

Abstract
Metal exposure is pervasive and not limited to sporadic poisoning events or toxic waste sites. Hundreds of millions of people around the globe are affected by chronic metal exposure, which is associated with serious health concerns including cancer, as demonstrated in a variety of studies at the molecular, systemic, and epidemiologic levels. Metal-induced toxicity and carcinogenicity is sophisticated and complex in nature. This review provides a broad context and holistic view of currently available studies on the mechanisms of metal-induced carcinogenesis. Specifically, we focus on the five most prevailing carcinogenic metals: arsenic, nickel, cadmium, chromium, and beryllium, and their potential to drive carcinogenesis in humans. A comprehensive understanding of the mechanisms behind the development of metal-induced cancer can provide valuable insights for potential cancer therapeutics.
Keywords: Carcinogenesis, Metals, Mechanisms, Epigenetics, Exposure
Introduction
Metal toxicity and carcinogenicity is not a recent concern. Exposure to toxic and carcinogenic metals such as arsenic can be traced back 2,400 years as part of traditional Chinese Medicine. Arsenic compounds such as Salvarsan was used as an antibacterial agent before the discovery of sulpha drugs and penicillin. Even though many toxic metals are not considered carcinogenic, we continue to risk human exposure by using these metals to manufacture end products that are used by humans resulting in exposure. The use of metals such as lead (Pb) in paints has poisoned children who eat the Pb-containing paint chips, as well as adults who restore old furniture and houses painted with Pb-containing paint. Even today we make Pb crystal for decanters and glasses to hold wine, resulting in the leaching of the Pb from the container into the wine that is ingested. We continue to make and use “silver” amalgam fillings to fill decayed teeth and these silver fillings contain about 50% by weight mercury metal. The mercury evaporates during gum chewing, is inhaled and reaches the central nervous system (1; 2). This practice has consequences: one study used the Beck Depression Inventory and compared 25 women who had amalgams to 23 women without amalgams (3). Women with amalgams had significantly higher depression scores and reported more symptoms of fatigue and insomnia. They also had higher anger scores, expressing anger without provocation. Anxiety scores showed the women with amalgams scored significantly less pleasant, satisfied, happy, secure, and steady, and had a more difficult time making decisions (3). The women with amalgams also had significantly higher levels of mercury in the oral cavity before and after chewing gum. This study suggests that mercury amalgam may be an etiological factor in depression, excessive anger, and anxiety because mercury can produce such symptoms by affecting the neurotransmitters in the brain (3).
Metals are persistent environmental contaminants because they are not broken down by microorganisms as are organic pollutants. Thus they accumulate and bio-concentrate in our ecosystems. The only way to get rid of contamination with metals is to remove them and this can be quite costly. Jersey City, NJ, once had large chromate refineries to produce hexavalent Chromate from ore that was shipped into Jersey City. The chromate ore residue after refining, were attractive brown rocks that were used for landfill throughout Jersey City and the surrounding areas. A substantial portion of Jersey City contained many feet of landfill with these brown rocks. Water passing over the rocks extracted yellow hexavalent Chromate and spread throughout Jersey City resulting in substantial human exposure to carcinogenic hexavalent chromate. The water containing carcinogenic chromate even seeped into basements and destroyed brick and cement walls. Buildings built on these mine tailings sometimes collapsed. In the last several decades many millions of dollars were spent getting rid of the chromate contaminated soil and water from Jersey City by digging it up and moving it to toxic waste dump sites. In these instances it was fortuitous that the hexavalent chromate was converted to the nontoxic trivalent Chromiumin organisms and plants that were exposed, making it safe to eat vegetables, fish and game. Exposure to carcinogenic metals poses serious public health risks, and this review will examine the mechanisms of carcinogenesis induced by the five most prominent metals: Cadmium, Arsenic, Nickel, Beryllium, and Chromium.
Cadmium
Cadmium is a toxic and non-essential transition metal that first gained recognition in the 19th century as the causative agent for Ita-itai (“ouch ouch” in Japanese) disease, which results from consuming contaminated rice (4; 5). The loss of calcium causes damage to the kidneys and bones. In fact, the bones become so brittle that they would break during episodes of coughing. Although cadmium is a naturally occurring element, human activities have substantially magnified its presence in nature. Today, the main sources of occupational cadmium contamination come from zinc/lead smelters as well as handling and assembling industrial products such as cadmium-nickel batteries, mobile phones and computer circuit boards (6). Cadmium is heavily concentrated in tobacco leaves and 50% of inhaled cadmium through smoking can be absorbed into the body (7), making it the highest toxic metal found in cigarette smoke. With an exceptionally long biological half-life of 15-20 years, cadmium is considered a cumulative toxin and can pose great health risks including neurological disorder and reproductive system defects (8-10). Moreover, cadmium carcinogenicity has long been established (11-13). In 2004, cadmium was officially categorized as a Class I human lung carcinogen based on epidemiological studies. Cadmium also plays an important role in prostate, renal, liver, bladder, and stomach cancers (14).
Due to weak DNA binding activity, cadmium is considered weakly genotoxic (15;16). Cadmium causes chromosomal aberrations and DNA damage (17). Cadmium can induce both genotoxicity and mutagenicity at levels that induce apoptosis in 50% of exposed cells, making it unlikely that cellular mutations will occur (14). There are four major Cd-induced carcinogenesis mechanisms: 1) oxidative stress, 2) attenuation of apoptosis, 3) inhibition of DNA damage repair, and 4) alterations in gene expression (11;12; 18). In addition, it may exert carcinogenic effects by endocrine disruption, cell proliferation, and aberrant DNA methylation (19-21). In fact the presence of Zinc finger motifs in Steroid hormone receptors and other DNA binding molecules offers targets for Cd binding and subsequent alterations in their function, see Figure 1. In some cases such as with the Estrogen receptor, Cd interaction will actually activate the receptor. The similarity of Cd to Zn is a major driver of the toxic effects of Cd. In fact the requirement for Zn in the coordination center of histone demethylases has been described as a mechanism for Cd inhibition of this family of enzymes. (22) ( Figure 1)
Figure 1.
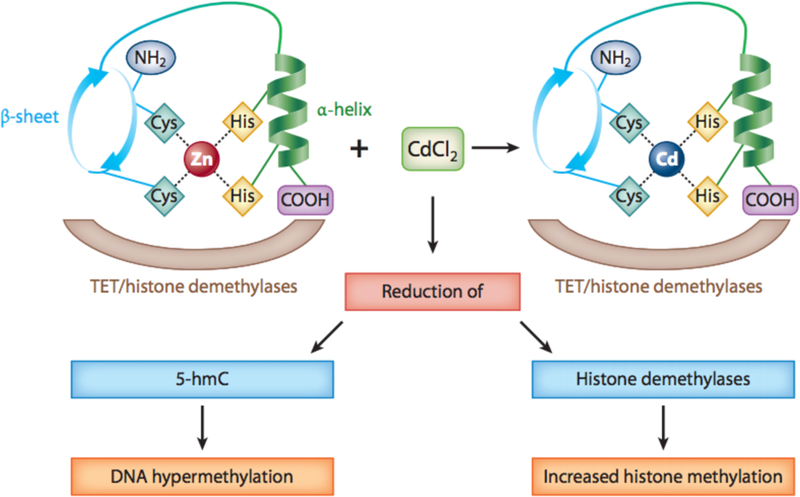
Model illustrating CdCl2 displacing Zn in TET proteins and histone demethylases. Similarities in physical and chemical properties between Cd and Zn enable them to antagonize each other. Cd has the potential to displace Zn in the Zn finger DNA binding domain of TET protein, which can lead to changes in conformation and activity. Abbreviations: 5-hmC, 5-hydroxymethylcytosine; Cd, cadmium; Cl, chlorine; COOH, carboxylic acid; Cys, cysteine; His, histidine; TET, ten-eleven translocation; Zn, zinc.
