

Abstract
Background and Objectives
Autosomal recessive non-syndromic hearing loss (ARNSHL) with genetic origin is common (1/2000 births). ARNSHL can be associated with mutations in gap junction protein beta 2 (GJB2). To this end, this cohort investigation aimed to find the contribution of GJB2 gene mutations with the genotype-phenotype correlations in 45 ARNSHL cases in the Kurdish population.
Subjects and Methods
Genomic DNA was extracted from a total of 45 ARNSHL families. The linkage analysis with 3 short tandem repeat markers linked to GJB2 was performed on 45 ARNSHL families. Only 9 of these families were linked to the DFNB1 locus. All the 45 families who took part were sequenced for confirmation linkage analysis (to perform a large project).
Results
A total of three different mutations were determined. Two of which [c.35delG and c.-23+1G>A (IVS1+1G>A)] were previously reported but (c.299-300delAT) mutation was novel in the Kurdish population. The homozygous pathogenic mutations of GJB2 gene was observed in nine out of the 45 families (20%), also heterozygous genotype (c.35delG/N)+(c.-23+1G>A/c.-23+1G>A) were observed in 4/45 families (8.8%). The degree of hearing loss (HL) in patients with other mutations was less severe than patients with c.35delG homozygous mutation (p<0.001).
Conclusions
Our data suggest that GJB2 mutations constitute 20% of the etiology of ARNSHL in Iran; moreover, the c.35delG mutation is the most common HL cause in the Kurdish population. Therefore, these mutations should be included in the molecular testing of HL in this population.
Keywords: Hearing loss, Autosomal recessive non-syndromic hearing loss, Gap junction protein beta 2
Introduction
Hearing loss (HL) is a common sensorineural disorder, with an incidence of 1 in every 500-1,000 children [1]. At least half of the HL cases can be attributed to genetic factors and more than two-third of these individuals are classified as autosomal recessive non-syndromic hearing loss (ARNSHL) [2]. Non-syndromic or syndromic HL may be caused by environmental factors in configuration with a certain genotype. But it should be noted that the environmental factors are almost ineffective on the gap junction protein beta 2 (GJB2) gene. Because HL is extremely heterogeneous, studying large-sized families from different ethnicities such as the Middle East populations are helpful [3].
At least 42 genetic loci are associated with syndromic forms and over 163 genetic loci are associated with non-syndromic HL (http://hereditaryhearingloss.org/). The inheritance of the disease may be autosomal recessive, autosomal dominant, X-linked recessive and mitochondrial [4]. Based on different investigations, mutations of GJB2 gene are the most common cause of ARNSHL worldwide (e.g. Iran).
The connexin 26 (Cx26) gap junctional proteins are encoded by GJB2 gene. Gap junctions are clusters of intercellular channels for cell-to-cell diffusion of small molecules [5]. These proteins exist in different organs and tissues. For example, the second messenger inositol 1,4,5-trisphosphate (IP3) are in cochlea which transmit signals to intracellular compartments and are essential in perceiving sounds, originally assigned to chromosome 13q12. It accounts for about 50% of this common type of HL [6].
The c.35delG mutation that has been tested in recombinant expression systems confirmed loss of function due to altered sorting, also this mutation, is the most frequent recessive Cx26 mutation in Caucasoid families, which results in a frameshift and premature termination of the protein. The pattern of GJB2 allele distribution has a considerable diversity among different races and ethnic groups. A variant-specific, the c.35delG mutation, accounts for >70% of the disease alleles in the northern and southern Europe as well as American Caucasian, carrier frequencies range from 1.3% to 2.8% [7].
However, the high spectrum and prevalence of GJB2 mutations in exon 2 (coding) exists but the prevalence of this mutations are also significantly evident in exon 1 (non-coding) [8]. The c.-23+1G>A and c.299-300delAT mutations were already detected respectively in exon 1 and exon 2 of GJB2, the c.-23+1G>A is a splice site mutation [9], and the c.299-300delAT mutation was reported to be the severe phenotype of ARNSHL in this population.
The c.299-300delAT mutation caused that gap junction proteins cannot form in the plasma membrane. These disabled proteins were remained in the endoplasmic reticulum (ER), it proposed that ER stress (ERS) and followed of ERS-induced cell death may be responsible for HL [10].
