

Abstract

Developing PROTACs to redirect the ubiquitination activity of E3 ligases and potently degrade a target protein within cells can be a lengthy and unpredictable process, and it remains unclear whether any combination of E3 and target might be productive for degradation. We describe a probe-quality degrader for a ligase–target pair deemed unsuitable: the von Hippel–Lindau (VHL) and BRD9, a bromodomain-containing subunit of the SWI/SNF chromatin remodeling complex BAF. VHL-based degraders could be optimized from suboptimal compounds in two rounds by systematically varying conjugation patterns and linkers and monitoring cellular degradation activities, kinetic profiles, and ubiquitination, as well as ternary complex formation thermodynamics. The emerged structure–activity relationships guided the discovery of VZ185, a potent, fast, and selective degrader of BRD9 and of its close homolog BRD7. Our findings qualify a new chemical tool for BRD7/9 knockdown and provide a roadmap for PROTAC development against seemingly incompatible target–ligase combinations.
Introduction
Targeted protein degradation is an emerging strategy to use small molecules to knock down a protein by hijacking the ubiquitin–proteasome system.1,2 PROTACs (proteolysis targeting chimeras) are bifunctional degrader molecules composed of a ligand for the target protein and a ligand for E3 ligase recruitment, connected by a linker.3,4 Upon formation of a ternary complex target:degrader:E3,5−7 the protein of interest is ubiquitinated and degraded by the proteasome. Compared to target blockade, post-translational protein degradation more closely phenocopies genetic approaches to target validation and can lead to a more sustained cellular effect with more extended duration of action. An attractive feature of PROTACs is their catalytic mode of action, as any one molecule may perform multiple rounds of target ubiquitination and degradation.8 A consequence of this feature is that degraders can function at substoichiometric receptor occupancies, meaning they exhibit degradation activities at concentrations that can be orders of magnitude lower than their binary dissociation constants (Kd) from the target, alleviating the requirement for full target engagement.9 Moreover, PROTAC molecules can add a layer of target selectivity beyond that expected from the constitutive binding ligands, thus providing highly selective degraders with reduced off-target effect.5,10−12 The mounting interest in PROTAC drug discovery is also motivated by the promise to target proteins considered “undruggable” via conventional medicinal chemistry approaches.13 To date, different target classes have been successfully degraded, including epigenetic targets such as bromodomain-containing proteins BRD2, BRD3, and BRD4,5,10,11,14−17 BRD9,18 TRIM24,19 SIRT2,20 PCAF/GNC5,21 protein kinases,8,12,22−26 nuclear receptors,27,28 and E3 ubiquitin ligases to self-degrade.29,30
To fulfill the potential of targeted protein degradation, a general methodology for an efficient PROTAC design would be desirable. However, the development of active PROTAC degraders is often a laborious and unguided process. The choice of E3 ligase and the selection of target ligands and their conjugation are all potential optimization variables that expand the chemical space to be exploited by medicinal chemists. Properties of the linker, such as length, composition, and site of attachment, are known to be important but often their impact on activity vary in a target- and context-dependent fashion.11,24,29,31,32 Moreover, small-molecule binders for both the protein of interest and the E3 ligase are required. Despite the large number of human E3 ubiquitin ligases postulated to function in cells, only a few have good-quality ligands33 that have been successfully used for PROTACs.34 The most common ligases recruited are the von Hippel–Lindau (VHL) protein complex CRL2VHL and the cereblon (CRBN) complex CRL4CRBN. Studies have shown that PROTACs made of the same target ligand but either VHL or CRBN ligands can exhibit different degradation selectivity and efficacy.11,14,18,22 In some systems, CRBN-based degraders show a more active profile than VHL-based molecules. Potential greater flexibility of the Cullin4 based CRL4CRBN compared to CRL2VHL has been invoked to suggest more productive ubiquitination of the accessible lysine residues on the target protein.1,32,35 These observations would suggest that the development of VHL-based degraders might require more exploration in the PROTAC design than those based on CRBN. Even if degradation of a given target protein can be readily obtained by recruiting one E3 ligase, emerging evidence suggests that it could be beneficial to develop a parallel chemical series hijacking other E3 ligases. For example, chemical liabilities on a particular ligase ligand could be readily circumvented by switching to a different compound. The hijacked E3 ligase expression and intrinsic activity may be context-dependent, and vary widely among different cells and tissue types.36 Furthermore, resistance mechanisms could potentially arise from the loss of the hijacked E3 ligase, as demonstrated by the correlation between level of CRBN and response to CRBN-recruiting drugs in multiple myeloma.37 Switching the hijacked ligase can thus aid targeted protein degradation. It however remains unclear whether optimal target–E3 pairs exist or indeed whether any combination of E3 ligase and target protein might be tractable.
Here, we demonstrate the development of probe-quality PROTACs for a ligase–target pair previously considered incompatible: VHL and the protein BRD9.18 BRD9 and its close homolog BRD7 (85% sequence identity38) are bromodomain-containing subunits of the BAF (BRG-/BRM-associated factor) and PBAF (polybromo-associated BAF) complexes, respectively.39,40 BAF and PBAF represent two variants of the SWI/SNF complex, one of the four mammalian ATP-dependent chromatin remodeling complexes. The SWI/SNF complexes control gene expression, DNA replication, and DNA repair by modulating access to promoters and coding regions of DNA through modification of the degree of compactness of chromatin.41−43 Mounting evidence from genetics and sequencing of cancer-associated mutations have spurred efforts to unravel yet largely elusive physiological roles of BAF/PBAF subunits and to develop targeted therapeutics in cancer and other human diseases.39 In particular, BRD9 is overexpressed in several malignancies, such as cervical cancer and in non-small-cell lung cancer (NSCLC).44,45 In contrast, BRD7 gene has been proposed as candidate tumor suppressor gene,46−49 as it regulates breast cancer cell metabolism50 and acts as negative regulator of aerobic glycolysis essential for tumor progression.51 BRD7 also promotes X-box binding protein 1 (XBP1) nuclear translocation, which prevents the development of insulin-resistance disorders.52 In contrast to these roles, it has been recently shown that inactivation of the BRD7 gene sensitizes tumor cells to T cell-mediated killing, suggesting that knockdown of BRD7 could be an attractive target for cancer immunotherapy.53 Potent and selective inhibitors that bind to the BRD7/9 bromodomains have recently emerged from structure-guided medicinal chemistry campaigns, including compounds I-BRD9,38 LP99,54 ketone “compound 28”,55 BI-7273 and BI-956456 (1a,b, Figure 1), and GNE-375.57 These BRD7/9 inhibitors have been used in cells to help clarify the roles of the BRD7/9 bromodomains in oncogenesis and other disease states. For example, pharmacological studies of inhibitors 1a and 1b in combination with domain-swap protein engineering revealed that an active bromodomain of BRD9 is required to sustain MYC transcription and proliferation of leukemic cells.56,58 These findings and availability of bromodomain ligands prompted us to initiate a PROTAC medicinal chemistry campaign to target BRD7 and BRD9 proteins for degradation.
Figure 1.
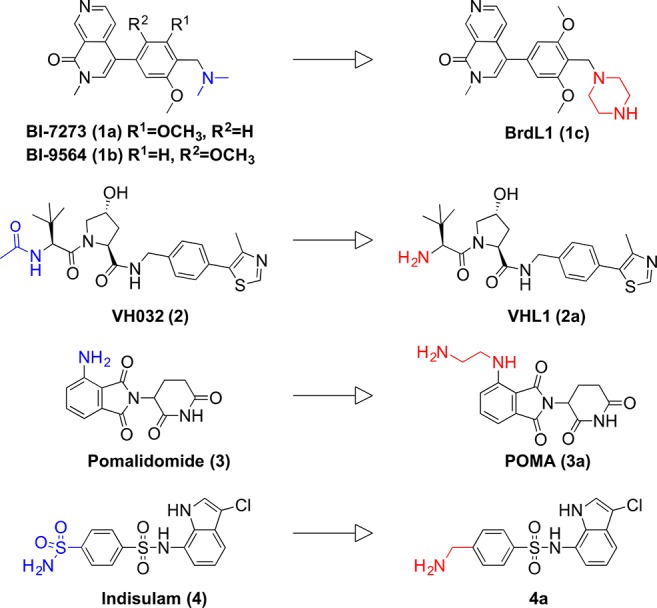
Chemical structures of parent (1a, 1b, 2–4) and modified (1c, 2a–4a) BRD7/9 and E3 ligase ligands. Functional groups selected for conjugation are shown in blue on parent ligands and in red on modified ligands.
Results and Discussion
First Generation of BRD7 and BRD9 Degraders
We began our investigation by designing a small set of PROTACs aimed to induce BRD7/9 degradation by recruiting three different E3 ubiquitin ligases: VHL, CRBN, and DCAF15.59,60 We aimed to leverage available E3 ligase ligands and to maximize the opportunity for complementary surfaces between the bromodomain and the ligase within the ternary complex. As BRD7/9 bromodomain ligands, we selected compounds 1a,b (Figure 1),56 on the basis of their high binding affinity56 and of their superiority as BRD9 chemical probes over other ligands.58 To design the first generation of degraders, we inspected the crystal structure of 1a bound to BRD9 (PDB code 5EU1)56 to identify suitable attachment points and vectors for linker conjugation, important considerations for PROTAC design as known to greatly influence degradation activities.11,29 The dimethylamine group of the molecule was identified as a solvent-exposed group not involved in interactions with the protein. For synthetic reasons, the dimethylamine group of compound 1a was replaced by a piperazine group (BrdL1 (1c), Figure 1 and Supporting Information Figure S1), providing a convenient isosteric handle. As E3 ligase recruiting moieties, VH032 (2, VHL ligand, Figure 1)61−63 and pomalidomide (3, CRBN ligand, Figure 1)64 were selected and modified to afford compounds VHL1 (2a) and 3a (Figure 1 and Supporting Information Figure S1). The amino terminal groups of 2a and 3a were conjugated via amide bond to the linker without perturbing the interaction with the E3 ligases, as previously demonstrated.8,10,15,22 To expand the arsenal of E3 ligase ligands being explored, we designed conjugates containing indisulam (4, Figure 1), a small molecule recently reported to bind to the E3 ligase DCAF15 and to redirect the activity of the CRL4DCAF15 complex toward the neosubstrate CAPERα (also known as RBM39).59,60 As the binding mode of 4 is not known, we leveraged information on the activity of a biotinylated photoactive analogue probe to guide our conjugation strategy.60 Accordingly, a para-benzylamine analogue of 4 (derivative 4a, Figure 1 and Supporting Information Figure S1) was designed as conjugatable ligase ligand. To generate a first set of compounds, we decided to use PEG linkers composed of two or four PEG units to connect the two warheads (compounds 5–10, Scheme 2).
Scheme 2. Synthesis of the First Generation of Degraders.

Reagents and conditions: (a) oxalyl chloride, DMSO, DCM, then TEA, −78 °C to rt, 2 h; (b) 1c, NaBH(OAc)3, TEA, DMF, rt, overnight; (c) TFA, DCM, rt, 2 h; (d) E3 ligand (2a, 3a, or 4a), HATU, HOAt, DIPEA, DMF, rt, 2 h.
Compounds 5–10 were obtained as reported in Schemes 1 and 2. Briefly, reductive amination between the commercially available 4-bromo-2,6-dimethoxybenzaldehyde (11) and boc-piperazine led to the formation of compound 12 which was then cross-coupled with 14 (obtained by methylation of 13) under the Myaura–Suzuki condition (one-pot two steps); the cleavage of tert-butyloxycarbonyl protecting group in acidic conditions afforded 1c in quantitative yield (Scheme 1). The primary alcohol functionality of the linkers (15 and 16) was oxidized to aldehyde group using a Swern reaction and condensed with the terminal secondary amine of 1c to afford the tert-butyl ester intermediates 17 and 18 that were converted into the corresponding acids by trifluoroacetic acid (TFA) treatment (Scheme 2). Compounds 19 and 20 were then conjugated with the conjugatable E3 ligands (2a and 3a prepared as previously described,61,65 and 4a synthesis in Supporting Information Synthetic Procedures) using HATU as coupling reagent yielding final compounds 5–10.
Scheme 1. Synthesis of the BRD7/9 Ligand 1c.

Reagents and conditions: (a) 1-Boc-piperazine, NaBH(OAc)3, THF, rt, overnight, yield 97%; (b) NaH, CH3I, DMF, 0 °C, 5 h, yield 95%. (c) Step 1: 12, B2pin2, KOAc, Pd(dppf)Cl2, 1,4 dioxane, microwave, 140 °C, 40 min. Step 2: 14 and K2CO3 (aq) are added to step 1; microwave, 120 °C, 30 min, yield 55%. (d) HCl 4 M in dioxane, DCM, rt, 1 h, quantitative yield.
To assess the degradation activity of the first generation of PROTACs, HeLa cells were treated with compounds 5–10 at fixed concentration of 1 μM, for 4 and 16 h before harvesting (Figure 2A). CRBN-based PROTACs 7 and 8 demonstrated strong degradation of BRD9 already after 4 h of treatment, whereas no BRD7 degradation was observed (Figure 2A). In contrast, VHL-based degraders 5 and 6 showed weak activity against both BRD7 and BRD9, inducing at most 20% degradation after 4 h and 30% after 16 h, and even weaker activity was observed with the indisulam-based PROTACs 9 and 10 (Figure 2A). siRNA knockdown experiments (Figure 2B) validated the specificity of the bands observed by Western blot.
Figure 2.

Screening of first generation of degraders. (A) Western blot analysis of BRD9 and BRD7 levels after treatment of HeLa cells with 1 μM compounds for 4 and 16 h before harvesting. Degradation activity is reported below each lane as % of protein abundance relative to 0.1% DMSO vehicle. (B) Western blot analysis of BRD9 and BRD7 levels after 48 h transfection with 1.25 nM siRNA targeting respectively BRD7, BRD9, or negative control (N.C.) siRNA. Intensity values are quantified as described in the Experimental Section.
While this research was underway, Remillard et al.18 disclosed the BRD9 degrader dBRD9 (21, Figure 3) confirming the strong degradation activity of our CRBN-recruiting PROTACs. Compounds 7 and 21 share the same target and E3 ligands, which, however, were differently modified to attach two distinct linkers (Figure 3). CRBN-based PROTACs, however, can exhibit off-target degradation of non-PROTAC-targets such as IKZF1/3 and GSPT1 due to the neomorphic activity of the CRBN ligand alone.18,66
Figure 3.

Structures of compounds 7 and 21.18
We therefore turned our attention to VHL-based PROTACs. Encouraged by the degradation, albeit partial, of both BRD7 and BRD9 induced by our initial VHL-based PROTACs, we decided to characterize the binding of 5 biophysically and structurally. To determine the binding mode of the conjugated bromodomain ligand, compound 5 was cocrystallized with the BRD9 bromodomain (BRD9-BD). In the BRD9-BD:5 complex, the bromodomain ligand was clearly observed within its binding pocket, and its binding mode closely recapitulates that of 1a (rmsd = 0.569 Å) (Figure 4A,B). The alkylated piperazine is favorably accommodated within the binding site, with the linker directed toward solvent, as desired (Figure 4A,B).
Figure 4.

Ternary complex formation and analysis of binding mode for compound 5. (A) Cocrystal structure of BRD9-BD and compound 5. The warhead component of the degrader (purple and cyan; one from each protomer in the ASU) recapitulates the binding of inhibitor 1a (magenta, PDB code 5EUI),56 whereas the alkylated piperazine used to attach the linker is solvent-exposed and does not form any unfavorable interactions with the protein. (B) Fo – Fc omit map of compound 5 (contoured at 2.5σ) showing electron density for the alkylated piperazine ring. (C) BRD9-BD titrated into 5 alone. (D) VCB titrated into 5 alone. (E) VCB titrated into BRD9-BD:5 binary complex. VCB binds more weakly to the binary complex BRD9-BD:5 (Kd = 73 nM) compared to compound 5 alone (Kd = 33 nM), indicating negative cooperativity.
To characterize biophysically the interplay between VCB (VHL-ElonginC-ElonginB complex), compound 5, and the BRD9 bromodomain, we applied our previously developed ITC assay5,11 that measures the thermodynamics and cooperativities of ternary complex formation. Previous work has demonstrated that VHL is capable of forming highly stable and cooperative ternary complexes, as seen with MZ1-Brd4BD2 (ref (5)) and with the homo-PROTAC dimerizer CM11 (ref (29)). Titrations of protein into PROTAC alone revealed a binary binding affinity for VCB (Kd = 33 ± 2 nM, Figure 4D and Table 1) within 2-fold of that measured on previously characterized VHL-based BET degrader MZ1 (Kd = 70 nM, refs (5) and (11)). Similarly, the binary affinity (Kd = 15 ± 3 nM) and binding enthalpy (ΔH = −12 kcal/mol) for BRD9-BD (Figure 4C and Table 1) were comparable to that of inhibitor 1a,56 consistent with the conserved binding modes of 5 and inhibitor 1a observed crystallographically (Figure 4A). The titrations of VCB into BRD9-BD:5 complex (Figure 4E) showed that VCB binds more weakly to the binary complex BRD9-BD:5 (Kd = 73 nM) than to compound 5 alone (Kd = 33 nM), indicating negative cooperativity (α < 1, Table 1). Negative cooperativity has previously been observed with tetrahydroquinoline-based BET PROTACs, where it was demonstrated that linker length and exit vector can significantly influence ternary complex formation.11 On the basis of these considerations, we reasoned to explore variations on the linker as well as the point of derivatization.
Table 1. Thermodynamic Parameters of Formation of Binary and Ternary Complexes between VCB, BRD9 Bromodomain, and Compound 5 Measured by Isothermal Titration Calorimetry (ITC)a.
| syringe | cell | Kd (nM) | ΔH (kcal/mol) | ΔG (kcal/mol) | –TΔS (kcal/mol) | N | α |
|---|---|---|---|---|---|---|---|
| BRD9-BD | 5 | 15 ± 3 | –12.5 ± 0.4 | –10.7 ± 0.1 | 1.8 ± 0.5 | 0.753 ± 0.004 | |
| VCB | 5 | 33 ± 2 | –15.2 ± 0.1 | –10.2 ± 0.1 | 4.9 ± 0.1 | 0.60 ± 0.01 | |
| BRD9-BD:5 | 73 ± 2 | –6.4 ± 0.1 | –9.7 ± 0.1 | –3.3 ± 0.1 | 0.75 ± 0.05 | 0.45 |
All ITC titrations were performed at 25 °C. Values reported are the mean ± SEM from at least three independent measurements (n ≥ 3).
Second Generation of BRD7 and BRD9 Degraders
To improve ternary complex formation and degradation activity of PROTACs 5 and 6, we explored the impact of varying their derivatization point, linker length, and composition (compounds 22–31, Table 2). First, to expand the conjugation pattern between VHL1 and BrdL1 (Figure 1), the number of PEG units was modified (3 units for 24; 5 units for 22), a more lipophilic 11-atoms chain was inserted (compound 23), and a different attachment to BrdL1 moiety via amide conjugation was explored (compound 25) (Table 2). Furthermore, we explored a conjugation vector via a phenolic position (VHL2 (2b), compounds 28 and 31, Table 2 and Supporting Information Figure S1), as previously described.29,67 To improve binary binding affinities to VHL and potentially fine-tune intermolecular interactions in the ternary complex,5 we replaced the VHL2 acetyl group with either a cyanocyclopropyl group (VHL3 (2c), derivatives 27 and 30, Table 2 and Supporting Information Figure S1)29,62 or a fluorocyclopropyl group (VHL4 (2d), derivatives 26 and 29, Table 2 and Supporting Information Figure S1).63 As assessed by crystallographic studies (for 2c, PDB code 5LLI; for 2d, PDB code 5NVX), these modifications accommodate snugly on the VHL protein surface and enhance binding affinity.62,63 To allow direct comparison with the compounds from the first series, compounds 26–28 and 29–31 were designed to contain PEG2 and PEG4 linkers, respectively.
Table 2. SAR of Second Generation of PROTACsa.


Footnote: *Degradation activity reported as % of total protein remaining after 1 μM compound treatment relative to 0.1% DMSO vehicle as quantified by Western blotting of HeLa cell lysates.
Compounds 22 and 24 were obtained via the same synthetic route used for the first generation (synthesis of compounds 22, 24, and 25 detailed in the Supporting Information). The synthesis of 23 (Scheme 3) involved nucleophilic attack of diethylene glycol (PEG2) to 6-bromo-1-hexene to afford derivative 33 that was oxidized to carboxylic acid 34 by treatment with TEMPO and bis-acetoxyiodobenzene (BAIB). After coupling with 2a, Lemieux–Johnson oxidation of the double bond yielded the aldehyde intermediate that was reacted with amine 1c to yield compound 23 in 54% yield. A similar synthetic strategy was applied for the synthesis of degraders 26–31 (Scheme 4). Briefly, after nucleophilic substitution of polyethylene glycol (PEG2 and PEG4) with allyl bromide, the alcoholic group of 38 and 39 was activated as mesylate and reacted with the phenolic group present in the VHL ligand scaffold affording derivatives 40–45. The alkene moiety was then oxidized, and the resulting aldehyde derivative was directly used for the final reductive amination step with 1c to afford the final products 26–31.
Scheme 3. Synthesis of Compound 23.