Oxidative stress is a factor important in cadmium toxicity (23). However, because Cd cannot cause Fenton-like reactions, it is unable to directly produce reactive oxygen species (ROS), but oxidative stress such as lipid peroxidation can occur by the loss of reduced glutathione and protein-bound sulfhydryl groups as well as inhibition of antioxidant enzymes (4; 14; 24; 25). Elevated oxidative stress has been shown to promote tumor development through mutagenesis and effects on cell cycle (26). A functional DNA repair system removes errors produced by metabolism and environmental carcinogens. However, inadequate repair mechanisms allow the accumulation of DNA damage, which promote cancer (27). Evidence suggests that cadmium is capable of inhibiting DNA repair including mismatch, base excision, and nucleotide excision (28;29). Lack of functional DNA repair allows for an accumulation of cells with DNA damage which following cell division will produce mutations. In addition, cadmium exposure can lead to the activation of oncogenes associated with cell proliferation (i.e. c-myc, c-jun, and c-fos) as well as inhibition of tumor suppressor genes such as p53 and p27 (30; 31). Some studies suggest that Cd can inhibit DNA methyltransferases, thereby inducing DNA hypomethylation (14; 32). It has been reported that short-term Cd exposure reduced DNA methylation, by inhibition of DNMT1 (33; 34). Without the activity of this enzyme, methylated Cytosines in DNA will be lost due to passive DNA demethylation, since this enzyme preserves parental cytosine methylation on the daughter strand. On the other hand, chronic Cd exposure can also elevate DNMT1 activation resulting in hypermethylation (15; 20; 32; 33; 35). Cd exposure can induce aneuploidy and loss of expression of tumor suppressor genes such as RASSF1A and P16 by promoter hypermethylation (18; 36). The discrepancy between the opposite effects of long or short-term Cd exposure on global DNA methylation is not understood, although a changing landscape of chromatin methylation in either direction may initiate signaling pathways responsible for many processes. (37). Notably, 60 key signaling pathways including important cancer-related pathways such as HIF-1 α, NF-κB, RAS, and PI3K-Akt have been activated following Cd-exposure (38). And although previously unreported, signaling pathways involving TGF-β, a membrane receptor-binding protein implicated in various types of malignant cell transformation, also exhibited increased activity following Cd exposure (38; 39). As a prominent tumor suppressor gene, Cd-induced inactivation of p53 has been shown to disrupt cell cycle arrest and apoptosis (4; 37). Specifically, Cd is able to alter p53 structure and function by displacing zinc. The acquisition of apoptotic resistance through p53 inactivation may be critical for Cd-induced carcinogenesis. Current studies have not explored other types of epigenetic mechanisms including histone posttranslational modifications (40). The role of miRNA deregulation in Cd-induced carcinogenesis has not been well studied.
Arsenic
Arsenic (As) is a naturally occurring ubiquitous metalloid and a Class I human Carcinogen. Approximately 200 million people around the world are exposed to unsafe levels of arsenic. Chronic exposure to arsenic has been correlated with cancers of the lung, liver, bladder, kidneys, skin, as well as non-carcinogenic diseases such as skin lesions, cardiovascular disease, reproductive defects, neurological injuries, and diabetes mellitus (41).
Although arsenic lacks direct mutagenic activity, it is capable of inducing mutations by inhibiting DNA repair and causing chromosomal aberrations (42). One of the most studied mechanisms of arsenic carcinogenicity is the production of ROS. Evidence suggests that ROS react with DNA and induce structural DNA damage leading to genetic defects. Moreover, overexpression of antioxidant enzymes in response to As-induced oxidative stress will desensitize cells to apoptosis, which allows cancer cells to persist instead of die (43; 44). Of note, arsenic can cause oxidative stress either through direct Fenton-type reactions to produce ROS, or indirect depletion of important antioxidants such as glutathione (45-47). Furthermore, As is capable of mediating chromosomal instability through generation of DNA double-strand breaks and impairment of proteins necessary for DNA repair. As-induced chromosomal instability often occurs at the centromeres, which can lead to aneuploidy and micronuclei formation (48-50).
Epigenetic alterations including changes in DNA methylation, histone PTMs, and miRNA expression can hinder chromatin accessibility to regulatory factors and thereby affect gene expression at transcription initiation and gene splicing. (43). As-induced global epigenetic alterations were first discovered in the 1980s (51). DNA methylation is an integral component of gene regulation as well as overall genomic integrity (52; 53). Studies have provided evidence that As promotes both DNA hyper-and hypomethylation. The effect of DNA methylation depends on the site and type of the regulatory element, which may include global hypomethylation and gene-specific hypermethylation (43). Furthermore, as an integral part of arsenic biotransformation, S-adenosylmethionine (SAM) is used to methylate arsenic and facilitate its excretion from the body. SAM deficiency caused by the production of methylated arsenic species has been implicated in DNA hypomethylation by a reduction in the amount of available methyl group donors in the cell (54). The lack of available SAM may explain the occurrence of global hypomethylation when arsenic levels reach higher than 500 ug/L. However in most instances, the levels of As in the cell do not exceed several uM while SAM levels are about 80uM with a robust reserve available if SAM falls below 80uM.These facts argue against the hypothesis that As at environmentally relevant concentrations will deplete cellular SAM levels. Methylated As is more rapidly excreted from the body because it is less tightly bound to protein compared to unmethylated arsenite.
Genome-wide association studies have identified 2919 genes with differential DNA methylation at transcriptional start sites following arsenic exposure (55). In fact, arsenic has been shown to promote hypermethylation in important tumor suppressor genes such as p53 and p16 (36). Aside from gene-specific DNA, methylation of transposable elements such as long interspersed nuclear elements-1 (LINE-1) and Alu repeats is caused by As exposure (56). LINE-1 are dispersed throughout the genome and have been correlated with multiple human diseases including colon cancer, β-thalassemia, and oculomotor apraxia. In fact, hypomethylation of LINE-1 appears to be specifically associated with arsenic exposure (57; 58).
Since the discovery of arsenic-induced aberrant DNA methylation, new evidence suggests that arsenic-mediated epigenetic changes span from DNA methylation to histone post-translational modifications, as well as changes in miRNA expression (56). Post-translational modification of histone N-terminal tails encompasses a range of events including glycosylation, carbonylation, ubiquitylation, biotinylation, sumoylation, citrullination, ADP-ribosylation, N-formylation, crotonylation, propionylation, butyrylation, proline and aspartic acid isomerization, as well as more common modifications such as methylation, acetylation, and phosphorylation (59). Multiple studies have illustrated global changes in histone PTMs after arsenic exposure (60-62). Changes in histone marks are mediated by histone kinases such as the nuclear mitogen and stress-activated protein kinase 1 (MSK1), which are both activated upon arsenic exposure (63). In response to stress, MSK1 activation can lead to both demethylation of H3K9, which increases transcriptional activity, and increased phosphorylation of H3S10, which induces activation of proto-oncogenes such as c-fos and c-jun (13; 64). In addition to canonical histones, emerging evidence of modifications on highly conserved histone variants has provided new insights. Examples of altered histone variants after arsenic exposure include H2B1K, H2B1C, H2B1D, and H2B1B(65). Furthermore, arsenic has also been found to induce histone H3.1 polyadenylation through degradation of stem-loop binding protein (SLBP) (66). The presence of a poly(A) tail increases mRNA stability by preventing degradation and subsequently promotes accumulation of higher levels of polyadenylated H3.1 mRNA outside of S phase. Polyadenylated H3.1 alone can induce genomic instability, Mitotic blockade, and cell transformation. Increased SLBP degradation and subsequent polyadenylation of H3.1 can interrupt and block histone H3.3 function, which in turn promotes overall genomic instability and subsequent aberrant gene expression.
Recent studies have also demonstrated that arsenic-induced aberrant alternative splicing (AS) may be associated with cancer development (67). Alternative splicing of pre-mRNA is important for the biodiversity of proteins and occurs in 95% of multi-exon genes (43). Dysregulation of normal splicing events is known to occur in various human cancers and is correlated with angiogenesis, carcinogenesis, and EMT (68-70). In addition to possible changes in the kinetics of polymerase elongation, arsenic-mediated DNA methylation and histone PTMs may be responsible for exon-selection. Notably, epigenetic silencing of poly(ADP) ribose polymerase (PARP1) or CCCTC-binding factor (CTCF) can alter splicing decisions(71; 72).
Besides histone PTMs, arsenic has also been found to alter the regulation of non-coding RNAs (ncRNAs). Since only 2% of the transcribed human genome is translated into protein, the role of non-coding RNAs, with special interest in small ncRNAs such as microRNAs (miRNAs), endogenous small interfering RNAs (endo-siRNAs), and PIWI-interacting RNAs (piRNAs) has emerged as a potential factor in cancer etiology (73). Evidence suggests that arsenic is able to stimulate global changes in miRNA expression (74). In fact, one particular study showed that arsenic exposure can lead to the deregulation of 36 different miRNAs (72). Likewise, piRNAs are known epigenetic regulators and are implicated in various tumors due to their ability to regulate gene expression and genome instability.
Nickel
Nickel (Ni) is the 2nd most abundant element in the Earth’s inner core and occurs naturally from forest fire, rock erosion, and volcanic emission. Despite natural abundance of Ni, anthropogenic activities such as fossil fuel combustion and industrial disposal can release roughly 180,000 tons of nickel into the environment every year (75). For reasons unknown, oil burning has a specific signature of Nickel and Vanadium release. Due to its exceptional physiochemical properties, nickel has been heavily used in welding, nickel plating, manufacturing of stainless steel, etc. Alloys with high Ni content are most resistant to any insult (Chen et al., 2016). Interestingly, because the greatest deposit of Ni ore is found in the mantle of the earth, rendering its location too deep for mining, all the Ni mined in the world actually came from outer space, when meteors fell to earth. Since meteors are fragments from planets and other large objects in space they can contain deposits of Ni, which can be mined. Sudbury Canada is an example of a location where Ni is mined from a large meteor that fell onto Earth’s surface many millions of years ago. Today a significant portion of Ni produced in the world comes from this meteor.
