

Abstract
Background/Aim: Pancreatic cancer (PC) is currently the fourth leading cause of cancer-related mortality worldwide. Peripheral blood mononuclear cells (PBMCs) is a subpopulation of accessible and functional immune cells. Comparative analysis of the proteome of PBMCs can help us elucidate the mechanism of disease and find potential biomarkers for diagnosis. Materials and Methods: PBMCs were collected from healthy individuals, patients with benign diseases, and pancreatic cancer. iTRAQ-2DLC-MS/MS and SWATH methodologies were applied to make a comparative proteomics analysis of PBMCs. Results: A total of 3,357 proteins with a false discovery rate (FDR) <1% were identified, of which 114 proteins were found dysregulated in the PC group. An extensive SWATH library was constructed which showed a potential application for large scale clinical sample analysis. Conclusion: A PBMCs proteome with extensive protein representation was achieved, which will potentially allow the identification of novel biomarkers for PC.
Keywords: PBMC, blood, iTRAQ, SWATH, pancreatic cancer
Pancreatic cancer (PC) is an aggressive malignancy, characterized by invasiveness, rapid progression and profound resistance to treatments (1-3). To date, there are no clinically validated screening methods for PC in the curative stage (4,5). There is an urgent need for additional studies, applying modern technologies, on PC to increase our understanding of this disease, and improve the ability to diagnose PC in asymptomatic patients.
In our previous work, surgically resected fresh PC tissues and adjacent non-tumor tissues were investigated and novel prognostic predictors of PC and PC-associated diabetes mellitus were discovered (6,7). Given the asymptomatic nature of PC, a blood-based assay is preferred because it would be feasible and minimally invasive (8). Peripheral blood mononuclear cells (PBMCs), including monocytes, T-cells, B cells, and natural killer (NK) cells (9), play important roles in the function of the immune system and in monitoring immune-relevant events. They are readily accessible from routinely collected blood. Furthermore, it is known that the immune system is involved not only in the pathogenesis of autoimmune and infectious diseases, but also in cancer (10,11), metabolic syndrome (12), atherosclerosis (13) and many other diseases (14). The PBMCs represent a biological sample which closely reflect the response of the body to pathogens and diseases like cancer. Therefore, they may allow for discovery of potentially pathology-relevant biomolecules of PC, such as DNA, RNA and proteins (15).
Baine et al. reported an in-depth comparison of global gene expression profiles in PBMCs of PC patients and healthy controls, revealing that 383 genes were significantly different between PC and healthy controls (16). However, it is unknown whether these alterations have been transferred into the proteome of PBMCs in PC. Proteomic analysis provides large-scale determination of gene and cellular function at the protein level, so that it is quintessential to understanding health and disease of any organ (17).
Comparative proteomic technologies allow for identification and quantitation of thousands of proteins in complex biological systems, indicating tens and hundreds of candidate protein biomarkers. Currently, the most common strategy for peptide identification and quantitation remains the data-dependent acquisition (DDA) approach, in which the instrument sequentially surveys the peptide ions that are eluted from the LC column at a particular time (18). DDA-based methods have achieved remarkable progress providing proteome-wide analysis in complex samples (19,20). Isobaric tags for relative and absolute quantitation incorporate with two-dimensional liquid chromatography-tandem mass spectrometry (iTRAQ-2DLC-MS/MS), one of the typical DDA-based comparative proteomics method, offer a multiplex strategy to simultaneously determine protein abundance in up to 8 groups of biological samples (21) and are widely used in biomarker discovery (22-25).
Recent improvements in mass spectrometry (MS) instrumentation have enabled alternative workflows to DDA, namely, data-independent acquisition (DIA) methods. In DIA mode, the instrument collects tandem mass spectrometry (MS2) spectra for the overall expected tryptic peptides within a predefined mass-to-charge (m/z) range (15,18,23,24) by co-isolating and fragmenting multiple peptide precursors. Unlike DDA-based method of stochastic precursor-ion selection, DIA can be recorded independently and then be perpetually re-mined using the targeted analysis strategy (21). Besides, in terms of data consistency and reproducibility, DIA-based methods show a powerful potential to serve as a digital recording of the peptides in multiple samples (26-30). Sequential window acquisition of all theoretical mass spectra (SWATH) approach is one of the exemplified DIA-based methods.