The c.299-300delAT mutation is a frameshift that caused an altered amino acid sequence. Studies showed that c.-23+1G>A produced non-functional mRNA [11]. The c.-23+1G>A mutation was shown in different frequencies between various populations including Mongolian, Yakut, Czech, Hungarian, Turkish and Iranian. However, they do not provide a sufficient data on the frequency of GJB2 mutations with genotype-phenotype correlation, in certain ethnic groups such as Kurds [12]. This study focuses on assessing the contribution of GJB2 mutations (exons 1 and 2) for developing an early onset of HL. Moreover, it aims to determine the mutation profile with genotype-phenotype correlation in hearing-impaired Kurdish participants.
Subjects and Methods
Subjects
In this study 45 Kurdish families with the ARNSHL were selected from a genetic counseling clinic in Iran. ARNSHL individuals were selected by screening the family pedigrees, the patients’ audiograms, the medical records, and also the individual information of each patient in these families. They were, therefore, 45 non-related families. All the family members were interviewed after signing a written consent form. The criteria for choosing individuals to take part in the study were as follows: 1) Autosomal recessive pattern of inheritance should be suspected; 2) Clinical and demographic information should be provided; 3) There must be no syndromic (signs and symptoms other than HL) or environmental evidence (infections such as rubella, meningitis, and ototoxic drugs); and 4) At least two patients should exist in the family.
The Review Board of Shahrekord University of Medical Sciences, Shahrekord, Iran, confirmed this study in 2016 (IR.SKUMS.REC.1395.105). After filling out an informed consent form, about 5 mL of peripheral blood was collected in tubes containing EDTA (0.5 M) from all ARNSHL family members. Also in this study, we collected samples of three generations in each family (grandparents and cousins were included).
DNA extraction
DNA was extracted using a DNA extraction kit (DNPTM, CinnaGen, Tehran, Iran). The quality and quantity of DNA samples can be checked with the Nanodrop spectrophotometer (Thermo Scientific NanoDrop 1000 Spetrophotometer, Thermo Scienific, Wilmington, NC, USA).
Genetic analysis of GJB2
Two methods of mutation detection including genetic linkage analysis followed by DNA sequencing of GJB2 were used. Altogether 45 families participated in parallel in this study.
Amplification of the short tandem repeat markers by linkage analysis
PCR reaction of three primer pairs for three DFNB1 short tandem repeat (STR) markers was performed (Table 1). The amplified conditions were as follows: reaction volume was 15 μL with a final concentration of: ddH2O 7.5 μL, master mix 5.5 μL, Primer F 0.5 μL (20 pmol/μL), Primer R 0.5 μL (20 pmol/μL), and template genomic DNA 1 μL (100 ng/μL).
Table 1.
The short tandem repeat markers of DFNB1 (GJB2) and their primer sequences
| Primer name | Type of primer | Primer sequence (5’ → 3’) | Fragment size (bp) |
|---|---|---|---|
| D13S1236 | Primer R | AAGGGGCTGGCTCTTCA | 108-132 |
| Primer F | GCACTTGGCCTGGGTAA | ||
| D13S1275 | Primer R | CCAGCATGACCTTTACCAG | 180-214 |
| Primer F | ATCACTTGAATAAGAAGCCATTTG | ||
| D13S175 | Primer R | TGCATCACCTCACATAGGTTA | 101-113 |
| Primer F | TATTGGATACTTGAATCTGCTG |
F: Forward, R: Reverse
The PCR-reactions were performed in a thermal cycler (Program Temp Control System PC-700, ASTEC, Fukuoka, Japan) with the following conditions: 95°C for 3 minutes, 30 cycles including 95°C for 30 seconds, 57°C for 30 seconds, 72°C for 30 seconds, and finally 72°C for 6 minutes. PCR amplicons were analyzed on a 15% polyacrylamide gel electrophoresis [13] (Fig. 1). Haplotype and LOD score analysis were used to infer genetic linkage to DFNB1 as previously described (Fig. 1).
Fig. 1.