Reagents and conditions: (a) NaH, 6-bromo-1-hexene, DMF/THF, rt, overnight; (b) BAIB, TEMPO, ACN/H2O, rt, overnight; (c) 2a, HATU, HOAt, DIPEA, DMF, rt, 2 h; (d) OsO4 4 wt % in H2O, NaIO4, pyridine, dioxane/H2O, rt; (e) 1c, NaBH(OAc)3, TEA, DMF, rt, overnight.
Scheme 4. Synthesis of Compounds 26–31.

Reagents and conditions: (a) NaOH, allyl bromide, dioxane, rt, overnight; (b) MsCl, TEA, DCM, rt, 3 h; (c) VHL ligands 2b–d, K2CO3, DMF, 70 °C, overnight; (d) OsO4, NaIO4, pyridine, dioxane/H2O, rt; (e) 1c, NaBH(OAc)3, TEA, DMF, rt, overnight.
All second-generation compounds were profiled for BRD9 and BRD7 degradation after treatment for 4 and 16 h (Figure 5A and Supporting Information Figures S2 and S3). The two VHL-based PROTACs 5 and 6 were included as reference compounds. Derivative 26 (Figure 5B) demonstrated marked and selective (around 90%) depletion of BRD9 over BRD7 already after 4 h treatment in HeLa cells (Figure 5A, Table 2). By comparing and contrasting the degradation profiles of related compounds, structure–activity relationships began to emerge (Table 2). As expected, the length of linkers influenced degradation profiles, with more pronounced BRD9 degradation observed with shorter linkers for given matched pairs (compare 26 and 29; 27 and 30; 5 with 24, 6, and 22). We also observed that for a given linker length, replacement of an oxygen atom with a methylene group was beneficial to degradation activity (compare 23 vs 24). Among conjugates derivatized at the phenolic position of the VHL ligand, the nature of the terminal capping group also influenced degradation potency. Within each series of conjugates of fixed linker, compounds bearing the fluorocyclopropyl moiety (VHL4) were more potent degraders than those containing cyanocyclopropyl (VHL3) and even more so than acetyl group (VHL2) (26 vs 27 and 28; and 29 vs 30 and 31, respectively) (Table 2). These trends are consistent with the binary binding affinity values measured for the corresponding VHL ligands.63 To account for potential different protein expression levels, compounds 22–31 were tested across a panel of different cancer cell lines (Supporting Information Figures S2 and S3). From these degradation screens, 26 was confirmed as the most active compound, consistently across all assays and cell lines.
Figure 5.

Compound 26 induces rapid and profound depletion of BRD9 in cells. (A) Western blot analysis of BRD9, BRD7, and β-actin after treatment of HeLa cells with 1 μM compounds for 4 h before harvesting (data reported in Table 2). (B) Chemical structure of 26. (C) Quantification of BRD9 protein levels after treatment of HeLa with six different concentrations of 26 at 30 min and 4 h before harvesting. (D) Quantification of BRD9 protein levels after treatment with 1 μM 26 in HeLa at the desired time points. Intensity values, DC50, and half-lives were quantified as described in the Experimental Section.
Compound 26 was further characterized by profiling its concentration-dependent activity at early time points (30 min and 4 h before harvesting; Figure 5C and Supporting Information Figure S4). PROTAC 26 induced preferential degradation of BRD9 over BRD7, resulting in a half-degrading concentration (DC50) of 560 nM against BRD9 (Figure 5C and Supporting Information Figure S4). More than 60% of degradation of BRD9 was observed at 1 μM after 4 h, while maximal degradation (Dmax around 80%) was reached at 10 μM. At higher doses, the characteristic “hook effect” was observed, consistent with 26 acting preferentially as inhibitor over degrader at high concentrations. Rapid and selective target degradation was confirmed by profiling cellular activities over time (Figure 5D and Supporting Information Figure S4). Treatments of HeLa cells at fixed concentration (1 μM) of 26 at varying time points revealed rapid degradation of BRD9 with an apparent half-life of 3.5 h, resulting in more than 50% degradation already after 4 h treatment and achieving the highest level of depletion after 8 h (Figure 5D).
To determine to what extent a correlation could be seen between ternary complex formation and the improved degradation properties of 26, we turned to measurements of thermodynamic parameters and cooperativity by ITC (Figure 6A and Supporting Information Figure S5). Titrating VCB into 26 alone or BRD9-BD:26 complex indicated a cooperativity α = 1, an over 2-fold improvement over 5. At the binary level compared to 5, we observed negligible difference between 26 and 5 in binding affinity for BRD9-BD, but a slight loss in affinity for VCB (Kd = 87 ± 5 nM for 26 compared to 33 ± 2 nM for 5) (Supporting Information Table S1).
Figure 6.

Improved ternary complex formation by series-2 degraders. (A) Overlay of compound 26 titrations shows no difference between binary (VCB into 26 alone; black) and ternary (VCB into BRD9-BD:26 binary complex; green) titrations. (B) Fluorescence polarization measurements for compound 5 in the presence (red) and absence (black) of BRD9-BD. The rightward shift in the presence of BRD9-BD indicates negative cooperativity. Each point is the mean ± SEM of at least two individual experiments performed in triplicate. (C) Fluorescence polarization measurements for compound 26 in the presence (green) and absence (black) of BRD9-BD. Each point is the mean ± SEM of at least two individual experiments performed in triplicate. (D) AlphaLISA assay showing increased ternary complex formation for 26 (green) compared to 5 (red). Each point is the mean ± SEM of at least one experiment performed in quadruplicate.
To confirm these findings, we repurposed an existing fluorescence polarization (FP) assay that involves the competitive displacement of a FAM-labeled HIF-1α peptide.68 By comparing PROTAC-induced peptide displacement in the presence and absence of BRD9-BD, it is possible to determine the cooperativity (Figure 6B,C). FP measurements for compound 5 showed a rightward shift in the IC50 curve in the presence of BRD9-BD, indicating negative cooperativity, whereas compound 26 showed no shift. Back-calculation of Kd values63 produced affinity values that were comparable to those obtained by ITC (Supporting Information Table S1 and Figure S5).
Previous studies with VCB and BET bromodomains have demonstrated that the amount of ternary complex formed is well correlated to the cooperativity of the system.5 Therefore, we employed an AlphaLISA proximity assay to determine the effect of increasing cooperativity on ternary complex formation (Figure 6D). Indeed, there was a 50% increase in the maximum α intensity measured for VCB:26:BRD9-BD compared to VCB:5:BRD9-BD, suggesting that the increased cooperativity of 26 compared to 5 resulted in increased population of ternary complex.
Third Generation of BRD7 and BRD9 Degraders
The pronounced degradation of BRD9 observed with 26 motivated us to design a third generation of PROTACs with the goal to further optimize our degrader. On the basis of the SAR previously acquired, the design strategy was to keep VHL4 moiety fixed and to focus on systematically varying the linker length and composition, as well as the substitution and conjugation chemistry at the BRD7/9 warhead. The focus on the linker concentrated on exploring varying ratios between hydrophilic and lipophilic portions within the context of three different lengths: 5, 8, and 11 atoms (Table 3). We reasoned that varying the balance between hydrophilicity and lipophilicity might influence the conformational equilibria and intrinsic folding propensity of the linker, with direct impact on the process of ternary complex formation as well as potentially cell permeability. Linkers were connected to four different analogues of BRD7/9 ligands. Guided by the structure–activity relationships of BRD7/9 bromodomain inhibitors developed by Martin et al.,56 we introduced two structural variations, single or in combination. First, we arranged the methoxy groups on the phenyl ring in either meta (BrdL1, 1c) or para (BrdL2, 1d) (Table 3 and Supporting Information Figure S1) relative to each other. Second, we replaced the piperazine moiety with an azetidine group (BrdL3, 1e and BrdL4, 1f, Table 3 and Supporting Information Figure S1). For conjugation between the bromodomain ligand and the linker, either tertiary amines (via reductive amination reactions) or amide bonds were contemplated. This design yielded a set of 19 new PROTACs (Table 3).
Table 3. SAR of Third Generation of PROTACsa.


Footnote: *Degradation activity reported as % of total protein remaining after 1 μM compound treatment relative to 0.1% DMSO vehicle as quantified by Western blotting of HeLa and RI-1 cell lysates.
To circumvent the need to use the hazardous reagent osmium tetroxide previously applied in the Lemieux–Johnson reaction, some key modifications and optimization to the synthesis were made (Scheme 5 and Supporting Information Synthetic Procedures). Mesylation of the free hydroxy group of the linkers allowed nucleophilic attack by the phenolic group of 2d (Scheme 5). The lipophilic compound 66, instead, was conjugated to 2d via alkylation (Scheme 5). Then, the diacetal functional group on the linker was hydrolyzed to aldehyde under acidic conditions to allow reductive amination with 1c,d (synthesis of 1d is described in the Supporting Information) warheads, leading to the formation of compounds 46–54. To conjugate the linker to 1e,f via amide bond, VHL-linker aldehyde intermediates were oxidized to acid through a Pinnick reaction. Then, HATU-mediated coupling with 1e,f (synthesis described in the Supporting Information) was applied to obtain the final compounds 55–64 (Scheme 5 and Supporting Information Synthetic Procedures).
Scheme 5. General Synthetic Routes for Third-Generation Compounds.

Reagents and conditions: (a) MsCl, TEA, DCM, rt, 3 h; (b) 2d, K2CO3, DMF, 70 °C, overnight; (c) K2CO3, DMF, 70 °C, overnight; (d) HCl 1 N, THF (1:1), 50 °C, 2 h; (e) 1c,d, NaBH(OAc)3, TEA, DMF, rt, overnight; (f) Na2HPO4, NaClO2, 2-methyl-2-butene, t-BuOH, H2O, rt, 4 h ; (g) 1e,f, HATU, HOAt, DIPEA, DMF, rt, 2 h.
Degraders 46–64 were screened at 1 μM in HeLa cells following 4 and 16 h of treatment (Table 3, Figure 7A and Supporting Information Figure S6). A large proportion of compounds induced efficient degradation of both BRD7 and BRD9 in HeLa cells, with less than 10% total protein remaining after 4 h treatment (Table 3, Figure 7A). Overall, within matched pairs, greater degradation activity was observed for compounds containing meta (BrdL1 and BrdL3) over para (BrdL2 and BrdL4) dimethoxy substituents (as representative examples: 46 vs 47, 26 vs 48, 51 vs 52, Figure 7A). To assess consistency of cellular activity, compound screening was repeated in RI-1 (DLBCL) cells after 2 h of treatment (Table 3, Figure 7B). This cell line was chosen because it is sensitive to BRD9 inhibition,58 it is of clinical relevance, and it also expresses both VHL and CRBN ligases (Supporting Information Figure S7). The compounds that induced greatest protein degradation activity after 2 h were 46 and 52 (on BRD9), and 51 (on BRD7) (Figure 7B). Compounds 46 and 51 differ only by one atom in the center of the linker, which is oxygen or carbon, respectively (Table 3). Compound 52 is the analogue of 51 but with BrdL2 instead of BrdL1 (Table 3). 46 and 51 showed enhanced degradation of BRD9 compared to 26, and similar levels of degradation compared to CRBN-based degrader 7 in RI-1 cells after 8 h treatment. In addition, 46 and 51 achieved PROTAC-induced degradation of BRD7 in addition to BRD9 (Figure 7C).
Figure 7.

Screening of third generation of degraders. (A) Western-blot analysis of BRD9 and BRD7 levels after treatment of HeLa cells with 1 μM compounds for 4 h before harvesting. (B) Western blot analysis of BRD9 and BRD7 levels after treatment of RI-1 cells with 1 μM compounds for 2 h before harvesting. (C) Western blot analysis of BRD9 and BRD7 levels after treatment of RI-1 cells with 1 μM 7, 26, 46, and 51 for 2 and 8 h before harvesting. Intensity values were quantified as described in the Experimental Section.
We next decided to assess the concentration-dependent activity of 46 and 51 in RI-1 cells at two time points (2 and 8 h) after treatments at different concentrations (Figure 8A,C). This experiment revealed 51 to be the most potent of the two, achieving maximal protein degradation of BRD9 in the 10–100 nM window, and of BRD7 between 0.1–1 μM, with the hook effect observed at higher concentrations with both proteins (Figure 8A). In contrast, 46 required concentrations of about 10-fold higher than 51 to achieve its Dmax, and consequently longer treatment times to achieve similar degradation levels of both BRD9 and BRD7 (Figure 8A). Dose-dependent degradation profiles of 51 at the 8 h time-point gave half-degrading concentrations (DC50) of 1.76 nM and 4.5 nM against BRD9 and BRD7, respectively (Figure 8C). The pronounced hook effect observed on BRD9 protein levels upon treatment with 1 μM of 51 (Figure 8A,C) explains its apparent weaker degradation activity in the initial screening (Figure 7B). The potent and rapid protein degradation induced by 51 was confirmed by assessing its activity in RI-1 cells treated with 10 nM or 100 nM of compound at varying time points (Figure 8D). Levels of both BRD7 and BRD9 decreased by more than 50% already after 30 min of treatment at 100 nM, reaching more than 90% of degradation after 4 h. No protein recovery was observed through 48 h of treatment. At the lower concentration used (10 nM) 51 was seen to degrade preferentially BRD9 over BRD7, consistent with the lower DC50 value for BRD9 versus BRD7. Together, the results of the stepwise design and optimization of VHL-based degraders allowed us to identify 51 as our most potent degrader, with DC50 in the single-digit nanomolar range and profound Dmax greater than 90% (Figure 8). These data qualified PROTAC 51 as a potent dual BRD9 and BRD7 degrader, hence afterward referred to as VZ185.
Figure 8.

VZ185 induces strong and rapid degradation in a time- and dose-dependent manner. (A) Western blot analysis of BRD9, BRD7, and β-actin after treatment of RI-1 cells with six different concentrations of compounds 46 and 51 for 2 and 8 h before harvesting. (B) Chemical structures of 51 (VZ185) and 46. (C) Quantification of protein levels relative to DMSO control after treatment with different concentration of VZ185 and DC50 values. (D) Time-dependent experiment in RI-1 cells after treatment with 10 nM and 100 nM 51 at the desired time points. Intensity values and DC50 were quantified as described in the Experimental Section.
To confirm the potent and rapid degradation activity of VZ185 and its superiority over close analogues, we employed orthogonal kinetic degradation studies using live cell luminescent monitoring of BRD7 and BRD9 endogenously tagged with HiBiT in HEK293 cells using CRISPR/Cas9.69 Degradation profile experiments carried out over 24 h confirmed the superior degradation activity of VZ185 over compounds 26 and 46 at a fixed concentration of 1 μM (Supporting Information Figure S8). Treatment across a range of concentrations of VZ185 of both HiBiT-BRD7 and HiBiT-BRD9 revealed differential degradation profiles (Figure 9A) and allowed the calculation of Dmax, DC50 values, and initial rates of degradation (Figure 9B,C). Indeed, rapid degradation was observed within a few hours by VZ185 showing preference for BRD9 as compared to BRD7 (BRD9-DC50 = 4 nM; BRD7-DC50 = 34 nM) (Figure 9B). Degradation rate was directly proportional to the concentration of VZ185 except at high concentration for BRD9 where the curve reaches a plateau (Figure 9C). This trend was in line with the strong “hook effect” previously observed with VZ185. Degradation activity was also confirmed within matched-pairs 26–48 and VZ185-52 with CRISPR/Cas9-mediated HiBiT endogenous tagging of BRD9 and BRD7 in HEK293 cells (Supporting Information Figure S8). In addition, degradation analysis in a panel of other human cancer cell lines (EOL-1, A-204) confirmed the potency of VZ185, showing a DC50 between 2 and 8 nM for BRD9 (Supporting Information Figure S9).
Figure 9.

Quantitative live-cell kinetics of VZ185 induced degradation of BRD7 and BRD9. Degradation profile (A), calculation of DC50 values (B), and initial degradation rate (C) across concentration series indicated of VZ185 using continual luminescent reading of CRISPR/Cas9 endogenously tagged HiBiT-BRD7 or HiBiT-BRD9 in HEK293 cells. Error bars are expressed as SEM taken from n = 3 experiments.
Following its identification as the best degrader, we subjected VZ185 to a thorough biophysical characterization in order to dissect the molecular basis underpinning its potent degradation activity. Both the ITC and FP data indicated that the PROTAC was not more cooperative than 26 (Table 4 and Supporting Information Figures S5 and S10). Consistent with this, the amount of ternary complex estimated by AlphaLISA was also not significantly different from that measured for 26 (Table 4 and Supporting Information Figure S10). However, the binary affinity for both VCB (Kd = 26 ± 9 nM) and the BRD9 bromodomain (Kd = 5.1 ± 0.6 nM) was significantly greater than 26. The pronounced binary affinity for each respective protein provides some basis for the strong “hook effect” observed in the degradation assays, consistent with previous reports that a high binary affinity can direct PROTACs to function preferentially as inhibitors at higher concentrations and exhibit strong hook effects.11 Despite the absence of a significant boost in cooperativity over 26, the total ΔG° for ternary complex formation increased to −21.7 kcal/mol, which is greater compared to those of 26 (−20.7 kcal/mol) and 5 (−20.4 kcal/mol) (Table 4) and in line with that of our potent Brd4 degrader MZ1 (−22.2 kcal/mol).5 VZ185 and its analogues 26 and 46 all showed low cell permeability in PAMPA assay, suggesting that permeability is unlikely to be the main driver of the enhanced degradation activity of VZ185 (Table S3). The data together suggest that the thermodynamically more stable ternary complex formed by VZ185 helps to drive its more potent and rapid degradation activity, in spite of its low cell permeability.
Table 4. Biophysical Comparison between BRD9 Degraders from Three Generations.
| ITCa |
FPb |
AlphaLISA | ||||||
|---|---|---|---|---|---|---|---|---|
| compd | binary Kd (nM) | ternary Kd (nM) | α | total ΔG (kcal/mol) | binary Kd (nM) | ternary Kd (nM) | α | max intensity |
| 5 | 33 ± 2 | 73 ± 2 | 0.45 | –20.4 | 24 ± 6 | 98 ± 2 | 0.24 | 1.0 × 106 |
| 26 | 87 ± 5 | 83 ± 2 | 1.05 | –20.7 | 70 ± 14 | 60 ± 5 | 1.17 | 1.56 × 106 |
| VZ185 | 26 ± 9 | 27 ± 3 | 0.96 | –21.7 | 35 ± 5 | 35 ± 6 | 1.00 | 1.47 × 106 |
All ITC titrations were performed at 25 °C. Values reported are the mean ± SEM from at least two independent measurements (n ≥ 2).
Kd values are reported as the mean ± SEM of at least two individual experiments performed in triplicate.
We next studied the functional mechanism of VZ185 by confirming proteasome and CRL2VHL involvement and ubiquitination of BRD7 and BRD9 (Figure 10). Combined treatment of the active degrader with the proteasome inhibitor MG132 and the neddylation inhibitor MLN4924, which blocks the activity of CRL2VHL,70 suppressed the degradation of BRD7 and BRD9, clearly indicating that the degradation is proteasome- and CRL2VHL-dependent (Figure 10A). Moreover, to investigate levels of target ubiquitination, NanoBRET experiments were performed using the endogenously tagged HiBiT-BRD7 and BRD9 as energy donors and a polyclonal-Ub primary in conjunction with a fluorescently labeled secondary antibody as an energy acceptor (Figure 10B). Increases in BRET can be observed over time after treatment with VZ185 for both proteins, indicating PROTAC-induced target ubiquitination. The observed signal was stronger for BRD9 as compared to BRD7, consistent with the trends observed for VZ185 inducing preferential degradation of BRD9 compared to BRD7 (Figure 10B). Additionally, to confirm the involvement of VHL in the mechanism of action of VZ185, a competition assay with VHL inhibitor VH298 (ref (62)) was carried out. RI-1 cells were treated with 100 μM VH298 30 min before adding VZ185 (100 nM), and the cells were incubated for 4 h before cell lysis (Figure 10A). As anticipated, pretreatment with VHL inhibitor blocked the degradation activity of VZ185. Moreover, since the trans stereochemistry of the hydroxyl group of the hydroxyproline moiety on VHL is essential for the binding to VHL,62,71 RI-1 cells were treated with cisVZ185 (100 nM) as inactive negative control (synthesis detailed in the Supporting Information) for 4 h before harvesting. As expected, no degradation of BRD7 or BDR9 was observed with cisVZ185 (Figure 10A).
Figure 10.

Mechanistic characterization of VZ185 mode of action. (A) VZ185 activity is proteasome and CRL2VHL-dependent. RI-1 cells were treated in two replicates with MG132, MLN4924, VH298, and cisVZ185 in the presence of absence of VZ185 (100 nM) at desired time points before harvesting. (B) NanoBRET experiments in HEK293 cells to monitor ubiquitination of HiBiT endogenously tagged BRD9 (purple) or BRD7 (green) using monoclonal-Ub primary and Alexa594 secondary antibodies at times indicated after treatment with 1 μM VZ185. Error bars expressed as SEM of n = 3 experiments.
We next assessed the impact of compound-induced BRD7/9 degradation on the viability of cancer cell lines. EOL-1 (acute myeloid eosinophilic leukemia) and A-204 (malignant rhabdoid tumor) cell lines were chosen because they are sensitive to BRD9 inhibition/degradation18,56 and dependent on an active BAF complex,72 respectively. Cellular ATP presence was quantified as signal of metabolically active cells (Figure 11). VZ185 was cytotoxic in both cell lines, with EC50 of 3 nM (EOL-1) and 40 nM (A-402), and proved to be equipotent to CRBN-based degrader 21 (dBRD9, EC50 of 5 and 90 nM, respectively) (Figure 11). Differential cytotoxicity of BRD7/9 degradation by VZ185 over and above BRD7/9 bromodomain inhibition was clearly observed (EC50 of 90–340 nM and 370–3550 nM for compounds 1a and 1b, respectively). The activity in A-204 cells is of particular relevance as malignant rhabdoid tumors are rare, chemoresistant cancers with poor survival rate (<25%) that are distinctly characterized by biallelic inactivation of SMARCB1, a core subunit of the BAF complex, suggesting specific vulnerabilities.86
Figure 11.