To take full advantage of the strength of both DDA-based method and DIA-based approaches, the proteome of PBMCs from healthy individuals (normal control, NC), patients with benign diseases (BD) and PC patients were analyzed using both strategies. The DDA analysis afforded a deep sequence of proteome identification of PBMCs, and the DIA analysis showed the potential to screen biomarker panels individually. Combining the DDA and DIA results, a total of 3,357 proteins were detected and 114 proteins were found to be altered. These proteins were highly related to immune functions according to bioinformatics annotations. Among these, AOPA1 and CCS were significantly up and down-regulated and showed a potential to be biomarker candidates.
Materials and Methods
Preparation of PBMC lysates. A total of 27 blood samples were collected (using informed consent) at the Huashan Hospital of Fudan university, and categorized as follows: NC (n=9), BD (n=9, pancreatitis (PT, n=3), pancreatic cysts (CY, n=3), benign tumors (BT, n=3)), and PC (n=9). The research followed the tenets of the Declaration of Helsinki and was approved by the Ethics Committee of the Fudan University Shanghai Huashan hospital. Blood samples were collected by venipuncture into EDTA-coated tubes in the morning after an overnight fast. PBMCs were separated and purified from 10 ml blood via density-gradient centrifugation method using Histopaque-1077 (Sigma-Aldrich, Prague, Czech Republic) according to the manufacturer’s instructions. The cells were washed in PBS extensively and centrifuged at 1,000 ×g for three times. Then, the cell pellets were resuspended in lysis buffer (8 M urea/4 mM CaCl2/0.2 M Tris-HCl, pH 8.0), followed by ultrasonic lysis for protein extraction. Protein concentration of each sample was quantified with the BCA Protein Assay Kit (Bio-Rad, Hercules, CA, USA). The sample was stored at −80˚C for further analysis.
Protein digestion. For protein digestion, 200 μg protein of each sample was reduced in 50 mM tris-(2-carboxyethyl) phosphine (TCEP) at 56˚C for 1 h and alkylated in 200 mM methyl methanethiosulfonate (MMTS) at room temperature for 1 h. The reduced and alkylated protein mixtures were precipitated by adding 5× volume of chilled acetone at –20˚C overnight. After centrifugation at 30,000 g for 30 min at 4˚C, the pellet was dissolved in 0.5 M tetraethyl-ammonium bromide (TEAB) and sonicated in ice. Samples were then proteolyzed with 1 μg trypsin:20 μg protein at 37˚C for 16 h and centrifuged at 12,000 rpm for 20 min.
iTRAQ-labeling proteomic analysis. Each group of digested peptides (100 μg) were labeled with iTRAQ reagent at room temperature for 2 h according to the manufacturer’s instructions. In total, a 6-plex group (NC with 113 and 114 tags, BD with 115 and 116 tags, and PC with 117, 118,) and an 8-plex group (NC with 113 and 114 tags, BD with 115 and 116 tags, and PC with 117, 118, 119 and 121 tags) were constructed for 4 biological replicates. The labeled peptides of two groups were mixed separately and dried on a rotation vacuum concentrator (Christ, Germany) for further analysis.
Peptide fractionation with high-pH reverse-phase liquid chromatography. The labeled peptides were fractionated using high pH reversed-phase liquid chromatography on a UPLC system (Waters, Milford, MA, USA). The peptides were resuspended with loading buffer (5 mM Ammonium formate containing 2% acetonitrile, pH 10) and separated on a C-18 column (2.1×250 mm X Bridge BEH300). The gradient elution was performed by 0-25% B (5 mM Ammonium formate containing 98% acetonitrile, pH 10, 5-35 min) and 25-45% B (35-48 min) on high pH RPLC column (Waters, Xbridge C18 3.5 μm, 150×2.1 mm) with a flow rate of 300 μl/min. A total of 20 fractions were collected and then mixed to 10 fractions for each iTRAQ group set. All the fractions were dried on a rotation vacuum concentrator (Christ, Germany).
Liquid chromatography/tandem mass spectrometry (LC-MS/MS) analysis. Each fraction was dissolved in loading buffer (2% acetonitrile with 0.1% FA) and analyzed on a Nanoeasy system (Thermo Fisher Scientific, Waltham, MA, USA) connected to a Q Exactive hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific). Peptides were separated on a 25-cm-long column (75 μm id × 25-cm-long, packed with 2 μm id 100 Å pore size, C18 packing material, Thermo Fisher Scientific). The elution gradient was set from 5% B (98% acetonitrile with 0.1% formic acid) to 40% B in 90 min within a total analysis time of 120 min. Q Exactive mass spectrometer was operated in the data-dependent mode to switch automatically between MS and MS/MS acquisition. Full-scan MS spectra (m/z 300-1200) were acquired with a mass resolution of 70K, followed by ten sequential high-energy collisional dissociation (HCD) MS/MS scans with a resolution of 17.5K. The dynamic exclusion was set as 15 sec. For MS/MS analysis, precursor ions were activated using 27% normalized collision energy.
