

Abstract
DNA repair defects are inborn errors of immunity that result in increased apoptosis and oncogenesis. DNA Ligase 4-deficient patients suffer from a wide range of clinical manifestations since early in life, including: microcephaly, dysmorphic facial features, growth failure, developmental delay, mental retardation; hip dysplasia, and other skeletal malformations; as well as a severe combined immunodeficiency, radiosensitivity, and progressive bone marrow failure; or, they may present later in life with hematological neoplasias that respond catastrophically to chemo- and radiotherapy; or, they could be asymptomatic. We describe the clinical, laboratory, and genetic features of five Mexican patients with LIG4 deficiency, together with a review of 36 other patients available in PubMed Medline. Four out of five of our patients are dead from lymphoma or bone marrow failure, with severe infection and massive bleeding; the fifth patient is asymptomatic despite a persistent CD4+ lymphopenia. Most patients reported in the literature are microcephalic females with growth failure, sinopulmonary infections, hypogammaglobulinemia, very low B-cells, and radiosensitivity; while bone marrow failure and malignancy may develop at a later age. Dysmorphic facial features, congenital hip dysplasia, chronic liver disease, gradual pancytopenia, lymphoma or leukemia, thrombocytopenia, and gastrointestinal bleeding have been reported as well. Most mutations are compound heterozygous, and all of them are hypomorphic, with two common truncating mutations accounting for the majority of patients. Stem-cell transplantation after reduced intensity conditioning regimes may be curative.
Keywords: DNA repair defects, ligase IV deficiency, primary immunodeficiency, inborn error of immunity, case series, clinical spectrum
Introduction
Inborn errors of immunity are a group of over 350 individually rare congenital diseases that result from mutations in one or more genes coding for proteins of the immune system. DNA repair defects result from mutations in any of several genes coding for proteins in a complex nuclear machinery that detect and fix double strand DNA breaks. Every cell in our body is exposed to 10 to 50 DNA double strand breaks per day, from intracellular mechanisms (meiosis, isotype class switch, gene recombination) and extracellular insults (free radicals, ionizing radiation, drugs) (1). In lymphocytes, a crucial process is the generation of diversity in T-cell receptors and immunoglobulins through V(D)J somatic recombination, which starts with the introduction of double strand breaks in the genes by RAG1 and RAG2 (2). Defects in the DNA repair machinery proteins (ATM, RAD50, NHEJ1, Artemis, NBN, SBDS, LIG4, MRE11, POLE) result in increased apoptosis, and oncogenesis. Most patients with DNA repair defects share a syndrome of neurologic deficits, combined (T and B-cell) immunodeficiency, bone marrow failure and/or hematologic neoplasia (3).
DNA Ligase IV is a 911-aminoacid protein, a nuclear enzyme with a DNA binding domain and two BRCT motifs, involved in double strand DNA repair through the non-homologous end-joining (NHEJ) pathway, the major repair mechanism in mammalian cells, which consists of at least 5 proteins. Specifically, the LIG4/XRCC4 complex oversees the final ligation step of the process (making ends of the repaired strands meet). Knockout models of the gene LIG4, located in human chromosome 13 and consisting of a single long exon, are lethal to mouse embryos because of massive neuronal apoptosis (4); all known human mutations are thus hypomorphic and result in a wide clinical spectrum, ranging from normal to severely compromised immune system with microcephaly, growth failure, facial dysmorphism, mental retardation, hypogonadism, progressive bone marrow failure, and leukemia or lymphoma. The physiopathology at a cellular level includes mutagenesis, apoptosis and oncogenesis, despite some residual function of LIG4, and despite a redundant DNA repair machinery. The resulting immune defect might be a radiosensitive T-B-NK+ severe-combined immunodeficiency. Even with a clinically normal phenotype, chances are that lymphocytes will have a restricted receptor repertoire, and that hematopoietic cells be susceptible to ionizing radiation and other insults, with cumulative DNA damage leading to bone marrow failure and hematologic malignancies (5).
Here, we describe five cases of DNA Ligase 4 deficiency (MIM phenotype number #606593) in two unrelated families from Mexico, whose clinical presentations ranged from lymphoma and early death, to mild disease. We reviewed the literature to further document the varying clinical presentations.
Written informed consent was obtained from the parents of all patients, for the publication of this case series, and any potentially identifying information, including face pictures and family trees.
Case Reports
Family A
A one-year-old boy from Northern Mexico was referred to UMAE-25 to rule out primary immunodeficiency. The third of four siblings, he was born at term to non-consanguineous parents.
An elder brother had died of Non-Hodgkin lymphoma of the brain at age 18 months old. He received BCG vaccination at birth, in 2005. At age 4 months he was hospitalized for viral encephalitis, and at 12 months for pneumonia and sepsis, with positive Cytomegalovirus (CMV) viral load (2,020 copies/ml); he suffered recurrent upper respiratory, gastrointestinal, and urinary infections; he developed oral candidiasis and a perianal ulcer caused by Pseudomonas spp; laboratory workup showed anemia (Hb 8.13 g/dL) and thrombocytopenia (80,000 per microliter), with normal leukocyte numbers and serum immunoglobulin levels: IgG 760 mg/dL, IgM 49 mg/dL, IgA 83 mg/dL; flow cytometry of lymphocyte subsets reported low CD4+ (110 cells/μl) and CD8+ (307 cells/μl) T-cells, with 1982 B-cells (43%), and 130 NK cells (9%); HIV infection was ruled out. A bone marrow aspirate showed myeloid maturation arrest with dysplastic forms. He was admitted with neurologic decay, fever, and systemic inflammation shortly before his death. The autopsy confirmed a diffuse large B-cell Non-Hodgkin lymphoma. Immunohistochemical staining for Epstein-Barr virus (EBV) was not available.
A second sister died at age 9 months of lung lymphoma. She started at 4 months old, with three episodes of pneumonia and one of sepsis; she suffered from milk protein allergy, vulvovaginal candidiasis, and diaper area dermatitis; her laboratory workup found: transient peripheral lymphopenia, low T-cells subsets (CD4+ 8 to 403 cells/mm3, CD8+ 6 to 232/mm3) with normal B and NK cells; normal serum immunoglobulin levels (IgG 853mg/dL, IgM 41, IgA 64 mg/dL, IgE < 18 IU/ml); positive CMV viral load, Acinetobacter baumanii cultured from blood, Enterococcus faecium from urine, and Enterobacter aerogenes from bronchial aspirate. Cystic fibrosis, HIV infection, tuberculosis, and gastroesophageal reflux disease were ruled out; a nitroblue tetrazolium (NBT) reduction assay was normal at 81%. The chest X-ray showed a paravertebral mediastinal mass; a chest computed tomography (CT) confirmed a well-delimited, right retrocardiac rounded mass shortly before her death. She deteriorated abruptly with metabolic acidosis, progressive respiratory distress and heart failure; she was admitted to the intensive care unit and received mechanical ventilation support, broad-spectrum antibiotic, milrinone, and cyclophosphamide, without improvement. The autopsy confirmed a diffuse large B-cell lymphoma; EBV staining was not performed.