Effect of BRD7/9 degradation on viability of BRD9-sensitive cancer cell lines. Cell proliferation activity assessed in EOL-1 (A) and A-204 (B) cell lines after treatment with 1a, 1b, VZ185, 7, 21 (dBRD9), and doxorubicin for 7 days.
Finally, to assess the cellular selectivity of VZ185 for BRD7/9 degradation and identify potential degradation off-targets, multiplexed isobaric tagging mass spectrometry proteomic experiments were performed to monitor protein levels in a quantitative and unbiased manner. RI-1 cells were treated in triplicate with DMSO, 100 nM VZ185, or 100 nM cisVZ185 for 4 h. Among 6273 proteins quantified in this analysis, of those that met the criteria for a statistically significant change in abundance (p-value y < 0.001; fold change {x < −20% U, x > 20%}, Figure 12 and Supporting Information Figures S11 and S12), markedly selective degradation of BRD7 and BRD9 was observed. As expected, BRD7/9 proteins were not depleted by treatment with negative control cisVZ185 (Supporting Information Figures S11 and S12). Protein levels of other bromodomain-containing proteins or other BAF/PBAF subunits remained unaffected. To confirm selectivity over key potential off-target proteins within the bromodomain protein family, live cell kinetic analyses of endogenously tagged BRD2/3/4 and SMARCA4 proteins expressing LgBiT were performed (Supporting Information Figure S13). These results together with the proteomic data confirmed VZ185 as an effective and highly selective degrader of BRD7/9 proteins in cells. In vitro PK data further showed high stabilities of VZ185 in both plasma and microsomes from both human and mouse species, as well as high aqueous kinetic solubility (up to ∼100 μM, Table S3). Together, the data qualify VZ185 as a novel high-quality degrader probe for cellular and potentially in vivo investigations.
Figure 12.
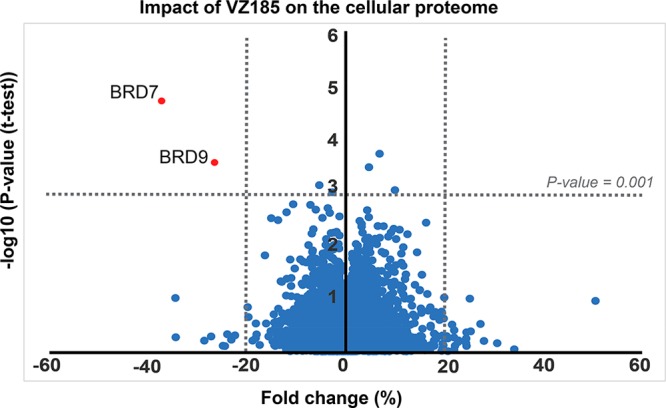
Impact of VZ185 on the cellular proteome after treatment of RI-1 cells with 100 nM compound for 4 h before harvesting. Data are plotted as fold change (%) versus −log10 of p-value (t test) for a total of 6273 proteins, expressed as the mean of the replicates. For quantification, see Experimental Section.
Conclusion
We describe the development of a new series of PROTACs against BRD9, a target thought as not degradable through recruitment of the E3 ligase VHL. In spite of starting from unimpressive degradation profiles of initial compounds, VHL-based degraders could be optimized by systematically varying the conjugation patterns and monitoring cellular degradation activities and formation of ternary complexes. Throughout the campaign, we revealed important structure–activity relationships that proved invaluable to guide the optimization search space and led to significant improvements in degradation activities. We thoroughly characterize VZ185 as a highly selective, potent, and rapid dual degrader with a slight preference for BRD9 over BRD7. Our findings thus qualify VZ185 as a new high-quality chemical probe that will be valuable to explore the biology and therapeutic potential of degrading these two proteins.
Biophysical and mechanistic studies suggest that increased ternary complex stability correlates with improved degradation profiles. The absence of positive cooperativity in VZ185 allows much scope for optimizing the thermodynamics of ternary complex formation, which in turn should warrant enhanced degradation activities of future compounds.5,11,32 Further investigation of the contributing factors of single-target selectivity would be important and could also allow rational design of single-target selective degraders, for example, for BRD7 over BRD9. More generally, this successful campaign exemplifies a broadly applicable approach to arrive at degraders that are effective for any target–ligase pairs, even those that might have been considered unproductive based on negative studies with initial compounds. It is thus tempting to speculate that there may not exist a preferred E3 ligase for “PROTACing” a given target protein. Any E3 ligase might, in principle, be hijacked for productive target ubiquitination and degradation, provided the combinatorial chemical space is adequately explored.
Experimental Section
Chemistry. Synthesis
Chemicals, commercially available, were purchased from Apollo Scientific, Sigma-Aldrich, Fluorochem, or Manchester Organics and used without any further purification. All reactions were carried out using anhydrous solvents. Analytical thin-layer chromatography (TLC) was performed on precoated TLC plates (layer 0.20 mm silica gel 60 with fluorescent indicator (UV 254: Merck)). The TLC plates were air-dried and revealed under UV lamp (254/365 nm). Flash column chromatography was performed using prepacked silica gel cartridges (230–400 mesh, 40–63 mm; SiliCycle) using a Teledyne ISCO Combiflash Companion or Combiflash Retrieve using the solvent mixtures stated for each synthesis as mobile phase. Liquid chromatography–mass spectrometry (LC–MS) analyses were performed with either an Agilent HPLC 1100 series connected to a Bruker Daltonics MicroTOF or an Agilent Technologies 1200 series HPLC connected to an Agilent Technologies 6130 quadrupole spectrometer or a Waters 2795 connected to a Waters ZQ Micromass spectrometer; all instruments were connected to a diode array detector. All of the final compounds used in all of the experiments were evaluated after preparative LC–MS separations with a Waters X-bridge C18 column (50 mm × 2.1 mm × 3.5 mm particle size); flow rate, 0.5 mL/min with a mobile phase of water/MeCN + 0.1% NH4OH or water/MeCN + 0.1% CHOOH; 95/5 water/MeCN was initially held for 0.5 min followed by a linear gradient from 95/5 to 5/95 water/MeCN over 3.5 min which was then held for 2 min. The purity of all the compounds was evaluated using the analytical LC–MS system described before, and purity was >95%. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance II 500 spectrometer (1H at 500.1 MHz, 13C at 125.8 MHz) or on a Bruker DPX-400 spectrometer (1H at 400.1 MHz, 13C at 101 MHz). Chemical shifts (δ) are expressed in ppm reported using residual solvent as the internal reference in all cases. Signal splitting patterns are described as singlet (s), doublet (d), triplet (t), multiplet (m), or a combination thereof. Coupling constants (J) are quoted to the nearest 0.1 Hz.
General Method A
To a mixture of aryl bromide (1 equiv) in dioxane (0.2 M), Pd(dppf)Cl2 (0.1 equiv), bis(pinacolato)diboron (1.2 equiv), and KOAc (3 equiv) were added. The mixture was heated under microwave conditions at 140 °C for 40 min. Then aryl iodide 14 (1 equiv) and a degassed solution of K2CO3 2 M (2 equiv) were added. The reaction mixture was heated in microwave at 120 °C for 30 min. The resulting mixture was filtered through Celite and washed several times with DCM. The organic phase was washed with H2O and brine, dried over MgSO4, filtered, and evaporated in vacuum. The crude was purified by flash column chromatography using a gradient from 0% to 20% of MeOH in DCM to obtain the desired compound. Then the intermediate was taken up in a mixture 1:1 HCl 4 N in dioxane/DCM and stirred at rt for 3 h. The solvents were evaporated to dryness to obtain the desired compound as hydrochloride salt.
General Method B
To a solution of acid (1 equiv) in DMF (0.2 M), HATU (1 equiv), HOAt (1 equiv), amine (1 equiv), and DIPEA (5 equiv) were added. The reaction mixture was stirred at rt for 2 h. The solvent was evaporated under reduced pressure to give the corresponding crude, which was chromatographically purified to yield the final compound.
General Method C
A mixture of aldehyde (1 equiv), amine (HCl salt, 1.1 equiv), and TEA (1.1 equiv) in DMF (0.2 M) was stirred at rt. After 15 min, NaBH(OAc)3 (1.5 equiv) was added, and the reaction was stirred overnight at rt under nitrogen. The solvent was evaporated under reduced pressure to give the corresponding crude, which was purified accordingly to yield the desired compound.
General Method D
To a stirred solution of oxalyl chloride (1.5 equiv) in DCM (0.3 M), DMSO (2 equiv) was added dropwise at −78 °C. After 10 min at −78 °C, the alcohol starting material (1 equiv) in DCM (0.3 M) was added. The reaction was stirred at −78 °C for 1 h, TEA (10 equiv) was added dropwise, and temperature was increased to rt. After 1 h, the reaction mixture was diluted with DCM, washed with a saturated solution of ammonium chloride. The organic phases were combined, washed with brine, dried over MgSO4, filtered, and evaporated to dryness. Then the crude was dissolved in DMF (0.05 M); 1c (1 equiv) and TEA (1.1 equiv) were added. After 15 min, NaBH(OAc)3 was added and the reaction mixture was stirred overnight at rt under nitrogen. The solvent was evaporated under reduced pressure to give the corresponding crude, which was purified by preparative HPLC to yield the desired compound.
General Method E
To a solution of the alcohol starting material (1 equiv) and trimethylamine (1.5 equiv) in DCM (0.2 M), methansulfonyl chloride (1.2 equiv) was added at 0 °C. The reaction mixture was stirred at 0 °C for 3 h. The reaction was quenched with a saturated solution of ammonium chloride and extracted with DCM. The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness. The crude (1.5 equiv) was suspended in DMF (0.2 M), and 2b–d inhibitor derivative (1 equiv) and K2CO3 (3 equiv) were added. The mixture was heated overnight at 70 °C. The reaction mixture was diluted with water and extracted with DCM. The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness. The crude was purified by flash column chromatography using a gradient from 0% to 20% of MeOH in DCM to yield the desired compound.
General Method F
A 1 M aqueous solution of HCl (1 mL) was added to the VHL-acetal linker (0.06 mmol) in THF (1 mL). The reaction mixture was heated at 50 °C for 2 h. The solvent was evaporated under reduced pressure, and the product was extracted with DCM/MeOH (9:1). The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness. The crude was used directly without any further purification.
General method G
Aldehyde (1 equiv) was taken up in t-BuOH and water (4:1) (0.3 M). Then 2-methyl-2-butene 2 M in THF (4 equiv) was added, followed by sodium phosphate dibasic (1 equiv) and sodium chlorite (2 equiv). The reaction mixture was stirred at rt for 4 h. A 1 M aqueous solution of HCl was added. The product was extracted with DCM. The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness. The crude was used directly without any further purification.
4-(3,5-Dimethoxy-4-(piperazin-1-ylmethyl)phenyl)-2-methyl-2,7-naphthyridin-1(2H)-one (1c)
Following general method A, compound 1c was obtained from compound 12 and 14 as brown powder. Yield: 50 mg, 55%. 1H NMR (400 MHz, MeOD) δ: 9.67 (s, 1H), 8.76 (d, J = 6.9 Hz, 1H), 8.27 (s, 1H), 8.21 (d, J = 6.9 Hz, 1H), 6.95 (s, 2H), 4.62 (s, 2H), 4.03 (s, 6H), 3.81 (s, 3H), 3.73 (s, 8H). 13C NMR (101 MHz, CDCl3) δ: 161.5, 161.4, 148.6, 146.3, 145.5, 141.9, 139.7, 122.9, 117.9, 107.1, 105.8, 57.2, 50.8, 49.7, 41.9, 37.9. MS m/z calcd for C22H26N4O3 394.2, found 395.3 [M + H+].
(2S,4R)-1-((S)-2-(2-(2-(2-(4-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)ethoxy)ethoxy)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (5)
Following general method B, compound 5 was obtained from 19 and 2a (synthesized accordingly to literature61). The crude was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid to obtain 5 as white powder. Yield: 6.3 mg, 44%. 1H NMR (400 MHz, MeOD) δ: 9.44 (s, 1H), 8.80 (s, 1H), 8.59 (d, J = 5.5 Hz, 1H), 7.66 (s, 1H), 7.54 (d, J = 5.9 Hz, 1H), 7.37 (q, J = 7.8 Hz, 4H), 6.73 (s, 2H), 4.66 (s, 1H), 4.52–4.26 (m, 4H), 4.05–3.95 (m, 4H), 3.86–3.72 (m, 8H), 3.69–3.59 (m, 9H), 2.96–2.73 (m, 10H), 2.40 (s, 3H), 2.21–2.14 (m, 1H), 2.06–1.97 (m, 1H), 0.99 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.3, 172.0, 171.7, 163.0, 161.0, 152.8, 151.7, 151.2, 149.1, 143.5, 140.3, 139.0, 133.3, 131.6, 130.5, 130.4, 129.5, 129.0, 121.7, 119.2, 119.0, 106.6, 72.2, 71.3, 71.1, 71.0, 69.1, 60.9, 58.2, 58.1, 57.9, 56.6, 52.8, 52.4, 43.7, 39.0, 37.4, 37.2, 27.0, 15.8. HRMS m/z calcd for C50H64N8O9S 952.45, found 953.4728 [M + H+].
(2S,4R)-1-((S)-2-(tert-Butyl)-17-(4-(2,6-dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)-4-oxo-6,9,12,15-tetraoxa-3-azaheptadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (6)
Following general method B, compound 6 was obtained from 20 and 2a (synthesized accordingly to literature61). The crude was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid to obtain 6 as white powder. Yield: 4.9 mg, 30%. 1H NMR (400 MHz, MeOD) δ: 9.44 (s, 1H), 8.81 (s, 1H), 8.60 (d, J = 5.6 Hz, 1H), 7.67 (s, 1H), 7.54 (d, J = 5.6 Hz, 1H), 7.37 (q, J = 9.0 Hz, 4H), 6.73 (s, 2H), 4.63 (s, 1H), 4.53–4.26 (m, 4H), 4.02–3.94 (m, 4H), 3.84–3.72 (m, 8H), 3.66–3.50 (m, 17H), 2.89–2.67 (m, 10H), 2.41 (s, 3H), 2.20–2.14 (m, 1H), 2.06–1.97 (m, 1H), 0.98 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.4, 172.0, 171.7, 163.0, 161.0, 152.9, 151.7, 151.2, 149.1, 143.5, 140.3, 139.0, 138.6, 133.4, 131.5, 130.5, 130.4, 129.5, 129.0, 121.7, 119.2, 119.1, 106.6, 72.3, 71.7, 71.6, 71.5, 71.4, 71.1, 71.0, 68.9, 60.9, 58.2, 58.0, 56.6, 52.8, 43.7, 39.0, 37.4, 37.1, 27.0, 15.9. HRMS m/z calcd for C54H72N8O11S 1040.50, found 1041.5287 [M + H+].
2-(2-(2-(4-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)ethoxy)ethoxy)-N-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)acetamide (7)
Following general method B, compound 7 was obtained from compound 19 and 3a (synthesized accordingly to literature73). The crude was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid to obtain compound 7 as yellow powder. Yield: 5.6 mg, 33%. 1H NMR (400 MHz, MeOD) δ: 9.54 (d, J = 0.8 Hz, 1H), 8.70 (d, J = 6.0 Hz, 1H), 8.11 (s, 1NH), 7.84 (s, 1H), 7.73 (dd, J = 0.6, 6.0 Hz, 1H), 7.55–7.49 (m, 1H), 7.10–7.03 (m, 2H), 6.87 (s, 2H), 5.08 (dd, J = 5.5, 12.7 Hz, 1H), 4.38 (s, 2H), 4.05 (s, 2H), 3.96 (s, 6H), 3.77–3.67 (m, 9H), 3.51–3.46 (m, 4H), 3.39 (s, 4H), 3.20 (s, 4H), 3.01 (t, J = 4.8 Hz, 2H), 2.94–2.67 (m, 4H). 13C NMR (101 MHz, MeOD) δ: 174.6, 173.4, 171.6, 170.6, 169.1, 162.7, 161.1, 150.9, 149.8, 147.9, 144.0, 140.1, 137.2, 133.9, 121.8, 119.6, 118.5, 117.9, 112.2, 111.4, 106.7, 71.7, 71.3, 71.0, 67.7, 57.5, 56.8, 51.8, 51.1, 50.2, 50.0, 42.6, 39.4, 37.5, 32.2, 23.8. HRMS m/z calcd for C43H50N8O10 838.36, found 839.3796 [M + H+].
14-(4-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)-N-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)-3,6,9,12-tetraoxatetradecanamide (8)
Following general method B, compound 8 was obtained from compound 20 and 3a (synthesized accordingly to literature73). The crude was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid to obtain compound 8 as yellow powder. Yield: 10 mg, 54%. 1H NMR (400 MHz, MeOD) δ: 9.53 (s, 1H), 8.70 (d, J = 5.7 Hz, 1H), 8.15 (s, 1NH), 7.77 (s, 1H), 7.64 (d, J = 5.7 Hz, 1H), 7.59–7.53 (m, 1H), 7.17–7.13 (m, 1H), 7.08–7.04 (m, 1H), 6.86 (s, 2H), 5.07 (dd, J = 5.5, 12.5 Hz, 1H), 4.29 (s, 2H), 4.01 (s, 2H), 3.96 (s, 6H), 3.72–3.63 (m, 17H), 3.54 (s, 4H), 3.25 (s, 4H), 3.02 (s, 4H), 2.94–2.67 (m, 6H). 13C NMR (101 MHz, MeOD) δ: 174.6, 173.5, 171.5, 170.6, 169.2, 163.0, 161.0, 151.7, 151.2, 148.0, 143.4, 139.8, 139.2, 137.3, 133.9, 121.7, 119.1, 118.7, 118.1, 112.1, 111.4, 108.1, 106.7, 72.0, 71.4, 71.1, 68.5, 57.6, 56.8, 52.4, 51.6, 50.2, 49.9, 42.5, 39.4, 37.5, 32.2, 23.8. HRMS m/z calcd for C47H58N8O12 926.42, found 927.4396 [M + H+].
N-(4-(N-(3-Chloro-1H-indol-7-yl)sulfamoyl)benzyl)-2-(2-(2-(4-(2,6-dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)ethoxy)ethoxy)acetamide (9)
Following general method B, compound 9 was obtained from compound 19 and 4a (synthesis detailed in the Supporting Information). The crude was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid to obtain compound 9 as white powder. Yield: 10.8 mg, 34%. 1H NMR (400 MHz, MeOD) δ: 9.54 (s, 1H), 8.69 (d, J = 5.8 Hz, 1H), 8.27 (s, 1NH), 7.75 (s, 1H), 7.68–7.62 (m, 3H), 7.39 (d, J = 8.3 Hz, 2H), 7.33 (dd, J = 0.9, 8.1 Hz, 1H), 7.26 (s, 1H), 6.91 (t, J = 7.8 Hz, 1H), 6.85 (s, 2H), 6.70 (d, J = 7.7 Hz, 1H), 4.49 (s, 2H), 4.28 (s, 2H), 4.11 (s, 2H), 3.94 (s, 6H), 3.76–3.66 (m, 7H), 3.63 (t, J = 5.3 Hz, 2H), 3.22 (s, 4H), 2.88 (s, 4H), 2.73 (t, J = 5.1 Hz, 2H). 13C NMR (101 MHz, MeOD) δ: 173.0, 166.4, 163.0, 161.0, 151.7, 151.2, 145.5, 143.3, 140.0, 139.4, 139.1, 132.0, 128.8, 128.7, 128.2, 123.4, 122.7, 121.6, 120.9, 119.4, 119.1, 118.8, 117.0, 107.5, 106.7, 106.2, 71.9, 71.4, 71.3, 68.7, 57.6, 56.8, 52.5, 51.3, 49.9, 43.0, 37.5. HRMS m/z calcd for C43H48ClN7O8S 857.30, found 858.3111 [M + H+].
N-(4-(N-(3-Chloro-1H-indol-7-yl)sulfamoyl)benzyl)-14-(4-(2,6-dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)-3,6,9,12-tetraoxatetradecanamide (10)