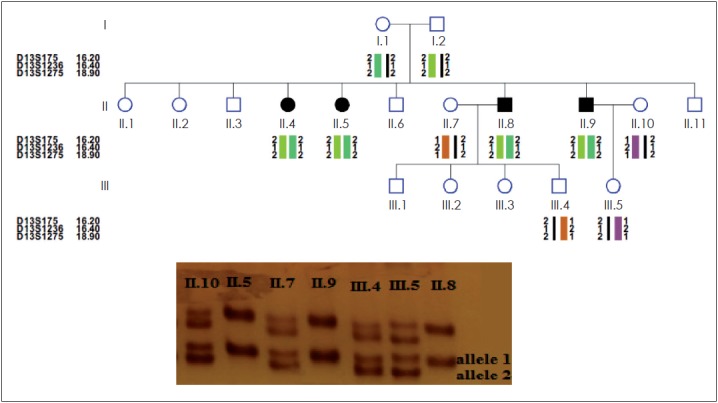
Haplotype analysis of the Iran-Saberi pedigree was shown in up figure and being linked to the D13S 1236 marker of GJB2 gene was detected by the 15% polyacrylamide gel at this pedigree in down figure. Healthy individuals were heterozygous (1/2); three patients were homozygous (1/1).
The 9 families linked to DFNB1 locus were further analyzed for mutation detection but GJB2 was sequenced in each of the tests performed on 45 families to confirm linkage analysis (to carry out a large project and to investigate other effective genes in HL by linkage analysis). The mutations were detected in the 9 families linked and so our results confirm the accuracy of linkage analysis.
Mutation analysis of GJB2 gene in patients by sequence analysis
The GJB2 gene sequencing was performed on one patient in each of the families. Genomic DNA was PCR amplified for sequencing using the following primer pairs: F2: 5' CTC CCT GTT CTG TCC TAG CT 3', R2: 5' CTC ATC CCT CTC ATG CTG TC 3' covering the exon 2 of GJB2. To amplify the exon 1 of GJB2 gene, the following primers were used: F1: 5' CGT CTT TGG GGG TGT TGC TT 3', R1: 5' CAT GAA GAG GGC GTA CAA GTT AGA A 3'.
The amplification conditions were as follows: reaction volume was 25 μL with a final concentration of: ddH2O 18 μL, MgCl2 1.7 μL (100 mM), dNTPs 0.5 μL (10 mM), PCR buffer 2.5 μL, Taq polymers 0.3 μL (3 units/μL), primer F 0.5 μL (20 pmol/μL), primer R 0.5 μL (20 pmol/μL), and template genomic DNA 1 μL (100 ng/μL). The PCR-reaction was performed in a thermal cycler (Program Temp Control System PC-700, ASTEC) at 95°C for 4 minutes, 35 cycles including 95°C for 30 seconds, 60°C for 30 seconds, 72°C for 24 seconds, and finally 72°C for 5 minutes. Sequencing was carried out in SEQLAB (Sequence Laboratories, Gottingen, Germany).
The audiological study of GJB2 mutation positive patients
For each patient (3 to 35 years old), a complete database of the patient’s medical conditions was reviewed to understand the cause of HL, no environmental factors such as maternal to fetal infections, prenatal complications, bacterial or viral infection (meningitis), prenatal and postnatal ototoxic drugs such as aminoglycosides were involved in HL. All the affected family members underwent an ear examination and a general health check to determine the type of HL.
Individuals were also screened for vision assessment, proteinuria, and haematuria. Hearing sensitivity was measured by pure-tone audiometric test (PTA) for air and bone conduction at different frequencies (250-8,000 Hz) in all patients. The degree of HL was classified according to the PTA average at 500, 1,000, 2,000 and 4,000 Hz: mild ≥21 dB, moderate ≥46 dB, moderately severe ≥61 dB, severe ≥76 dB and profound ≥97 dB [14].
Statistical analysis
The data was analyzed by SPSS 11.5 (SPSS Institute, Chicago, IL, USA). The χ2 or Fisher’s exact tests was used to detect differences in Genotype-phenotype with GJB2 mutations. A p value <0.05 was accepted as statistically significant.
Results
In this research 45 Kurdish families were selected to determine the genetic and molecular basis of hearing impairment. HL state was autosomal recessive in this research. HL occurrences may vary in range from mild to profound. Linkage analysis was performed with 3 STR markers (Table 1). Linkage analysis and sequencing were performed for all 45 families who participated in the study but only 9 families were linked to the locus DFNB1 and their mutations were detected by sequencing analysis. So our results confirm the accuracy of linkage analysis.
The c.35delG mutation was detected in 17.7% (8/45) of Kurdish population and the allele frequency was 13.33% (12 positives in 90 alleles). In the 45 families analyzed, the homozygous state was observed for this mutation in 8.8% of the Kurdish population, the heterozygous state was observed in 8.8% and 82.4% of these individual have no c.35delG mutation.