Our patient, the third sibling, received the BCG vaccine at birth, without complications. Before 1 year of age he was treated for uncomplicated pharyngitis and avascular necrosis of the femoral head (Legg-Calvé-Perthes disease). Given his family history, he was started on oral trimethoprim/sulfamethoxazole (TMP/SMZ) and sent to our hospital for evaluation.
Other than an initially positive CMV viral load (440 copies/ml, age 1 year, treated with ganciclovir for 90 days), his first laboratory workup was unremarkable. A CT scan from head to abdomen was normal. Serum immunoglobulin levels and absolute lymphocyte counts were normal at age 1, 4, and 7 years old. In contrast, the CD4+ T-cell subset count has remained steadily low (163–657 cells/mm3, or 7–23%). The Mantoux tuberculin skin test was reactive at 11 mm of induration, at age 7 months. However, CFSE lymphocyte proliferation assay was low at age 9 months, and absent 3 years later, under: PMA/ionomycin, phytohemagglutinin, concanavalin, and anti-CD3/CD28 stimuli (Figure 3).
Figure 3.

Carboxyfluorescein succinimidyl ester lymphoproliferation assay, plot (upper panel), and indexes (lower panel).
Whole-exome sequencing, performed at Texas Children's Hospital, revealed a compound heterozygous pathogenic variant (missense and small 5bp deletion) in LIG4: c.833G>A (p.R278H)/c.1271_1275delAAAGA (p.K424RfsX20). The p.K424R variant encodes a truncated protein missing 482 aminoacids; its frequency is lower than 0.03% for all populations according to ExAc (exac.broadinstitute.org). The p.R278H variant is predicted to be deleterious by all: DANN, GERP, dbNSFP, FATHMM, LRT, MetaLR, MetaSVM, MutationAssessor, MutationTaster, and PROVEAN in silico predictors. Both variants have been previously reported in patients with LIG4 deficiency and predicted to be deleterious by both SIFT and PolyPhen (transcript NM_206937.1, LIG4base, www.ensembl.org).
In time, the patient, now 6 years old, developed obesity with normal stature and head circumference (see Figure 1), and no other manifestation. A younger brother, born in 2017, is also asymptomatic, in apparent good health (Figure 2).
Figure 1.
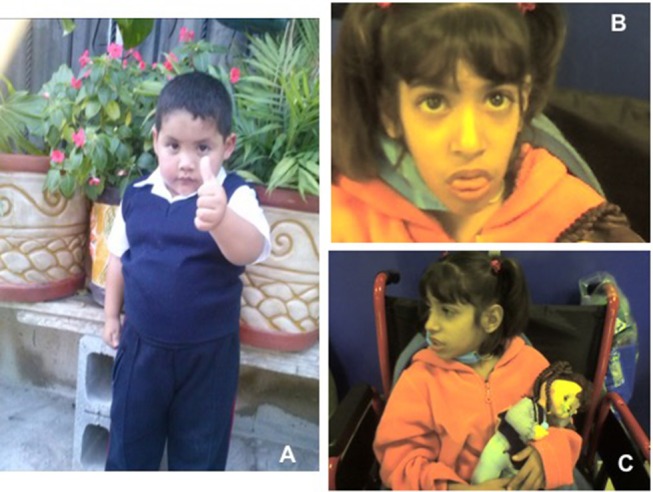
(A) Short stature with overweight in normocephalic school-age boy. Contrast with (B,C) Facial dysmorphisms seen in two twin sisters with LIG4 deficiency. Note prominent middle third of the face, long nose, micrognathia, long ears, jaundice, protruding tongue. (Face photos published with permission from their mothers).
Figure 2.

Family trees. In Family A, the elder siblings of the proband died young of lymphoma. The proband has a history of avascular necrosis of the femoral head, short stature and isolated CD4+ deficiency; his younger brother is asymptomatic. In Family B, teenage twin sisters died after a long history of recurrent infections, chronic liver disease, and progressive bone marrow failure (The pedigrees were built at pedigree.progenygenetics.com; they are included after written informed consent from the mothers).
Family B
Two teenage girl twins from Western Mexico were referred to INER for recurrent respiratory infections. They had a history of microcephaly and facial dysmorphism, with growth and psychomotor delay. Born pre-term (at 32 weeks of gestation) to non-consanguineous parents, the patients had three healthy elder siblings, and a pet dog.
During their first months of life, they suffered from 3 to 4 pneumoniae, gastroenteritis, and urinary tract infections. By 13 years of life, when they were referred to the National Institute of Pediatrics, they had developed moderate generalized jaundice with hepatosplenomegaly.
On physical examination, short stature (below percentile 10), low weight, mild to moderate mental retardation and speech delay were noted, with microcephaly below p3, a prominent mid-face with long nose, and micrognathia with a protruding tongue (see Figure 1). They had primary amenorrhea with absent secondary sexual features. A chest CT documented diffuse bilateral bronchiectasis; abdominal ultrasound and a barium swallow revealed portal hypertension with esophageal varices.
Laboratory workup showed persistent mild leukopenia with lymphopenia. Since around age twelve, they developed progressive pancytopenia: Hb 8g/dL, lymphocytes 210–300/mm3, less than 500 neutrophils, and 120,000 platelets; as well as elevated liver enzymes: increased conjugated bilirubin, ALT, AST, GGT, alkaline phosphatase, high ferritin (over 600 ng/ml), low fibrinogen (100 mg/dL), and high triglycerides (300 mg/dl). All serum immunoglobulins and lymphocyte subsets (CD3+, CD4+, CD8+, CD19+, CD16/56+) were also low: IgG 105–169 mg/dL, IgM 8.96–26.6 mg/dL, IgA < 6.67 mg/dL, IgE 1.1–1.5 IU/mL. A bone marrow aspirate reported medullary aplasia. Cellular radiosensitivity (radiomimetic assay) was negative to bleomycin; serologic tests for HepA, HepB, HepC, CMV, and EBV came back negative. Liver biopsy reported portal fibrosis with perivascular cell infiltration, suggestive of obstructive cirrhosis.
A provisional diagnosis of Nijmegen breakage syndrome (NBS) with bone marrow failure and sclerosing cholangitis was given. They continued treatment with prophylactic antibiotics and monthly intravenous gammaglobulin (IVIG), as well as ursodeoxycholic acid, molgramostin (GM-CSF), and oral transfer factor (leukocyte dialysate). They died 2 years apart at ages 14 and 16, at their hometown hospital, one twin of sudden massive gastrointestinal bleeding, and the other of sepsis and multiorgan failure (with pneumonia, anuria, and a distended abdomen). No autopsy was performed.
NBN was amplified and sequenced in Berlin with no mutations found. Years later, a targeted exome panel (Illumina TruSight) performed at INMEGEN in Mexico, revealed a compound heterozygous mutation in LIG4, consisting of the same 5 base-pair deletion as that of family A (c.1271_1275delAAAGA) in allele 1 (p. K424RfsX20), and a missense point mutation (c.745A>G) in allele 2 (p.M249V); both previously reported in patients with LIG4 deficiency (transcript NM_206937.1; LIG4base, www.ensembl.org), and predicted to be deleterious/probably damaging by both SIFT and PolyPhen. The p.M249V variant is absent from population databases such as gnomAD exomes; it is also predicted to be deleterious/probably damaging by all: DANN, GERP, dbNSFP, FATHMM, LRT, MetaLR, MetaSVM, MutationAssessor, MutationTaster, and PROVEAN in silico predictors.