Following general method B, compound 10 was obtained from compound 20 and 4a (synthesis detailed in the Supporting Information). The crude was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid to obtain compound 10 as white powder. Yield: 11.1 mg, 37%. 1H NMR (400 MHz, MeOD) δ: 9.54 (d, J = 0.6 Hz, 1H), 8.69 (d, J = 5.7 Hz, 1H), 8.30 (s, 1NH), 7.75 (s, 1H), 7.68–7.62 (m, 3H), 7.41 (d, J = 8.2 Hz, 2H), 7.34 (dd, J = 0.8, 8.1 Hz, 1H), 7.27 (s, 1H), 6.92 (t, J = 7.8 Hz, 1H), 6.84 (s, 2H), 6.72 (dd, J = 0.9, 7.5 Hz, 1H), 4.50 (s, 2H), 4.26 (s, 2H), 4.09 (s, 2H), 3.94 (s, 6H), 3.73–3.67 (m, 7H), 3.65–3.59 (m, 4H), 3.58–3.53 (m, 6H), 3.22 (s, 4H), 2.96 (s, 4H), 2.82 (t, J = 5.3 Hz, 2H). 13C NMR (101 MHz, MeOD) δ: 173.1, 166.7, 163.1, 161.0, 151.7, 151.2, 145.5, 143.4, 139.7, 139.2, 139.0, 131.9, 128.9, 128.6, 128.2, 123.4, 122.7, 121.6, 121.0, 119.3, 119.2, 118.9, 117.0, 108.0, 106.7, 106.2, 72.0, 71.3, 71.3, 71.2, 68.3, 57.5, 56.8, 52.3, 51.5, 49.9, 42.9, 37.6. HRMS m/z calcd for C47H56ClN7O10S 945.35, found 946.3676 [M + H+].
tert-Butyl 4-(4-Bromo-2,6-dimethoxybenzyl)piperazine-1-carboxylate (12)
Following general method C, compound 12 was obtained from 4-bromo-2,6-dimethoxybenzaldehyde (11) and boc-piperazine (both commercially available). The reaction was quenched with saturated solution of NaHCO3, extracted with DCM, washed with water and brine. The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness to give the desired compound without any further purification as sticky oil. Yield: 495 mg, 97%. 1H NMR (500 MHz, CDCl3) δ: 6.69 (s, 2H), 3.78 (s, 6H), 3.62 (s, 2H), 3.38 (t, J = 5.0 Hz, 4H), 2.41 (t, J = 4.4 Hz, 4H), 1.43 (s, 9H). 13C NMR (125 MHz, CDCl3) δ: 159.9, 154.7, 122.8, 107.7, 79.8, 56.1, 51.8, 48.5, 31.0, 28.5. MS m/z calcd for C18H27BrN2O4 414.12, found 415.2 [M + H+].
4-Iodo-2-methyl-2,7-naphthyridin-1(2H)-one (14)
A mixture of 4-iodo-2,7-naphthyridin-1(2H)-one 13 (synthesized accordingly to literature74) (1 equiv) and NaH (2 equiv) was stirred 30 min at 0 °C in DMF (0.2 M). Then CH3I (1.6 equiv) was added and the reaction mixture was stirred at 0 °C for 5 h. Water was added, and the precipitate formed was filtered and dried in vacuum. Yield: 300 mg, 95%. 1H NMR (400 MHz, DMSO) δ: 9.27 (s, 1H), 8.82 (d, J = 5.6 Hz, 1H), 8.26 (s, 1H), 7.47 (d, J = 5.5 Hz, 1H), 3.52 (s,3H). 13C NMR (101 MHz, DMSO) δ: 160.3, 152.0, 150.3, 144.8, 143.0, 122.4, 120.7, 67.9, 36.2. MS m/z calcd for C9H7IN2O 285.96, found 287.1 [M + H+].
tert-Butyl 2-(2-(2-(4-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)ethoxy)ethoxy)acetate (17)
Following general method D, compound 17 was obtained from tert-butyl 2-(2-(2-hydroxyethoxy)ethoxy)acetate 15 and 1c after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of ammonia as white powder. Yield: 8.8 mg, 32%. 1H NMR (500 MHz, CDCl3) δ: 9.69 (d, J = 0.8 Hz, 1H), 8.70 (d, J = 5.7 Hz, 1H), 7.43 (dd, J = 0.8, 5.6 Hz, 1H), 7.26 (s, 1H), 6.52 (s, 2H), 4.00 (s, 2H), 3.81 (s, 6H), 3.70–3.60 (m, 11H), 2.65–2.56 (m, 10H), 1.46 (s, 9H). 13C NMR (125 MHz, CDCl3) δ: 170.0, 161.8, 160.0, 152.0, 151.3, 142.1, 136.1, 135.5, 120.8, 118.4, 117.8, 113.9, 105.7, 81.9, 71.0, 70.7, 69.3, 58.1, 56.2, 53.7, 52.6, 48.9, 37.4, 28.4. MS m/z calcd for C32H44N4O7 596.32, found 597.34 [M + H+].
tert-Butyl 14-(4-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)-3,6,9,12-tetraoxatetradecanoate (18)
Following general method D, compound 18 was obtained from tert-butyl 14-hydroxy-3,6,9,12-tetraoxatetradecanoate 16 and 1c after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of ammonia as white powder. Yield: 10.7 mg, 34%. 1H NMR (500 MHz, CDCl3) δ: 9.69 (d, J = 0.8 Hz, 1H), 8.70 (d, J = 5.6 Hz, 1H), 7.43 (d, J = 5.6 Hz, 1H), 7.26 (s, 1H), 6.52 (s, 2H), 4.00 (s, 2H), 3.81 (s, 6H), 3.73–3.57 (m, 19H), 2.67–2.55 (m, 10H), 1.46 (s, 9H). 13C NMR (125 MHz, CDCl3) δ: 170.0, 161.8, 160.0, 152.0, 151.3, 142.1, 136.2, 120.8, 118.4, 117.8, 105.7, 81.9, 71.1, 70.9, 70.7, 69.4, 69.2, 58.0, 56.3, 53.8, 52.4, 48.9, 37.5, 28.5. MS m/z calcd for C36H52N4O9 684.37, found 685.39 [M + H+].
2-(2-(2-(4-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)ethoxy)ethoxy)acetic Acid (19)
A mixture of compound 17 (0.015 mmol), TFA (0.5 mL), and DCM (0.5 mL) was stirred at rt for 3 h. Then the solvent was evaporated; the crude was dried under high pressure overnight and used directly in the next step without any further purification. Quantitative yield. MS m/z calcd for C28H36N4O7 540.26, found 541.4 [M + H+].
17-(4-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)-3,6,9,12,15-pentaoxaheptadecanoic Acid (20)
A mixture of compound 18 (0.034 mmol), TFA (0.5 mL), and DCM (0.5 mL) was stirred at rt for 3 h. Then the solvent was evaporated; the crude was dried under high pressure overnight and used directly without any further purification. Quantitative yield. MS m/z calcd for C34H48N4O10 672.34, found 673.36 [M + H+].
(2R,4S)-1-((R)-2-(2-(2-((5-(4-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)pentyl)oxy)ethoxy)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (23)
A mixture of 35 (1 equiv), osmium tetroxide 4% in H2O (0.2 equiv), sodium periodate (4 equiv), pyridine (2 equiv) in dioxane/H2O (3:1) was stirred at rt for 48 h. Then H2O was added to the reaction and the product was extracted with DCM. The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness. The crude was dissolved in DCE, and a mixture of 1c (1 equiv) and TEA (1.1 equiv) in DCE (0.02 M) was added. After 15 min, NaBH(OAc)3 was added and the reaction mixture was stirred at rt for 4 h under nitrogen. The solvent was evaporated under reduced pressure to give the corresponding crude, which was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of ammonia. Compound 23 was obtained as a white powder. Yield: 6.9 mg, 54%. 1H NMR (400 MHz, CDCl3) δ: 9.68 (s, 1H), 8.69 (d, J = 5.6 Hz, 1H), 8.66 (s, 1H), 7.41 (dd, J = 0.6, 5.6 Hz, 1H), 7.37–7.32 (m, 4H), 7.27 (s, 1H), 6.53 (s, 2H), 4.72 (t, J = 8.0 Hz, 1H), 4.59–4.49 (m, 3H), 4.35–4.29 (m, 1H), 4.07–3.95 (m, 3H), 3.85–3.80 (m, 8H), 3.67–3.55 (m, 8H), 3.43 (t, J = 6.4 Hz, 2H), 2.74–2.37 (m, 14H), 2.15–2.09 (m, 1H), 1.60–1.47 (m, 4H), 1.36–1.27 (m, 2H), 0.94 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 171.5, 170.9, 170.5, 168.6, 161.6, 159.8, 151.8, 151.1, 150.4, 148.6, 141.9, 138.3, 136.0, 131.7, 131.1, 129.6, 128.3, 120.6, 118.1, 117.6, 105.5, 71.4, 71.3, 70.6, 70.1, 58.6, 58.2, 57.2, 56.9, 56.1, 52.5, 51.7, 48.6, 43.4, 37.3, 36.1, 35.2, 29.4, 26.6, 26.0, 24.2, 16.2. HRMS m/z calcd for C53H70N8O9S 994.50, found 995.5178 [M + H+].
(2S,4R)-N-(2-(2-(2-(2-(4-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)ethoxy)ethoxy)ethoxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (26)
A mixture of 40 (1 equiv), osmium tetroxide 4% in H2O (0.2 equiv), sodium periodate (4 equiv), pyridine (2 equiv) in dioxane/H2O (3:1) was stirred at rt for 48 h. Then H2O was added to the reaction and the product was extracted with DCM. The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness. Then, following general method A, compound 26 was synthesized from the aldehyde derivative and 1c. The solvent was removed in vacuum to give the corresponding crude, which was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid to yield compound 26 as white powder. Yield: 7 mg, 45%. 1H NMR (400 MHz, MeOD) δ: 9.54 (s, 1H), 8.90 (s, 1H), 8.70 (d, J = 5.8 Hz, 1H), 8.58 (s, 1NH), 7.75 (s, 1H), 7.63 (dd, J = 0.6, 5.7 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.10–7.04 (m, 2H), 6.79 (s, 2H), 4.78 (s, 1H), 4.66 (t, J = 8.4 Hz, 1H), 4.55–4.42 (m, 3H), 4.32–4.23 (m, 2H), 3.97–3.77 (m, 14H), 3.73–3.66 (m, 7H), 2.85–2.68 (m, 10H), 2.52 (s, 3H), 2.30–2.24 (m, 1H), 2.16–2.09 (m, 1H), 1.46–1.27 (m, 4H), 1.07 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.2, 171.7, 171.5, 171.3, 170.2, 163.0, 161.0, 158.0, 152.8, 151.7, 151.2, 149.1, 143.6, 138.9, 138.1, 133.5, 132.9, 130.0, 128.4, 122.9, 121.7, 119.2, 113.8, 106.5, 80.3, 78.0, 71.8, 71.4, 71.0, 70.8, 69.4, 69.3, 60.8, 58.7, 58.3, 58.2, 56.5, 53.3, 53.0, 39.5, 39.0, 37.4, 37.3, 26.9, 16.0, 14.1, 14.0, 13.9. MS m/z calcd for C54H69FN8O10S 1040.48, found 1041.4430 [M + H+].
(2S,4R)-1-((S)-2-(1-Cyanocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-N-(2-(2-(2-(2-(4-(2,6-dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)ethoxy)ethoxy)ethoxy)-4-(4-methylthiazol-5-yl)benzyl)-4-hydroxypyrrolidine-2-carboxamide (27)
A mixture of 42 (1 equiv), osmium tetroxide 4% in H2O (0.2 equiv), sodium periodate (4 equiv), pyridine (2 equiv) in dioxane/H2O (3:1) was stirred at rt for 48 h. Then H2O was added to the reaction and the product was extracted with DCM. The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness. Then, following general method A, from the aldehyde derivative and 1c compound 27 was obtained after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid as white powder. Yield: 11 mg, 44%. 1H NMR (400 MHz, MeOD) δ: 9.55 (s, 1H), 8.90 (s, 1H), 8.69 (d, J = 5.8 Hz, 1H), 8.58 (s, 1NH), 7.75 (s, 1H), 7.63 (d, J = 5.6 Hz, 1H), 7.50 (d, J = 7.7 Hz, 1H), 7.10–7.03 (m, 2H), 6.79 (s, 2H), 4.71–4.63 (m, 2H), 4.56–4.42 (m, 3H), 4.33–4.23 (m, 2H), 3.98–3.66 (m, 21H), 2.86–2.69 (m, 10H), 2.52 (s, 3H), 2.30–2.23 (m, 1H), 2.16–2.08 (m, 1H), 1.70–1.55 (m, 4H), 1.06 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.1, 171.5, 170.1, 167.1, 163.0, 161.0, 158.1, 152.8, 151.7, 151.2, 149.1, 143.5, 138.9, 138.1, 133.4, 132.9, 130.1, 128.4, 122.8, 121.7, 120.8, 119.2, 119.2, 113.8, 106.5, 71.8, 71.4, 71.0, 70.8, 69.4, 69.2, 60.8, 59.5, 58.3, 58.1, 56.5, 53.2, 53.0, 39.5, 38.9, 37.7, 37.4, 26.8, 18.1, 18.0, 16.0, 14.8. HRMS m/z calcd for C55H69N9O10S 1047.49, found 1048.4493 [M + H+].
(2S,4R)-1-((S)-2-Acetamido-3,3-dimethylbutanoyl)-N-(2-(2-(2-(2-(4-(2,6-dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)ethoxy)ethoxy)ethoxy)-4-(4-methylthiazol-5-yl)benzyl)-4-hydroxypyrrolidine-2-carboxamide (28)
A mixture of 44 (1 equiv), osmium tetroxide 4% in H2O (0.2 equiv), sodium periodate (4 equiv), pyridine (2 equiv) in dioxane/H2O (3:1) was stirred at rt for 48 h. Then H2O was added to the reaction and the product was extracted with DCM. The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness. Then, following general method A, from the aldehyde derivative and 1c compound 28 was obtained after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid as white powder. Yield: 3.3 mg, 17%. 1H NMR (400 MHz, MeOD) δ: 9.55 (d, J = 0.7 Hz, 1H), 8.90 (s, 1H), 8.70 (d, J = 5.9 Hz, 1H), 7.76 (s, 1H), 7.64 (dd, J = 0.7, 5.9 Hz, 1H), 7.52 (d, J = 7.7 Hz, 1H), 7.09 (d, J = 1.5 Hz, 1H), 7.03 (dd, J = 1.7, 7.7 Hz, 1H), 6.79 (s, 2H), 4.65–4.60 (m, 2H), 4.54–4.42 (m, 3H), 4.28–4.25 (m, 2H), 3.96–3.65 (m, 21H), 2.86–2.67 (m, 10H), 2.52 (s, 3H), 2.27–2.21 (m, 1H), 2.15–2.08 (m, 1H), 2.04 (s, 3H), 1.05 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.4, 173.1, 172.3, 163.1, 161.0, 158.0, 152.8, 151.7, 151.2, 149.1, 143.6, 138.9, 133.5, 132.9, 130.0, 128.4, 122.8, 121.7, 119.2, 113.7, 106.5, 71.8, 71.4, 71.1, 70.8, 69.4, 69.2, 60.7, 59.2, 58.2, 58.0, 56.5, 53.2, 53.0, 49.8, 39.4, 38.9, 37.4, 36.5, 27.0, 22.3, 16.0. HRMS m/z calcd for C52H68N8O10S 996.48, found 997.5008 [M + H+].
(2S,4R)-N-(2-((14-(4-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)-3,6,9,12-tetraoxatetradecyl)oxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (29)
A mixture of 41 (1 equiv), osmium tetroxide 4% in H2O (0.2 equiv), sodium periodate (4 equiv), pyridine (2 equiv) in dioxane/H2O (3:1) was stirred at rt for 48 h. Then H2O was added to the reaction and the product was extracted with DCM. The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness. Then, following general method A, from the aldehyde derivative and 1c compound 29 was obtained after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of ammonia as white powder. Yield: 9.5 mg, 46%. 1H NMR (400 MHz, MeOD) δ: 9.54 (s, 1H), 8.90 (s, 1H), 8.70 (d, J = 5.8 Hz, 1H), 7.75 (s, 1H), 7.64 (dd, J = 0.6, 5.9 Hz, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.10–7.03 (m, 2H), 6.77 (s, 2H), 4.78 (s, 1H), 4.69–4.63 (m, 1H), 4.55–4.42 (m, 3H), 4.31–4.23 (m, 2H), 3.97–3.57 (m, 29H), 2.71–2.60 (m, 10H), 2.52 (s, 3H), 2.30–2.24 (m, 1H), 2.17–2.08 (m, 1H), 1.43–1.29 (m, 4H), 1.07 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.2, 171.6, 171.3, 163.0, 161.0, 158.0, 152.8, 151.7, 151.2, 149.1, 143.6, 138.8, 137.3, 133.5, 132.9, 130.0, 128.4, 122.8, 121.7, 119.4, 119.3, 113.8, 113.2, 106.5, 80.3, 78.0, 73.7, 71.8, 71.6, 71.5, 71.4, 71.0, 70.8, 69.6, 69.4, 62.2, 60.8, 58.7, 58.6, 58.2, 56.3, 54.0, 53.3, 39.5, 38.9, 37.4, 37.3, 26.9, 16.0, 14.1, 14.0, 13.9. HRMS m/z calcd for C58H77FN8O12S 1128.54, found 1129.5445 [M + H+].
(2S,4R)-1-((S)-2-(1-Cyanocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-N-(2-((14-(4-(2,6-dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)-3,6,9,12-tetraoxatetradecyl)oxy)-4-(4-methylthiazol-5-yl)benzyl)-4-hydroxypyrrolidine-2-carboxamide (30)
A mixture of 43 (1 equiv), osmium tetroxide 4% in H2O (0.2 equiv), sodium periodate (4 equiv), pyridine (2 equiv) in dioxane/H2O (3:1) was stirred at rt for 48 h. Then H2O was added to the reaction and the product was extracted with DCM. The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness. Then, following general method A, from the aldehyde derivative and 1c compound 30 was obtained after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of ammonia as white powder. Yield: 9 mg, 39%. 1H NMR (400 MHz, MeOD) δ: 9.55 (d, J = 0.7 Hz, 1H), 8.89 (s, 1H), 8.70 (d, J = 5.9 Hz, 1H), 7.74 (s, 1H), 7.64 (dd, J = 0.7, 5.8 Hz, 1H), 7.49 (d, J = 7.7 Hz, 1H), 7.09–7.02 (m, 2H), 6.76 (s, 2H), 4.69–4.63 (m, 2H), 4.55–4.42 (m, 3H), 4.34–4.23 (m, 2H), 3.95 (t, J = 4.5 Hz, 2H), 3.89 (s, 6H), 3.84–3.75 (m, 6H), 3.73–3.70 (m, 5H), 3.69–3.58 (m, 9H), 2.68–2.56 (m, 10H), 2.52 (s, 3H), 2.32–2.22 (m, 1H), 2.17–2.09 (m, 1H), 1.71–1.55 (m, 4H), 1.06 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.1, 171.5, 167.2, 163.1, 161.0, 158.1, 152.8, 151.7, 151.2, 149.2, 143.7, 138.8, 137.2, 133.5, 133.0, 130.2, 128.4, 122.8, 121.7, 120.7, 119.5, 119.3, 113.9, 113.7, 106.5, 71.8, 71.6, 71.6, 71.4, 71.1, 70.9, 69.7, 69.4, 60.8, 59.5, 58.7, 58.1, 56.3, 54.2, 53.4, 39.6, 38.9, 37.4, 37.3, 26.8, 18.1, 18.0, 16.0, 14.9. HRMS m/z calcd for C59H77N9O12S 1035.54, found 1036.5169 [M + H+].
14-(4-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)-N-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)-3,6,9,12-tetraoxatetradecanamide (31)
A mixture of 45 (1 equiv), osmium tetroxide 4% in H2O (0.2 equiv), sodium periodate (4 equiv), pyridine (2 equiv) in dioxane/H2O (3:1) was stirred at rt for 48 h. Then H2O was added to the reaction and the product was extracted with DCM. The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness. Then, following general method A, from the aldehyde derivative and 1c compound 31 was obtained after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid as white powder. Yield: 3.7 mg, 17%. 1H NMR (400 MHz, MeOD) δ: 9.55 (d, J = 0.7 Hz, 1H), 8.90 (s, 1H), 8.70 (d, J = 5.8 Hz, 1H), 7.76 (s, 1H), 7.64 (dd, J = 0.6, 5.8 Hz, 1H), 7.52 (d, J = 7.7 Hz, 1H), 7.08–7.01 (m, 2H), 6.81 (s, 2H), 4.65–4.60 (m, 2H), 4.53–4.41 (m, 3H), 4.27–4.25 (m, 2H), 4.02–3.61 (m, 29H), 2.96–2.76 (m, 10H), 2.51 (s, 3H), 2.27–2.21 (m, 1H), 2.15–2.08 (m, 1H), 2.04 (s, 3H), 1.06 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.4, 173.1, 172.3, 163.0, 161.0, 158.0, 152.8, 151.8, 151.3, 151.2, 149.1, 143.5, 139.0, 133.5, 132.8, 130.0, 128.4, 122.8, 121.7, 119.2, 119.1, 113.7, 106.6, 71.9, 71.6, 71.5, 71.4, 71.1, 70.9, 69.4, 60.7, 59.2, 58.1, 58.0, 56.6, 52.8, 49.8, 39.4, 38.9, 37.4, 36.5, 27.0, 22.3, 16.0. HRMS m/z calcd for C56H76N8O12S 1084.53, found 1085.5658 [M + H+].
2-(2-(Hex-5-en-1-yloxy)ethoxy)ethan-1-ol (33)
To a suspension of NaH (2.5 equiv) in DMF (1 M) and THF (1 M), diethylene glycol (5 equiv) was added at 0 °C under nitrogen. After 45 min 6-bromo-1-hexene (1 equiv) was added dropwise at 0 °C. Then the ice bath was removed, and the reaction mixture was stirred overnight at rt under nitrogen. Distillate water was added, and the reaction mixture was acidified with HCl 1 M up to pH 2. The product was extracted with CHCl3. The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness. The resulting oil was purified by flash column chromatography using a gradient from 50% to 100% of ethyl acetate in heptane to obtain the desired compound 33 as an oil. Yield: 552 mg, 49%. 1H NMR (400 MHz, CDCl3) δ: 5.85–5.74 (m, 1H), 5.03–4.96 (m, 1H), 4.96–4.91 (m, 1H), 3.75–3.56 (m, 8H), 3.46 (t, J = 6.7 Hz, 2H), 2.09–2.03 (m, 2H), 1.65–1.56 (m, 2H), 1.48–1.39 (m, 2H). 13C NMR (101 MHz, CDCl3) δ: 138.8, 114.7, 72.6, 71.5, 70.6, 70.3, 62.0, 33.6, 29.2, 25.5.
2-(2-(Hex-5-en-1-yloxy)ethoxy)acetic Acid (34)
A mixture of 30 (1 equiv), BAIB (2.2 equiv), TEMPO (0.22 equiv) in ACN/H2O (1:1) (0.5 M) was stirred overnight at rt. The day after the solvent was evaporated, the crude was resuspended in DCM and washed with H2O. The organic phases were combined, dried over MgSO4, filtered, and evaporated to dryness. The resulting product was purified by flash column chromatography using a gradient from 0% to 20% of MeOH in DCM to obtain compound 34 as an oil. Yield: 472 mg, 81%. 1H NMR (400 MHz, CDCl3) δ: 5.84–5.73 (m, 1H), 5.03–4.97 (m, 1H), 4.96–4.92 (m, 1H), 4.15 (s, 2H), 3.76–3.73 (m, 2H), 3.63–3.59 (m, 2H), 3.52 (t, J = 6.6 Hz, 2H), 2.10–2.03 (m, 2H), 1.65–1.57 (m, 2H), 1.49–1.40 (m, 2H). 13C NMR (101 MHz, CDCl3) δ: 173.2, 138.7, 114.8, 71.7, 71.6, 69.7, 69.1, 33.6, 28.9, 25.4.
(2R,4S)-1-((R)-2-(2-(2-(Hex-5-en-1-yloxy)ethoxy)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (35)