Furthermore, in the next step of the study all individuals who were not c.35delG positive and 4 heterozygous patients for this mutation were analyzed to detect other mutations in the entire coding and exon 1 (noncoding) of GJB2 gene using direct genomic DNA sequencing (Fig. 2). In 8.8% (4/45) of the families, homozygosity for the c.23+1G>A mutation was found in exon 1, also these 4 families had heterozygosity for the c.35delG mutation (Table 2). In addition, 2.2% (1/45) of the families had homozygosity for the c.299-300delAT mutation in exon 2 (Fig. 2). This is the first reported mutation in Kurdish population.
Fig. 2.
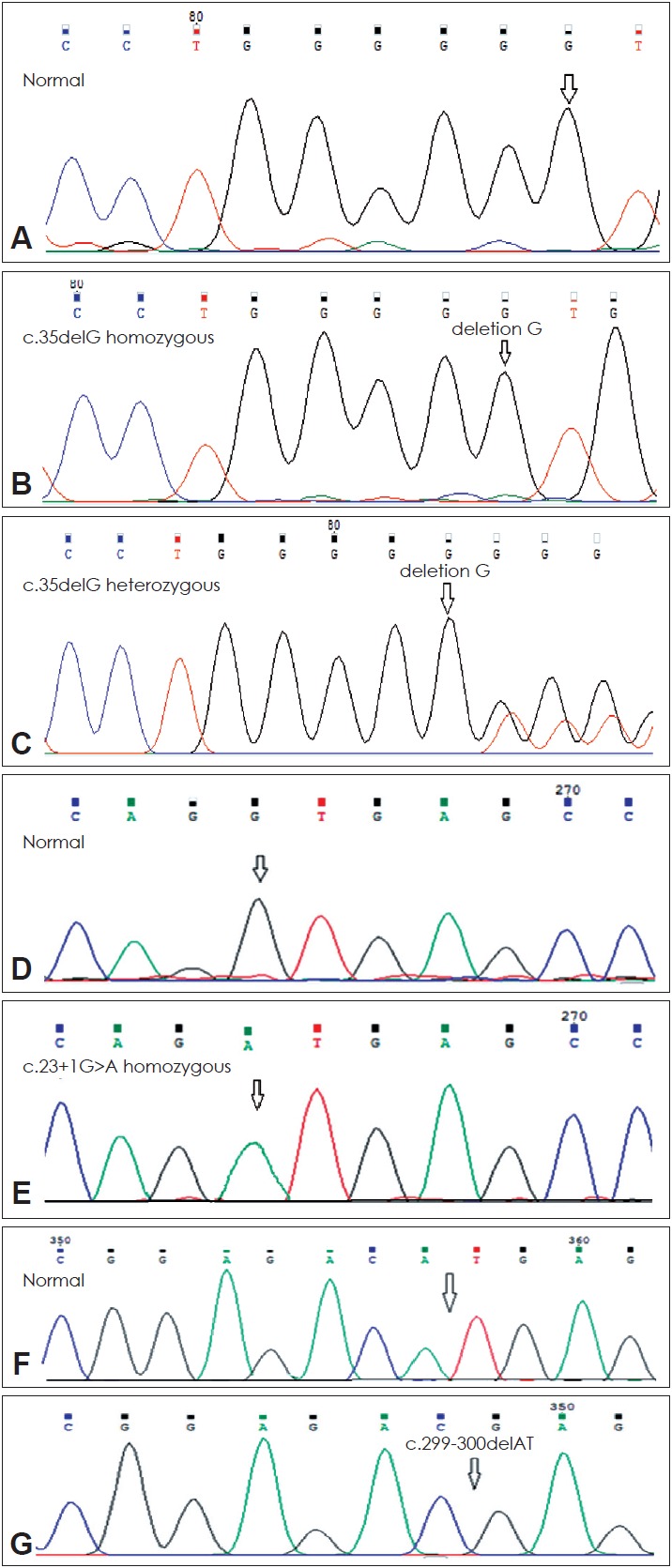
The electropherograms of the GJB2 sequance normal form (A), the c.35delG homozygous mutation (B), the c.35delG heterozygous mutation (C), the GJB2 sequance normal form (D), the c.23+1G>A homozygous mutation (E), the GJB2 sequance normal form (F) and the c.299-300delAT homozygous mutation (G), genotypes are shown. Arrows show the location of the base change.
Table 2.