Database searching and statistical analysis. Protein identification and quantitation was performed on Proteome Discovery software (PD, version 1.3, Thermo Scientific) using MASCOT search engine. All the raw data were searched against Swiss-Prot human database (20,238 entries). Searching parameters were set as following: Quantitation: iTRAQ-8plex; Enzyme: Trypsin; Variable modification: Oxidation (Met); Fixed modification: Methyl methanethiosulfonate (Cys). MS1 tolerance: 20ppm; MS/MS tolerance: 20 mmu; with one missed cleavage on trypsin was allowed.
In this study, a false discovery rate (FDR) lower than 1% was used to control both peptide and protein level identification based on the target-decoy strategy. Proteins identified with at least one unique peptide (confidence was higher than 95%) were used for quantitation. Student’s t-test (PC vs. control (NC and BD)) was applied to compare the protein expression levels between the PC group and the control groups. The mean value of the protein ratio in each group was used to calculate the fold change. Proteins with a fold change larger than 1.2 or less than 0.8 with a Student’s t-test p-value <0.05 were selected as differentially expressed proteins.
Creation of SWATH spectral library. Equal amounts of digested peptides of 6 samples (3 NCs vs. 3 PCs) were pooled together to obtain a good representation of the peptides. Two-microgram peptide mixtures were analyzed in a standard DDA mode on a platform with 1D Plus nano LC system (Eksigent, Dublin, CA, USA) coupled with Triple TOF 5600 system (AB SCIEX, Redwood City, CA, USA) using a 15-cm-long column (75 μm id × 15-cm-long, packed with 2 μm id 100 Å pore size, C18 packing material (Thermo scientific). Peptides were subsequently eluted using the following gradient conditions with phase B (98% acetonitrile with 0.1% formic acid): 5-7% B (0-5 min), 7-23% B (5-40 min) and 23-45% B (40-75 min), and the total flow rate was maintained at 300 nl/min. Triple TOF 5600 mass spectrometer was operated in information-dependent (IDA) mode to switch automatically between MS and MS/MS acquisition. MS spectra were acquired across the mass range of 350-1250 m/z in high resolution mode using 250 ms accumulation time per spectrum. Tandem mass spectral scanned from 100-1250 m/z in high sensitivity mode with rolling collision energy. The 20 most intense precursors were selected for fragmentation per cycle with dynamic exclusion time of 15 s. Database searching was performed on ProteinPilot software (Version 4.5) with similar parameters mentioned in section 2.3.4.
SWATH analysis. 3 NC samples and 3 PC samples were analyzed separately using the same platform and LC gradient described in 2.4.1 in a SWATH-MS mode. The mass spectrometry was specifically tuned to allow a quadrupole resolution of 25-m/z mass selection. Using an isolation width of 26 m/z (containing 1 m/z for the window overlap), a set of 32 continuous MS/MS scans was constructed covering the entire precursor mass range of 400-1250 m/z. An accumulation time of 250 msec was used for the survey scans in high-resolution mode acquired at the beginning of each cycle, followed by a 100 msec accumulation time for all fragment-ion scans in high-sensitivity mode, resulting in a ~3.45 sec total cycle time.
Data analysis. Fragment-ion chromatograms extraction of each precursor in SWATH-MS data was performed using the Peakview software (Verion 2.2) with SWATH Acquisition MicroApp. The reference spectral library constructed before was imported with following restrictions: 1) up to 6 peptides per protein and 6 fragments from one peptide with a confidence above 95% were imported; 2) Any shared and modified peptides were excluded from the extraction. Additionally, the retention time of all SWATH-MS runs were normalized based on the six most abundant peptides of identification. Peak groups from the extracted fragment-ion chromatograms were formed and scored according to their elution profiles and similarity to the target assay in terms of RT, relative fragment-ion intensity and pattern. Only peptides with an FDR less than 5% were used for further protein quantitation. Signal normalization between different SWATH-MS runs was performed using MarkerView (version 1.2.1, Sciex) software based on the total area of each sample. Then the summarized peak areas of the corresponding fragment ions of each peptide were used to calculate the protein abundance ratio. The proteins with Student’s T-test p<0.05 and fold change >1.2 or <0.8 were considered to be differentially expressed.