Following general method B, compound 35 was obtained from compounds 34 and 2a (synthesized accordingly to literature61). The crude was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid to obtain 35 as white powder. Yield: 14.7 mg, 30%. 1H NMR (400 MHz, CDCl3) δ: 8.64 (s, 1H), 7.28–7.23 (m, 4H), 5.74–5.63 (m, 1H), 4.93–4.82 (m, 2H), 4.65 (t, J = 7.9 Hz, 1H), 4.49–4.21 (m, 4H), 4.03–3.85 (m, 3H), 3.59–3.47 (m, 5H), 3.40–3.34 (m, 2H), 2.47–2.39 (m, 4H), 2.06–1.91 (m, 3H), 1.54–1.45 (m, 2H), 1.37–1.29 (m, 2H), 0.87 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 171.6, 170.8, 150.7, 148.1, 138.7, 138.4, 132.0, 130.7, 129.6, 128.3, 114.8, 71.5, 71.4, 70.4, 70.3, 69.8, 58.6, 57.5, 56.7, 43.3, 36.0, 34.9, 33.6, 29.1, 26.5, 25.4, 16.0. MS m/z calcd for C32H46N4O6S 614.31, found 615.4 [M + H+].
2-(2-(Allyloxy)ethoxy)ethan-1-ol (38)
To a mixture of NaOH (1 equiv) in dioxane (0.3 M), diethylene glycol (2 equiv) and allyl bromide (1 equiv) were added dropwise. The mixture was heated overnight at 55 °C. Then the reaction mixture was dried over MgSO4, filtered, and evaporated in vacuum. The crude was purified by flash column chromatography using a gradient from 50% to 100% of ethyl acetate in heptane to obtain compound 38 as an oil. Yield: 329 mg, 54%. 1H NMR (400 MHz, CDCl3) δ: 5.90–5.80 (m, 1H), 5.21 (qd, J = 1.6, 17.2 Hz, 1H), 5.14–5.10 (m, 1H), 3.96 (td, J = 1.4, 5.7 Hz, 2H), 3.68–3.59 (m, 4H), 3.56–3.52 (m, 4H). 13C NMR (101 MHz, CDCl3) δ: 134.5, 117.3, 72.6, 72.2, 70.4, 69.4, 61.7.
3,6,9,12-Tetraoxapentadec-14-en-1-ol (39)
Following the same procedure applied for compound 38, from tetraethylene glycol and allyl bromide compound 39 was obtained as oil. Yield: 3.29 g, 68%. Analytical data matched those previously reported.75
(2S,4R)-N-(2-(2-(2-(Allyloxy)ethoxy)ethoxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (40)
Following general method E, compound 40 was obtained from compounds 38 and 2d (synthesis detailed in the Supporting Information). The crude was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid. Yield: 21.7 mg, 42%. 1H NMR (500 MHz, MeOD) δ: 8.91 (s, 1H), 8.52 (t, J = 5.9 Hz, 1H), 7.55 (dd, J = 3.4, 9.4 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.10 (d, J = 1.6 Hz, 1H), 7.06 (dd, J = 1.6, 7.8 Hz, 1H), 5.98–5.90 (m, 1H), 5.30 (ddt, J = 1.7, 6.9, 8.6 Hz, 1H), 5.18 (ddt, J = 1.4, 4.5, 5.2 Hz, 1H), 4.79 (d, J = 9.6 Hz, 1H), 4.66 (t, J = 8.3 Hz, 1H), 4.54–4.43 (m, 3H), 4.31–4.26 (m, 2H), 4.07–4.04 (m, 2H), 3.97–3.94 (m, 2H), 3.90–3.82 (m, 2H), 3.80–3.76 (m, 2H), 3.69–3.66 (m, 2H), 2.53 (s, 3H), 2.29–2.24 (m, 1H), 2.17–2.10 (m, 1H), 1.42–1.30 (m, 4H), 1.07 (s, 10H). MS m/z calcd for C33H45FN4O7S 660.30, found 661.3 [M + H+].
(2S,4R)-N-(2-((3,6,9,12-Tetraoxapentadec-14-en-1-yl)oxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (41)
Following general method E, compound 41 was obtained from compounds 39 and 2d (synthesis detailed in the Supporting Information). The crude was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid. Yield: 26.9 mg, 46%. 1H NMR (500 MHz, MeOD) δ: 8.91 (s, 1H), 8.51 (t, J = 6.1 Hz, 1H), 7.56 (dd, J = 3.2, 9.6 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.10 (d, J = 1.6 Hz, 1H), 7.06 (dd, J = 1.7, 7.7 Hz, 1H), 5.97–5.89 (m, 1H), 5.30 (ddt, J = 1.7, 6.9, 8.7 Hz, 1H), 5.19–5.16 (m, 1H), 4.79 (d, J = 9.1 Hz, 1H), 4.66 (t, J = 8.2 Hz, 1H), 4.56–4.43 (m, 3H), 4.30–4.26 (m, 2H), 4.04–4.02 (m, 2H), 3.97–3.94 (m, 2H), 3.90–3.82 (m, 2H), 3.80–3.77 (m, 2H), 3.73–3.70 (m, 2H), 3.69–3.64 (m, 7H), 3.62–3.59 (m, 2H), 2.54 (s, 3H), 2.30–2.25 (m, 1H), 2.16–2.11 (m, 1H), 1.42–1.31 (m, 4H), 1.07 (s, 9H). MS m/z calcd for C37H53FN4O9S 748.35, found 749.4 [M + H+].
(2S,4R)-N-(2-(2-(2-(Allyloxy)ethoxy)ethoxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-cyanocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (42)
Following general method E, compound 42 was obtained from compounds 38 and 2c (synthesized as previously reported29). The crude was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid. Yield: 15.8 mg, 61%. 1H NMR (500 MHz, MeOD) δ: 8.91 (s, 1H), 8.50 (t, J = 5.9 Hz, 1H), 7.50 (d, J = 7.9 Hz, 1H), 7.10 (d, J = 1.8 Hz, 1H), 7.06 (dd, J = 1.7, 8.0 Hz, 1H), 5.98–5.90 (m, 1H), 5.30 (ddt, J = 1.8, 6.9, 8.6 Hz, 1H), 5.20–5.16 (m, 1H), 4.71–4.64 (m, 2H), 4.54–4.44 (m, 4H), 4.31–4.27 (m, 2H), 4.06 (td, J = 1.4, 5.6 Hz, 2H), 3.97–3.95 (m, 2H), 3.87–3.78 (m, 5H), 3.69–3.66 (m, 3H), 2.53 (s, 4H), 2.28–2.23 (m, 1H), 2.16–2.10 (m, 1H), 1.71–1.56 (m, 4H), 1.06 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.3, 171.6, 167.3, 158.2, 152.9, 149.2, 136.2, 133.6, 133.0, 130.2, 128.5, 122.9, 120.8, 117.4, 113.9, 73.2, 71.9, 71.1, 71.0, 70.7, 69.5, 60.9, 59.7, 59.6, 39.8, 39.0, 37.4, 26.9, 18.2, 18.1, 16.0, 15.0, 14.9. MS m/z calcd for C34H45N5O7S 667.30, found 668.4 [M + H+].
(2S,4R)-N-(2-((3,6,9,12-Tetraoxapentadec-14-en-1-yl)oxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-cyanocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (43)
Following general method E, compound 43 was obtained from compounds 39 and 2c (synthesized as previously reported29). The crude was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid. Yield: 15.3 mg, 52%. 1H NMR (500 MHz, MeOD) δ: 8.91 (s, 1H), 8.49 (t, J = 6.1 Hz, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.10 (d, J = 1.6 Hz, 1H), 7.06 (dd, J = 1.6, 7.7 Hz, 1H), 5.97–5.89 (m, 1H), 5.30 (ddt, J = 1.6, 6.9, 8.6 Hz, 1H), 5.19–5.16 (m, 1H), 4.71–4.64 (m, 2H), 4.55–4.43 (m, 3H), 4.31–4.26 (m, 2H), 4.03 (td, J = 1.4, 5.6 Hz, 2H), 3.97–3.94 (m, 2H), 3.87–3.77 (m, 4H), 3.73–3.71 (m, 2H), 3.69–3.64 (m, 7H), 3.62–3.59 (m, 2H), 2.54 (s, 3H), 2.30–2.25 (m, 1H), 2.16–2.10 (m, 1H), 1.71–1.56 (m, 4H), 1.06 (s, 9H). MS m/z calcd for C38H53N5O9S 755.36, found 756.5 [M + H+].
(2S,4R)-1-((S)-2-Acetamido-3,3-dimethylbutanoyl)-N-(2-(2-(2-(allyloxy)ethoxy)ethoxy)-4-(4-methylthiazol-5-yl)benzyl)-4-hydroxypyrrolidine-2-carboxamide (44)
Following general method E, compound 44 was obtained from compounds 38 and 2b (synthesized accordingly to literature29). The crude was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid. Yield: 12.7 mg, 53%. 1H NMR (400 MHz, MeOD) δ: 8.91 (s, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.08 (s, 1H), 7.04 (dd, J = 1.4, 7.8 Hz, 1H), 5.99–5.88 (m, 1H), 5.30 (dq, J = 1.7, 6.9 Hz, 1H), 5.18 (dq, J = 1.3, 4.5 Hz, 1H), 4.67–4.60 (m, 2H), 4.55–4.42 (m, 3H), 4.30–4.25 (m, 2H), 4.05 (dt, J = 1.4, 5.6 Hz, 2H), 3.97–3.90 (m, 3H), 3.85–3.77 (m, 3H), 3.68–3.65 (m, 2H), 2.53 (s, 3H), 2.28–2.20 (m, 1H), 2.16–2.07 (m, 1H), 2.03 (s, 3H), 1.06 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.4, 173.1, 172.3, 158.0, 152.8, 149.1, 136.1, 133.5, 132.8, 130.0, 128.4, 122.8, 117.3, 114.0, 113.8, 73.1, 71.8, 70.9, 70.6, 69.4, 60.7, 59.1, 57.9, 39.4, 38.9, 36.5, 27.0, 22.3, 15.9. MS m/z calcd for C31H44N4O7S 616.29, found 617.4 [M + H+].
(2S,4R)-N-(2-((3,6,9,12-Tetraoxapentadec-14-en-1-yl)oxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-acetamido-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (45)
Following general method E, compound 45 was obtained from compounds 39 and 2b (synthesized accordingly to literature29). The crude was purified by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid. Yield: 13.8 mg, 53%. 1H NMR (400 MHz, MeOD) δ: 8.92 (s, 1H), 7.52 (d, J = 7.9 Hz, 1H), 7.08 (s, 1H), 7.04 (dd, J = 1.5, 7.8 Hz, 1H), 6.01–5.88 (m, 1H), 5.29 (dq, J = 1.7, 6.9 Hz, 1H), 5.17 (dq, J = 1.3, 4.5 Hz, 1H), 4.67–4.60 (m, 2H), 4.56–4.39 (m, 2H), 4.29–4.25 (m, 2H), 4.03 (dt, J = 1.4, 5.7 Hz, 2H), 3.97–3.90 (m, 3H), 3.85–3.76 (m, 3H), 3.73–3.63 (m, 9H), 3.62–3.58 (m, 2H), 2.53 (s, 3H), 2.28–2.21 (m, 1H), 2.16–2.08 (m, 1H), 2.04 (s, 3H), 1.06 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.4, 173.1, 172.2, 158.0, 152.8, 149.1, 136.1, 133.5, 132.8, 130.0, 128.4, 122.8, 117.2, 113.7, 73.1, 71.8, 71.6, 71.5, 71.1, 70.8, 70.6, 69.4, 60.7, 59.1, 58.0, 39.4, 38.9, 36.5, 27.0, 22.3, 15.9. MS m/z calcd for C35H52N4O9S 704.35, found 705.4 [M + H+].
(2S,4R)-N-(2-(2-(2-(4-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)ethoxy)ethoxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (46)
Following general method C, from 69 (0.033 mmol) and 1c (0.033 mmol) compound 46 was obtained after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid as white powder. Yield: 5.3 mg, 16%. 1H NMR (400 MHz, MeOD) δ: 9.61 (s, 1H), 8.95 (s, 1H), 8.71 (d, J = 6.2 Hz, 1H), 7.97 (s, 1H), 7.85 (d, J = 6.0 Hz, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.10–7.07 (m, 2H), 6.90 (s, 2H), 4.78 (s, 1H), 4.66 (t, J = 8.5 Hz, 1H), 4.54–4.44 (m, 3H), 4.32–4.29 (m, 2H), 4.00–3.81 (m, 14H), 3.76 (s, 3H), 3.55 (s, 4H), 3.45 (s, 4H), 3.30–3.27 (m, 2H), 2.53 (s, 3H), 2.32–2.26 (m, 1H), 2.18–2.10 (m, 1H), 1.44–1.30 (m, 4H), 1.08 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.2, 171.8, 164.4, 162.2, 161.2, 157.8, 153.0, 149.0, 146.4, 142.3, 140.0, 133.5, 132.9, 129.9, 128.2, 122.9, 122.3, 120.8, 118.2, 113.5, 106.8, 80.3, 78.0, 71.0, 70.9, 69.1, 67.2, 60.9, 58.8, 58.7, 58.2, 57.5, 56.9, 50.7, 50.1, 39.4, 39.0, 37.6, 37.3, 26.9, 15.8, 14.1, 14.0, 14.0, 13.9. HRMS m/z calcd for C52H65FN8O9S 996.46, found 997.4810 [M + H+].
(2S,4R)-N-(2-(2-(2-(4-(2,5-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)ethoxy)ethoxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (47)
Following general method C, from 69 (0.033 mmol) and 1d (synthesis detailed in the Supporting Information) (0.033 mmol) compound 47 was obtained after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid as white powder. Yield: 5.3 mg, 5%. 1H NMR (400 MHz, MeOD) δ: 9.52 (s, 1H), 8.91 (s, 1H), 8.62 (d, J = 5.7 Hz, 1H), 7.65 (s, 1H), 7.51 (d, J = 7.9 Hz, 1H), 7.23–7.19 (m, 2H), 7.10 (d, J = 1.2 Hz, 1H), 7.06 (dd, J = 1.6, 7.7 Hz, 1H), 6.98 (s, 1H), 4.78 (s, 1H), 4.66 (t, J = 8.5 Hz, 1H), 4.55–4.45 (m, 3H), 4.32–4.28 (m, 2H), 3.95–3.89 (m, 2H), 3.88–3.80 (m, 7H), 3.76–3.71 (m, 8H), 2.78–2.71 (m, 10H), 2.53 (s, 3H), 2.30–2.24 (m, 1H), 2.17–2.11 (m, 1H), 1.42–1.31 (m, 4H), 1.07 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.1, 171.7, 163.3, 158.0, 153.7, 152.8, 151.4, 150.6, 144.2, 139.2, 133.5, 133.0, 128.4, 124.1, 122.8, 121.5, 120.3, 115.8, 113.9, 80.3, 71.0, 70.8, 69.8, 69.3, 60.8, 58.7, 58.2, 56.7, 56.5, 54.3, 53.5, 39.4, 38.9, 37.4, 37.3, 26.9, 15.9, 14.1, 14.0, 13.9. HRMS m/z calcd for C52H65FN8O9S 996.46, found 997.4712 [M + H+].
(2S,4R)-N-(2-((5-(4-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)pentyl)oxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (51, VZ185)
Following general method C, from 70 (0.02 mmol) and 1c (0.02 mmol) compound 51 was obtained after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid as white powder. Yield: 4.8 mg, 24%. 1H NMR (400 MHz, MeOD) δ: 9.56 (d, J = 0.8 Hz, 1H), 8.92 (s, 1H), 8.71 (d, J = 5.8 Hz, 1H), 7.78 (s, 1H), 7.65 (dd, J = 0.7, 5.8 Hz, 1H), 7.52 (d, J = 7.7 Hz, 1H), 7.07–7.03 (m, 2H), 6.86 (s, 2H), 4.79 (s, 1H), 4.67 (t, J = 12.4 Hz, 1H), 4.55–4.47 (m, 3H), 4.18 (s, 2H), 4.14 (t, J = 6.0 Hz, 2H), 3.96–3.82 (m, 8H), 3.74 (s, 3H), 3.18–2.78 (m, 10H), 2.53 (s, 3H), 2.32–2.25 (m, 1H), 2.19–2.12 (m, 1H), 1.98–1.91 (m, 2H), 1.79–1.71 (m, 2H), 1.68–1.61 (m, 2H), 1.42–1.29 (m, 4H), 1.08 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.2, 171.8, 171.4, 163.1, 161.0, 158.0, 152.8, 151.7, 151.2, 149.1, 143.6, 138.8, 133.6, 132.8, 129.7, 128.0, 122.5, 121.7, 119.2, 113.1, 106.6, 80.3, 78.0, 71.0, 69.2, 60.8, 59.2, 58.7, 58.1, 56.4, 53.1, 49.8, 39.3, 38.9, 37.4, 37.3, 30.1, 26.9, 25.1, 15.9, 14.1, 14.0, 13.8. HRMS m/z calcd for C53H67FN8O8S 994.48, found 995.5024 [M + H+].
(2S,4R)-N-(2-((5-(4-(2,5-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)piperazin-1-yl)pentyl)oxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (52)
Following general method C, from 70 (0.02 mmol) and 1d (0.02 mmol) compound 52 was obtained after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid as white powder. Yield: 4 mg, 20%. 1H NMR (400 MHz, MeOD) δ: 9.53 (s, 1H), 8.92 (s, 1H), 8.62 (d, J = 5.7 Hz, 1H), 7.65 (s, 1H), 7.53 (d, J = 7.4 Hz, 1H), 7.23–7.19 (m, 2H), 7.05 (s, 2H), 7.02 (s, 1H), 4.79 (s, 1H), 4.68 (t, J = 8.4 Hz, 1H), 4.55–4.47 (m, 3H), 4.15 (t, J = 5.9 Hz, 2H), 3.92–3.82 (m, 7H), 3.75 (s, 3H), 3.72 (s, 3H), 3.18–2.91 (m, 10H), 2.53 (s, 3H), 2.32–2.26 (m, 1H), 2.19–2.12 (m, 1H), 2.00–1.93 (m, 2H), 1.87–1.78 (m, 2H), 1.70–1.62 (m, 2H), 1.43–1.30 (m, 4H), 1.08 (s, 9H). 13C NMR (101 MHz, MeOD) δ:174.2, 171.8, 163.3, 157.9, 153.7, 152.8, 151.4, 150.5, 149.2, 144.1, 139.3, 133.6, 132.9, 129.6, 127.9, 122.6, 121.5, 120.1, 116.0, 115.6, 113.1, 80.4, 78.1, 71.0, 69.0, 60.9, 58.7, 58.4, 58.2, 56.8, 56.5, 56.2, 53.1, 51.8, 39.3, 38.9, 37.4, 37.3, 29.8, 26.9, 24.7, 15.9, 14.1, 14.0, 13.9. HRMS m/z calcd for C53H67FN8O8S 994.48, found 995.4771 [M + H+].
(2S,4R)-N-(2-(2-(2-((1-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)azetidin-3-yl)amino)-2-oxoethoxy)ethoxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (55)
Following general method B, from the 71 (0.02 mmol) and 1e (0.02 mmol) compound 55 was obtained after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of ammonia as white powder. Yield: 2.4 mg, 12%. 1H NMR (400 MHz, MeOD) δ: 9.56 (d, J = 0.6 Hz, 1H), 8.93 (s, 1H), 8.71 (d, J = 6.0 Hz, 1H), 7.80 (s, 1H), 7.63 (dd, J = 0.6, 5.8 Hz, 1H), 7.55 (d, J = 8.1 Hz, 1H), 7.12–7.08 (m, 2H), 6.89 (s, 2H), 4.80–4.64 (m, 3H), 4.60–4.31 (m, 11H), 4.23 (s, 2H), 4.06 (t, J = 4.0 Hz, 2H), 3.98 (s, 6H), 3.91–3.80 (m, 2H), 3.74 (s, 3H), 2.53 (s, 3H), 2.31–2.25 (m, 1H), 2.18–2.10 (m, 1H), 1.42–1.30 (m, 4H), 1.07 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.3, 171.8, 166.7, 160.7, 157.8, 152.9, 151.8, 151.3, 149.2, 143.3, 140.4, 139.2, 133.4, 133.0, 130.0, 128.2, 123.0, 121.7, 119.1, 118.6, 113.4, 106.8, 71.5, 71.5, 71.0, 69.2, 60.9, 60.9, 58.7, 58.2, 56.9, 49.5, 39.4, 38.9, 37.5, 37.3, 29.5, 26.9, 15.9, 13.9. HRMS m/z calcd for C51H61FN8O10S 996.42, found 997.4452 [M + H+].
(2S,4R)-N-(2-(2-(2-((1-(2,5-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)azetidin-3-yl)amino)-2-oxoethoxy)ethoxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (56)
Following general method B, from 71 (0.02 mmol) and 1f (synthesis detailed in the Supporting Information) (0.02 mmol) compound 56 was obtained after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of ammonia as white powder. Yield: 3.3 mg, 17%. 1H NMR (400 MHz, MeOD) δ: 9.39 (s, 1H), 8.80 (s, 1H), 8.49 (d, J = 5.7 Hz, 1H), 7.54 (s, 1H), 7.42 (d, J = 8.2 Hz, 1H), 7.10 (s, 1H), 7.04 (d, J = 5.6 Hz, 1H), 6.99 (s, 1H), 6.98–6.94 (m, 2H), 4.66 (s, 1H), 4.60–4.51 (m, 2H), 4.45–4.32 (m, 5H), 4.27–4.15 (m, 6H), 4.10 (s, 2H), 3.93 (t, J = 4.1 Hz, 2H), 3.81 (s, 3H), 3.78–3.67 (m, 2H), 3.63 (s, 3H), 3.58 (s, 3H), 2.40 (s, 3H), 2.19–2.10 (m, 1H), 2.05–1.98 (m, 1H), 1.30–1.16 (m, 4H), 0.94 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.2, 173.2, 171.8, 167.1, 166.6, 163.2, 157.8, 153.4, 153.0, 152.9, 151.4, 150.6, 149.2, 143.8, 139.5, 133.4, 133.0, 130.0, 128.2, 127.1, 123.0, 121.5, 120.9, 120.0, 116.1, 115.6, 113.4, 80.3, 78.0, 71.5, 71.5, 71.0, 69.2, 69.1, 60.9, 58.7, 58.2, 56.8, 56.6, 55.9, 49.5, 49.5, 40.7, 39.4, 38.9, 37.4, 37.3, 26.9, 15.9, 14.0. HRMS m/z calcd for C51H61FN8O10S 996.42, found 997.4435 [M + H+].
(2S,4R)-N-(2-((5-((1-(2,6-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)azetidin-3-yl)amino)-5-oxopentyl)oxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (61)
Following general method B, from 72 (0.025 mmol) and 1e (0.025 mmol) compound 61 was obtained after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid as white powder. Yield: 4.5 mg, 18%. 1H NMR (400 MHz, MeOD) δ: 9.45 (d, J = 0.5 Hz, 1H), 8.81 (s, 1H), 8.61 (d, J = 5.7 Hz, 1H), 7.68 (s, 1H), 7.54 (d, J = 5.5 Hz, 1H), 7.41 (d, J = 7.6 Hz, 1H), 6.96–6.91 (m, 2H), 6.74 (s, 2H), 4.69 (s, 1H), 4.58 (t, J = 8.5 Hz, 1H), 4.54–4.32 (m, 5H), 4.24 (s, 2H), 4.11–4.01 (m, 4H), 3.85 (s, 6H), 3.81–3.70 (m, 3H), 3.63 (s, 3H), 2.42 (s, 3H), 2.28 (t, J = 6.5 Hz, 2H), 2.21–2.15 (m, 1H), 2.08–2.01 (m, 1H), 1.85–1.78 (m, 4H), 1.32–1.20 (m, 4H), 0.96 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 176.1, 174.3, 171.7, 163.0, 160.7, 157.9, 152.8, 151.7, 151.3, 149.1, 148.6, 143.4, 139.4, 139.1, 133.6, 132.8, 129.7, 128.2, 128.0, 122.5, 121.7, 120.9, 119.2, 118.9, 113.0, 106.7, 80.3, 78.0, 71.0, 68.8, 61.1, 60.8, 58.7, 58.2, 56.7, 56.1, 40.9, 39.3, 38.9, 37.4, 37.3, 36.4, 29.8, 29.5, 26.9, 23.4, 15.9, 14.1, 14.0, 13.9. HRMS m/z calcd for C52H63FN8O9S 994.44, found 995.4634 [M + H+].
(2S,4R)-N-(2-((5-((1-(2,5-Dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)azetidin-3-yl)amino)-5-oxopentyl)oxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (62)
Following general method B, from 72 (0.025 mmol) and 1f (0.025 mmol) compound 62 was obtained after purification by HPLC using a gradient of 5% to 95% v/v acetonitrile in 0.1% aqueous solution of formic acid as white powder. Yield: 5.8 mg, 23%. 1H NMR (400 MHz, MeOD) δ: 9.51 (d, J = 0.7 Hz, 1H), 8.91 (s, 1H), 8.62 (d, J = 6.0 Hz, 1H), 7.64 (s, 1H), 7.52 (d, J = 7.5 Hz, 1H), 7.19 (dd, J = 0.8, 5.8 Hz, 1H), 7.14 (s, 1H), 7.06–7.02 (m, 2H), 7.01 (s, 1H), 4.79 (s, 1H), 4.69 (t, J = 8.5 Hz, 1H), 4.58–4.43 (m, 4H), 4.13 (s, 2H), 4.01–3.81 (m, 10H), 3.74 (s, 3H), 3.71 (s, 3H), 3.47 (s, 1H), 2.52 (s, 3H), 2.36 (t, J = 6.5 Hz, 2H), 2.31–2.24 (m, 1H), 2.20–2.11 (m, 1H), 1.92 (t, J = 2.6 Hz, 4H), 1.46–1.29 (m, 4H), 1.08 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 175.8, 174.3, 171.7, 171.6, 171.4, 163.2, 157.9, 153.2, 152.8, 152.8, 151.3, 150.5, 149.1, 144.1, 139.3, 133.6, 132.8, 129.7, 128.0, 124.7, 122.5, 121.5, 120.2, 116.1, 115.6, 115.0, 113.0, 80.3, 78.0, 71.1, 68.8, 61.8, 60.8, 58.7, 58.2, 57.2, 56.6, 56.5, 41.4, 39.3, 38.9, 37.4, 37.3, 36.4, 29.8, 26.9, 23.5, 15.9, 14.1, 14.0, 13.9. HRMS m/z calcd for C52H63FN8O9S 994.44, found 995.4664 [M + H+].