Despite its usage in various industries, certain Ni compounds such as Ni Subsulphide and Ni Oxides are considered Class I human carcinogens. Exposure to Ni can cause health effects including asthma, cardiovascular disease, dermatitis, lung fibrosis, and respiratory tract cancer (76; 77). In 1949 the National Insurance of Great Britain and Minister of Pensions established lung and nose cancers as industrial diseases caused by prolonged occupational exposure to nickel in refinery workers (78). While Ni can enter the body via many routes, inhalation is the most common form of exposure in occupational settings.
Ni is known to mimic gene expression patterns similar to hypoxia, a condition commonly found in many tumors. Ni inhibits HIF-prolyl-and asparaginyl-hydroxylases and promotes the stabilization of HIFα proteins and HIF-1-dependent transcription, see Figure 2 (79; 80). HIF-1α/HIF-1β complex is only observable under hypoxic conditions or when exposed to proteasomal inhibitors (81). While cell proliferation is attenuated and apoptosis is promoted under hypoxic stress, tumor cells may be able to thrive by selecting cells that are damaged and resistant to apoptosis. In other words, the Ni-induced hypoxia like state under normal oxygen tension may be a mechanism for promoting cancer development (82). The ability of Ni ions to inhibit the prolyl hydroxylase was extended to other dioxygenases where Ni was found to inhibit these enzymes by displacing the iron (Fe) from their active site (coordination of Fe with two histidines and a carboxylate acid facial triad), see Figure 3 (83-86). Upon Fe displacement, Ni ions were coordinated to the same ligands as Fe except that Fe was penta-coordinated allowing for Oxygen to bind whereas Ni ions were hexa-coordinated resulting in an inactive enzyme (85). The major targets of Ni in the cell are these dioxygenase enzymes, since the binding constants of Ni ions for these enzymes are very low (IC-50 is in the low 20uM range whereas mM amounts of Ni are required to inhibit Aconitase where Fe is coordinated by Sulphurs (1;84). Ni ions are not highly toxic to cells and most cell types can tolerate 1mM Ni ion exposure for 24 hours with little toxicity (84).
Figure 2.
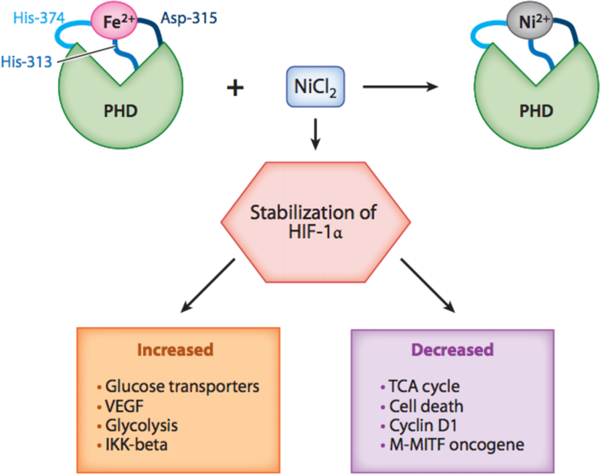
Model illustrating NiCl2 displacing Fe in prolyl hydroxylase domain. By displacing Fe in the PHD, Ni is able to inhibit HIF-prolyl- and asparaginyl-hydroxylases and promote the stabilization of HIFα proteins and HIF-1-dependent transcription. Abbreviations: Cl, chlorine; Fe, iron; HIF −1α, hypoxia-inducible factor 1α; His, histidine; IKKβ, nuclear factor kappa-B kinase subunit β inhibitor; M-MITF, microphthalmia-associated transcription factor type M; Ni, nickel; PHD, prolyl hydroxylase domain protein; TCA, tricarboxylic acid; TET, ten-eleven translocation; VEGF, vascular endothelial growth factor.
Figure 3.
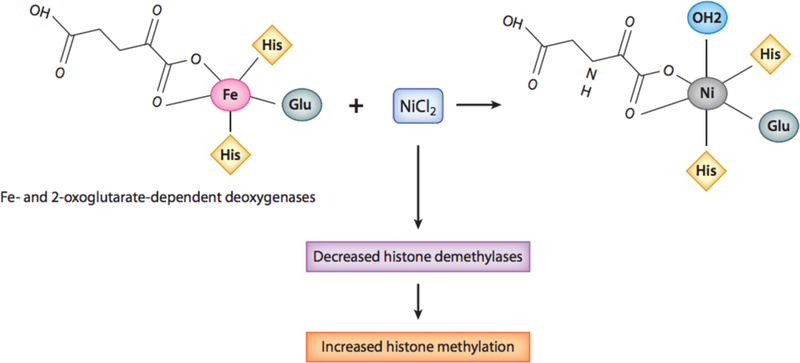
Model illustrating NiCl2 displacing Fe in 2-oxoglutarate-dependent oxygenases. Ni can displace Fe from its active site of Fe-2-oxoglutarate-dependent deoxygenases, which consists of two histidines and a carboxylate acid facial triad. Upon Fe displacement, Ni ions are able to coordinate with the same ligands as Fe, except that Fe is pentacoordinated, allowing for oxygen to bind, whereas Ni is hexacoordinated, resulting in an inactive enzyme. Abbreviations: Cl, chlorine; Fe, iron; Glu, glutamic acid; His, histidine; Ni, nickel.
Several studies have demonstrated nickel’s ability to cause deletion mutations, DNA-protein crosslinks, and chromosomal aberrations (87-89). However, weak mutagenic potential in mammalian mutation assays and especially in prokaryotic tests suggest that Ni may preferentially exert its carcinogenic effect through epigenetic changes, rather than mutation (33). Nickel-mediated epigenetic alterations include DNA methylation, histone post-translational modifications, and miRNAs. Notably, global DNA hypo-methylation and gene-specific hyper-methylation are found in nickel carcinogenesis (90). DNA methylation and inactivation of the gpt gene by Ni was first shown in Chinese hamster G12 and G10 cells (91). In G12 cells, gpt is located near heterochromatin while in G10 cells the gene is situated in a region of euchromatin. After Ni exposure, gpt was found to be silenced in G12 cells but not in G10, suggesting the ability of nickel to induce heterochromatin spreading, a phenomenon in which the chromatin condenses and pulls nearby genes into heterochromatic regions, thus silencing their expression (91; 92). One potential explanation suggests that Ni is able to displace magnesium in heterochromatic complexes, which triggers chromatin condensation through de novo DNA methylation (91; 93). In other words, these findings suggest that nickel is capable of initiating gene silencing through heterochromatin spreading, which will cause tumor suppressor genes near heterochromatin to be silenced. Ni has been shown to promote hyper-methylation of tumor suppressor genes such as E-cadherin and p16, which may be important in triggering carcinogenesis (94). In addition to DNA methylation, Ni exposure has also been found to alter global histone modifications both in vitro and in vivo (Chen et al., 2016). Chronic Ni exposure has led to changes in acetylation of histone H4, methylation of histone H3, phosphorylation of H3S10, and ubiquitination of histones H2A and H2B (33; 95). Similar to As, Ni exposure is also characterized by global histone hypoacetylation, which has been found to be a result of a dose-dependent inhibition of histone acetyltransferase (96). Moreover, one study observed that carcinogenic nickel is able to induce alpha helical conformation in histone H4 N-terminal tails. Since the new conformation is similar in structure to lysine acetylation, this interferes with acetyl groups being transferred to lysine residues and may prevent activation of potential tumor suppressor genes. Akin to As carcinogenesis, Ni has also been found to induce H3.1 polyadenylation through loss of SLBP (97). The study hypothesized that nickel may reduce SLBP mRNA by increasing DNA methylation and or decreasing histone acetylation in its promoter region, as well as promoting the degradation of the protein.
Ni has also been shown to influence miRNA regulation. Dysregulation of miRNA in tumor cells was first reported in 2002 (98). Since miRNAs are seemingly non-specific and can bind to multiple mRNAs, changes in these small non-coding RNAs may affect a range of gene expressions. As the most commonly up-regulated miRNA in cancers of the lung, stomach, colon, ovarian, etc., miRNA-21 has been found to be elevated in nickel-induced lung cancer (99). Other examples of differentially expressed miRNAs include miR152, −222, and −203, with miR-222 being a regulator of important tumor suppressor genes such as p27, p57, and PTEN (100; 101).