Review of the Literature
The first patient with LIG4 deficiency, a 14-year-old teenage boy with acute lymphoblastic leukemia who died 8 months after receiving prophylactic cranial radiotherapy, was characterized clinically in 1990 (6), and genetically in 1999 (7). Tables 1–3 summarize the clinical, laboratorial and genetic findings of all published patients available in PubMed Medline (https://www.ncbi.nlm.nih.gov/pubmed/) as of August 2018. Clinical and immunological manifestations vary widely, even among siblings with the same mutation. Table 4 summarizes the prevalence of neurological, developmental, infectious, and hematological components of LIG4 deficiency. The degree of protein truncation (early, mid, and late) seems to correlate with clinical, hematological, and immunological severity (mild, moderate, and severe).
Table 1.
Demographic and clinical features of published patients with LIG4 deficiency.
| Patient | Origin | Sex | Family | Age at dx (years) | Onset | Clinical features | Status | References |
|---|---|---|---|---|---|---|---|---|
| 1 (180BR) | Turkish-cypriot | Male | 14 | B-symptoms | Weight loss, anorexia, lethargy, pallor, lymphadenopathy, hepatosplenomegaly: Acute lymphoblastic leukemia | Died 8 months after radiotherapy | 6, 7 | |
| 2 | USA | Fem | 26 | Microcephaly and microsomy, lethargy and dizziness | Seckel syndrome. Cough, fever, pallor. | Died of pneumonia 2 months after chemotherapy | (8) | |
| 3 (411BR) | Germany | Male | 9 | Microcephaly | Dysmorphism, developmental delay and plantar warts. | Died at age 23 of lymphoma | (4, 9, 10) | |
| 4 (2303) | USA | Male | Sibling to 2304 | 46 | Microcephaly | Chronic skin conditions, photosensitivity, telangiectasia, sinusitis | NR | (9) |
| 5 (2304) | USA | Fem | Sibling to 2303 | 48 | Microcephaly | Dysmorphism, growth failure, respiratory infections, psoriasis | NR | (9) |
| 6 (99P0149) | Germany | Fem | Non-consang. | 9 | Microcephaly | Dysmorphism, dwarfism, mental delay, psoriasis | Alive and well 5 years after RIC HSCT from HLA-identical brother | (9, 11) |
| 7 (3703) | Canada | Male | First cousin died of brain tumor in early childhood | 4.75 | Acute leukemia | Microcephaly, dysmorphism, developmental delay, hypogonadism, leukemia | Died shortly after chemotherapy | (12) |
| 8 (SC2) | Turkish | Fem | Consang., heterozygous | 1.5 | Respiratory infections | Respiratory infections, candidiasis, chronic diarrhea, failure to thrive, fever | Died after conditioning, possibly due to veno-occlusive disease. | (13) |
| 9 (P2) | German | Fem | Sister to 10 | 0 | Microcephaly | Alive after HSCT | (14) | |
| 10 (P1) | German | Fem | Sister to 9 | 2 | Recurrent infections | Chronic diarrhea, recurrent infections, failure to thrive, autoimmune cytopenia | Died shortly after chemotherapy | (14) |
| 11 (P-1) | Morocco | Fem | Sister to 12 | 1.5 | Repeated infections since 3 months old | Microcephaly, otitis, bronchiolitis, pneumonia and sepsis | Died of EBV PTLD 50 days after HSCT | (2) |
| 12 (P-2) | Morocco | Fem | Sister to 11 | 0 | Microcephaly | Died of VOD 2 months after HSCT | (2) | |
| 13 | Japan | Fem | 14 | Mouth tumor, fever | Microcephaly, short stature, polydactyly, | Died after chemotherapy | (15) | |
| 14 | Canada | Fem | Non-consang. | 0.1 | Omenn syndrome | Microcephaly, low weight, rash, hepatosplenomegaly, lymphadenopathy, diarrhea. | Alive and well 3.5 y after full conditioning HSCT | (16) |
| 15 | Turkish | Fem | Consang., sister to 16 | 10 | Microcephaly, respiratory infections | Sinopulmonary recurrent infections | Alive and well after 2 RIC HSCTs | (8) |
| 16 | Turkish | Male | Consang., brother to 15 | 6 | Ecchymoses | Low weight, upper respiratory and urinary infections | Alive | (8) |
| 17 (F10) | Netherlands | Male | 0.25 | Microcephaly, dysmaturity, dysmorphism | Feeding difficulties, diarrhea, failure to thrive, icterus, tubulopathy, erythema | Died at 6 months of sepsis, respiratory insufficiency and severe gastrointestinal bleeding | (17, 18) | |
| 18 | USA | Fem | 34 | Dubowitz syndrome | Rectorrhagia | Died of metastatic anal cancer | (18, 19) | |
| 19 (F1.1) | Canada | Fem | Sister to 20 | 17.5 | Microcephaly | Small cerebral aneurysm, primary ovarian failure | Alive | (18) |
| 20 (F1.2) | Canada | Fem | Sister to 19 | 11.75 | Microcephaly | Atrial-ventricular septal defect, atrofphic kidney, rib hypoplasia, fusion of carpal bones, copper beaten skull, platybasia, abnormal C1 vertebrae, primary ovarian failure. | Alive | (18) |
| 21 (F2) | USA | Fem | 7.8 | Microcephaly, malformations | Anal atresia with rectovaginal fistula, esotropia. | Alive | (18) | |
| 22 (F3) | Australia | Fem | 2.1 | Microcephaly, hip dysplasia | Unilateral congenital hip dysplasia, cutis marmorata. | Alive. BMT | (18) | |
| 23 (F4) | UK | Fem | 2.5 | Microcephaly, psoriasis | Psoriasis | Alive | (18) | |
| 24 (F5) | USA | Male | 2 | Microcephaly, hip dysplasia | Unilateral congenital hip dysplasia. | Alive | (18) | |
| 25 (F6) | Germany | Fem | 2 | Microcephaly, hip dysplasia | Congenital hip dysplasia, 2/3 toe syndactyly, excessive vomiting | Alive | (18) | |
| 26 (F7) | USA | Fem | 3.67 | Microcephaly, growth failure | None | Alive | (18) | |
| 27 (F8) | UK | Fem | 1.75 | Microcephaly, growth failure | None | Alive | (18) | |
| 28 (F9) | Turkey | Male | 5.5 | Microcephaly, hypopigmentation | Hypopigmentation, hypermobile knees, single palmar crease, 2/3 toe syndactyly, sandal gap | Alive | (18) | |
| 29 (P2) | North America | Fem | Sibling to P1 and P3 | 13 | Recurrent pneumonias and sinusitis | Recurrent respiratory infections, microcephaly, short stature. | Alive | (5) |
| 30(P1) | North America | Male | Sibling to P2 and P3 | NA | NA | One walking pneumonia | Alive | (5) |
| 31 (P3) | North America | Fem | Sibling to P1 and P2 | NA | NA | Asymptomatic | Alive | (5) |
| 32 | Belarus | Fem | 2 | Stomatitis | Ulcerated stomatitis, encephalitis, lung and brain lymphoma. | Died of lymphoma | (20) | |
| 33 (C1) | African/Asian descent | Fem | 7 | Dysmorphic features, developmental delay | Respiratory infections, pancytopenia | Alive and well after HSCT | (21) | |
| 34 (C2) | Morocco | Male | Elder sister died at 18 mo. with pancytopenia | 9 | Chronic diarrhea | Chronic diarrhea, recurrent respiratory infections, hemorrhagic syndrome | Died of aplasia at age 10 | (21) |
| 35 | Syria | Fem | Consang. | 7 | Dysmorphic features and developmental delay | Recurrent sinopulmonary infections, urosepsis, urofacial syndrome | Died of pneumonia | (22) |
| 36 | Italy | Male | 6 | Complicated pneumonia | Recurrent upper respiratory infections since age 1, dysgammaglobulinemia | Alive | (23) | |
| 37 LRL | Mexico | Fem | Two brothers. 3 first cousins died of infections before age 2 | 0.75 | Pneumonia at 4 months | Died of lymphoma | This report | |
| 38 PRL | Mexico | Male | Two siblings | 1.5 | Pneumonia at 12 months | Died of lymphoma | This report | |
| 39 FRL | Mexico | Male | Two siblings | 1 | Pharyngoamigdal. at 2 months | Avascular necrosis of femoral head, overweight. | Alive and well | This report |
| 40 MGP | Mexico | Fem | Twin sister | 10 | Recurrent respiratory infections | Microcephaly, dysmorphism, developmental delay, recurrent infections, jaundice | Died | This report |
| 41 BGP | Mexico | Fem | Twin sister | 10 | Recurrent respiratory infections | Microcephaly, dysmorphism, developmental delay, recurrent infections, jaundice | Died | This report |
NR, Not reported.