(2S,4R)-N-(2-(2-(2,2-Diethoxyethoxy)ethoxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (67)
Following general method E, compound 67 was obtained after reaction between 2-(2,2-diethoxyethoxy)ethan-1-ol (synthesized accordingly to literature76) and 2d as white powder. Yield: 92 mg, 87%. 1H NMR (400 MHz, CDCl3) δ: 8.66 (s, 1H), 7.31 (d, J = 7.7 Hz, 1H), 6.95 (dd, J = 1.2, 7.7 Hz, 1H), 6.88 (s, 1H), 4.66 (t, J = 7.6 Hz, 2H), 4.55–4.40 (m, 4H), 4.23–4.13 (m, 2H), 3.97–3.87 (m, 3H), 3.71–3.49 (m, 7H), 2.50 (s, 3H), 2.44–2.34 (m, 1H), 2.13–2.05 (m, 1H), 1.35–1.26 (m, 4H), 1.18 (t, J = 7.1 Hz, 6H), 0.93 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 170.8, 170.3, 170.1, 156.9, 150.4, 148.6, 132.4, 131.8, 129.8, 126.9, 122.1, 112.9, 101.0, 79.5, 72.0, 70.3, 70.1, 68.0, 62.5, 62.3, 58.7, 57.5, 56.7, 39.1, 36.5, 35.6, 26.4, 16.2, 15.4, 13.9, 13.8, 13.7. MS m/z calcd for C34H49FN4O8S 692.33, found 693.3 [M + H+].
(2S,4R)-N-(2-((5,5-Dimethoxypentyl)oxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-2-(1-fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamide (68)
A mixture of 2d (0.075 mmol, 1 equiv), 5-bromo-1,1-dimethoxypentane 66 (synthesized accordingly to literature77) (0.112 mmol, 1.8 equiv), and K2CO3 (0.225 mmol, 3 equiv) in DMF (0.5 mL) was stirred at 70 °C overnight. The reaction mixture was diluted with water and extracted with DCM, dried over MgSO4, filtered and evaporated to dryness. The crude was purified by flash column chromatography using a gradient from 0% to 20% of methanol in DCM to obtain the desired compound as white powder. Yield: 23 mg, 55%. 1H NMR (400 MHz, MeOD) δ: 8.91 (s, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.05 (d, J = 1.5 Hz, 1H), 7.03 (s, 1H), 4.80 (d, J = 8.6 Hz, 1H), 4.68 (t, J = 8.3 Hz, 1H), 4.57–4.42 (m, 4H), 4.11 (t, J = 6.2 Hz, 2H), 3.81–3.62 (m, 2H), 3.36 (s, 6H), 2.53 (s, 3H), 2.31–2.25 (m, 1H), 2.21–2.14 (m, 1H), 1.95–1.88 (m, 2H), 1.76–1.68 (m, 2H), 1.67–1.58 (m, 2H), 1.38–1.29 (m, 4H), 1.08 (s, 9H). 13C NMR (101 MHz, MeOD) δ: 174.2, 171.7, 171.4, 158.0, 152.8, 149.1, 133.6, 132.8, 129.6, 127.9, 122.4, 113.0, 106.2, 80.3, 71.1, 69.2, 60.8, 58.1, 55.8, 53.5, 43.8, 39.3, 38.9, 37.3, 33.4, 30.1, 26.9, 22.3, 15.9, 14.1, 14.0, 13.9, 13.2. MS m/z calcd for C33H47FN4O7S 662.31, found 663.5 [M + H+].
(2S,4R)-1-((S)-2-(1-Fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)-2-(2-(2-oxoethoxy)ethoxy)benzyl)pyrrolidine-2-carboxamide (69)
Following general method F, compound 69 was obtained from 67 (0.066 mmol) and directly used in the next step without any further purification. MS m/z calcd for C30H39FN4O7S 618.15, found 619.3 [M + H+].
(2S,4R)-1-((S)-2-(1-Fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)-2-((5-oxopentyl)oxy)benzyl)pyrrolidine-2-carboxamide (70)
Following general method F, compound 70 was obtained from 68 (0.04 mmol) and directly used in the next step without any further purification. MS m/z calcd for C31H41FN4O6S 616.27, found 617.3 [M + H+].
2-(2-(2-(((2S,4R)-1-((S)-2-(1-Fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamido)methyl)-5-(4-methylthiazol-5-yl)phenoxy)ethoxy)acetic Acid (71)
Following general method G, compound 71 was obtained from compound 69 (0.04 mmol) and was used in the next step without any further purification. Quantitative yield. MS m/z calcd for C30H39FN4O8S 634.25, found 635.3 [M + H+].
5-(2-(((2S,4R)-1-((S)-2-(1-Fluorocyclopropane-1-carboxamido)-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamido)methyl)-5-(4-methylthiazol-5-yl)phenoxy)pentanoic Acid (72)
Following general method G, compound 72 was obtained from compound 70 (0.05 mmol) and was used in the next step without any further purification. Quantitative yield. MS m/z calcd for C31H41FN4O7S 632.27, found 633.3 [M + H+].
Biology. Cell Culture
Human cell lines HeLa and Hek293 were purchased from ATCC and propagated in DMEM medium (Gibco), supplemented with 10% fetal bovine serum (FBS) (Gibco), l-glutamine (Gibco), 100 μg mL–1 of penicillin/streptomycin. RI-1 cells, purchased from DSMZ, were propagated in RPMI medium (Gibco), supplemented with 10% fetal bovine serum (FBS) (Gibco), l-glutamine, 100 μg mL–1 of penicillin/streptomycin. Cells were kept at 37 °C and 5% of CO2. EOL-1 and A-204 cell lines were purchased from ATCC and DSMZ. EOL-1 cells were grown in RPMI supplemented with 10% fetal calf serum. A-204 cells were grown in McCoy’s medium supplemented with 10% fetal calf serum. All cell lines were routinely tested for mycoplasma contamination using MycoAlert kit from Lonza.
Testing Compounds in Cells
HeLa and Hek293 cells were seeded at either 0.3 × 106 or 0.5 × 106 cells per well in 6-well plate in 2 mL of medium. At 80% confluency, cells were treated with compounds at the desired concentration, with final DMSO concentration of 0.1% v/v. Cells were incubated at 37 °C and 5% of CO2 for the desired time before harvesting. For protein extraction from adherent cells, cells were washed twice with DPBS (Gibco) and lysed with RIPA buffer (Sigma-Aldrich), supplemented with cOmplete Mini EDTA-free protease inhibitor cocktail (Roche). Insoluble material was removed by centrifugation at 14 000 rpm for 15 min at 4 °C. Supernatant was collected and protein concentration was quantified by Bradford assay (Thermo Scientific no. 23200, mean of two replicates). For RI-1 cells, (1–1.5) × 106 cells/mL were seeded in 6-well plate in 2 mL of medium. The day after, cells were treated with compounds at the desired concentration, with final DMSO concentration of 0.1% v/v. Before harvesting, cells were incubated at 37 °C and 5% CO2 for the desired time. Cells were washed twice with DPBS (Gibco) and lysed with RIPA buffer (Sigma-Aldrich), supplemented with cOmplete Mini EDTA-free protease inhibitor cocktail (Roche). Lysates were sonicated (10 s) and centrifuged at 14 000 rpm for 20 min at 4 °C. Supernatant was collected, and protein concentration was quantified by Bradford assay (mean of two replicates). For EOL-1 and A-204 cell line, 20 000 cells were seeded in 300 μL per well in a 24-well plate and incubated at 37 °C overnight. Compounds were added from DMSO stock solution using a digital dispenser D300 (Tecan), normalizing for added DMSO, and cells were incubated at 37 °C for 18 h. Medium was removed, and cells were washed with PBS and lysed in 80 μL of lysis buffer (1% Triton, 350 mM KCl, 10 mM Tris, pH 7,4, phosphatase-protease inhibitor cocktail (Thermo Scientific no. 1861281), 10 mM DTT, Benzonase 0.5 μL/ml (Novagen no. 70746 10KU, 25 U/μL)) for 30 min on ice before insoluble debris was pelleted by centrifugation.
Small Interfering RNA
HeLa cells were seeded at 0.3 × 106 cells per well in 6-well plates in 2 mL of medium, with a goal to achieve 70% of confluence on the day of transfection. BRD7, BRD9, and CRBN targeting siRNA (L-020297-00-0005 ON-TARGETplus human BRD7, L-014250-02-0005 ON-TARGETplus human BRD9, and L-021086-00-0005 ON-TARGETplus human CRBN SMARTpool, 5 nmol, Dharmacon) were prepared as a 20 μM solution in RNase-free 1× siRNA buffer (Dharmacon). Nontargeting siRNA (On-TARGETplus control pool, nontargeting pool D-001810-10-05, 5 nmol, Dharmacon) was used as negative control. Medium was replaced on the day of transfection. siRNA solution (5 μL) of BRD7, BRD9, or CRBN targeting siRNA, negative control, and vehicle control (1× siRNA buffer) were added to 250 μL of Opti-mem (Gibco), prepared in duplicate. Lipofectamine RNAiMax (5 μL, Thermo Fisher Scientific) was added to 250 μL of Opti-mem in another tube, also in duplicate. The two solutions were combined, incubated for 20 min at rt, and added to the wells. Plates were incubated at 37 °C and 5% CO2 for 48 h before harvesting as described above.
Mechanistic Evaluation
Cells were seeded in 6-well plates with 5 × 105 cells per well in 2 mL of complete medium to aim for 80% confluence on the day of treatment. At time zero, ML4924 was added at final concentration of 3 μM with 0.1% v/v of DMSO. DMSO was added to the remaining wells in order to match concentration of vehicle in all wells. After 3 h, MG132 (50 μM) or VH298 (100 μM) was added to the designated wells at a final concentration of 0.1% v/v of DMSO. Again, DMSO was added to the remaining wells to match vehicle concentration. At t = 3.5 h, VZ185 or cisVZ185 was added at 100 nM and 0.1% v/v of DMSO, matching the DMSO concentration in the remaining wells (final concentration 0.3% v/v). Plates were incubated for an additional 4 h at 37 °C and 5% CO2 before harvesting as described before. The experiment was performed in duplicate.
Immunoblotting
Proteins were resolved by SDS–PAGE on NuPage 4–12% Bis-Tris Midi Gel (Invitrogen) and transferred to Amersham Protran 0.45 NC nitrocellulose membrane (GE Healthcare) using wet transfer. The membrane was blocked with 5% w/v milk in Tris-buffered saline (TBS) with 0.1% w/v Tween-20. The following primary antibodies at the given concentration were used: anti β-actin (Cell Signaling Technology, 4970S, 13E5) 1:2000, anti-Brd9 (Bethyl A303-781A) 1:1000, anti-Brd7 (Bethyl A302-304A) 1:1000, VHL rabbit Ab (Cell Signaling Technology, no. 68547S) 1:1000, anti-CRBN (Novus Bio, NBP 1-9-1810) 1:1000, anti-actin hFAB rhodamine antibody, Bio-Rad, no. 12004164. Following incubation with horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology) or IRDye secondary antibody (Licor), bands were developed using Amersham ECL Prime Western blotting detection kit on Amersham Hyperfilm ECL film (Amersham) or ChemiDoc imaging system (Bio-Rad). ImageJ software was used for band quantification, and the last was reported as relative amount as ratio of each protein band relative to the lane’s loading control. Then the values obtained were normalized to 0.1% DMSO vehicle control. Alternatively, protein levels were determined on a WES capillary electrophoresis instrument (Proteinsimple) using a BRD9 antibody (Bethyl A303-781A) and a GAPDH antibody (Abcam no. ab9485) for normalization. DC50 and half-lives were determined by assuming a linear model between the two data points across 50% protein level mark.
Proliferation Assays
A-204 cells (1 × 103) were seeded per well of 384-well plates. After overnight incubation, compounds were added to the cells at logarithmic dose series using the HP digital dispenser D300 (Tecan), normalizing for added DMSO. 1 day and 8 days after seeding cellular ATP content was measured using CellTiterGlo (Promega). Measurements after 8 days were divided by the measurement after 1 day (i.e., the T0 plate) to derive fold proliferation. Data were analyzed with GraphPad Prism software to obtain EC50 values.
MS Proteomics
Sample Preparation
RI-1 cells were seeded at 2 × 106 cells/mL in a 10 cm plate 12 h before treatment. Cells were treated with 0.1% DMSO as vehicle control and with 100 nM VZ185 and 100 nM cisVZ185 as negative control. Cells were incubated at 37 °C and 5% CO2 for 4 h before harvesting. Cells were washed twice with DPBS (Gibco) and lysed with 0.5 mL of 100 mM Tris, pH 8.0, 4% (w/v) SDS, supplemented with cOmplete Mini EDTA-free protease inhibitor cocktail (Roche). Lysates were sonicated (2 × 10 s) and centrifuged at 14 000 rpm for 20 min at 4 °C. The supernatant fraction of the cell extract was collected, and protein concentration was quantified by BCA assay (Thermo Fisher Scientific). Further sample processing, digestion, desalting, TMT 10-plex isobaric labeling were performed as previously described.5 After labeling, the peptides from the 9 samples were pooled together in equal proportion. The pooled sample was fractionated into 20 discrete fractions using high pH reverse-phase chromatography on an XBridge peptide BEH column (130 Å, 3.5 μm, 2.1 mm × 150 mm, Waters) on an Ultimate 3000 HPLC system (Thermo Scientific/Dionex). A mixture of buffer A (10 mM ammonium formate in water, pH 9) and B (10 mM ammonium formate in 90% CH3CN, pH 9) was used over a linear gradient of 5% to 60% buffer B over 60 min at a flow rate of 200 μL/min. The peptides eluted from the column were collected in 80 fractions before concatenation into 20 fractions based on the UV signal of each fraction. All the fractions were dried in a Genevac EZ-2 concentrator and resuspended in 1% formic acid for MS analysis.
nLC–MS/MS Analysis
The fractions were analyzed sequentially on a Q Exactive HF hybrid quadrupole-Orbitrap mass spectrometer (Thermo Scientific) coupled to an UltiMate 3000 RSLCnano UHPLC system (Thermo Scientific) and EasySpray column (75 μm × 50 cm, PepMap RSLC C18 column, 2 μm, 100 Å, Thermo Scientific). A mix of buffer A (0.1% formic acid in H2O) and B (0.08% formic acid in 80% CH3CN) was used over a linear gradient from 5% to 35% buffer B over 125 min using a flow rate of 300 nL/min. Column temperature was set at 50 °C. The Q Exactive HF hybrid quadrupole-Orbitrap mass spectrometer was operated in data dependent mode with a single MS survey scan from 335 to 1600 m/z followed by 15 sequential m/z dependent MS2 scans. The 15 most intense precursor ions were sequentially fragmented by higher energy collision dissociation (HCD). The MS1 isolation window was set to 0.7 Da and the resolution set at 120 000. MS2 resolution was set at 60 000. The AGC targets for MS1 and MS2 were set at 3e6 ions and 1e5 ions, respectively. The normalized collision energy was set at 32%. The maximum ion injection times for MS1 and MS2 were set at 50 and 200 ms, respectively.
Peptide and Protein Identification and Quantification
The raw MS data files for all 20 fractions were merged and searched against the Uniprot-sprot-Human-Canonical database by Maxquant software 1.6.0.16 for protein identification and TMT reporter ion quantitation. The identifications were based on the following criteria: enzyme used trypsin/P; maximum number of missed cleavages equal to 2; precursor mass tolerance equal to 10 ppm; fragment mass tolerance equal to 20 ppm. Variable modifications: oxidation (M), acetyl (N-term), deamidation (NQ), Gln → pyro-Glu (Q N-term). Fixed modifications: carbamidomethyl (C). The data were filtered by applying a 1% false discovery rate followed by exclusion of proteins with less than two unique peptides. Quantified proteins were filtered if the absolute fold-change difference between the three DMSO replicates was ≥1.5.
Protein Expression and Purification
Human proteins VHL (UniProt accession number P40337), ElonginC (Q15369), and ElonginB (Q15370) and the bromodomain (residues 134–239) for BRD9 (Q9H8M2) were used for all protein expression. His6-tagged constructs were transformed into Escherichia coli BL21(DE3), and expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG). E. coli cells were lysed using a pressure cell homogenizer (Stansted Fluid Power); lysates were clarified by centrifuge and loaded onto a HisTrap FF affinity column (GE Healthcare). Following elution, His-tags were removed with TEV protease and samples were loaded onto a second Ni affinity column to obtain tag-free protein. Following dialysis in a low-salt buffer, BRD9 bromodomain and VCB complex proteins were further purified using cation exchange (Resource S; GE Healthcare) or anion exchange (Resource Q; GE Healthcare) chromatography, respectively. This was followed by a final polish by size-exclusion chromatography using a Superdex-75 16/600 column (GE Healthcare) equilibrated with 20 mM HEPES, pH 7.5, 100 mM sodium chloride, and 1 mM TCEP.
Crystallization and Structure Solution of BRD9 Bromodomain Binary Complex
The binary complex of Brd9-BD:5 was generated by incubating 500 μM Brd9 bromodomain with 750 μM compound 5 (from a 100 mM stock in DMSO). Crystals were grown using the hanging-drop vapor diffusion method by mixing equal volumes of binary complex solution and a crystallization solution containing 24% PEG 3350 and 0.2 M NH4F. Small needle crystals appeared within 48 h but took approximately 2 weeks to reach a suitable size for harvesting. Crystals were flash-frozen in liquid nitrogen using 20% ethylene glycol in liquor solution as a cryoprotectant. Diffraction data were collected at Diamond Light Source beamline I24 using a Pilatus 6M-F detector at a wavelength of 0.98962 Å. Data were indexed and integrated using XDS,78 and scaling and merging were performed with AIMLESS79 in CCP4i.80 The structure was solved by molecular replacement using MOLREP81 and a search model derived from a BRD9 bromodomain structure (PDB entry 5I40). The initial model underwent iterative rounds of model building and refinement with COOT82 and REFMAC5,83 respectively. Compound 5 geometry restraints for refinement were prepared with the PRODRG84 server. Model geometry and steric clashes were validated using the MOLPROBITY server;85 Ramachandran plots indicate that 97.7% of backbone torsion angles are in the favored region and there are no outliers. The structure has been deposited in the Protein Data Bank (PDB) with accession code 6HM0; data collection and refinement statistics are presented in Supporting Information Table S1.
AlphaLISA Proximity Assay