Beryllium
Beryllium is the lightest of all metals on the periodic table. Interestingly, it has a very high melting point, of 1287 °C. It also has a low density and is extremely strong even though it is light. One adverse property is that, despite its strength alone, and strengthening capacity when alloyed, it is a very brittle metal. Beryllium is at least 40% more rigid than steel and can conduct heat and electricity very well. In regards to radiation and nuclear capacity, Be is permeable to X-rays and can reflect and scatter neutrons when bombarded with radiation. Remarkably, the chemical properties of Be are unique and unlike the other members of group IIA. However, it does share some properties in common with aluminum, including a high affinity for atomic oxygen and has the ability to serve as either an acid or base in chemical reactions.
As stated above, Be is used in various industries to manufacture a wide variety of products and tools, from golf clubs to ballistic missiles and nuclear weapons. Beryllium’s desirable chemical properties make for a good component for metal alloys. It is estimated by the US Geological Survey that 80% of Be is made into workable alloy, the most common being beryllium-copper. The two most common beryllium containing alloys are beryllium-aluminum (Be.Al) and the aforementioned beryllium copper (Be.Cu). Beryllium’s abilities to withstand extreme temperatures, oxidation, corrosion and radiation make it suitable for materials such as military-grade weapons and armor, computer parts and communication devices, as well as nuclear industry shields. The James Webb Space Telescope (JWST) set to be launched in 2018 as a successor to Hubble, has been fitted with honeycomb mirrors made from Be and coated with nanoscale gold. These mirrors will be able to withstand the extreme conditions of space for a long time (about 10-year mission), and allow scientists at NASA to see deep into space. It is an infrared telescope, allowing for observation of newly formed galaxies, which are moving away from ours so quickly that they emit infrared light.
Beryllium that is mined here on Earth is usually refined to its metal component from a mineral source such as beryl or bertrandite (102). Beryl has an interesting use, as it is a component of some very popular gemstones. Beryl containing traces of other metals, such as chromium, iron, or vanadium produce colored gems. For example, the emerald gemstone is beryl mineral with traces of chromium. Currently, the United States leads the world with Beryllium production from raw material. Utah contains an estimated 85% of the worldwide supply of beryllium. Approximately two thirds of the mined and refined beryllium is taken by the US government for military and defense development.
Beryllium is found naturally in the Earth’s crust and is a byproduct of many manufacturing and energy producing industries. Because of this, there are both natural and anthropogenic sources of Be in the environment that lead to human exposure. Beryllium can enter water or soil by natural weathering of rock, and can further enter the food chain by plant uptake. However, ingestion of Beryllium is non-toxic, and is not the route of exposure that results in the adverse health outcomes that are of concern. In addition, soluble beryllium compounds such as BeCl2 have been extensively studied, while there is limited information regarding in lab investigations pertaining to exposure to beryllium metal. It is thought that ionization of beryllium salts in aqueous solutions such as cytosol, lead to the cytotoxic effects that are observed with beryllium exposure (103). This is due to the solubility aspect of salts, versus the uptake of metal particles by cells. This is important to account for, as lung clearance and deposition of particles in the parts of the lung is an important aspect of inhalation toxicology.
Inhalation of beryllium containing dust is the main route of exposure leading to disease outcomes. The International Association of Cancer Research (IARC) has classified beryllium and Be containing compounds as group 1 carcinogens. Many studies performed using a rat model have led to the formation of tumors following exposure to beryllium compounds. Some of these studies relied on intraperitoneal, pleural, or intramuscular injection of a Be solution. Though tumor development was detected, this is not a true mimic of a beryllium exposure situation. Inhalation studies in rats have also led to lung tumor formation (104). However, because inhalation of Be is the main route leading to disease, occupational exposures are the leading concern, rather than general population exposure. Beryllium-containing particles can be introduced into the environment from sources such as coal-fire power plants and nuclear waste effluents, but workers that manufacture Be containing products are at the highest risk. Interestingly, there is little conclusive evidence to suggest that exposure to either Be-salts or Be-metal particles leads to genotoxicity or any kind of genetic event(103). Ames tests and mammalian HPRT expression assays have been utilized to explore potential genotoxicity associated with beryllium exposure (103; 105). The results of these investigations have not generated conclusive evidence to suggest that Be is mutagenic, or induces significant DNA repair events in cells (105).
Currently, the US Occupational Safety and Health Administration (OSHA) has a set permissible exposure limit of 0.2 ug/m3 for beryllium. This standard was set based on lung cancer in workers handling beryllium. A recent epidemiological study has reviewed OSHA’s guidelines based on data from a cohort of workers exposed from 1940 to 2005. It was found that the majority of beryllium being used in the workplace was a water insoluble form. Following this data set, the findings concluded that there was a monotonic change in the lung cancer incidence between samples, suggesting that all forms of beryllium (soluble and insoluble) are carcinogenic when inhaled, furthering the classification and regulations set by US regulatory agencies (106). There is limited recent data because the adverse health effects of beryllium are often not observed until up to 40 years after exposure (102).
Another disease outcome associated with occupational beryllium exposure is chronic beryllium disease (CBD). It is a life-long allergic sensitization and subsequent autoimmune-like response to beryllium exposure, that usually manifests sometime after removal from exposure (106; 107). The exposure of beryllium dust and development of CBD is evident by the development of a type of granulomatous pneumoconiosis (108). However, further testing is required to confirm that these granulomas are a direct result of beryllium exposure, as CBD manifests identically to sarcoidosis, and is often-times misdiagnosed (109) (108). A diagnostic test known as the beryllium lymphocyte proliferation assay is required to confirm CBD-induced granuloma (109). Beryllium cations (Be2+) have a high charge capacity and are thought to easily interact with electron donor molecules, which include many cell receptors and molecules involved in biological redox reactions (110). Because of Beryllium’s unique properties, it was found to bind in an MHC-peptide complex and change the properties of this immune response molecule. The changes in this complex caused by Be binding allowed for it to be recognized by T-cell receptors and initiate a hypersensitivity response (107; 111). This research has changed the thoughts on the way Be causes this response, as it was initially thought that Be acted similar to a traditional hapten (112). Rather, genetic screening and molecular-based studies have demonstrated that individuals possessing a genetic variant in the class II MHC allele; HLA-DP2 are highly susceptible to beryllium hypersensitivity, and if exposed, the polymorphism present in this protein elicits this cascading response (102;112; 113). The changes in protein structure present an acidic domain that is wide open for metal cation binding. This allows for the complexation of Be and its subsequent detection and interaction with CD4+ T-cells, invoking an immune response (107). Interestingly, this Be-CD4+ T cell interaction is highly similar to the reactions found in autoimmune diseases. There are very few studies that have addressed molecular mechanisms of Be toxicity and carcinogenicity aside from the SNPs associated with Be susceptibility.
Chromium
While chromium has several oxidation states, the two most important in regards to human health are trivalent (Cr(III)) and hexavalent (Cr(VI)) chromium. Chromium (III) is non-toxic and regarded as a beneficial metal in regards to nutrition and health, while Cr(VI) is extremely toxic and classified as a group I carcinogen by the health regulatory agencies in the United States and the world. It is well established that inhalation of chromate is associated with a wide range of respiratory diseases including lung cancer.
Environmental exposure to chromate is a major human health concern due to its use in occupational settings, and its presence as an environmental contaminant in water sources. According to the US EPA, chromium is released into the environment from industrial sources such as, but not limited to electrochrome plating facilities, leather tanning facilities, and coal-fire power plants. Any type of stainless steel or chrome plated objects, such as cookware, sink faucets, or automobile parts, are coated with chromium to improve the appearance and add a layer of protection from wear and tear. Nutritional benefits from Cr(III) are gained through eating plants, due to bioaccumulation (114). Regarding Cr(VI), as with any toxic substance used in the manufacturing of materials or products, occupational hazard is a substantial risk and appropriate standards and regulations are put into place to limit human exposure. In the work place, both OSHA and NIOSH have set standards for workday exposure via the air to the three predominant forms of chromium. OSHA’s standards are; Cr(0): 1.0 mg/m3, Cr(III): 0.5 mg/m3, and Cr(VI): 0.005 mg/m3. NIOSH standards are; 0.5 mg/m3 for chromium metal and Cr(III). For Cr(VI) the standard is0.001 mg/m3. These air standards are for a normal work day.
While inhalation of chromium is a main route of exposure and the reason why this metal has been classified as a group 1 carcinogen, chromium can also be dispersed in bodies of water as well. The United States EPA has set a water standard for chromium species of 0.1 mg/L for total chromium. This standard is set for total chromium, rather than individual species due to the chemical nature of the chromium ion. The oxidation state of chromium can change under particular environmental conditions. For example, Cr(VI) found in an acidic, aqueous environment with substantial organic matter can undergo Fenton-like reduction reactions, resulting in the step-wise reduction to Cr(III) (115; 116). The California public health goal for Cr(VI) is 0.02 ppb.