Bioinformatics analysis. All the dysregulated proteins were submitted to DAVID (26) (https://david.ncifcrf.gov/) for gene ontology (GO) term enrichment annotation, and Ingenuity Pathway Analysist (27) (IPA, Ingenuity® Systems, CA, USA) was applied for the pathway analysis. The physical and functional interactions of differentially expressed proteins were visualized by String database (https://string-db.org/) using the criteria of high confidence (0.7)
Western blot analysis. Apolipoprotein A-I (APOA1) and Copper chaperone for superoxide dismutase (CCS) were validated and semi-quantitated using Western blot. Equal amounts of protein extracted from the PBMCs (3NCs ,3 BDs and 3 PCs) were applied for Western blot analysis. Upon electrophoretic separation by 5-12% SDS-PAGE, the proteins were electro-blotted onto NC/PVDF membranes (Millipore, Billerica, MA, USA). The blots were then blocked using 5% (w/v) nonfat dry milk in PBS overnight at 4˚C prior to immune-probing with antibodies diluted in PBS with 5% (w/v) milk for 2 h each at room temperature. The membranes were incubated with rabbit anti-APOA1 4 (1:1,000), rabbit anti-CCS (1:1,000) obtained from Santa Cruz Biotechnology. HRP-conjugated anti-rabbit IgG (1:4,000) were obtained from Sigma-Aldrich and used as secondary antibodies. Subsequent visualization was performed using SuperSignal West Femto Maximun Sensitivity Substrate (Thermo Scientific) with ACT1 levels used as the loading control. The relative expression levels of APOA1 and CCS in different samples were evaluated based on the gray value of each band which was normalized based on the corresponding ACT1 level. The p-value of Student’s T-test was calculated based on the grey value of each western blot band.
Results
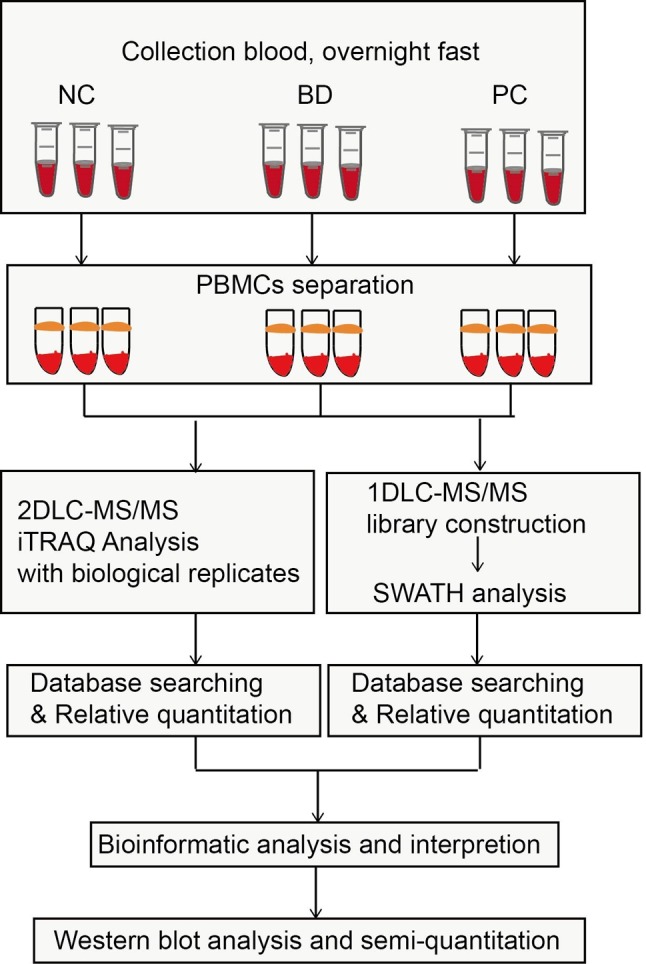
Protein identification. The overall scheme of comprehensive proteome analysis of PBMCs and candidate biomarker identification of PC is shown in Figure 1. Eighteen PBMCs samples were used for proteome identification and quantitation and 9 were applied for validation of candidate biomarkers. The samples were labeled differently to provide both technical and biological replicates. To provide an extensive proteome coverage of PBMCs, an iTRAQ-2D-LC-MS/MS strategy was executed twice to analyze both 6-plex and 8-plex iTRAQ labeled peptides. An extra 1D-LC-MS/MS analysis was carried out to construct a library for SWATH quantitation. As a result, 2430 and 2733 and 1447 proteins were identified from 8-plex ,6-plex iTRAQ analysis and SWATH library construction with FDR<1%. The three analyses added up to a total of 3,357 identified proteins.
Figure 1. The overall work scheme of the comprehensive proteome of PBMCs for candidate biomarker identification.