All assays were performed at room temperature in 384-well plates with a final assay volume of 25 μL per well, as described previously.5 All reagents were prepared as 5× stocks diluted in 50 mM HEPES, pH 7.5, 100 mM NaCl, 0.1% (w/v) bovine serum albumin, and 0.02% (w/v) 3-[(cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS). Plates were sealed by transparent film and briefly centrifuged at 100g between additions of reagents. Biotinylated VCB (100 nM final) and His6-BRD9-bromodomain (100 nM final) were incubated with a range of PROTAC concentrations (0.5–2000 nM; two-in-one serial dilution) for 1 h. Streptavidin-coated donor beads and nickel chelate acceptor beads (PerkinElmer) were added to a final concentration of 10 μg mL–1, and plates were incubated for another hour. Plates were read on a PHERAstar FS (BMG Labtech) using an optic module with an excitation wavelength of 680 nm and emission wavelength of 615 nm. Intensity values were plotted against PROTAC concentration on a log10 scale using GraphPad Prism, version 7. To obtain biotinylated-VCB, protein was mixed with EZ-link NHS-PEG4-biotin (Thermo Scientific) in a 1:1 molar ratio and incubated at room temperature for 1 h. The reaction was quenched using 1 M Tris-HCl, pH 7.5, and unreacted NHS-biotin was removed with a PD-10 MiniTrap desalting column (GE Healthcare) equilibrated with 20 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM DTT.
Fluorescence Polarization
All measurements were taken using a PHERAstar FS (BMG LABTECH) with fluorescence excitation and emission wavelengths (λ) of 485 and 520 nm, respectively. FP competitive binding assays were run in triplicate in 384-well plates (Corning 3575) using a total well volume of 15 μL (ref (63)). Each well solution contained 15 nM VCB protein, 10 nM FAM-labeled HIF-1α peptide (FAM-DEALAHypYIPMDDDFQLRSF, Kd = 3 nM as measured by a direct FP titration), and decreasing concentrations of PROTAC (14-point serial 2-fold dilutions starting from 50 μM PROTAC) or PROTAC:bromodomain (14-point serial 2-fold dilutions starting from 50 μM PROTAC:100 μM bromodomain into wells containing an additional 1 μM bromodomain) in 100 mM Bis-Tris propane, 100 mM NaCl, 1 mM TCEP, pH 7, with a final DMSO concentration of 1%. Control wells containing VCB and peptide with no compound (zero displacement), or peptide in the absence of protein (maximum displacement), were also included. Control values were used to obtain the percentage of displacement which was graphed against log[PROTAC]. Average IC50 values and the standard error of the mean (SEM) were determined for each titration using Prism 7. Dissociation constants Kd were back-calculated from the measured IC50 values using a displacement binding model, as described previously.68
Isothermal Titration Calorimetry
ITC experiments were performed in an ITC200 microcalorimeter (GE Healthcare) as described previously.5 Titrations were carried out at 25 °C while stirring at 750 rpm and were performed as reverse mode (protein in the syringe and the ligand in the cell). Compounds were diluted in ITC buffer (20 mM Bis-Tris propane, 100 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine (TCEP), pH 7.5) from 10 mM DMSO stock solutions to a final concentration of 20 μM (0.2% DMSO). Each run consisted of 19 injections of 2 μL of protein solution (ITC buffer with 0.2% DMSO) at a rate of 0.5 μL/s at 120 s time intervals. An initial injection of 0.4 μL was made and discarded during data analysis. BRD9 bromodomain (200 μM, in the syringe) was first titrated into the PROTAC (20 μM, in the cell); at the end of the titration, the excess of solution was removed from the cell and the syringe was washed and dried. VCB complex (168 μM, in the same buffer) was loaded in the syringe and titrated into the complex PROTAC-bromodomain. The concentration of the complex in the cell (C) after the first titration (16.8 μM), was calculated as follows:
where C0 is the initial concentration of the PROTAC in the cell (20 μM), Vcell is the volume of the sample cell (200.12 μL), and Vinj is the volume of titrant injected during the first titration (38.4 μL). Titrations for the binary complex PROTAC-VCB were performed by first adding buffer (38.4 μL) to the solution of PROTAC (20 μM, in the cell) by a single ITC injection. After removal of the excess solution from the cell, VCB complex (168 μM, in the same buffer) was loaded into the (washed and dried) syringe and titrated into the diluted PROTAC solution. The data were fitted to a single binding-site model using the MicroCal PEAQ-ITC analysis software provided by the manufacturer to obtain the stoichiometry n, the dissociation constant Kd, and the enthalpy of binding ΔH.
Live Cell Kinetic Analysis of BRD7 and BRD9 Degradation
HEK293 cells stably expressing LgBiT protein cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (Seradigm) and maintained at 37 °C and 5% CO2 were genome-edited using CRISPR/Cas9 to generate endogenous HiBiT-BRD7 or HiBiT-BRD9 fusions. Clonal populations were isolated, and cells were plated in 96-well tissue culture plates at a density of 2 × 104 cells per well in 100 μL of growth medium. Following overnight incubation at 37 °C and 5% CO2 medium was replaced with CO2-independent medium (Gibco) containing 20 μM Endurazine, an extended time-released substrate (Promega), and plates were incubated at 37 °C, 5% CO2, for 2.5 h before addition of a 3-fold serial dilution of 1 μM final concentration VZ185 compound. Plates retaining the plate lids were then read every 5 min for a period of 24 h on a GloMax Discover (Promega) set to 37 °C. Degradation traces for each concentration were plotted in GraphPad Prism, and the degradation portion of each curve was fitted to a one-component exponential decay model to obtain degradation parameters, rate, and Dmax, as previously described.69
NanoBRET Ubiquitination of BRD7 and BRD9
Clonal populations of edited HEK293 cells expressing HiBiT-BRD7 or HiBiT-BRD9 were plated in tissue 96-well tissue culture plates at a density of 2 × 104 cells per well in 100 μL of growth medium and incubated overnight at 37 °C, 5% CO2. Following treatment with 1 μM VZ185 for the indicated time frames, medium was replaced with Opti-MEM (Gibco) containing 200 μg/mL digitonin, 1:200 dilution of primary anti-Ub antibody (Enzo Life Sciences, BML-PW8810), 1:500 dilution of secondary anti-mouse Alexa 594 antibody (Cell Signaling Technologies, 8890), and 20 μM NanoGlo (Promega) substrate. Additional control wells received no antibodies (control for background NanoBRET) or no primary antibody (control for specificity). Plates were placed on an orbital shaker for 10 min, and NanoBRET measurements were collected on a CLARIOstar (BMG Labtech). Background subtracted NanoBRET ratios were generated by calculating the ratios of acceptor signal to donor signal in both the presence and absence of the Alexa594 antibody and computing the difference. Background subtracted BRET ratios were expressed in milliBRET units by multiplying by 1000.
Acknowledgments
We thank J. Wright and the staff of the MRC tissue culture facility for assistance with tissue culture; A. Atrih and D. Lamont for support with proteomics experiments; N. Trainor for assistance with proteomics data analysis; P. Fyfe for support with in-house X-ray facilities, and the Diamond Light Source for beamtime (BAG Proposal 14980) and beamline support at beamline I24; and Kevin Read and his team at the Dundee Drug Discovery Unit for in vitro DMPK data. This project has received funding from the European Research Council (ERC) under the European Union’s Seventh Framework Programme (FP7/2007–2013) as a Starting Grant to A.C. (Grant Agreement ERC-2012-StG-311460 DrugE3CRLs). Biophysics and drug discovery activities are supported by Wellcome Trust strategic awards to Dundee (Grants 100476/Z/12/Z and 094090/Z/10/Z, respectively). V.Z. was supported by the “Scuola di Dottorato in Scienze e Tecnologie della Chimica e dei Materiali” of the University of Genova, Italy.
Glossary
Abbreviations Used
- BET
bromodomain and extra-terminal
- BRD2/3/4/7/9
bromodomain-containing protein 2/3/4/7/9
- CRBN
cereblon
- DLBCL
diffuse large B cell lymphoma
- DIPEA
N,N-diisopropylethylamine
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- HOAt
1-hydroxy-7-azabenzotriazole solution
- MsCl
methanesulfonyl chloride
- PROTAC
proteolysis-targeting chimera
- NaHMDS
sodium bis(trimethylsilyl)amide
- NaH
sodium hydride
- TEMPO
2,2,6,6-tetramethyl-1-piperidinyloxy
- VCB
VHL-ElonginC-ElonginB complex
- VHL
von Hippel–Lindau
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01413.
Additional Western Blots and quantification of protein levels in HeLa, Hek293 and RI-1 cells; ITC titration curves and results; protein level profile of HiBiT-BRD2/3/4/7/9 and HiBiT-SMARCA4 after treatment with desired compounds; concentration-dependency evaluation of VZ185 and dBRD9 activity in EOL-1 and A-204 cells; AlphaLISA ternary complex formation for 5, 26, and VZ185 and cooperativity with VZ185 with FP; proteomic supplementary blots and graph for cisVZ185; physicochemical properties and in vitro PK data on compounds 26, 46, VZ185, and cisVZ185; original blot images; NMR spectra; HPLC traces of VZ185 and cisVZ185 (PDF)
Molecular formula strings (CSV)
Proteomics raw data (XLS)
Accession Codes
Atomic coordinates and structure factors for BRD9-BD:5 have been deposited to the Protein Data Bank (PDB) under accession number 6HM0. Authors will release the atomic coordinates and experimental data upon article publication.
Author Present Address
# C.M.: Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, United Kingdom.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): The Ciulli laboratory receives sponsored research support from Boehringer Ingelheim and Nurix Inc. A.C. is a scientific founder, director, and shareholder of Amphista Therapeutics, a company that is developing targeted protein degradation therapeutic platforms.
Supplementary Material
References
- Lai A. C.; Crews C. M. Induced Protein Degradation: An Emerging Drug Discovery Paradigm. Nat. Rev. Drug Discovery 2017, 16 (2), 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins I.; Wang H.; Caldwell J. J.; Chopra R. Chemical Approaches to Targeted Protein Degradation Through Modulation of the Ubiquitin–Proteasome Pathway. Biochem. J. 2017, 474 (7), 1127–1147. 10.1042/BCJ20160762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K. M.; Kim K. B.; Kumagai A.; Mercurio F.; Crews C. M.; Deshaies R. J. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (15), 8554–8559. 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottis P.; Crews C. M. Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol. 2017, 12 (4), 892–898. 10.1021/acschembio.6b01068. [DOI] [PubMed] [Google Scholar]
- Gadd M. S.; Testa A.; Lucas X.; Chan K.-H.; Chen W.; Lamont D. J.; Zengerle M.; Ciulli A. Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 2017, 13 (5), 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak R. P.; DeAngelo S. L.; Buckley D.; He Z.; Donovan K. A.; An J.; Safaee N.; Jedrychowski M. P.; Ponthier C. M.; Ishoey M.; Zhang T.; Mancias J. D.; Gray N. S.; Bradner J. E.; Fischer E. S. Plasticity in Binding Confers Selectivity in Ligand-Induced Protein Degradation. Nat. Chem. Biol. 2018, 14 (7), 706–714. 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes S. J.; Ciulli A. Molecular Recognition of Ternary Complexes: A New Dimension in the Structure-Guided Design of Chemical Degraders. Essays Biochem. 2017, 61 (5), 505–516. 10.1042/EBC20170041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 2015, 11 (8), 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham T. B.; Blanco M.-J. Target Engagement in Lead Generation. Bioorg. Med. Chem. Lett. 2015, 25 (5), 998–1008. 10.1016/j.bmcl.2014.12.076. [DOI] [PubMed] [Google Scholar]
- Zengerle M.; Chan K.-H.; Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10 (8), 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.-H.; Zengerle M.; Testa A.; Ciulli A. Impact of Target Warhead and Linkage Vector on Inducing Protein Degradation: Comparison of Bromodomain and Extra-Terminal (BET) Degraders Derived from Triazolodiazepine (JQ1) and Tetrahydroquinoline (I-BET726) BET Inhibitor Scaffolds. J. Med. Chem. 2018, 61 (2), 504–513. 10.1021/acs.jmedchem.6b01912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Smith B. E.; Burslem G. M.; Buhimschi A. D.; Hines J.; Jaime-Figueroa S.; Wang J.; Hamman B. D.; Ishchenko A.; Crews C. M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25 (1), 78–87. 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Dixit V. M. Drugging the Undruggables: Exploring the Ubiquitin System for Drug Development. Cell Res. 2016, 26 (4), 484–498. 10.1038/cr.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348 (6241), 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22 (6), 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C.; Hu Y.; Zhou B.; Fernandez-Salas E.; Yang C.-Y.; Liu L.; McEachern D.; Przybranowski S.; Wang M.; Stuckey J.; Meagher J.; Bai L.; Chen Z.; Lin M.; Yang J.; Ziazadeh D. N.; Xu F.; Hu J.; Xiang W.; Huang L.; Li S.; Wen B.; Sun D.; Wang S. Discovery of QCA570 as an Exceptionally Potent and Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of the Bromodomain and Extra-Terminal (BET) Proteins Capable of Inducing Complete and Durable Tumor Regression. J. Med. Chem. 2018, 61 (15), 6685–6704. 10.1021/acs.jmedchem.8b00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K.; Lu J.; Qian Y.; Altieri M.; Gordon D.; Rossi A. M. K.; Wang J.; Chen X.; Dong H.; Siu K.; Winkler J. D.; Crew A. P.; Crews C. M.; Coleman K. G. PROTAC-Induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (26), 7124–7129. 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remillard D.; Buckley D. L.; Paulk J.; Brien G. L.; Sonnett M.; Seo H.-S.; Dastjerdi S.; Wühr M.; Dhe-Paganon S.; Armstrong S. A.; Bradner J. E. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew. Chem. 2017, 129 (21), 5832–5837. 10.1002/ange.201611281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gechijian L. N.; Buckley D. L.; Lawlor M. A.; Reyes J. M.; Paulk J.; Ott C. J.; Winter G. E.; Erb M. A.; Scott T. G.; Xu M.; Seo H. S.; Dhe-Paganon S.; Kwiatkowski N. P.; Perry J. A.; Qi J.; Gray N. S.; Bradner J. E. Functional TRIM24 Degrader via Conjugation of Ineffectual Bromodomain and VHL Ligands. Nat. Chem. Biol. 2018, 14 (4), 405–412. 10.1038/s41589-018-0010-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiedel M.; Herp D.; Hammelmann S.; Swyter S.; Lehotzky A.; Robaa D.; Oláh J.; Ovádi J.; Sippl W.; Jung M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61 (2), 482–491. 10.1021/acs.jmedchem.6b01872. [DOI] [PubMed] [Google Scholar]
- Bassi Z. I.; Fillmore M. C.; Miah A. H.; Chapman T. D.; Maller C.; Roberts E. J.; Davis L. C.; Lewis D. E.; Galwey N. W.; Waddington K. E.; Parravicini V.; Macmillan-Jones A. L.; Gongora C.; Humphreys P. G.; Churcher I.; Prinjha R. K.; Tough D. F. Modulating PCAF/GCN5 Immune Cell Function Through a PROTAC Approach. ACS Chem. Biol. 2018, 13 (10), 2862–2867. 10.1021/acschembio.8b00705. [DOI] [PubMed] [Google Scholar]
- Lai A. C.; Toure M.; Hellerschmied D.; Salami J.; Jaime-Figueroa S.; Ko E.; Hines J.; Crews C. M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem., Int. Ed. 2016, 55 (2), 807–810. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.-T.; Dobrovolsky D.; Paulk J.; Yang G.; Weisberg E. L.; Doctor Z. M.; Buckley D. L.; Cho J.-H.; Ko E.; Jang J.; Shi K.; Choi H. G.; Griffin J. D.; Li Y.; Treon S. P.; Fischer E. S.; Bradner J. E.; Tan L.; Gray N. S. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-Kinase Degrader. Cell Chem. Biol. 2018, 25 (1), 88–99. 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crew A. P.; Raina K.; Dong H.; Qian Y.; Wang J.; Vigil D.; Serebrenik Y. V.; Hamman B. D.; Morgan A.; Ferraro C.; Siu K.; Neklesa T. K.; Winkler J. D.; Coleman K. G.; Crews C. M. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J. Med. Chem. 2018, 61 (2), 583–598. 10.1021/acs.jmedchem.7b00635. [DOI] [PubMed] [Google Scholar]
- Olson C. M.; Jiang B.; Erb M. A.; Liang Y.; Doctor Z. M.; Zhang Z.; Zhang T.; Kwiatkowski N.; Boukhali M.; Green J. L.; Haas W.; Nomanbhoy T.; Fischer E. S.; Young R. A.; Bradner J. E.; Winter G. E.; Gray N. S. Pharmacological Perturbation of CDK9 Using Selective CDK9 Inhibition or Degradation. Nat. Chem. Biol. 2017, 14 (2), 163–170. 10.1038/nchembio.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb C. M.; Contreras J. I.; Kour S.; Taylor M. A.; Abid M.; Sonawane Y. A.; Zahid M.; Murry D. J.; Natarajan A.; Rana S. Chemically Induced Degradation of CDK9 by a Proteolysis Targeting Chimera (PROTAC). Chem. Commun. 2017, 53 (54), 7577–7580. 10.1039/C7CC03879H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata N.; Nagai K.; Morita Y.; Ujikawa O.; Ohoka N.; Hattori T.; Koyama R.; Sano O.; Imaeda Y.; Nara H.; Cho N.; Naito M. Development of Protein Degradation Inducers of Androgen Receptor by Conjugation of Androgen Receptor Ligands and Inhibitor of Apoptosis Protein Ligands. J. Med. Chem. 2018, 61 (2), 543–575. 10.1021/acs.jmedchem.7b00168. [DOI] [PubMed] [Google Scholar]