While Cr(VI) in itself is considered non-mutagenic or interactive with biomolecules in the cells, the detoxification processes that it undergoes is what produces the toxicity that leads to various DNA lesions, cytotoxicity and tumor development (117;118). In the extracellular microenvironment, Cr(VI) can be reduced to Cr(III), which has poor membrane permeability and cannot easily enter the cell (119-121). This renders the Cr non-toxic before interacting with the molecular scaffolding and networks inside the cell itself. However, in aqueous environments hexavalent chromium predominantly exists as the chromate anion (CrO4−2). Chromate closely resembles the sulfate and phosphate anions (SO4−2, PO4−2, respectively) in structure, and therefore can readily pass through the cell membrane via anion transporters (75; 117). One of the major mechanisms by which Cr(IV) induces cellular damage is through the generation of ROS. The presence of Cr(VI) in the cell is detected by ROS scavengers such as glutathione and vitamin C (116;122). These molecules will bind Cr(VI) and reduce it to Cr(III). Interestingly, in vivo studies have found that vitamin C is the predominant scavenger to reduce Cr(VI), while in contrast, in vitro studies demonstrate that glutathione is the main interactor. This is due to the fact that cultured cells have significantly less vitamin C in their media and (117;119) supplemented serum compared to animals. In in vitro environments the concentration of ascorbate is extremely low, about 50 uM, and is provided solely by the supplementation of animal serum into cell culture media. This is a small fraction of the mM concentrations found in vivo, which is sustained due to nutritional uptake. Zhitkovich and colleagues discovered that this cell culture situation highly underestimates the cellular response to chromium exposure. Furthermore, addition of ascorbate back into the cell culture medium to improve the intercellular concentration decreases overall oxidative stress, but induces DNA double strand breaks by the interaction of DNA with Asc-Cr crosslinks (118; 123; 124). Though reduction of xenobiotics in the cell is considered a detoxifying process, this step-wise reaction will lead to the activation of Cr intermediates (117; 122). Reduction processes will generate free radicals such as hydroxide radical (•OH), and due to an imbalance of scavengers, ROS can accumulate and cause damage. In addition, because these detoxification reactions are step-wise, highly reactive Cr species such as Cr(V) and Cr(IV) are generated, which can in turn cause DNA damage as well as other cellular insults (125).
The scope of the damage induced by reductive products is DNA damage, including irreparable adducts, which can lead to mutations. Interestingly, in vitro studies on genotoxicity of Cr(VI) have found that Cr detoxification products form binary Cr-DNA adducts, but are weakly mutagenic, and easily repaired. However, the major adducts that cause lasting damage are those that have Cr conjugated to the ROS scavengers. Bulky adducts have been detected such as GSH-Cr-DNA and Vitamin C-Cr-DNA, as well as some amino acid residues of protein, such as histidine or cysteine (118). These large adducts can block replication, and lead to mutation (126). Experimental evidence suggests that these adducts are repaired by large patch nucleotide excision repair, as NER deficient cells were unable to repair many Cr-induced adducts, compared to control (127). Chromium can also induce changes in the epigenome, altering chromatin state via changes in histone modifications, and even change the DNA methylation landscape (128; 129). The extensive cross-talk between the ‘layers’ of epigenetic signals creates a complex network of gene expression within the cell.
Metal-induced changes in gene expression via epigenetic alterations at the chromatin and DNA level have been extensively studied and are thought to be one of the main contributing factors to the adverse responses and disease outcomes associated with chronic exposure to metals in the environment (75). Exposure to Cr(VI) is associated with changes in various histone marks including, decreased H3K27Me3, and increased levels of H3K4Me3 as well as H3K9Me2 and H3K9Me3 (130). Interestingly, Cr appears to be linked to a positive feedback look with a few interacting molecules that result in changes in the expression of the mismatch repair gene MLH1. Exposure to Cr induces the expression of the histone methyltransferase G9a, which is responsible for adding posttranslational methylation to H3K9. Increased level of H3K9Me2 have been detected in A549 cells exposed to Cr(VI). This results in a heterochromatin state and in turn will decrease the expression of the MLH1 gene, which limits the DNA repair capacity of the cell (130). As stated earlier, bulky Cr-DNA lesions are not well repaired and shutting down the repair machinery is adding to the threat of persistent mutations. It is estimated that about half of the US superfund sites across the country have chromium contamination (131). These wide range environmental dispersal along with the many different types of cellular damage associated with chronic exposure to chromate make it a dangerous and high priority environmental contaminant.
Acknowledgments
Funding: This research was funded by the following NIH grants: ES000260, ES022935, ES023174, ES02613
Contributor Information
Qiao Yi Chen, Department of Environmental Medicine, Dept of Biochemistry and Molecular Pharmacology, New York University School of Medicine, 341 East 25 Street, New York, NY 10010, Telephone: 316-300-5760 qyc203@nyu.edu.
Thomas DesMarais, Department of Environmental Medicine, Dept of Biochemistry and Molecular Pharmacology, New York University School of Medicine, 341 East 25 Street, New York, NY 10010, Telephone: 845-416-2404 tdm311@nyu.edu.
Max Costa, Department of Environmental Medicine, Dept of Biochemistry and Molecular Pharmacology, New York University School of Medicine, 341 East 25 Street, New York, NY 10010, Telephone: 845-731-3515, Fax: 845-351-2118 Max.Costa@nyumc.org.
References
- 1.Chen H, Giri NC, Zhang R, Yamane K, Zhang Y, et al. 2017. Nickel ions inhibit histone demethylase JMJD1A and DNA repair enzyme ABH2 by replacing the ferrous iron in the catalytic centers. JBiol Chem 292:10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen QY, Costa M. 2017. A comprehensive review of metal-induced cellular transformation studies. Toxicol Appl Pharmacol 331:33–40 [DOI] [PubMed] [Google Scholar]
- 3.Pinto VS, Marques SCR, Rodrigues P, Barros MT, Costa ML, et al. 2017. An electrospray ionization mass spectrometry study of azidoacetic acid/transition metal complexes. Rapid communications in mass spectrometry: RCM 31:1001–13 [DOI] [PubMed] [Google Scholar]
- 4.Bishak YK, Payahoo L, Osatdrahimi A, Nourazarian A. 2015. Mechanisms of cadmium carcinogenicity in the gastrointestinal tract. Asian Pac J Cancer Prev 16:9–21 [DOI] [PubMed] [Google Scholar]
- 5.Oudeh M, Khan M, Scullion J. 2002. Plant accumulation of potentially toxic elements in sewage sludge as affected by soil organic matter level and mycorrhizal fungi. Environ Pollut 116:293–300 [DOI] [PubMed] [Google Scholar]
- 6.Schoeters G, Den Hond E, Zuurbier M, Naginiene R, van den Hazel P, et al. 2006. Cadmium and children: exposure and health effects. Acta Paediatr Suppl 95:50–4 [DOI] [PubMed] [Google Scholar]
- 7.Sahmoun AE, Case LD, Jackson SA, Schwartz GG. 2005. Cadmium and prostate cancer: a critical epidemiologic analysis. Cancer Invest 23:256–63 [DOI] [PubMed] [Google Scholar]
- 8.Zalups RK, Ahmad S. 2003. Molecular handling of cadmium in transporting epithelia. Toxicol Appl Pharmacol 186:163–88 [DOI] [PubMed] [Google Scholar]
- 9.Johnson MD, Kenney N, Stoica A, Hilakivi-Clarke L, Singh B, et al. 2003. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nat Med 9:1081–4 [DOI] [PubMed] [Google Scholar]
- 10.Viaene MK, Masschelein R, Leenders J, De Groof M, Swerts LJ, Roels HA. 2000. Neurobehavioural effects of occupational exposure to cadmium: a cross sectional epidemiological study. Occup Environ Med 57:19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feki-Tounsi M, Hamza-Chaffai A. 2014. Cadmium as a possible cause of bladder cancer: a review of accumulated evidence. Environ Sci Pollut Res Int 21:10561–73 [DOI] [PubMed] [Google Scholar]