Table 3.
Immunological and genetic features of published patients with LIG4 deficiency.
| Pat | Germs (isolates) | Serum immunoglobulins | Other (comment) | Mutation type | Allele 1 | Allele 2 | References |
|---|---|---|---|---|---|---|---|
| 1 (180BR) | NR | Not reported | Prophylactic cranial radiation: ulcers, lethargy, tetraparesis | hom, miss | c. 833 G>A (p. R278H) | c. 833 G>A (p. R278H) | (6, 7) |
| 2 | NR | Not reported | Chromosomal abberrations. Chemotherapy: severe toxicity | NR | ND | ND | (8) |
| 3 (411BR) | NR | Low | Microcephaly at birth, not evident at age 9. Null B-cells, low T. T-B-NK+ CID | hom, miss, (+2 polymorphisms) | c. 833 G>A/8 C>T/26 C>T (p. R278H/A3V/T9I) | c. 833 G>A/8 C>T/26 C>T (p. R278H/A3V/T9I) | (4, 9, 10) |
| 4 (2303) | NR | Not reported | Hypothyroidism, type 2 diabetes, hypogonadism | comp het, nons | c. 1738 C>T (p.R580X) | c. 2440C>T (p.R814X) | (9) |
| 5 (2304) | NR | Not reported | Hypothyroidism, amenorrhea | comp het, nons | c. 1738 C>T (p.R580X) | c. 2440C>T (p.R814X) | (9) |
| 6 (99P0149) | NR | All normal | Multiple psoriasiform erythrodermic, squamous skin patches, atypical bone maturation, low CD3 and CD19 | comp het, miss/nons | c. 1406 G>A (p.G469E) | c. 2440C>T (p.R814X) | (9, 11) |
| 7 (3703) | NR | Not reported | Low birth weight, criptorchidism, hypogonadism, clinodactyly | hom, nons | c.2440C>T (p.R814X) | c. 2440C>T (p.R814X) | (12) |
| 8 (SC2) | Candida | Low IgG, IgA and IgM | T-B-NK+ SCID, reduced lymphoproliferation (PHA, aCD3) | hom, small del | g.5333_5335delCAA (p. Q433del) | g.5333_5335delCAA (p. Q433del) | (13) |
| 9 (P2) | No | Low IgM, absent IgA | T-B-NK+ SCID, isohemagglutinins present | comp het, miss/nons | c.1118A>T (p. H282L) | c.1544_1548delAAAGA (p.D423fs442X) | (14) |
| 10 (P1) | S. pneumoniae, HHV6, Norwalk, EBV, Aspergillus | High IgG and IgM, normal IgA | T-B-NK+ SCID, small thymus, autoimmune thrombocytopenia, no tetanus, diphteria or pneumococcus titers | comp het, miss/nons | c.1118A>T (p. H282L) | c.1544_1548delAAAGA (p.D423fs442X) | (14) |
| 11 (P-1) | S. pneumoniae | Absent IgA, Low IgG and IgM | T-B-NK+ SCID, reduced lymphoproliferation | comp het, miss/small del | c.1544_1548del5bp (p.K424fs20X) | c.1112A>G (p.Q280R) | (2) |
| 12 (P-2) | No | Low IgM, absent IgA (maternal IgG) | T-B-NK+ SCID, normal lymphoproliferation | comp het, miss/small del | c.1544_1548del5bp (p.K424fs20X) | c.1112A>G (p.Q280R) | (2) |
| 13 | Aspergillosis after neutropenia | Low IgM and IgG | T-B-NK+ CID | comp het, miss/small del | c.745A>G (p.M249V) | c.1270_1274del5bp (p.K424fs20X) | (15) |
| 14 | EBV after HSCT | Low IgM and IgA | Low TRECs, B cells, lymphoproliferation, GVHD | comp het, miss/small del | c.845A>T(p.H282L);c.26C>T (SNP) | c.1747_1751del5bp (p.R581fsX) | (16) |
| 15 | NR | Low IgM | Spontaneous chromosomal breakages increased, normal bone marrow cellularity | hom, small del | c.1762delAAG (p.K588del) | c.1762delAAG (p.K588del) | (8) |
| 16 | Low IgG and IgM | Inguinal hernia, spontaneous chromosomal breakage, normal BM cellularity | hom, small del | c.1762delAAG (p.K588del) | c.1762delAAG (p.K588del) | (8) | |
| 17 (F10) | P aeruginosa, E faecalis, P jiroveci, rhinovirus, norovirus, astrovirus, C difficile, Candida | Low IgG and B-cells | Polydactyly, dysplastic kidneys, corpus callosum dysgenesia, very low B-cells | comp het, SN del trunc/trunc | c.613delT (p.S205LfsX29) | c. 1904delA (p.Lys635ArgfsX10) | (17, 18) |
| 18 | No | NR | Radiotherapy: desquamative skin injury | comp het, SN del trunc/nons | c.613delT (p.S205LfsX232) | c.2440C>T (p.R814X) | (18, 19) |
| 19 (F1.1) | NR | Low IgG | Low CD4+, null CD19+ | comp het, miss/small del | c.2440C>T (p.R814X) | c.1271_1275delAAAGA (p.K424RfsX20) | (18) |
| 20 (F1.2) | NR | Low IgG | Low CD4+, null CD19+, very low naïve T cells | comp het, miss/small del | c.2440C>T (p.R814X) | c.1271_1275delAAAGA (p.K424RfsX20) | (18) |
