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013;9:1057–1069. doi: 10.7150/ijbs.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risau W., Drexler H., Mironov V., Smits A., Siegbahn A., Funa K., Heldin C.H. Platelet-derived growth factor is angiogenic in vivo. Growth Factors. 1992;7:261–266. doi: 10.3109/08977199209046408. [DOI] [PubMed] [Google Scholar]
- Robertson I.B., Rifkin D.B. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb. Perspect. Biol. 2016;8:a021907. doi: 10.1101/cshperspect.a021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochon E.R., Menon P.G., Roman B.L. Alk1 controls arterial endothelial cell migration in lumenized vessels. Development. 2016;143:2593–2602. doi: 10.1242/dev.135392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangpairoj K., Vivithanaporn P., Apisawetakan S., Chongthammakun S., Sobhon P., Chaithirayanon K. RUNX1 regulates migration, invasion, and angiogenesis via p38 MAPK pathway in human glioblastoma. Cell. Mol. Neurobiol. 2017;37:1243–1255. doi: 10.1007/s10571-016-0456-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano T., Kawata K., Ohno S., Yugi K., Kakuda H., Kubota H., Uda S., Fujii M., Kunida K., Hoshino D. Selective control of up-regulated and down-regulated genes by temporal patterns and doses of insulin. Sci. Signal. 2016;9:ra112. doi: 10.1126/scisignal.aaf3739. [DOI] [PubMed] [Google Scholar]
- Shanley L.J., McCaig C.D., Forrester J.V., Zhao M. Insulin, not leptin, promotes in vitro cell migration to heal monolayer wounds in human corneal epithelium. Invest. Ophthalmol. Vis. Sci. 2004;45:1088–1094. doi: 10.1167/iovs.03-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X., Guo L.W., Seedial S.M., Si Y., Wang B., Takayama T., Suwanabol P.A., Ghosh S., DiRenzo D., Liu B., Kent K.C. TGF-β/Smad3 inhibit vascular smooth muscle cell apoptosis through an autocrine signaling mechanism involving VEGF-A. Cell Death Dis. 2014;5:e1317. doi: 10.1038/cddis.2014.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahergorabi Z., Khazaei M. Imbalance of angiogenesis in diabetic complications: the mechanisms. Int. J. Prev. Med. 2012;3:827–838. doi: 10.4103/2008-7802.104853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi C.M., Emanuelli B., Kahn C.R. Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Tsujii M., Kawano S., Tsuji S., Sawaoka H., Hori M., DuBois R.N. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- Van Geest R.J., Klaassen I., Vogels I.M., Van Noorden C.J., Schlingemann R.O. Differential TGF-β signaling in retinal vascular cells: a role in diabetic retinopathy? Invest. Ophthalmol. Vis. Sci. 2010;51:1857–1865. doi: 10.1167/iovs.09-4181. [DOI] [PubMed] [Google Scholar]
- van Meeteren L.A., ten Dijke P. Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res. 2012;347:177–186. doi: 10.1007/s00441-011-1222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varewijck A.J., Janssen J.A. Insulin and its analogues and their affinities for the IGF1 receptor. Endocr. Relat. Cancer. 2012;19:F63–F75. doi: 10.1530/ERC-12-0026. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe S.R., Alvania R.S., Ramanan N., Wood J.N., Mandai K., Ginty D.D. Serum response factor mediates NGF-dependent target innervation by embryonic DRG sensory neurons. Neuron. 2008;58:532–545. doi: 10.1016/j.neuron.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley S.R., Cassiano L., Lofton T., Davis-Smith T., Winkles J.A., Lindner V., Liu H., Daniel T.O., Smith C.A., Fanslow W.C. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity. 2001;15:837–846. doi: 10.1016/s1074-7613(01)00232-1. [DOI] [PubMed] [Google Scholar]
- Wu F., Song H., Zhang Y., Mu Q., Jiang M., Wang F., Zhang W., Li L., Li H., Wang Y. Irisin induces angiogenesis in human umbilical vein endothelial cells in vitro and in zebrafish embryos in vivo via activation of the ERK signaling pathway. PLoS One. 2015;10:e0134662. doi: 10.1371/journal.pone.0134662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Derynck R. Essential role of TGF-β signaling in glucose-induced cell hypertrophy. Dev. Cell. 2009;17:35–48. doi: 10.1016/j.devcel.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi S., Kawakami T., Fujimori H., Yonekura H., Tanaka N., Yamamoto Y., Urayama H., Watanabe Y., Yamamoto H. Insulin stimulates the growth and tube formation of human microvascular endothelial cells through autocrine vascular endothelial growth factor. Microvasc. Res. 1999;57:329–339. doi: 10.1006/mvre.1999.2145. [DOI] [PubMed] [Google Scholar]
- Yang E.Y., Moses H.L. Transforming growth factor β1-induced changes in cell migration, proliferation, and angiogenesis in the chicken chorioallantoic membrane. J. Cell Biol. 1990;111:731–741. doi: 10.1083/jcb.111.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa M., Senzaki K., Yokomizo T., Takahashi S., Ozaki S., Shiga T. Runx1 selectively regulates cell fate specification and axonal projections of dorsal root ganglion neurons. Dev. Biol. 2007;303:663–674. doi: 10.1016/j.ydbio.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Lv L. Effect of local insulin injection on wound vascularization in patients with diabetic foot ulcer. Exp. Ther. Med. 2016;11:397–402. doi: 10.3892/etm.2015.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M., Hu Y., Jin J., Yu Y., Zhang S., Cao J., Zhai Y., Wei R., Shou J., Cai W. Interleukin 37 promotes angiogenesis through TGF-β signaling. Sci. Rep. 2017;7:6113. doi: 10.1038/s41598-017-06124-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z., Leister W.H., Robinson R.G., Barnett S.F., Defeo-Jones D., Jones R.E., Hartman G.D., Huff J.R., Huber H.E., Duggan M.E., Lindsley C.W. Discovery of 2,3,5-trisubstituted pyridine derivatives as potent Akt1 and Akt2 dual inhibitors. Bioorg. Med. Chem. Lett. 2005;15:905–909. doi: 10.1016/j.bmcl.2004.12.062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data comparing insulin treatment and control treatment are organized by lowest adjusted p-value and p < 0.05 was considered as a statistically significant difference.
Data comparing SB431542 treatment and control treatment are organized by lowest adjusted p-value, and p < 0.05 was considered as a statistically significant difference.
Data comparing insulin + SB431542 treatment and insulin treatment are organized by lowest adjusted p-value, and p < 0.05 was considered to indicate a statistically significant difference.
A positive or negative z-score value indicates that a function is predicted to be increased or decreased when compared to the indicated group.
Primer sequences are given in 5′ to 3′ direction.