Relative quantitation of proteins. Protein expression ratio in three independent experiments was calculated according to corresponding algorithms. For iTRAQ quantitation, the peptide for quantification was automatically selected by PD software (at least one unique peptide with 95% confidence) to calculate the ratio of reporter peak area and variabilities. For SWATH analysis, summarized peak area of peptides was used for ratio calculation of the corresponding proteins. All the results were then exported into Microsoft Excel for manual data interpretation.
To compare the protein expression level in different sample groups, both NC and BD groups were set as control groups compared with PC group. The mean value of the protein ratio in each group was used to calculate the protein expression ratio between PC and the control. Students’ T-test (p<0.05) and fold change (FC, FC>1.2 or FC<0.8) of proteins between PC and NC groups were set as the criteria to classify differentially expressed proteins. Missing value handle followed the principles :1) If there were two missing values of ratio within a group, the corresponding data was deleted and not used for further quantitation; 2) if there was only one missing value, mean imputation strategy was applied. As a result, among the commonly detected proteins in the three experiments, 71 proteins were up- and down-regulated in PC compared to the controls. With further analysis of proteins identified only in the two iTRAQ sets, 43 additional proteins were altered in abundance. On the whole, a total of 114 proteins were shown to be significantly different between PC and NC groups, including 35 up-regulations and 79 down-regulations.
Bioinformatics analysis. To gain global insights of the 114 differentially expressed proteins, Gene Ontology (GO,23) and Ingenuity Pathway Analysis tool (IPA, Ingenuity® Systems, Redwood City, CA) (24) were used to analyze the dysregulated proteins to reveal the molecular and cellular functions, as well as canonical pathways altered in PBMCs during PC tumor development. Functional protein association networks were visualized using String database (https://string-db.org/).
Three independent ontologies are accessible using GO analysis: biological process (BP), molecular functions (MF) and cellular components (CC). For BP ontology, altered proteins were annotated into the positive regulation of cellular metabolic process, innate immune response and regulation of response to external stimulus. Besides, the up and down-regulated proteins were enriched in different BP ontologies as shown in Figure 2. Proteins associated with innate immune and receptor-mediated endocytosis diminished in a significant rate comparing with PC group and the control group. Taking an insight to MF ontology, the key function of changed proteins was associate with binding, such as small molecule binding, large molecule binding, receptor and antigen bindings.
Figure 2. Gene Ontology analysis results of differentially expressed proteins in PC. Results of biological process classification (A) and molecular function enrichment (B) of the altered proteins are illustrated.

According to IPA analysis, the most enriched canonical pathways are acute phase response signaling (p-value<0.001). Besides, the second and third prominent protein class identified were grouped to FXR/RXR activation (p-value<0.01) and LXR/RXR activation (p-value<0.01) (Figure 3A). Protein association networks of targeted proteins were visualized based on high confident results of String database. The protein–protein associations of dysregulated proteins function as complexes in the canonical pathways mentioned above (Figure 3B). These proteins are essential maintenance for cell signaling and homeostasis. The physiological system development and functions of these differentially expressed proteins were further analyzed. The top three enriched physiological system development and function were hematological system development and function, Immune cell trafficking and Lymphoid tissue structure and development. And a variety of proteins affected in PC have also been described in the context of connective tissue development and function, and organismal development (Table I).
Figure 3. IPA and String analysis results of differentially expressed proteins in PC. The top ten enriched canonical pathways of altered proteins by IPA analysis (A) and the protein–protein associations analyzed by the String software (B) are shown.

Table I. The top 5 enriched physiological system development categories and functions of differentially expressed proteins in PC.

Western blot. Among all the dysregulated proteins, APOA1 and CCS corresponding to significantly down- and up-regulated proteins were chosen to be validated using western blot. Actin was applied as the internal standard. The expression level of APOA1 is decreased in both BD groups (consisting with CY, BT and PS) and PC groups comparing to NC group (p<0.001, Figure 4A and B). CCS, Copper chaperone for superoxide dismutase, is up-regulated in PC group (p<0.001, Figure 4A and C). Based on the gray value of the western blot band, APOA1 expression level in the PC group is 0.7-fold greater than that in the NC group, while CCS is up-regulated 2-fold in the PC compared with the NC group. These two proteins can significantly differentiate PC group from BD and NC group which means that they might be clinically relevant biomarkers for PC in blood.
Figure 4. Western blot analysis of APOA1, CCS in NC, BD group and PC groups. Western blot result illustration (A). Group comparison based on gray value western blot result of APOA1 (B) and CCS (C). Actin was used as an internal standard.

Discussion
PBMCs, encompassing subpopulations of immune cells in blood, reach various body compartments and participate in a variety of processes underling the genesis and progression of cancer. So that they are considered as reporter cells of the process of cancer (22).