| 21 (F2) | NR | NR | NR | comp het, nons | c.2440C>T (p.R814X) | c.2094C>G (p.Y698X) | (18) |
| 22 (F3) | NR | Low IgG | Low CD3, CD8, very low CD19 | comp het, dup/nons | c.2440C>T (p.R814X) | c.2386_2389dupATTG (p.A797DfsX3) | (18) |
| 23 (F4) | NR | Low IgG | Low CD4+, very low CD19+ | comp het, nons/small del | c.2440C>T (p.R814X) | c.1271_1275delAAAGA (p.K424RfsX20) | (18) |
| 24 (F5) | NR | Low IgG | Low CD3, CD4, CD8, very low CD19 | comp het, nons/small del | c.2440C>T (p.R814X) | c.1271_1275delAAAGA (p.K424RfsX20) | (18) |
| 25 (F6) | NR | Low IgG | Very low CD19+ | comp het, nons/small del | c.2440C>T (p.R814X) | c.1271_1275delAAAGA (p.K424RfsX20) | (18) |
| 26 (F7) | NR | NR | NR | comp het, nons/small del | c.2440C>T (p.R814X) | c.1512_1513delTC (p.R505CfsX12) | (18) |
| 27 (F8) | NR | Low IgG | NR | comp het, nons/small del | c.2440C>T (p.R814X) | c.1246_1250dupGATGC (p.Leu418MetfsX3) | (18) |
| 28 (F9) | NR | Normal | Very low CD19+ | comp het, nons/small del | c.2440C>T (p.R814X) | c.1271_1275delAAAGA (p.K424RfsX20) | (18) |
| 29 (P2) | NR | Panhypogammaglobulinemia | Low CD3+, CD19+, naïve T | comp het, miss/nons | c.2440C>T (p.R814X) | c.1345A>C (p.K449Q) | (5) |
| 30(P1) | NR | Normal IgG | Low CD19+, naïve T cells | comp het, miss/nons | c.2440C>T (p.R814X) | c.1345A>C (p.K449Q) | (5) |
| 31 (P3) | No | Low IgG | Low CD3+, CD19+, naïve T | comp het, miss/nons | c.2440C>T (p.R814X) | c.1345A>C (p.K449Q) | (5) |
| 32 | EBV | Low IgA | Low T cells | comp het, small del/miss | c.2736+3delC | c.8C>T (p.A3V); c.26C>T(p.T9I) | (20) |
| 33 (C1) | NR | Low IgG and IgM | comp het, nons/small del | c.2440C>T (p.R814X) | c.1271_1275delAAAGA (p.K424RfsX20) | (21) | |
| 34 (C2) | HPV | NR | comp het, miss/small del | c.847A>G (p.K283E) | c.1271_1275delAAAGA (p.K424RfsX20) | (21) | |
| 35 | S. pneumoniae, H. influenzae | Low IgG2 and IgM | Low CD4+, null CD19+. Urofacial syndrome with hom. LRIG2 mut. | hom, miss | c.T1312C (p.Y438H) | c.T1312C (p.Y438H) | (22) |
| 36 | NR | Absent IgA and IgM. Normal IgG, low IgG3 | Very low T and B cells. Poor response to vaccines. Bronchiectasis | comp het, miss/small del | c. 833 G>A (p. R278H) | c.1271_1275delAAAGA (p.K424RfsX20) | (23) |
| 37 LRL | Acinetobacter baumanii, Enterococcus faecium, Enterobacter aerogenes, CMV | All normal | Low T cells | NR | ND | ND | This report |
| 38 PRL | Pseudomonas, CMV | All normal | Low T cells | NR | ND | ND | This report |
| 39 FRL | No | All normal | Legg-Calvé-Perthes disease, right hip. Low CD4+, low lymphoproliferation. | comp het, miss | c.1236T>A (p.N412K) | c.32C>G (p.A11G) | This report |
| 40 MGP | NR | Low IgG, IgA and IgM | Primary amenorrhea, sclerosing cholangitis | comp het, small del/miss | c.1271_1275delAAAGA (p.K424RfsX20) | c.745A>G (p.M249V) | This report |
| 41 BGP | NR | Low IgG, IgA and IgM | Primary amenorrhea, sclerosing cholangitis | NR | ND | ND | This report |
ND, Not done; NR, Not reported; NT, Not tested; IR, ionizing radiation; BLM, bleomycin; DEB, diepoxybutane; MITC, mitomycin C. All reported mutations are hypomorphic.
Table 4.
Prevalence of clinical and laboratory features found in 41 LIG4 deficiency patients.
| Female | 28/41 | 68% |
| Microcephaly | 32/40 | 80% |
| Dysmorphic facial features | 15/41 | 37% |
| Growth failure | 28/40 | 70% |
| Syndactyly/Polysindactyly | 4/41 | 10% |
| Other malformations | 3/41 | 7% |
| Congenital hip dysplasia | 4/41 | 10% |
| Infections (any) | 28/41 | 68% |
| Sinopulmonary infections | 23/41 | 56% |
| Skin conditions | 8/41 | 20% |
| Warts | 2/41 | 5% |
| Hypogammaglobulinemia | 25/30 | 83% |
| Very low CD19+ B-cells | 24/30 | 80% |
| Malignancy | 10/41 | 24% |
| Bone marrow failure | 17/39 | 44% |
| Radiosensitivity | 24/28 | 86% |
(100% radiosensitivity with ionizing radiation).
Table 2.
Key features of published patients with LIG4 deficiency.