- Salami J.; Alabi S.; Willard R. R.; Vitale N. J.; Wang J.; Dong H.; Jin M.; McDonnell D. P.; Crew A. P.; Neklesa T. K.; Crews C. M. Androgen Receptor Degradation by the Proteolysis-Targeting Chimera ARCC-4 Outperforms Enzalutamide in Cellular Models of Prostate Cancer Drug Resistance. Commun. Biol. 2018, 1, 100. 10.1038/s42003-018-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniaci C.; Hughes S. J.; Testa A.; Chen W.; Lamont D. J.; Rocha S.; Alessi D. R.; Romeo R.; Ciulli A. Homo-PROTACs: Bivalent Small-Molecule Dimerizers of the VHL E3 Ubiquitin Ligase to Induce Self-Degradation. Nat. Commun. 2017, 8 (1), 830. 10.1038/s41467-017-00954-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinebach C.; Lindner S.; Udeshi N. D.; Mani D. C.; Kehm H.; Köpff S.; Carr S. A.; Gütschow M.; Kronke J. Homo-PROTACs for the Chemical Knockdown of Cereblon. ACS Chem. Biol. 2018, 13 (9), 2771–2782. 10.1021/acschembio.8b00693. [DOI] [PubMed] [Google Scholar]
- Cyrus K.; Wehenkel M.; Choi E.-Y.; Lee H.; Swanson H.; Kim K.-B. Jostling for Position: Optimizing Linker Location in the Design of Estrogen Receptor-Targeting PROTACs. ChemMedChem 2010, 5 (7), 979–985. 10.1002/cmdc.201000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorba A.; Nguyen C.; Xu Y.; Starr J.; Borzilleri K.; Smith J.; Zhu H.; Farley K. A.; Ding W.; Schiemer J.; Feng X.; Chang J. S.; Uccello D. P.; Young J. A.; Garcia-Irrizary C. N.; Czabaniuk L.; Schuff B.; Oliver R.; Montgomery J.; Hayward M. M.; Coe J.; Chen J.; Niosi M.; Luthra S.; Shah J. C.; El-Kattan A.; Qiu X.; West G. M.; Noe M. C.; Shanmugasundaram V.; Gilbert A. M.; Brown M. F.; Calabrese M. F. Delineating the Role of Cooperativity in the Design of Potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (31), E7285–E7292. 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulatov E.; Ciulli A. Targeting Cullin–RING E3 Ubiquitin Ligases for Drug Discovery: Structure, Assembly and Small-Molecule Modulation. Biochem. J. 2015, 467 (3), 365–386. 10.1042/BJ20141450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas X.; Ciulli A. Recognition of Substrate Degrons by E3 Ubiquitin Ligases and Modulation by Small-Molecule Mimicry Strategies. Curr. Opin. Struct. Biol. 2017, 44, 101–110. 10.1016/j.sbi.2016.12.015. [DOI] [PubMed] [Google Scholar]
- Fischer E. S.; Scrima A.; Böhm K.; Matsumoto S.; Lingaraju G. M.; Faty M.; Yasuda T.; Cavadini S.; Wakasugi M.; Hanaoka F.; Iwai S.; Gut H.; Sugasawa K.; Thomä N. H. The Molecular Basis of CRL4DDB2/CSA Ubiquitin Ligase Architecture, Targeting, and Activation. Cell 2011, 147 (5), 1024–1039. 10.1016/j.cell.2011.10.035. [DOI] [PubMed] [Google Scholar]
- Petroski M. D.; Deshaies R. J. Function and Regulation of Cullin–RING Ubiquitin Ligases. Nat. Rev. Mol. Cell Biol. 2005, 6 (1), 9–20. 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- Zhu Y. X.; Braggio E.; Shi C.-X.; Bruins L. A.; Schmidt J. E.; Van Wier S.; Chang X.-B.; Bjorklund C. C.; Fonseca R.; Bergsagel P. L.; Orlowski R. Z.; Stewart A. K. Cereblon Expression is Required for the Antimyeloma Activity of Lenalidomide and Pomalidomide. Blood 2011, 118 (18), 4771–4779. 10.1182/blood-2011-05-356063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoulou N. H.; Bamborough P.; Bannister A. J.; Becher I.; Bit R. A.; Che K. H.; Chung C.; Dittmann A.; Drewes G.; Drewry D. H.; Gordon L.; Grandi P.; Leveridge M.; Lindon M.; Michon A. M.; Molnar J.; Robson S. C.; Tomkinson N. C.; Kouzarides T.; Prinjha R. K.; Humphreys P. G. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem. 2016, 59 (4), 1425–1439. 10.1021/acs.jmedchem.5b00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C.; Hargreaves D. C.; Hodges C.; Elias L.; Ho L.; Ranish J.; Crabtree G. R. Proteomic and Bioinformatic Analysis of Mammalian SWI/SNF Complexes Identifies Extensive Roles in Human Malignancy. Nat. Genet. 2013, 45 (6), 592–601. 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser M. D.; Aslanian A.; Dong M.-Q.; Yates J. R.; Emerson B. M. BRD7, a Novel PBAF-Specific SWI/SNF Subunit, is Required for Target Gene Activation and Repression in Embryonic Stem Cells. J. Biol. Chem. 2008, 283 (47), 32254–32263. 10.1074/jbc.M806061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar G. J.; Sundaramoorthy R.; Owen-Hughes T. Mechanisms and Functions of ATP-Dependent Chromatin-Remodeling Enzymes. Cell 2013, 154 (3), 490–503. 10.1016/j.cell.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann A. F.; Vakoc C. R. A Rationale to Target the SWI/SNF Complex for Cancer Therapy. Trends Genet. 2014, 30 (8), 356–363. 10.1016/j.tig.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens J. A. Evidence That Swi/Snf Directly Represses Transcription in S. Cerevisiae. Genes Dev. 2002, 16 (17), 2231–2236. 10.1101/gad.1009902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J. U.; Koo S. H.; Kwon K. C.; Park J. W.; Kim J. M. Gain at Chromosomal Region 5p15.33, Containing TERT, is the Most Frequent Genetic Event in Early Stages of Non-Small Cell Lung Cancer. Cancer Genet. Cytogenet. 2008, 182 (1), 1–11. 10.1016/j.cancergencyto.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Scotto L.; Narayan G.; Nandula S. V.; Subramaniyam S.; Kaufmann A. M.; Wright J. D.; Pothuri B.; Mansukhani M.; Schneider A.; Arias-Pulido H.; Murty V. V. Integrative Genomics Analysis of Chromosome 5p Gain in Cervical Cancer Reveals Target Over-Expressed Genes, Including Drosha. Mol. Cancer 2008, 7, 58. 10.1186/1476-4598-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Li Z.; Shen J. BRD7: A Novel Tumor Suppressor Gene in Different Cancers. Am. J. Transl. Res. 2016, 8 (2), 742–748. [PMC free article] [PubMed] [Google Scholar]
- Drost J.; Mantovani F.; Tocco F.; Elkon R.; Comel A.; Holstege H.; Kerkhoven R.; Jonkers J.; Voorhoeve P. M.; Agami R.; Del Sal G. BRD7 Is a Candidate Tumour Suppressor Gene Required for P53 Function. Nat. Cell Biol. 2010, 12 (4), 380–389. 10.1038/ncb2038. [DOI] [PubMed] [Google Scholar]
- Chiu Y.-H.; Lee J. Y.; Cantley L. C. BRD7, a Tumor Suppressor, Interacts with P85α and Regulates PI3K Activity. Mol. Cell 2014, 54 (1), 193–202. 10.1016/j.molcel.2014.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harte M. T.; O’Brien G. J.; Ryan N. M.; Gorski J. J.; Savage K. I.; Crawford N. T.; Mullan P. B.; Harkin D. P. BRD7, a Subunit of SWI/SNF Complexes, Binds Directly to BRCA1 and Regulates BRCA1-Dependent Transcription. Cancer Res. 2010, 70 (6), 2538–2547. 10.1158/0008-5472.CAN-09-2089. [DOI] [PubMed] [Google Scholar]
- Niu W.; Luo Y.; Wang X.; Zhou Y.; Li H.; Wang H.; Fu Y.; Liu S.; Yin S.; Li J.; Zhao R.; Liu Y.; Fan S.; Li Z.; Xiong W.; Li X.; Li G.; Ren C.; Tan M.; Zhou M. BRD7 Inhibits the Warburg Effect and Tumor Progression Through Inactivation of HIF1α/LDHA Axis in Breast Cancer. Cell Death Dis. 2018, 9 (5), 519. 10.1038/s41419-018-0536-7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gatenby R. A.; Gillies R. J. Why Do Cancers Have High Aerobic Glycolysis?. Nat. Rev. Cancer 2004, 4 (11), 891–899. 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Park S. W.; Herrema H.; Salazar M.; Cakir I.; Cabi S.; Basibuyuk Sahin F.; Chiu Y.-H.; Cantley L. C.; Ozcan U. BRD7 Regulates XBP1s’ Activity and Glucose Homeostasis Through its Interaction with the Regulatory Subunits of PI3K. Cell Metab. 2014, 20 (1), 73–84. 10.1016/j.cmet.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D.; Kobayashi A.; Jiang P.; Ferrari de Andrade L.; Tay R. E.; Luoma A. M.; Tsoucas D.; Qiu X.; Lim K.; Rao P.; Long H. W.; Yuan G. C.; Doench J.; Brown M.; Liu X. S.; Wucherpfennig K. W. A Major Chromatin Regulator Determines Resistance of Tumor Cells to T Cell-Mediated Killing. Science 2018, 359 (6377), 770–775. 10.1126/science.aao1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark P. G. K.; Vieira L. C. C.; Tallant C.; Fedorov O.; Singleton D. C.; Rogers C. M.; Monteiro O. P.; Bennett J. M.; Baronio R.; Müller S.; Daniels D. L.; Méndez J.; Knapp S.; Brennan P. E.; Dixon D. J. LP99: Discovery and Synthesis of the First Selective BRD7/9 Bromodomain Inhibitor. Angew. Chem., Int. Ed. 2015, 54 (21), 6217–6221. 10.1002/anie.201501394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D. A.; Rogers C. M.; Fedorov O.; Tallant C.; Martin S.; Monteiro O. P.; Müller S.; Knapp S.; Schofield C. J.; Brennan P. E. Design and Synthesis of Potent and Selective Inhibitors of BRD7 and BRD9 Bromodomains. MedChemComm 2015, 6 (7), 1381–1386. 10.1039/C5MD00152H. [DOI] [Google Scholar]
- Martin L. J.; Koegl M.; Bader G.; Cockcroft X.-L.; Fedorov O.; Fiegen D.; Gerstberger T.; Hofmann M. H.; Hohmann A. F.; Kessler D.; Knapp S.; Knesl P.; Kornigg S.; Müller S.; Nar H.; Rogers C.; Rumpel K.; Schaaf O.; Steurer S.; Tallant C.; Vakoc C. R.; Zeeb M.; Zoephel A.; Pearson M.; Boehmelt G.; McConnell D. Structure-Based Design of an in Vivo Active Selective BRD9 Inhibitor. J. Med. Chem. 2016, 59 (10), 4462–4475. 10.1021/acs.jmedchem.5b01865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford T. D.; Vartanian S.; Côté A.; Bellon S.; Duplessis M.; Flynn E. M.; Hewitt M.; Huang H.-R.; Kiefer J. R.; Murray J.; Nasveschuk C. G.; Pardo E.; Romero F. A.; Sandy P.; Tang Y.; Taylor A. M.; Tsui V.; Wang J.; Wang S.; Zawadzke L.; Albrecht B. K.; Magnuson S. R.; Cochran A. G.; Stokoe D. Inhibition of Bromodomain-Containing Protein 9 for the Prevention of Epigenetically-Defined Drug Resistance. Bioorg. Med. Chem. Lett. 2017, 27 (15), 3534–3541. 10.1016/j.bmcl.2017.05.063. [DOI] [PubMed] [Google Scholar]
- Hohmann A. F.; Martin L. J.; Minder J. L.; Roe J.-S.; Shi J.; Steurer S.; Bader G.; McConnell D.; Pearson M.; Gerstberger T.; Gottschamel T.; Thompson D.; Suzuki Y.; Koegl M.; Vakoc C. R. Sensitivity and Engineered Resistance of Myeloid Leukemia Cells to BRD9 Inhibition. Nat. Chem. Biol. 2016, 12 (9), 672–679. 10.1038/nchembio.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T.; Goralski M.; Gaskill N.; Capota E.; Kim J.; Ting T. C.; Xie Y.; Williams N. S.; Nijhawan D. Anticancer Sulfonamides Target Splicing by Inducing RBM39 Degradation via Recruitment to DCAF15. Science 2017, 356 (6336), eaal3755 10.1126/science.aal3755. [DOI] [PubMed] [Google Scholar]
- Uehara T.; Minoshima Y.; Sagane K.; Sugi N. H.; Mitsuhashi K. O.; Yamamoto N.; Kamiyama H.; Takahashi K.; Kotake Y.; Uesugi M.; Yokoi A.; Inoue A.; Yoshida T.; Mabuchi M.; Tanaka A.; Owa T. Selective Degradation of Splicing Factor CAPERα by Anticancer Sulfonamides. Nat. Chem. Biol. 2017, 13 (6), 675–680. 10.1038/nchembio.2363. [DOI] [PubMed] [Google Scholar]
- Galdeano C.; Gadd M. S.; Soares P.; Scaffidi S.; Van Molle I.; Birced I.; Hewitt S.; Dias D. M.; Ciulli A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction Between the Von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 2014, 57 (20), 8657–8663. 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost J.; Galdeano C.; Soares P.; Gadd M. S.; Grzes K. M.; Ellis L.; Epemolu O.; Shimamura S.; Bantscheff M.; Grandi P.; Read K. D.; Cantrell D. A.; Rocha S.; Ciulli A. Potent and Selective Chemical Probe of Hypoxic Signalling Downstream of HIF-α Hydroxylation via VHL Inhibition. Nat. Commun. 2016, 7, 13312. 10.1038/ncomms13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares P.; Gadd M. S.; Frost J.; Galdeano C.; Ellis L.; Epemolu O.; Rocha S.; Read K. D.; Ciulli A. Group-Based Optimization of Potent and Cell-Active Inhibitors of the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase: Structure–Activity Relationships Leading to the Chemical Probe (2S,4R)-1-((S)-2-(1-Cyanocyclopropanecarboxamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (VH298). J. Med. Chem. 2018, 61 (2), 599–618. 10.1021/acs.jmedchem.7b00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer E. S.; Böhm K.; Lydeard J. R.; Yang H.; Stadler M. B.; Cavadini S.; Nagel J.; Serluca F.; Acker V.; Lingaraju G. M.; Tichkule R. B.; Schebesta M.; Forrester W. C.; Schirle M.; Hassiepen U.; Ottl J.; Hild M.; Beckwith R. E.; Harper J. W.; Jenkins J. L.; Thomä N. H. Structure of the DDB1–CRBN E3 Ubiquitin Ligase in Complex with Thalidomide. Nature 2014, 512 (7512), 49–53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.; Hu J.; Xu F.; Chen Z.; Bai L.; Fernandez-Salas E.; Lin M.; Liu L.; Yang C.-Y.; Zhao Y.; McEachern D.; Przybranowski S.; Wen B.; Sun D.; Wang S. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem. 2018, 61 (2), 462–481. 10.1021/acs.jmedchem.6b01816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishoey M.; Chorn S.; Singh N.; Jaeger M. G.; Brand M.; Paulk J.; Bauer S.; Erb M. A.; Parapatics K.; Müller A. C.; Bennett K. L.; Ecker G. F.; Bradner J. E.; Winter G. E. Translation Termination Factor GSPT1 Is a Phenotypically Relevant Off-Target of Heterobifunctional Phthalimide Degraders. ACS Chem. Biol. 2018, 13 (3), 553–560. 10.1021/acschembio.7b00969. [DOI] [PubMed] [Google Scholar]
- Buckley D. L.; Raina K.; Darricarrere N.; Hines J.; Gustafson J. L.; Smith I. E.; Miah A. H.; Harling J. D.; Crews C. M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10 (8), 1831–1837. 10.1021/acschembio.5b00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Molle I.; Thomann A.; Buckley D. L.; So E. C.; Lang S.; Crews C. M.; Ciulli A. Dissecting Fragment-Based Lead Discovery at the Von Hippel-Lindau Protein:Hypoxia Inducible Factor 1α Protein-Protein Interface. Chem. Biol. 2012, 19 (10), 1300–1312. 10.1016/j.chembiol.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching K. M.; Mahan S.; Corona C. R.; McDougall M.; Vasta J. D.; Robers M. B.; Urh M.; Daniels D. L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13 (9), 2758–2770. 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
- Soucy T. A.; Smith P. G.; Milhollen M. A.; Berger A. J.; Gavin J. M.; Adhikari S.; Brownell J. E.; Burke K. E.; Cardin D. P.; Critchley S.; Cullis C. A.; Doucette A.; Garnsey J. J.; Gaulin J. L.; Gershman R. E.; Lublinsky A. R.; McDonald A.; Mizutani H.; Narayanan U.; Olhava E. J.; Peluso S.; Rezaei M.; Sintchak M. D.; Talreja T.; Thomas M. P.; Traore T.; Vyskocil S.; Weatherhead G. S.; Yu J.; Zhang J.; Dick L. R.; Claiborne C. F.; Rolfe M.; Bolen J. B.; Langston S. P. An Inhibitor of NEDD8-Activating Enzyme as a New Approach to Treat Cancer. Nature 2009, 458 (7239), 732–736. 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- Loenarz C.; Mecinović J.; Chowdhury R.; McNeill L. A.; Flashman E.; Schofield C. J. Evidence for a Stereoelectronic Effect in Human Oxygen Sensing. Angew. Chem., Int. Ed. 2009, 48 (10), 1784–1787. 10.1002/anie.200805427. [DOI] [PubMed] [Google Scholar]
- Wang X.; Sansam C. G.; Thom C. S.; Metzger D.; Evans J. A.; Nguyen P. T. L.; Roberts C. W. M. Oncogenesis Caused by Loss of the SNF5 Tumor Suppressor is Dependent on Activity of BRG1, the ATPase of the SWI/SNF Chromatin Remodeling Complex. Cancer Res. 2009, 69 (20), 8094–8101. 10.1158/0008-5472.CAN-09-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Han X.-R.; Yang X.; Jiang B.; Liu J.; Xiong Y.; Jin J. Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK). Eur. J. Med. Chem. 2018, 151, 304–314. 10.1016/j.ejmech.2018.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner R.; Ziegler D. S.; Cibu D.; Jakowetz A. C.; Auras F.; Bein T.; Knochel P. Preparation of Polyfunctional Naphthyridines by Cobalt-Catalyzed Cross-Couplings of Halogenated Naphthyridines with Magnesium and Zinc Organometallics. Org. Lett. 2017, 19 (23), 6384–6387. 10.1021/acs.orglett.7b03242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. A.; Chalker J. M.; Davis B. G. Olefin Cross-Metathesis on Proteins: Investigation of Allylic Chalcogen Effects and Guiding Principles in Metathesis Partner Selection. J. Am. Chem. Soc. 2010, 132 (47), 16805–16811. 10.1021/ja104994d. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Ren H.; Baker G. L. Synthesis of a Library of Propargylated and PEGylated α-Hydroxy Acids Toward “Clickable” Polylactides via Hydrolysis of Cyanohydrin Derivatives. J. Org. Chem. 2014, 79 (20), 9546–9555. 10.1021/jo5016135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara M.; Suzuki S.; Taniguchi N.; Fukumoto K. Deconjugation of α,β-Unsaturated Esters and an Intramolecular Michael Reaction of Bis-α,β-Unsaturated Esters with Trialkylsilyl Trifluoromethanesulfonate in the Presence of Tertiary Amine: Synthesis of (±)-Ricciocarpin A. J. Chem. Soc., Perkin Trans. 1 1993, (19), 2251–2258. 10.1039/P19930002251. [DOI] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (2), 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and Assessment of Data Quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62 (1), 72–82. 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67 (4), 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. MOLREP: An Automated Program for Molecular Replacement. J. Appl. Crystallogr. 1997, 30 (6), 1022–1025. 10.1107/S0021889897006766. [DOI] [Google Scholar]
- Emsley P.; Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60 (12), 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53 (3), 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Schüttelkopf A. W.; van Aalten D. M. F. PRODRG: A Tool for High-Throughput Crystallography of Protein–Ligand Complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60 (8), 1355–1363. 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (1), 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. H.; Roberts C. W. Mechanisms by Which SMARCB1 Loss Drives Rhabdoid Tumor Growth. Cancer Genet. 2014, 207 (9), 365–372. 10.1016/j.cancergen.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.