Proteome databases of biological samples appear especially useful for the interpretation of the differences between sample groups. To date, many researchers have aimed to provide a proteome database of PBMCs using various strategies. Recently, Koncarevic et al. performed 49 different LC-MS/MS runs to analyze PBMC proteome and identified 4,129 proteins (16). In the current work, based on a careful monitoring of each step, 2,430 and 2,733 proteins were detected from each of the 10 fractions of PBMCs’ peptides. Compared with the proteins detected in Koncarevic’s work, 790 additional proteins were observed in this study increasing the number of identified PBMCs proteins to 4,921 proteins. The newly identified proteins in our study correspond to 16.1% of the updated database.
Targeted proteomics such as SWATH typically relies on spectral reference libraries for peptide identification. Quality and coverage of these libraries are therefore of crucial importance for the performance of the methods (26-28). Carolina Silva (15) generated a SWATH-MS library by a comprehensive 2D-LC-MS/MS analysis (8 fractions) of a pooled sample of PBMCs from 6 donors resulting in an identification of 1,102 proteins allowing quantitation of 920 proteins. In our study, a SWATH spectrum library was generated using a simple and ordinary 1D-LC-MS/MS analysis and 1,447 proteins observed and quantified. Our work shows a feasible and easy-handling application in clinical practice of large sample analysis using SWATH as well. It will be beneficial to construct a digital database of PBMCs of PC which would assist further analysis of large-scale samples using SWATH strategy.
According to the bioinformatics interpretation, most significantly changed proteins were related to innate and adaptive responses of PBMCs in PC. According to the cancer immune editing hypothesis, immune system is thought to play a dual role in cancer: It can not only suppress tumor growth by destroying cancer cells or inhibiting their outgrowth, but also promote tumor progression either by selecting tumor cells that are more fit to survive in an immune competent host, or by establishing conditions within the tumor microenvironment that facilitate tumor growth (31). In our study, a series of proteins with the molecular function of binding were identified to be altered. These proteins have great dependence in the molecular recognitions and interactions which should be further analyzed to reveal the mechanism of the biological process of cancer. These results indicated that deep analysis of the proteome of PBMCs has the potential to increase our understanding of the pathophysiology of the development of PC.
Of all the quantitated proteins, APOA1 (1.47 times lower in PC vs. NC) is a key component of the reverse cholesterol transport pathway. Binding to prion inflammatory phospholipids, APOA1 gains anti-inflammatory properties (32,33). CCS (2.17 times higher in PC vs. NC) is essential to activate the mammalian Cu/Zn superoxide dismutase (SOD1) which plays an important inflammatory role in pathogenic processes (34,35). These proteins showed significant changes in the PC group vs. the NC and BD groups which indicates that they can potentially by biomarkers in PBMCs for the diagnosis of PC.
In conclusion, a comprehensive proteome profiling was applied to identify and interpret the differences in protein expression during the progress of PC. A highly efficient identification of PBMCs proteome was achieved resulting in an observation 3,357 proteins of which 114 proteins were altered in PC compared to NC and BD groups. According to bioinformatics analysis, these proteins were highly associated with the functions of innate and adaptive responses during cancer progression. Besides, because of the easy handling and high reproducibility, SWATH analysis of PBMCs showed a perspective for clinical application in large-scale sample analysis for blood biomarker discovery. The levels of AOPA1 and CCS were significantly altered in PC samples compared to NC and BD groups indicating that APOA1 and CCS have the potential to be blood biomarkers for PC.
Conflicts of Interest
The Authors declare that they have no competing interests.
Acknowledgements
This work was supported by the Special Project on Precision Medicine under the National Key R&D Program (2017YFC0906600), the Shanghai Municipal Planning Commission of Science and Research Fund (201840192), the Natural Science Foundation of China (31800691, 21874026 and 81472221), the National Key Research and Development Program of China (2017YFA0505100) and the National Basic Research Program of China (2013CB910802).