| Patient | Micro-cephaly | Dysmorphism | Growth failure | Developmental delay | Malignancy | Infections | Bone marrow failure | References |
|---|---|---|---|---|---|---|---|---|
| 1 (180BR) | No | No | No | No | AL Leukemia | None | No | (6, 7) |
| 2 | Yes | Receding forehead, hypertelorism, beaked nose, low set ears, micrognathia, downward palpebral fissures. | Yes | Mental retardation, high pitched voice. | AM Leukemia | Pneumonia (post chemo) | Yes (after chemo-therapy) | (8) |
| 3 (411BR) | Yes | Bird-like | No | Yes, global | Lymphoma (Diffuse large B-cell, nasopharyngeal) | Extensive plantar warts | Pancytopenia | (4, 9, 10) |
| 4 (2303) | Yes | Seckel-like | Yes | No | Myelodysplasia | Sinusitis | Pancytopenia | (9) |
| 5 (2304) | Yes | Seckel-like | Yes | No | None | Chronic respiratory infections | No | (9) |
| 6 (99P0149) | Yes | Seckel-like, Bird-like | Yes | Developmental/mental | None | Sinopulmonary recurrent | Pancytopenia, hypoplastic marrow | (9, 11) |
| 7 (3703) | Yes, brachy. | NBS-like | No | Yes, significant cognitive delay | AL Leukemia | Neutropenic sepsis after chemo. | Aplasia after chemo. | (12) |
| 8 (SC2) | No | No | No | No | None | Recurrent severe respiratory infections, candidiasis in diaper region, chronic diarrhea | Anemia, leukopenia | (13) |
| 9 (P2) | Yes | No | No | Yes, significant neurodevelopmental | None | Lymphopenia | (14) | |
| 10 (P1) | Yes | No | No | No | Brain and lung non-Hodgkin B cell lymphoma | Sepsis, diarrhea | Neutropenia, lymphopenia, thrombocytopenia | (14) |
| 11 (P-1) | Yes | No | Yes | No | None | Otitis, bronchiolitis, pneumonia, sepsis | Lymphopenia | (2) |
| 12 (P-2) | Yes | No | No | No | None | None | Lymphopenia | (2) |
| 13 | Yes | No | Yes | No | Lymphoma (B-cell, NH) in upper gingiva and hard palate | Pulmonary aspergillosis | Progressive | (15) |
| 14 | Yes | No | Yes | Yes | None | None | Marked eosinophilia, mild lymphopenia | (16) |
| 15 | Yes | Low hairline, prominent nasal bridge, bilateral epicanthi | Yes | No | None | Sinopulmonary, ear. | Pancytopenia, progressive | (24) |
| 16 | Yes | Prominent nasal bridge, bilateral epicanthi | Yes | No | None | No | (24) | |
| 17 (F10) | Yes | Hypotelorism, small viscerocranium, flat philtrum, thin upper lip, preaxial polydactyly, brachimesophalangy, partial syndactyly | Yes | Neurologic abnormalities | None | Urinary tract, sepsis | Thrombocytopenia | (17, 18) |
| 18 | Yes | Yes | NR | No | Anal cancer | None | Pancytopenia | (18, 19) |
| 19 (F1.1) | Yes | No | Yes | Yes, mild | None | Mild. Influenza | Pancytopenia, self-resolved. Thrombocytopenia | (18) |
| 20 (F1.2) | Yes | Malformations | Yes | Yes, mild-moderate | None | Mild recurrent (respiratory, skin, GI) | Pancytopenia, persistent. Thrombocytopenia | (18) |
| 21 (F2) | Yes | Malformation | Yes | Yes, mild | None | None | Pancytopenia, self-resolved. Thrombocytopenia | (18) |
| 22 (F3) | Yes | No | Yes | Yes, mild | None | Mild recurrent (respiratory, skin, GI) | Thrombocytopenia | (18) |
| 23 (F4) | Yes | No | Yes | No | None | None | Anemia, thrombocytopenia | (18) |
| 24 (F5) | Yes | No | Yes | No | None | None | Lymphopenia | (18) |
| 25 (F6) | Yes | Malformations | Yes | Yes, mild | None | Mild recurrent (respiratory, skin, GI) | Thrombocytopenia | (18) |
| 26 (F7) | Yes | No | Yes | No | None | None | Thrombocytopenia | (18) |
| 27 (F8) | Yes | No | Yes | Yes, mild | None | None | Thrombocytopenia | (18) |
| 28 (F9) | Yes | Malformations | Yes | Yes, mild | None | Mild recurrent (respiratory, skin, GI) | Thrombocytopenia | (18) |
| 29 (P2) | Yes | No | Yes | No | None | Pneumonia and sinusitis | Lymphopenia | (5) |
| 30(P1) | unknown | No | Small stature | None | Walking pneumonia | Mild neutropenia | (5) | |
| 31 (P3) | No | No | No | None | None | Lymphopenia, mild neutropenia | (5) | |
| 32 | No | No | No | No | Lymphoma, EBV-pos; diffuse large B-cell; lung and brain | EBV stomatitis/encephalitis | No | (20) |
| 33 (C1) | Yes | Yes | Yes | Yes, mild | None | Respiratory | Slowly progressive pancytopenia | (21) |
| 34 (C2) | Yes | Yes | Yes | Severe language delay, moderate mental retardation | None | Respiratory, warts | Yes | (21) |
| 35 | Yes | Yes | Yes | Yes, mild | Pneumonia, otitis media, sinusitis, oral candidiasis. Urosepsis | Pancytopenia | (22) | |
| 36 | No | No | No | No | None | Sinopulmonary recurrent, ear | No. Progressive lymphopenia | (23) |
| 37 LRL | No | No | No | No | Lung lymphoma | Pneumonia 3x | No | This report |
| 38 PRL | No | No | No | No | Brain B-cell lymphoma (Non-Hodgkin) | Pneumonia, Perianal ulcers, Sepsis | No | This report |
| 39 FRL | No | No | Yes | No | None | Mild upper respiratory. | No | This report |
| 40 MGP | Yes | Yes | Yes | Yes | None | Recurrent: sinopulmonary, gastrointestinal, urinary | Yes | This report |
| 41 BGP | Yes | Yes | Yes | Yes | None | Recurrent: sinopulmonary, gastrointestinal, urinary | Yes | This report |
NR, Not reported.
Discussion
We describe a series of five LIG4 deficiency patients with a wide spectrum of clinical manifestations, ranging from short stature and asymptomatic CD4+ lymphopenia, to liver failure with sudden massive bleeding.
Although relatively small, this is one of the largest case series to date on the subject of LIG4 deficiency; the genetic diagnosis was confirmed in both families by whole-exome sequencing only, but we also reviewed the literature for all published patients to date (our job was aided by a recent review by Altmann and Gennery (1) comprising 28 LIG4 deficient patients up until 2016).
Forty-one LIG4 deficiency patients have been described to date (In addition, Bluteau et al. recently found three cases of LIG4 deficiency among their cohort of bone marrow failure patients, although their features were not described in detail) (25). Their phenotypes are varied, with more clinical severity associated to early mutations and a truncated protein. The degree of severity is probably linked to the amount of residual function of the ligase, but confounded by the complex interactions and the intricate redundancy of the DNA repair defect machinery.
To our knowledge, our patients from family B are the first to develop sclerosing cholangitis (SC) in the context of LIG4 deficiency. SC has been reported mainly in X-linked Hyper-IgM syndrome due to CD40L deficiency, but also in other forms of combined immunodeficiencies (26). This autoimmune complication is thought to result from the colonization of the bile ducts by Cryptosporidium and microsporidium from the small intestine, eventually leads to biliary cirrhosis and liver failure, which greatly complicates prognosis. SC should be considered in patients with lymphocyte defects and cholestatic jaundice. All such patients should be warned against drinking unpurified water, and against having close contact with dog pets.
When there is a full-blown phenotype of combined immunodeficiency, we can fear the complications of malignancy and bone marrow failure. In contrast, it is unclear what the best approach is with asymptomatic patients carrying a deleterious variant in LIG4. In theory, they will accumulate DNA double-strand breaks and cell damage that result in increased apoptosis of hematopoietic cells, with the consequent risk of late-onset aplastic anemia and malignancy. Felgentreff et al. (5) recently reported immunological findings in two asymptomatic siblings who shared a LIG4 bi-allelic mutation (K449Q/R814X) with their sister (the proband), but not a history of infections; all three siblings with compound heterozygous mutations had: low mean corpuscular volume, B-cell lymphopenia, shortened telomeres, reduced naïve T-cells, and increased radiosensitivity, as well as some skewing of their lymphocytes' receptor repertoires. The immune defects are thus present but clinically silent, and environmental or infectious triggers might precipitate bone marrow failure or myelodysplasia.