- 12.Joseph P 2009. Mechanisms of cadmium carcinogenesis. Toxicol Appl Pharmacol 238:272–9 [DOI] [PubMed] [Google Scholar]
- 13.Cheng MB, Zhang Y, Cao CY, Zhang WL, Zhang Y, Shen YF. 2014. Specific phosphorylation of histone demethylase KDM3A determines target gene expression in response to heat shock. PLoS Biol 12:e1002026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waalkes MP. 2003. Cadmium carcinogenesis. Mutat Res 533:107–20 [DOI] [PubMed] [Google Scholar]
- 15.Suzuki M, Takeda S, Teraoka-Nishitani N, Yamagata A, Tanaka T, et al. 2017. Cadmium-induced malignant transformation of rat liver cells: Potential key role and regulatory mechanism of altered apolipoprotein E expression in enhanced invasiveness. Toxicology 382:16–23 [DOI] [PubMed] [Google Scholar]
- 16.Waalkes MP, Poirier LA. 1984. In vitro cadmium-DNA interactions: cooperativity of cadmium binding and competitive antagonism by calcium, magnesium, and zinc. Toxicol Appl Pharmacol 75:539–46 [DOI] [PubMed] [Google Scholar]
- 17.Filipic M, Fatur T, Vudrag M. 2006. Molecular mechanisms of cadmium induced mutagenicity. Hum Exp Toxicol 25:67–77 [DOI] [PubMed] [Google Scholar]
- 18.Inglot P, Lewinska A, Potocki L, Oklejewicz B, Tabecka-Lonczynska A, et al. 2012. Cadmium-induced changes in genomic DNA-methylation status increase aneuploidy events in a pig Robertsonian translocation model. Mutat Res 747:182–9 [DOI] [PubMed] [Google Scholar]
- 19.Benbrahim-Tallaa L, Liu J, Webber MM, Waalkes MP. 2007. Estrogen signaling and disruption of androgen metabolism in acquired androgen-independence during cadmium carcinogenesis in human prostate epithelial cells. Prostate 67:135–45 [DOI] [PubMed] [Google Scholar]
- 20.Benbrahim-Tallaa L, Waterland RA, Dill AL, Webber MM, Waalkes MP. 2007. Tumor suppressor gene inactivation during cadmium-induced malignant transformation of human prostate cells correlates with overexpression of de novo DNA methyltransferase. Environ Health Perspect 115:1454–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang D, Zhang Y, Qi Y, Chen C, Ji W. 2008. Global DNA hypomethylation, rather than reactive oxygen species (ROS), a potential facilitator of cadmium-stimulated K562 cell proliferation. Toxicol Lett 179:43–7 [DOI] [PubMed] [Google Scholar]
- 22.Xiao C, Liu Y, Xie C, Tu W, Xia Y, et al. 2015. Cadmium induces histone H3 lysine methylation by inhibiting histone demethylase activity. Toxicol Sci 145:80–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rani A, Kumar A, Lal A, Pant M. 2014. Cellular mechanisms of cadmium-induced toxicity: a review. IntJ Environ Health Res 24:378–99 [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Qu W, Kadiiska MB. 2009. Role of oxidative stress in cadmium toxicity and carcinogenesis. Toxicol Appl Pharmacol 238:209–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stohs SJ, Bagchi D, Hassoun E, Bagchi M. 2001. Oxidative mechanisms in the toxicity of chromium and cadmium ions. J Environ Pathol Toxicol Oncol 20:77–88 [PubMed] [Google Scholar]
- 26.Hartwig A 2010. Mechanisms in cadmium-induced carcinogenicity: recent insights. Biometals 23:951–60 [DOI] [PubMed] [Google Scholar]
- 27.Giaginis C, Gatzidou E, Theocharis S. 2006. DNA repair systems as targets of cadmium toxicity. Toxicol Appl Pharmacol 213:282–90 [DOI] [PubMed] [Google Scholar]
- 28.Jin YH, Clark AB, Slebos RJ, Al-Refai H, Taylor JA, et al. 2003. Cadmium is a mutagen that acts by inhibiting mismatch repair. Nat Genet 34:326–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertin G, Averbeck D. 2006. Cadmium: cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review). Biochimie 88:1549–59 [DOI] [PubMed] [Google Scholar]
- 30.Spruill MD, Song B, Whong WZ, Ong T. 2002. Proto-oncogene amplification and overexpression in cadmium-induced cell transformation. J Toxicol Environ Health A 65:2131–44 [DOI] [PubMed] [Google Scholar]
- 31.Fang MZ, Mar W, Cho MH. 2002. Cadmium affects genes involved in growth regulation during two-stage transformation of Balb/3T3 cells. Toxicology 177:253–65 [DOI] [PubMed] [Google Scholar]
- 32.Takiguchi M, Achanzar WE, Qu W, Li G, Waalkes MP. 2003. Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp Cell Res 286:355–65 [DOI] [PubMed] [Google Scholar]
- 33.Martinez-Zamudio R, Ha HC. 2011. Environmental epigenetics in metal exposure. Epigenetics 6:820–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doi T, Puri P, McCann A, Bannigan J, Thompson J. 2011. Epigenetic effect of cadmium on global de novo DNA hypomethylation in the cadmium-induced ventral body wall defect (VBWD) in the chick model. Toxicol Sci 120:475–80 [DOI] [PubMed] [Google Scholar]
- 35.Jiang G, Xu L, Song S, Zhu C, Wu Q, et al. 2008. Effects of long-term low-dose cadmium exposure on genomic DNA methylation in human embryo lung fibroblast cells. Toxicology 244:49–55 [DOI] [PubMed] [Google Scholar]
- 36.Brocato J, Costa M. 2013. Basic mechanics of DNA methylation and the unique landscape of the DNA methylome in metal-induced carcinogenesis. Crit Rev Toxicol 43:493–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luevano J, Damodaran C. 2014. A review of molecular events of cadmium-induced carcinogenesis. J Environ Pathol Toxicol Oncol 33:183–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen P, Duan X, Li M, Huang C, Li J, et al. 2016. Systematic network assessment of the carcinogenic activities of cadmium. Toxicol Appl Pharmacol 310:150–8 [DOI] [PubMed] [Google Scholar]
- 39.Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, et al. 1992. TGF beta signals through a heteromeric protein kinase receptor complex. Cell 71:1003–14 [DOI] [PubMed] [Google Scholar]
- 40.Vardabasso C, Hasson D, Ratnakumar K, Chung CY, Duarte LF, Bernstein E. 2014. Histone variants: emerging players in cancer biology. Cell Mol Life Sci 71:379–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bjorklund G, Aaseth J, Chirumbolo S, Urbina MA, Uddin R. 2017. Effects of arsenic toxicity beyond epigenetic modifications. Environ Geochem Health [DOI] [PubMed] [Google Scholar]
- 42.Mandal P 2017. Molecular insight of arsenic-induced carcinogenesis and its prevention. Naunyn Schmiedebergs Arch Pharmacol 390:443–55 [DOI] [PubMed] [Google Scholar]
- 43.Eckstein M, Eleazer R, Rea M, Fondufe-Mittendorf Y. 2017. Epigenomic reprogramming in inorganic arsenic-mediated gene expression patterns during carcinogenesis. Rev Environ Health 32:93–103 [DOI] [PubMed] [Google Scholar]
- 44.Zhang Z, Pratheeshkumar P, Budhraja A, Son YO, Kim D, Shi X. 2015. Role of reactive oxygen species in arsenic-induced transformation of human lung bronchial epithelial (BEAS-2B) cells. Biochem Biophys Res Commun 456:643–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tandon N, Roy M, Roy S, Gupta N. 2012. Protective Effect of Psidium guajava in Arsenic-induced Oxidative Stress and Cytological Damage in Rats. Toxicol Int 19:245–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta DK, Inouhe M, Rodriguez-Serrano M, Romero-Puertas MC, Sandalio LM. 2013. Oxidative stress and arsenic toxicity: role of NADPH oxidases. Chemosphere 90:1987–96 [DOI] [PubMed] [Google Scholar]
- 47.Vattanasit U, Navasumrit P, Khadka MB, Kanitwithayanun J, Promvijit J, et al. 2014. Oxidative DNA damage and inflammatory responses in cultured human cells and in humans exposed to traffic-related particles. Int J Hyg Environ Health 217:23–33 [DOI] [PubMed] [Google Scholar]
- 48.Xie H, Huang S, Martin S, Wise JP Sr., 2014. Arsenic is cytotoxic and genotoxic to primary human lung cells. Mutat Res Genet Toxicol Environ Mutagen 760:33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kesari VP, Kumar A, Khan PK. 2012. Genotoxic potential of arsenic at its reference dose. Ecotoxicol Environ Saf 80:126–31 [DOI] [PubMed] [Google Scholar]
- 50.Warner ML, Moore LE, Smith MT, Kalman DA, Fanning E, Smith AH. 1994. Increased micronuclei in exfoliated bladder cells of individuals who chronically ingest arsenic-contaminated water in Nevada. Cancer Epidemiol Biomarkers Prev 3:583–90 [PubMed] [Google Scholar]