References
- 1.Makohon-Moore A, Lacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016;16:553–565. doi: 10.1038/nrc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shelton CA, Umapathy C, Stello K, Yadav D, Whitcomb DC. Hereditary Pancreatitis in the United States: Survival and Rates of Pancreatic Cancer. Am J Gastroenterol. 2018;113:1376–1384. doi: 10.1038/s41395-018-0194-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strobel O, Neoptolemos J, Jäger D, Büchler MW. Optimizing the outcomes of pancreatic cancer surgery. Nat Rev Clin Oncol. 2018;19 doi: 10.1038/s41571-018-0112-1. https://doi.org/10.1038/s41571-018-0112-1. [DOI] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer Statistics. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 5.Erickson LA. Pancreatic Ductal Adenocarcinoma. Mayo Clin Proc. 2017;92:1461–1462. doi: 10.1016/j.mayocp.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Wang WS, Liu XH, Liu LX, Jin DY, Yang PY, Wang XL. Identification of proteins implicated in the development of pancreatic cancer-associated diabetes mellitus by iTRAQ-based quantitative proteomics. J Proteom. 2013;84:52–60. doi: 10.1016/j.jprot.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 7.Wang WS, Liu XH, Liu LX, Lou WH, Jin DY, Yang PY, Wang XL. iTRAQ-based quantitative proteomics reveals myoferlin as a novel prognostic predictor in pancreatic adenocarcinoma. J Proteom. 2013;91:453–465. doi: 10.1016/j.jprot.2013.06.032. [DOI] [PubMed] [Google Scholar]
- 8.Liu XH, Zheng WM, Wang WS, Shen HL, Liu LX, Lou WH, Wang XL, Yang PY. A new panel of pancreatic cancer biomarkers discovered using a mass spectrometry-based pipeline. Brit J Cancer. 2017;117:1846–1854. doi: 10.1038/bjc.2017.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kervezee L, Cuesta M, Cermakian N, Boivin DB. Simulated night shift work induces circadian misalignment of the human peripheral blood mononuclear cell transcriptome. PNAS. 2018;115:5540–5545. doi: 10.1073/pnas.1720719115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mantovani A. The inflammation – cancer connection. FEBS J. 2018;285:638–640. doi: 10.1111/febs.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD. Complement in cancer: untangling an intricate relationship. Nat Rev Immunol. 2018;18:5–18. doi: 10.1038/nri.2017.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taniguchi K, Karin M. NF-ĸB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18:309–324. doi: 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- 13.Sayour EJ, Mitchell DA. Manipulation of Innate and Adaptive Immunity through Cancer Vaccines. J Immunol Res. 2017;2017:3145742. doi: 10.1155/2017/3145742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gourbal B, Pinaud S, Beckers GJM, Van Der Meer JWM, Conrath U, Netea MG. Innate immune memory: An evolutionary perspective. Immunol Rev. 2018;1:21–40. doi: 10.1111/imr.12647. [DOI] [PubMed] [Google Scholar]
- 15.Silva C, Santa C, Anjo SI, Manadas B. A reference library of peripheral blood mononuclear cells for SWATH-MS analysis. Proteom Clin Appl. 2016;10:760–764. doi: 10.1002/prca.201600070. [DOI] [PubMed] [Google Scholar]
- 16.Končarević S, Lößner C, Kuhn K, Prinz T, Pike I, Zucht HD. In-depth profiling of the peripheral blood mononuclear cells proteome for clinical blood proteomics. Int J Proteom. 2014;2014:129–259. doi: 10.1155/2014/129259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schreiber RD, Old LJ, Smyth MJ. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 18.Roos BD, Duthie SJ, Polley ACJ, Mulholland F, Bouwman FG, Heim C, Rucklidge GJ, Johnson IT, Mariman EC, Daniel H, Elliott RM. Proteomic methodological recommendations for studies involving human plasma, platelets, and peripheral blood mononuclear cells. J Proteome Res. 2008;7:2280–2290. doi: 10.1021/pr700714x. [DOI] [PubMed] [Google Scholar]