The differential diagnosis of LIG4 deficiency includes Nijmegen Breakage Syndrome (NBS), RAD50 and NHEJ1 deficiencies; Fanconi anemia, and any other congenital disease presenting with: microcephaly, facial dysmorphisms (described as bird-like facies); hypogammaglobulinemia, and low B-cells; including IKAROS and NFKB2 deficiencies. Dubowitz and Seckel's are clinical syndromes of which LIG4 deficiency patients undoubtedly represent a considerable subset of cases (19). Of note, LIG4 deficient patients may first seek medical care for hematologic malignancies, but respond badly to chemotherapy and radiotherapy; the astute clinician should consider this diagnosis in patients with suggestive clinical features (12), and consider instead hematopoietic stem-cell transplantation (HSCT) after a reduced intensity conditioning (RIC) regime.
Not all patients have had microcephaly, but all had radiosensitivity when their cell cultures were exposed to ionizing gamma radiation. Our twin sisters' cells were reported normal or negative for the bleomycin (BLM) radiomimetic test, which has been described to have a strong positive correlation when compared to ionizing radiation with gamma rays. However, it has also been found to be more selective and less predictable than gamma radiation (27–29). BLM is an antibiotic obtained from Streptomyces verticillus, used in the treatment of cancer and considered a radiomimetic for its induction of single and double DNA strand breaks, in the presence of iron ions in cell cultures (27).
Given the dreaded fatal complications of hematologic neoplasia, bone marrow failure and chronic liver disease, the best chance LIG4-deficient patients with severe (hematological/immunological) phenotypes have of reaching adulthood, might be to receive an HSCT (11, 30) under the best possible conditions (HLA-identical donor, a reduced intensity conditioning regime with low-dose Cyclosporin A (24, 30, 31), infection-free, no organ failure). The neurological deficits and mental retardations are usually mild enough to allow for a happy and productive life in transplant survivors, capable of loving and being loved, and of learning a craft.
In conclusion, LIG4 deficient patients may present soon after birth or later in life, with, or without: a family history of consanguinity and affected siblings, microcephaly, beaked nose, T-B-NK+ SCID, Omenn syndrome, low serum immunoglobulins, and very low B-cells, worsening pancytopenia, radiosensitivity; and they may develop bone marrow failure, sepsis, severe bleeding, and/or hematologic malignancies. Early diagnosis is crucial, as hematologic and infectious complications are more frequent with increasing age, and because RIC HSCT may be curative.
Ethics Statement
The molecular and genetic diagnostic studies were approved by the Institutional Review Board (Comité de Investigación) at the National Institute of Pediatrics. Said studies were preceded by informed consent from the patients' next of kin. Face photographs are included with authorization from their mothers.
Author Contributions
AB coordinated the care of one patient, conceived the manuscript and helped collect data. IC supervised and performed the analysis of exome sequencing for one patient. CA-V supervised and analyzed exome sequencing for one patient, edited the manuscript, and participated in scientific discussions. MY-N coordinated the care of two patients, proposed differential diagnoses and discussed the manuscript. KC-S performed the WES for one of the patients. MG-C coordinated the care of two patients. MP participated in the genetic diagnosis of one patient, and discussed the manuscript. MG performed and analyzed lymphoproliferation assays. EM participated in the molecular and genetic diagnoses of one patient. DM participated in clinical data collection. LF was involved in the genetic diagnosis of one patient and reviewed the manuscript. FE-R facilitated the genetic diagnosis of one patient, and participated in the care of two patients. SE-P participated in the care, and coordinated the molecular and genetic diagnoses of one patient. JO coordinated the genetic diagnosis of one patient. SL conceived and wrote the manuscript, participated in the care of two patients, and built the tables. All the authors read and contributed to the final draft of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank James R. Lupski, Richard A. Gibbs, and Zeynep H. Coban-Akdemir for sequencing through the Baylor-Hopkins Center for Mendelian Genomics (NIH-NHGRI/NHLBI grant number UM1HG006542). Dr. Raymonda Varon-Mateeva and her team in Berlin amplified and sequenced NBN from patient 4, in search for mutations; we are grateful for her kind help. Edna Venegas Montoya helped collect and organize data from family A. The authors wish to acknowledge the valiant stoicism of our patients with LIG4 deficiency and their parents. We also thank the Mexican Foundation for girls and boys with Primary immunodeficiencies (FUMENI, AC) for funding this article publication fee and their continuous support to help diagnose and treat patients in Mexico.
Glossary
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BCG
Bacille Calmette-Guérin
- BCM, Baylor College of Medicine
Houston
- BLM
Bleomycin
- BRCT
C-terminal domain of breast cancer susceptibility protein
- CFSE
Carboxyfluorescein diacetate succinimidyl ester
- CMV
cytomegalovirus
- DNA
deoxyribonucleic acid
- GGT
Gamma-glutamyl transferase
- GM-CSF
granulocyte-macrophage colony stimulating factor
- IEI
Inborn errors of immunity
- INER, National Institute of Respiratory Diseases
Mexico
- INP, National Institute of Pediatrics
Mexico
- INMEGEN, National Institute of Genomic Medicine
Mexico
- IVIG
intravenous immunoglobulin
- LIG4
DNA Ligase 4
- NBN
Nibrin
- NBS
Nijmegen Breakage Syndrome
- NHEJ
Non-homologous end join
- PCR
Polymerase chain reaction
- SC
sclerosing cholangitis
- TMP/SMZ
trimethoprim/sulfamethoxazole
- UMAE 25
Unidad Médica de Alta Especialidad No.