Details of GJB2 gene mutations investigated in mutation taster
| Site | Clinical significance | Exon | Nucleotide change | Variant type | Protein change | Patients homozygous | Patients heterozygous | Origin |
|---|---|---|---|---|---|---|---|---|
| Mutation taster | Pathogenic | 2 | c.35delG (rs80338939) | Deletion | frameshift | 4 (8.8%) | 4 (8.8%) | Germ line |
| Mutation taster | Pathogenic | 1 | c.23+1G>A (rs80338940) | Splice site | Non-coding | 4 (8.8%) | - | Germ line |
| Mutation taster | Pathogenic | 2 | c.299-300delAT (rs111033204) | Deletion | Frameshift (leading to a stop at Codon) | 1 (2.2%) | - | Germ line |
Genotype-phenotype correlation of GJB2 patients
Patients with (c.35delG/N)+(c.-23+1G>A/c.-23+1G>A) genotype and c.35delG homozygous genotype were investigated audiologically. This analysis was carried out on 19 patients from 8 families with GJB2 mutations. Ten patients from these families with profound HL (≥97 dB) at 500, 1,000, 2,000 and 4,000 Hz had c.35delG homozygous mutation (4 families), and 9 other patients (4 families) with moderately severe HL (=61 dB) (Fig. 3) had (c.35delG/N)+(c.-23+1G>A/c.-23+1G>A) genotype (p<0.001) (Fig. 4). Fisher’s exact test was used to detect a positive genotype-phenotype correlation in two genotypes (Table 3). A patient with severe phenotype (≥76 dB) had the c.299-300delAT homozygous genotype.
Fig. 3.

Pure tone audiometries of profound and moderately severe with two GJB2 mutations [c.35delG homozygous and (c.35delG/N)+(c.-23+1G>A/c.-23+1G>A) genotype] respectively are in patient 2 and patient 1.
Fig. 4.

Distribution of two classes of HL: the c.35delG homozygous and the (c.35delG/N)+(c.-23+1G>A/c.-23+1G>A) genotype (p<0.001).
Table 3.
Comparison of our data and the literature for specific GJB2 genotype-phenotype correlations
| Mutation and genotype | No. of subjects | Frequency of moderately severe | Frequency of profound | p value* |
|---|---|---|---|---|
| c.35delG/c.35delG | 10 (52.6) | 0 (0) | 10 (100) | <0.001 |
| (c.35delG/N)+(c.-23+1G>A/c.-23+1G>A) | 9 (47.4) | 9 (100) | 0 (0) |
Values are presented as numbers (%).
p value calculated by comparing two genotypes to hearing sensitivity, Fisher’s exact tests were used to determine genotype-phenotype positive correlation in this study
Discussion
In the present research, the contribution of GJB2 mutations was associated to the HL in nine out of the 45 ARNSHL Kurdish families. We developed a simple, accurate method to determine the c.35delG mutation. Three GJB2 mutations were determined in the present research. The c.35delG mutation, c.-23+1G>A mutation, and c.299-300delAT mutation (first report in Kurdish population) have already been investigated [11]. GJB2 mutations were detected in 9 ARNSHL (20%) patients which was similar to the results of the previous investigations in other regions of Iran [4,15].
The severity of the HL in these 9 patients varied from moderately severe to profound. This study determined the correlation between the phenotype and the GJB2 mutations. Patients with c.35delG/N mutation may have a homozygous different pathogenic mutation in other places. Then after sequencing of the entire coding and noncoding sequence of GJB2 gene, homozygous c.299-300delAT (2.2%) and c.-23+1G>A (8.8%) mutations were observed. Based on these findings, the ARNSHL phenotype in families with c.35delG heterozygote state associated to c.-23+1G>A homozygous state was moderately severe, while families with homozygous c.35delG (8.8%) mutation had profound HL.
Our results detected the contribution of common GJB2 mutations in Kurdish population. Various investigations had determined that the c.35delG mutation is the most common in many ethnic groups, this mutation accounts for 85% of the mutations in GJB2 gene [6]. It is probable that the c.35delG mutation originated in ancient Greece and was subsequently propagated in the Mediterranean [16]. The c.35delG is the most common mutation in the Mediterranean [17], which shows genetic heterogeneity of HL.