- 19.Gholami AM, Hahne H, Wu Z, Auer FJ, Meng C, Wilhelm M, Kuster B. Global proteome analysis of the NCI-60 cell line panel. Cell Rep. 2013;4:609–620. doi: 10.1016/j.celrep.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 20.Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R. The quantitative proteome of a human cell line. Mol Syst Biol. 2011;7:549. doi: 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo T, Kouvonen P, Koh CC, Gillet LC, Wolski WE, Röst HL, Rosenberger G, Collins BC, Blum LC, Gillessen S, Joerger M, Jochum W, Aebersold R. Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat Med. 2015;21:407–413. doi: 10.1038/nm.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X, Zhang Y, Ni M, Cao H, Signer RAJ, Li D, Li M, Gu Z, Hu Z, Dickerson KE, Weinberg SE, Chandel NS, DeBerardinis RJ, Zhou F, Shao Z, Xu J. Regulation of mitochondrial biogenesis in erythropoiesis by mTORC1-mediated protein translation. Nat Cell Biol. 2017;19:626–638. doi: 10.1038/ncb3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang KL, Li SQ, Mertins P, Cao S, Gunawardena HP, Ruggles KV, Mani DR, Clauser KR, Tanioka M, Usary J, Kavuri SM, Xie L, Yoon C, Qiao JW, Wrobel J, Wyczalkowski MA, Erdmann-Gilmore P, Snider JE, Hoog J, Singh P, Niu B, Guo Z, Sun SQ, Sanati S, Kawaler E, Wang X, Scott A, Ye K, McLellan MD, Wendl MC, Malovannaya A, Held JM, Gillette MA, Fenyö D, Kinsinger CR, Mesri M Rodriguez H, Davies SR, Perou CM, Ma C, Reid Townsend R, Chen X, Carr SA, Ellis MJ, Ding L. Proteogenomic integration reveals therapeutic targets in breast cancer xenografts. Nat Commun. 2017;8:14864. doi: 10.1038/ncomms14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun SS, Shah P, Toghi Eshghi SD, Yang WM, Trikannad N, Yang S, Chen L, Aiyetan P, Höti N, Zhang Z, Chan DW, Zhang H. Comprehensive analysis of protein glycosylation by solid-phase extraction of N-linked glycans and glycosite-containing peptides. Nat Biotechnol. 2016;34:84–88. doi: 10.1038/nbt.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen TW, Lee CC, Liu H, Wu CS, Pickering CR, Huang PJ, Wang J, Chang IY, Yeh YM, Chen CD, Li HP, Luo JD, Tan BC, Chan TEH, Hsueh C, Chu LJ, Chen YT, Zhang B, Yang CY, Wu CC, Hsu CW, See LC, Tang P, Yu JS, Liao WC, Chiang WF, Rodriguez H, Myers JN, Chang KP, Chang YS. APOBEC3A is an oral cancer prognostic biomarker in Taiwanese carriers of an APOBEC deletion polymorphism. Nat Commun. 2017;8:465. doi: 10.1038/s41467-017-00493-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collins BC, Gillet LC, Rosenberger G, Röst HL, Vichalkovski A, Gstaiger M, Aebersold R. Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14-3-3 system. Nat Methods. 2013;10:1246–1253. doi: 10.1038/nmeth.2703. [DOI] [PubMed] [Google Scholar]
- 27.Navarro P, Kuharev J, Gillet LC, Bernhardt OM, MacLean B, Röst HL, Tate SA, Tsou CC, Reiter L, Distler U, Rosenberger G, Perez-Riverol Y, Nesvizhskii AI, Aebersold R, Tenzer S. A multicenter study benchmarks software tools for label-free proteome quantification. Nat Biotechnol. 2016;34:1130–1136. doi: 10.1038/nbt.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fuchs D, Vafeiadou K, Hall WL, Daniel H, Williams CM, Schroot JH, Wenzel U. Proteomic biomarkers of peripheral blood mononuclear cells obtained from postmenopausal women undergoing an intervention with soy isoflavones. Am J Clin Nutr. 2016;86:1369–1375. doi: 10.1093/ajcn/86.5.1369. [DOI] [PubMed] [Google Scholar]
- 29.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–457. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 30.Schubert OT, Gillet LC, Collins BC, Navarro P, Rosenberger G, Wolski WE, Lam H, Amodei D, Mallick P, MacLean B, Aebersold R. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat Protoc. 2015;10:426–441. doi: 10.1038/nprot.2015.015. [DOI] [PubMed] [Google Scholar]
- 31.Schreiber RD, Old LJ, Smyth MJ. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 32.Shi M, Movius J, Dator R, Aro P, Zhao Y, Pan C, Lin X, Bammler TK, Stewart T, Zabetian CP, Peskind ER, Hu SC, Quinn JF, Galasko DR, Zhang J. Cerebrospinal fluid peptides as potential Parkinson disease biomarkers: a staged pipeline for discovery and validation. Mol Cell Proteom. 2015;14:544–555. doi: 10.1074/mcp.M114.040576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saraswat M, Makitie A, Agarwal R, Joenvaara S, Renkonen S. Oral squamous cell carcinoma patients can be differentiated from healthy individuals with label-free serum proteomics. Brit J Cancer. 2017;117:376–384. doi: 10.1038/bjc.2017.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O-Halloran TV. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 35.Wong PC, Waggoner D, Subramaniam JR, Tessarollo L, Bartnikas TB, Culotta VC, Price DL, Rothstein J, Gitlin JD. Copper chaperone for superoxide dismutase is essential to activate mammalian Cu/Zn superoxide dismutase. PNAS. 2000;97:2886–2891. doi: 10.1073/pnas.040461197. [DOI] [PMC free article] [PubMed] [Google Scholar]