References
- 1.Altmann T, Gennery AR. DNA ligase IV syndrome; a review. Orphanet J Rare Dis. (2016) 11:1–7. 10.1186/s13023-016-0520-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buck D, Moshous D, de Chasseval R, Ma Y, le Deist F, Cavazzana-Calvo M, et al. Severe combined immunodeficiency and microcephaly in siblings with hypomorphic mutations in DNA ligase IV. Eur J Immunol. (2006) 36:224–35. 10.1002/eji.200535401 [DOI] [PubMed] [Google Scholar]
- 3.Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. (2017) 38:129–43. 10.1007/s10875-017-0465-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Girard PM, Kysela B, Härer CJ, Doherty AJ, Jeggo PA. Analysis of DNA ligase IV mutations found in LIG4 syndrome patients: the impact of two linked polymorphisms. Hum Mol Genet. (2004) 13:2369–76. 10.1093/hmg/ddh274 [DOI] [PubMed] [Google Scholar]
- 5.Felgentreff K, Baxi S, Lee YN, Dobbs K, Henderson LH, Csomos K, et al. Ligase-4 deficiency causes distinctive immune abnormalities in asymptomatic individuals. J Clin Immunol. (2016) 36:341–53. 10.1007/s10875-016-0266-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plowman PN, Bridges BA, Arlett CF, Hinney A, Kingston JE. An instance of clinical radiation morbidity and cellular radiosensitivity, not associated with ataxia-telangiectasia. Br J Radiol. (1990) 63:624–8. 10.1259/0007-1285-63-752-624 [DOI] [PubMed] [Google Scholar]
- 7.Riballo E, Critchlow SE, Teo SH, Doherty AJ, Priestley A, Broughton B, et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol. (1999) 9:699–702. 10.1016/S0960-9822(99)80311-X [DOI] [PubMed] [Google Scholar]
- 8.Hayani A, Suarez CR, Molnar Z, LeBeau M, Godwin J. Acute myeloid leukaemia in a patient with Seckel syndrome. J Med Genet. (1994) 31:148–9. 10.1136/jmg.31.2.148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell. (2001) 8:1175–85. 10.1016/S1097-2765(01)00408-7 [DOI] [PubMed] [Google Scholar]
- 10.Bacon CM, Wilkinson SJ, Spickett GP, Barge D, Lucraft HH, Jackson G, et al. Epstein-Barr virus-independent diffuse large B-cell lymphoma in DNA ligase 4 deficiency. J Allergy Clin Immunol. (2013) 131:1237–9. 10.1016/j.jaci.2012.10.027 [DOI] [PubMed] [Google Scholar]
- 11.Gruhn B, Seidel J, Zintl F, Varon R, Tönnies H, Neitzel H, et al. Successful bone marrow transplantation in a patient with DNA ligase IV deficiency and bone marrow failure. Orphanet J Rare Dis. (2007) 2:5. 10.1186/1750-1172-2-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Omran TI, Cerosaletti K, Concannon P, Weitzman S, Nezarati MM. A patient with mutations in DNA Ligase IV: clinical features and overlap with Nijmegen breakage syndrome. Am J Med Genet. (2005) 137 A:283–7. 10.1002/ajmg.a.30869 [DOI] [PubMed] [Google Scholar]
- 13.Burg M Van Der, Veelen LR Van, Verkaik NS, Wiegant WW, Hartwig NG, Barendregt BH, et al. A new type of radiosensitive T–B –NK+ severe combined immunodeficiency caused by a LIG4 mutation. J Clin. (2006) 116:137–45. 10.1172/JCI26121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Enders A, Fisch P, Schwarz K, Duffner U, Pannicke U, Nikolopoulos E, et al. A severe form of human combined immunodeficiency due to mutations in DNA ligase IV. J Immunol. (2006) 176:5060–8. 10.4049/jimmunol.176.8.5060 [DOI] [PubMed] [Google Scholar]
- 15.Toita N, Hatano N, Ono S, Yamada M, Kobayashi R, Kobayashi I, et al. Epstein-barr virus-associated B-cell lymphoma in a patient with DNA ligase IV (LIG4) syndrome. Am J Med Genet Part A. (2007) 143A:742–5. 10.1002/ajmg.a.31644 [DOI] [PubMed] [Google Scholar]
- 16.Grunebaum E, Bates A, Roifman CM. Omenn syndrome is associated with mutations in DNA ligase IV. J Aller Clin Immunol. (2008) 122:1219–20. 10.1016/j.jaci.2008.08.031 [DOI] [PubMed] [Google Scholar]
- 17.Ijspeert H, Warris A, van der Flier M, Reisli I, Keles S, Chishimba S, et al. Clinical spectrum of LIG4 deficiency is broadened with severe dysmaturity, primordial dwarfism, and neurological abnormalities. Hum Mutat. (2013) 34:1611–4. 10.1002/humu.22436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murray JE, Bicknell LS, Yigit G, Duker AL, van Kogelenberg M, Haghayegh S, et al. Extreme growth failure is a common presentation of ligase IV deficiency. Hum Mutat. (2014) 35:76–85. 10.1002/humu.22461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yue J, Lu H, Lan S, Liu J, Stein MN, Haffty BG, et al. Identification of the DNA repair defects in a case of dubowitz syndrome. PLoS ONE (2013) 8:54389. 10.1371/journal.pone.0054389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharapova SO, Chang EY, Guryanova IE, Proleskovskaya I V, Fedorova AS, Rutskaya EA, et al. Next generation sequencing revealed DNA ligase IV deficiency in a “developmentally normal” patient with massive brain Epstein-Barr virus-positive diffuse large B-cell lymphoma. Clin Immunol. (2016) 163:108–10. 10.1016/j.clim.2016.01.002 [DOI] [PubMed] [Google Scholar]
- 21.Dard R, Herve B, Leblanc T, de Villartay JP, Collopy L, Vulliami T, et al. DNA ligase IV deficiency : Immunoglobulin class deficiency depends on the genotype. Pediatr Allergy Immunol. (2017) 28:298–303. 10.1111/pai.12694 [DOI] [PubMed] [Google Scholar]
- 22.Fadda A, Butt F, Tomei S, Deola S, Lo B, Robay A, et al. Two hits in one: Whole genome sequencing unveils LIG4 syndrome and urofacial syndrome in a case report of a child with complex phenotype. BMC Med Genet. (2016) 17:84. 10.1186/s12881-016-0346-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cifaldi C, Angelino G, Chiriaco M, Di Cesare S, Claps A, Serafinelli J, et al. Late-onset combined immune-deficiency due to LIGIV mutations in a 12 years old patient. Pediatr Allergy Immunol. (2016) 28:203–6. 10.1111/pai.12684 [DOI] [PubMed] [Google Scholar]
- 24.Unal S, Cerosaletti K, Uckan-Cetinkaya D, Cetin M, Fatma G. A novel mutation in a family with DNA ligase IV deficiency syndrome. Pediatr Blood Cancer (2009) 53:482–4. 10.1002/pbc.22031 [DOI] [PubMed] [Google Scholar]
- 25.Bluteau O, Sebert M, Leblanc T, De Latour RP, Quentin S, Lainey E, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood (2018) 131:717–32. 10.1182/blood-2017-09-806489 [DOI] [PubMed] [Google Scholar]
- 26.Rodrigues F, Davies EG, Harrison P, McLauchlin J, Karani J, Portmann B, et al. Liver disease in children with primary immunodeficiencies. J Pediatr. (2004) 145:333–9. 10.1016/j.jpeds.2004.05.037 [DOI] [PubMed] [Google Scholar]
- 27.Kruszewski M, Zaim J, Gradzka I, Szumiel I. Comparison of the effects of bleomycin and ionizing radiation in two sublines of murine lymphoma L5178Y. Nukleonika (2001) 46:81–6. [Google Scholar]
- 28.Adema AD, Cloos J, Verheijen RHM, Braakhuis BJM, Bryant PE. Comparison of bleomycin and radiation in the G2 assay of chromatid breaks. Int J Radiat Biol. (2003) 79:655–61. 10.1080/09553000310001596968 [DOI] [PubMed] [Google Scholar]
- 29.Ostling O, Johanson KJ. Bleomycin, in contrast to gamma irradiation, induces extreme variation of DNA strand breakage from cell to cell. Int J Radiat Biol Relat Stud Phys Chem Med. (1987) 52:683–91. 10.1080/09553008714552201 [DOI] [PubMed] [Google Scholar]
- 30.Slack J, Albert MH, Balashov D, Belohradsky BH, Bertaina A, Bleesing J, et al. Outcome of hematopoietic cell transplantation for DNA double-strand break repair disorders. J Allergy Clin Immunol. (2018) 141:322–328.e10. 10.1016/j.jaci.2017.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Driscoll M, Jeggo PA. CsA can induce DNA double-strand breaks: implications for BMT regimens particularly for individuals with defective DNA repair. Bone Marrow Transplant. (2008) 41:983–9. 10.1038/bmt.2008.18 [DOI] [PubMed] [Google Scholar]