The frequency of the c.35delG mutation in the Oman deaf population is null [18], as in African Americans [19], Ghanaian [20], and Kenyan populations [21]. The frequency of this mutation is less than 4% in Australia [22] and Sudan (2.7%); in Iran it is similar to other Mediterranean populations. Also, the main mutations in the GJB2 gene in Dagestan were represented by three forms typical for West Asia: c.35delG mutation (22% of all mutant alleles), deltaE120 mutation (22%), and c.-23+1G>A (44%) were found in a compound heterozygous or a heterozygous state [23], and in Çukurova region of turkey the c.35delG mutation was known as the most frequent mutation of GJB2 gene and had the most frequently mutation (5.5%) in patients with HL [24].
Variation in frequency between different ethnic groups can be the result of human migrations; the high prevalence of the c.35delG mutation in Caucasian populations is possibly the result of a founder effect rather than a mutational hot spot [25]. The Iranian population is composed of various ethnic groups; the prevalence of this mutation differs between Iranian population with a higher slope of mutation frequency from south-tonorth (0% to 33%), and from east-to-west (8% to 8.8%) [12,15].
As a result, GJB2 gene related HL has a low portion of ARNSHL in our population comparing to other ones [26]. The high percentage in northwest and center of Iran may be due to immigration of people in some cities in Iran or may be due to the consanguinity or ascertainment among different ethnic groups. The observations support the presence of a founder effect in northern Iran (compared to 43% in Anatolia) [19]. Based on the population frequencies of the c.35delG mutation and tribes migrating from central Asia to Anatolia 10 centuries ago, we think that the c.35delG mutation probably arose in northwest of Iran [19].
The c.-23+1G>A and c.299-300delAT mutations are among the top ten GJB2 mutations responsible for HL within various populations [14]. Functional studies of c.-23+1G>A mutation have not obtained detectable GJB2 protein and mRNA [11]. Homozygous c.-23+1G>A mutation reported in Iran’s ethnic groups have a different percentage (2.5% average), in another study [27], which is lower than our observation (8.8%).
The (c.35delG/N)+(c.-23+1G>A/c.-23+1G>A) genotype was less severe than that of c.35delG homozygous (p<0.001). We conclude that c.-23+1G>A homozygous mutation is often restricted in the Kurdish population; that shows a founder effect for this mutation. Also, we could identify c.299-300delAT homozygous mutation in one Kurdish family for the first time in Iran who had severe phenotype ARNSHL. The c.299-300delAT mutation is a frameshift that caused an altered amino acid sequence from codon 100, leading to a stop at codon 113 (Fig. 5).
Fig. 5.

The location of the GJB2 mutations in the connexion protein.
This mutation was first reported as a heterozygote variant related to ARNSHL [28]. Another study found this mutation in homozygote form [29], and then reported it in Turkey [30], and Iran [12]. These findings suggested the involvement of other genetic or environmental causes playing an important role in manifesting any disorders in this population. So far, etiology of this disorder remains largely unknown; the presence of modifier gene or other connexin genes can be involved in HL.
Consequently, more investigations should be conducted to search the existence of pathogenic mutations in other loci. The high penetrance of GJB2 mutations (very much the c.35delG and c.-23+1G>A) in Kurdish population may increase the chance for determining newborns with the ARNSHL by genetic testing. So, it can prevent unnecessary diagnostic testing and improve its diagnosis and treatment in Iran. The Iranian population is composed of various ethnic groups.
Therefore, further studies on the basis of the ethnicity are necessary to identify the ethnicity-based gene profile associated with ARNSHL. So these observations can be helpful in clinical settings to prognosticate the outcomes of genetic testing and disease period to the individuals with HL in Kurdish population.
Acknowledgments
The authors gratefully acknowledge all families and the province of Kurdistan Social Welfare & Rehabilitation Center staff of the Cellular and Molecular Research Center who participated in this research. This study was financially supported by the research deputy of the Shahrekord University of Medial Sciences (grant: 2165).
Footnotes
Conflicts of interest: The authors have no financial conflicts of interest.