- 51.Arrigo AP. 1983. Acetylation and methylation patterns of core histones are modified after heat or arsenite treatment of Drosophila tissue culture cells. Nucleic Acids Res 11:1389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choudhury SR, Cui Y, Narayanan A, Gilley DP, Huda N, et al. 2016. Optogenetic regulation of site-specific subtelomeric DNA methylation. Oncotarget 7:50380–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Eijk KR, de Jong S, Boks MP, Langeveld T, Colas F, et al. 2012. Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMC Genomics 13:636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bustaffa E, Stoccoro A, Bianchi F, Migliore L. 2014. Genotoxic and epigenetic mechanisms in arsenic carcinogenicity. Arch Toxicol 88:1043–67 [DOI] [PubMed] [Google Scholar]
- 55.Rojas D, Rager JE, Smeester L, Bailey KA, Drobna Z, et al. 2015. Prenatal arsenic exposure and the epigenome: identifying sites of 5-methylcytosine alterations that predict functional changes in gene expression in newborn cord blood and subsequent birth outcomes. Toxicol Sci 143:97–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paul S, Bhattacharjee P, Giri AK, Bhattacharjee P. 2017. Arsenic toxicity and epimutagenecity: the new LINEage. Biometals 30:505–15 [DOI] [PubMed] [Google Scholar]
- 57.Lambrou A, Baccarelli A, Wright RO, Weisskopf M, Bollati V, et al. 2012. Arsenic exposure and DNA methylation among elderly men. Epidemiology 23:668–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tajuddin SM, Amaral AF, Fernandez AF, Rodriguez-Rodero S, Rodriguez RM, et al. 2013. Genetic and non-genetic predictors of LINE-1 methylation in leukocyte DNA. Environ Health Perspect 121:650–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gardner KE, Allis CD, Strahl BD. 2011. Operating on chromatin, a colorful language where context matters. J Mol Biol 409:36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chervona Y, Hall MN, Arita A, Wu F, Sun H, et al. 2012. Associations between arsenic exposure and global posttranslational histone modifications among adults in Bangladesh. Cancer Epidemiol Biomarkers Prev 21:2252–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou X, Li Q, Arita A, Sun H, Costa M. 2009. Effects of nickel, chromate, and arsenite on histone 3 lysine methylation. Toxicol Appl Pharmacol 236:78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou X, Sun H, Ellen TP, Chen H, Costa M. 2008. Arsenite alters global histone H3 methylation. Carcinogenesis 29:1831–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kannan-Thulasiraman P, Katsoulidis E, Tallman MS, Arthur JS, Platanias LC. 2006. Activation of the mitogen- and stress-activated kinase 1 by arsenic trioxide. J Biol Chem 281:22446–52 [DOI] [PubMed] [Google Scholar]
- 64.Winter S, Simboeck E, Fischle W, Zupkovitz G, Dohnal I, et al. 2008. 14-3-3 proteins recognize a histone code at histone H3 and are required for transcriptional activation. EMBO J 27:88–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rea M, Jiang T, Eleazer R, Eckstein M, Marshall AG, Fondufe-Mittendorf YN. 2016. Quantitative Mass Spectrometry Reveals Changes in Histone H2B Variants as Cells Undergo Inorganic Arsenic-Mediated Cellular Transformation. Mol Cell Proteomics 15:2411–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brocato J, Chen D, Liu J, Fang L, Jin C, Costa M. 2015. A Potential New Mechanism of Arsenic Carcinogenesis: Depletion of Stem-Loop Binding Protein and Increase in Polyadenylated Canonical Histone H3.1 mRNA. Biol Trace Elem Res 166:72–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Riedmann C, Ma Y, Melikishvili M, Godfrey SG, Zhang Z, et al. 2015. Inorganic Arsenic-induced cellular transformation is coupled with genome wide changes in chromatin structure, transcriptome and splicing patterns. BMC Genomics 16:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.David CJ, Chen M, Assanah M, Canoll P, Manley JL. 2010. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463:364–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nowak DG, Woolard J, Amin EM, Konopatskaya O, Saleem MA, et al. 2008. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J Cell Sci 121:3487–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sveen A, Kilpinen S, Ruusulehto A, Lothe RA, Skotheim RI. 2016. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 35:2413–27 [DOI] [PubMed] [Google Scholar]
- 71.Agirre E, Bellora N, Allo M, Pages A, Bertucci P, et al. 2015. A chromatin code for alternative splicing involving a putative association between CTCF and HP1alpha proteins. BMC Biol 13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matveeva E, Maiorano J, Zhang Q, Eteleeb AM, Convertini P, et al. 2016. Involvement of PARP1 in the regulation of alternative splicing. Cell Discov 2:15046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Le Thomas A, Toth KF, Aravin AA. 2014. To be or not to be a piRNA: genomic origin and processing of piRNAs. Genome Biol 15:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marsit CJ, Eddy K, Kelsey KT. 2006. MicroRNA responses to cellular stress. Cancer Res 66:10843–8 [DOI] [PubMed] [Google Scholar]
- 75.Chervona Y, Arita A, Costa M. 2012. Carcinogenic metals and the epigenome: understanding the effect of nickel, arsenic, and chromium. Metallomics 4:619–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Coogan TP, Latta DM, Snow ET, Costa M. 1989. Toxicity and carcinogenicity of nickel compounds. Crit Rev Toxicol 19:341–84 [DOI] [PubMed] [Google Scholar]
- 77.Kasprzak KS, Sunderman FW Jr., Salnikow K 2003. Nickel carcinogenesis. Mutat Res 533:67–97 [DOI] [PubMed] [Google Scholar]
- 78.Doll R, Mathews JD, Morgan LG. 1977. Cancers of the lung and nasal sinuses in nickel workers: a reassessment of the period of risk. Br JInd Med 34:102–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ke Q, Costa M. 2006. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol 70:1469–80 [DOI] [PubMed] [Google Scholar]
- 80.Maxwell P, Salnikow K. 2004. HIF-1: an oxygen and metal responsive transcription factor. Cancer Biol Ther 3:29–35 [DOI] [PubMed] [Google Scholar]
- 81.Salceda S, Caro J. 1997. Hypoxia-inducible factor lalpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 272:22642–7 [DOI] [PubMed] [Google Scholar]
- 82.Salnikow K, Zhitkovich A. 2008. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium. Chem Res Toxicol 21:28–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen H, Kluz T, Zhang R, Costa M. 2010. Hypoxia and nickel inhibit histone demethylase JMJD1A and repress Spry2 expression in human bronchial epithelial BEAS-2B cells. Carcinogenesis 31:2136–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen H, Giri NC, Zhang R, Yamane K, Zhang Y, et al. 2010. Nickel ions inhibit histone demethylase JMJD1A and DNA repair enzyme ABH2 by replacing the ferrous iron in the catalytic centers. J Biol Chem 285:7374–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Giri NC, Passantino L, Sun H, Zoroddu MA, Costa M, Maroney MJ. 2013. Structural investigations of the nickel-induced inhibition of truncated constructs of the JMJD2 family of histone demethylases using X-ray absorption spectroscopy. Biochemistry 52:4168–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carrington PE, Al-Mjeni F, Zoroddu MA, Costa M, Maroney MJ. 2002. Use of XAS for the elucidation of metal structure and function: applications to nickel biochemistry, molecular toxicology, and carcinogenesis. Environ Health Perspect 110 Suppl 5:705–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Patierno SR, Sugiyama M, Basilion JP, Costa M. 1985. Preferential DNA-protein cross-linking by NiCl2 in magnesium-insoluble regions of fractionated Chinese hamster ovary cell chromatin. Cancer Res 45:5787–94 [PubMed] [Google Scholar]
- 88.Sen P, Conway K, Costa M. 1987. Comparison of the localization of chromosome damage induced by calcium chromate and nickel compounds. Cancer Res 47:2142–7 [PubMed] [Google Scholar]
- 89.Conway K, Costa M. 1989. Nonrandom chromosomal alterations in nickel-transformed Chinese hamster embryo cells. Cancer Res 49:6032–8 [PubMed] [Google Scholar]
- 90.Yang LQ, Ji WD, Tao GH, Zhang WJ, Gong CM, et al. 2010. [Genome DNA hypomethylation in the process of crystalline nickel-induced cell malignant transformation]. Zhonghua Yu Fang YiXue Za Zhi 44:622–5 [PubMed] [Google Scholar]