REFERENCES
- 1.Yazdanpanahi N, Tabatabaiefar MA, Farrokhi E, Abdian N, Bagheri N, Shahbazi S, et al. Compound heterozygosity for two novel SLC26A4 mutations in a large Iranian pedigree with Pendred Syndrome. Clin Exp Otorhinolaryngol. 2013;6:201–8. doi: 10.3342/ceo.2013.6.4.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azadegan-Dehkordi F, Ahmadi R, Bahrami T, Yazdanpanahi N, Farrokhi E, Tabatabaiefar MA, et al. A novel variant of SLC26A4 and first report of the c.716T>A variant in Iranian pedigrees with non-syndromic sensorineural hearing loss. Am J Otolaryngol. 2018;39:719–25. doi: 10.1016/j.amjoto.2018.07.022. [DOI] [PubMed] [Google Scholar]
- 3.Rehman AU, Friedman TB, Griffith AJ. Unresolved questions regarding human hereditary deafness. Oral Dis. 2017;23:551–8. doi: 10.1111/odi.12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaleshtori MH, Farrokhi E, Shahrani M, Kheiri S, Dolati M, Rad LH, et al. High carrier frequency of the GJB2 mutation (35delG) in the north of Iran. Int J Pediatr Otorhinolaryngol. 2007;71:863–7. doi: 10.1016/j.ijporl.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Selli J, Unal D, Mercantepe F, Akaras N, Kabayel R, Unal B, et al. Protective effects of beta glucan in brain tissues of post-menopausal rats: a histochemical and ultra-structural study. Gynecol Endocrinol. 2016;32:234–9. doi: 10.3109/09513590.2015.1110139. [DOI] [PubMed] [Google Scholar]
- 6.Palmer CG, Boudreault P, Baldwin EE, Sinsheimer JS. Impact of genetic counseling and Connexin-26 and Connexin-30 testing on deaf identity and comprehension of genetic test results in a sample of deaf adults: a prospective, longitudinal study. PLoS One. 2014;9:e111512. doi: 10.1371/journal.pone.0111512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denoyelle F, Weil D, Maw MA, Wilcox SA, Lench NJ, Allen-Powell DR, et al. Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Hum Mol Genet. 1997;6:2173–7. doi: 10.1093/hmg/6.12.2173. [DOI] [PubMed] [Google Scholar]
- 8.Mani RS, Ganapathy A, Jalvi R, Srikumari Srisailapathy CR, Malhotra V, Chadha S, et al. Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss. Eur J Hum Genet. 2009;17:502–9. doi: 10.1038/ejhg.2008.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Angeli SI. Phenotype/genotype correlations in a DFNB1 cohort with ethnical diversity. Laryngoscope. 2008;118:2014–23. doi: 10.1097/MLG.0b013e31817fb7ad. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Wang J, Li L, Sun Y, Feng B. Three common GJB2 mutations causing non-syndromic hearing loss in Chinese populations are retained in the endoplasmic reticulum. Acta Otolaryngol. 2010;130:799–803. doi: 10.3109/00016480903443191. [DOI] [PubMed] [Google Scholar]
- 11.Shahin H, Walsh T, Sobe T, Lynch E, King MC, Avraham KB, et al. Genetics of congenital deafness in the Palestinian population: multiple connexin 26 alleles with shared origins in the Middle East. Hum Genet. 2002;110:284–9. doi: 10.1007/s00439-001-0674-2. [DOI] [PubMed] [Google Scholar]
- 12.Bazazzadegan N, Nikzat N, Fattahi Z, Nishimura C, Meyer N, Sahraian S, et al. The spectrum of GJB2 mutations in the Iranian population with non-syndromic hearing loss--a twelve year study. Int J Pediatr Otorhinolaryngol. 2012;76:1164–74. doi: 10.1016/j.ijporl.2012.04.026. [DOI] [PubMed] [Google Scholar]
- 13.Dehkordi FA, Rashki A, Bagheri N, Chaleshtori MH, Memarzadeh E, Salehi A, et al. Study of VSX1 mutations in patients with keratoconus in southwest Iran using PCR-single-strand conformation polymorphism/heteroduplex analysis and sequencing method. Acta Cytol. 2013;57:646–51. doi: 10.1159/000353297. [DOI] [PubMed] [Google Scholar]
- 14.Barashkov NA, Pshennikova VG, Posukh OL, Teryutin FM, Solovyev AV, Klarov LA. Spectrum and frequency of the GJB2 gene pathogenic variants in a large cohort of patients with hearing impairment living in a subarctic region of Russia (the Sakha Republic) PLoS One. 2016;11:e0156300. doi: 10.1371/journal.pone.0156300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koohiyan M, Hashemzadeh-Chaleshtori M, Salehi M, Abtahi H, Reiisi S, Pourreza MR, et al. GJB2 mutations causing autosomal recessive non-syndromic hearing loss (ARNSHL) in two Iranian populations: report of two novel variants. Int J Pediatr Otorhinolaryngol. 2018;107:121–6. doi: 10.1016/j.ijporl.2018.01.012. [DOI] [PubMed] [Google Scholar]
- 16.Lucotte G, Diéterlen F. The 35delG mutation in the connexin 26 gene (GJB2) associated with congenital deafness: European carrier frequencies and evidence for its origin in ancient Greece. Genet Test. 2005;9:20–5. doi: 10.1089/gte.2005.9.20. [DOI] [PubMed] [Google Scholar]
- 17.Duman D, Tekin M. Autosomal recessive non-syndromic deafness genes: a review. Front Biosci (Landmark Ed) 2012;17:2213–36. doi: 10.2741/4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simsek M, Al-Wardy N, Al-Khayat A, Shanmugakonar M, Al-Bulushi T, Al-Khabory M, et al. Absence of deafness-associated connexin-26 (GJB2) gene mutations in the Omani population. Hum Mutat. 2001;18:545–6. doi: 10.1002/humu.1233. [DOI] [PubMed] [Google Scholar]
- 19.Azadegan-Dehkordi F, Ahmadi R, Koohiyan M, Hashemzadeh-Chaleshtori M. Update of spectrum c.35delG and c.-23+1G>A mutations on the GJB2 gene in individuals with autosomal recessive non-syndromic hearing loss. Ann Hum Genet. 2018 Sep 3; doi: 10.1111/ahg.12284. [Epub]. [DOI] [PubMed] [Google Scholar]
- 20.Hamelmann C, Amedofu GK, Albrecht K, Muntau B, Gelhaus A, Brobby GW, et al. Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum Mutat. 2001;18:84–5. doi: 10.1002/humu.1156. [DOI] [PubMed] [Google Scholar]
- 21.Gasmelseed NM, Schmidt M, Magzoub MM, Macharia M, Elmustafa OM, Ototo B, et al. Low frequency of deafness-associated GJB2 variants in Kenya and Sudan and novel GJB2 variants. Hum Mutat. 2004;23:206–7. doi: 10.1002/humu.9216. [DOI] [PubMed] [Google Scholar]
- 22.Wilcox SA, Saunders K, Osborn AH, Arnold A, Wunderlich J, Kelly T, et al. High frequency hearing loss correlated with mutations in the GJB2 gene. Hum Genet. 2000;106:399–405. doi: 10.1007/s004390000273. [DOI] [PubMed] [Google Scholar]
- 23.Bozhkova VP, Khashaev ZKh, Umanskaia TM. [Frequency and the mutation spectrum of GJB2-related disorders of hearing in children from Dagestan as compared with the central European part of Russia] Biofizika. 2010;55:514–25. [PubMed] [Google Scholar]
- 24.Bozdoğan ST, Kuran G, Yüregir ÖÖ, Aslan H, Haytoğlu S, Ayaz A, et al. The prevalence of gap junction protein beta 2 (GJB2) mutations in non syndromic sensorineural hearing loss in Çukurova region. J Int Adv Otol. 2015;11:118–21. doi: 10.5152/iao.2015.1212. [DOI] [PubMed] [Google Scholar]
- 25.Banjara H, Mungutwar V, Swarnkar N, Patra P. Detection of connexion 26 GENE (GJB2) mutations in cases of congenital non syndromic deafness. Indian J Otolaryngol Head Neck Surg. 2016;68:248–53. doi: 10.1007/s12070-015-0950-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, et al. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet. 1998;62:792–9. doi: 10.1086/301807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snoeckx RL, Hassan DM, Kamal NM, Van Den Bogaert K, Van Camp G. Mutation analysis of the GJB2 (connexin 26) gene in Egypt. Hum Mutat. 2005;26:60–1. doi: 10.1002/humu.9350. [DOI] [PubMed] [Google Scholar]
- 28.Abe S, Usami S, Shinkawa H, Kelley PM, Kimberling WJ. Prevalent connexin 26 gene (GJB2) mutations in Japanese. J Med Genet. 2000;37:41–3. doi: 10.1136/jmg.37.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu BC, Bian PP, Liu XW, Zhu YM, Yang XL, Ma JL, et al. Analysis of common deafness gene mutations in deaf people from unique ethnic groups in Gansu Province, China. Acta Otolaryngol. 2014;134:924–9. doi: 10.3109/00016489.2014.927588. [DOI] [PubMed] [Google Scholar]
- 30.Fishelson M, Geiger D. Optimizing exact genetic linkage computations. J Comput Biol. 2004;11:263–75. doi: 10.1089/1066527041410409. [DOI] [PubMed] [Google Scholar]