- 91.Lee YW, Klein CB, Kargacin B, Salnikow K, Kitahara J, et al. 1995. Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: a new model for epigenetic carcinogens. Mol Cell Biol 15:2547–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Klein CB, Costa M. 1997. DNA methylation, heterochromatin and epigenetic carcinogens. Mutat Res 386:163–80 [DOI] [PubMed] [Google Scholar]
- 93.Ellen TP, Kluz T, Harder ME, Xiong J, Costa M. 2009. Heterochromatinization as a potential mechanism of nickel-induced carcinogenesis. Biochemistry 48:4626–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu CH, Tang SC, Wang PH, Lee H, Ko JL. 2012. Nickel-induced epithelial-mesenchymal transition by reactive oxygen species generation and E- cadherin promoter hypermethylation. JBiol Chem 287:25292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ke Q, Li Q, Ellen TP, Sun H, Costa M. 2008. Nickel compounds induce phosphorylation of histone H3 at serine 10 by activating JNK-MAPK pathway. Carcinogenesis 29:1276–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bal W, Kasprzak KS. 2002. Induction of oxidative DNA damage by carcinogenic metals. Toxicol Lett 127:55–62 [DOI] [PubMed] [Google Scholar]
- 97.Jordan A, Zhang X, Li J, Laulicht-Glick F, Sun H, Costa M. 2017. Nickel and cadmium-induced SLBP depletion: A potential pathway to metal mediated cellular transformation. PLoS One 12:e0173624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, et al. 2002. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 99:15524–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chiou YH, Liou SH, Wong RH, Chen CY, Lee H. 2015. Nickel may contribute to EGFR mutation and synergistically promotes tumor invasion in EGFR-mutated lung cancer via nickel-induced microRNA-21 expression. Toxicol Lett 237:46–54 [DOI] [PubMed] [Google Scholar]
- 100.Ji W, Yang L, Yuan J, Yang L, Zhang M, et al. 2013. MicroRNA-152 targets DNA methyltransferase 1 in NiS-transformed cells via a feedback mechanism. Carcinogenesis 34:446–53 [DOI] [PubMed] [Google Scholar]
- 101.Zhang J, Zhou Y, Wu YJ, Li MJ, Wang RJ, et al. 2013. Hyper-methylated miR-203 dysregulates ABL1 and contributes to the nickel-induced tumorigenesis. Toxicol Lett 223:42–51 [DOI] [PubMed] [Google Scholar]
- 102.Taylor TP, Ding M, Ehler DS, Foreman TM, Kaszuba JP, Sauer NN. 2003. Beryllium in the environment: a review. J Environ Sci Health A Tox Hazard Subst Environ Eng 38:439–69 [DOI] [PubMed] [Google Scholar]
- 103.Strupp C 2011. Beryllium metal I. experimental results on acute oral toxicity, local skin and eye effects, and genotoxicity. Ann Occup Hyg 55:30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.National Toxicology P 2011. Beryllium and beryllium compounds. Rep Carcinog 12:67–70 [PubMed] [Google Scholar]
- 105.Strupp C 2011. Beryllium metal II. a review of the available toxicity data. Ann Occup Hyg 55:43–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schubauer-Berigan MK, Couch JR, Deddens JA. 2017. Is beryllium-induced lung cancer caused only by soluble forms and high exposure levels? Occup Environ Med 74:601–3 [DOI] [PubMed] [Google Scholar]
- 107.Clayton GM, Wang Y, Crawford F, Novikov A, Wimberly BT, et al. 2014. Structural basis of chronic beryllium disease: linking allergic hypersensitivity and autoimmunity. Cell 158:132–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cullinan P, Munoz X, Suojalehto H, Agius R, Jindal S, et al. 2017. Occupational lung diseases: from old and novel exposures to effective preventive strategies. Lancet Respir Med 5:445–55 [DOI] [PubMed] [Google Scholar]
- 109.Mayer AS, Hamzeh N, Maier LA. 2014. Sarcoidosis and chronic beryllium disease: similarities and differences. Semin Respir Crit Care Med 35:316–29 [DOI] [PubMed] [Google Scholar]
- 110.Sutton M, Burastero SR. 2003. Beryllium chemical speciation in elemental human biological fluids. Chem Res Toxicol 16:1145–54 [DOI] [PubMed] [Google Scholar]
- 111.Dai S, Falta MT, Bowerman NA, McKee AS, Fontenot AP. 2013. T cell recognition of beryllium. Curr Opin Immunol 25:775–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Illing PT, Vivian JP, Purcell AW, Rossjohn J, McCluskey J. 2013. Human leukocyte antigen-associated drug hypersensitivity. Curr Opin Immunol 25:81–9 [DOI] [PubMed] [Google Scholar]
- 113.Kwo E, Christiani D. 2017. The role of gene-environment interplay in occupational and environmental diseases: current concepts and knowledge gaps. Curr Opin Pulm Med 23:173–6 [DOI] [PubMed] [Google Scholar]
- 114.Uddin AN, Burns FJ, Rossman TG, Chen H, Kluz T, Costa M. 2007. Dietary chromium and nickel enhance UV-carcinogenesis in skin of hairless mice. Toxicol Appl Pharmacol 221:329–38 [DOI] [PubMed] [Google Scholar]
- 115.Luo H, Lu Y, Shi X, Mao Y, Dalal NS. 1996. Chromium (IV)-mediated fenton-like reaction causes DNA damage: implication to genotoxicity of chromate. Ann Clin Lab Sci 26:185–91 [PubMed] [Google Scholar]
- 116.Sugden KD, Stearns DM. 2000. The role of chromium(V) in the mechanism of chromate-induced oxidative DNA damage and cancer. J Environ Pathol Toxicol Oncol 19:215–30 [PubMed] [Google Scholar]
- 117.Zhitkovich A 2011. Chromium in drinking water: sources, metabolism, and cancer risks. Chem Res Toxicol 24:1617–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhitkovich A 2005. Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium(VI). Chem Res Toxicol 18:3–11 [DOI] [PubMed] [Google Scholar]
- 119.Standeven AM, Wetterhahn KE. 1992. Ascorbate is the principal reductant of chromium(VI) in rat lung ultrafiltrates and cytosols, and mediates chromium-DNA binding in vitro. Carcinogenesis 13:1319–24 [DOI] [PubMed] [Google Scholar]
- 120.Suzuki Y, Fukuda K. 1990. Reduction of hexavalent chromium by ascorbic acid and glutathione with special reference to the rat lung. Arch Toxicol 64:169–76 [DOI] [PubMed] [Google Scholar]
- 121.Suzuki T, Miyata N, Horitsu H, Kawai K, Takamizawa K, et al. 1992. NAD(P)H-dependent chromium (VI) reductase of Pseudomonas ambigua G-1: a Cr(V) intermediate is formed during the reduction of Cr(VI) to Cr(III). J Bacteriol 174:5340–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Arita A, Costa M. 2009. Epigenetics in metal carcinogenesis: nickel, arsenic, chromium and cadmium. Metallomics 1:222–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Martin BD, Schoenhard JA, Hwang JM, Sugden KD. 2006. Ascorbate is a pro-oxidant in chromium-treated human lung cells. Mutat Res 610:74–84 [DOI] [PubMed] [Google Scholar]
- 124.Reynolds M, Stoddard L, Bespalov I, Zhitkovich A. 2007. Ascorbate acts as a highly potent inducer of chromate mutagenesis and clastogenesis: linkage to DNA breaks in G2 phase by mismatch repair. Nucleic Acids Res 35:465–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jomova K, Valko M. 2011. Advances in metal-induced oxidative stress and human disease. Toxicology 283:65–87 [DOI] [PubMed] [Google Scholar]
- 126.Quievryn G, Peterson E, Messer J, Zhitkovich A. 2003. Genotoxicity and mutagenicity of chromium(VI)/ascorbate-generated DNA adducts in human and bacterial cells. Biochemistry 42:1062–70 [DOI] [PubMed] [Google Scholar]
- 127.Reynolds M, Peterson E, Quievryn G, Zhitkovich A. 2004. Human nucleotide excision repair efficiently removes chromium-DNA phosphate adducts and protects cells against chromate toxicity. J Biol Chem 279:30419–24 [DOI] [PubMed] [Google Scholar]
- 128.Arita A, Shamy MY, Chervona Y, Clancy HA, Sun H, et al. 2012. The effect of exposure to carcinogenic metals on histone tail modifications and gene expression in human subjects. Journal of trace elements in medicine and biology : organ of the Society for Minerals and Trace Elements 26:174–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chervona Y, Costa M. 2012. The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free radical biology & medicine 53:1041–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sun H, Zhou X, Chen H, Li Q, Costa M. 2009. Modulation of histone methylation and MLH1 gene silencing by hexavalent chromium. Toxicol Appl Pharmacol 237:258–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Aldrich MV, Gardea-Torresdey JL, Peralta-Videa JR, Parsons JG. 2003. Uptake and reduction of Cr(VI) to Cr(III) by mesquite (Prosopis spp.): chromateplant interaction in hydroponics and solid media studied using XAS. Environ Sci Technol 37:1859–64 [DOI] [PubMed] [Google Scholar]