

Abstract
The aetiopathogenesis of inflammatory bowel diseases (IBD) involves the complex interaction between a patient’s genetic predisposition, environment, gut microbiota and immune system. Currently, however, it is not known if the distinctive perturbations of the gut microbiota that appear to accompany both Crohn’s disease and ulcerative colitis are the cause of, or the result of, the intestinal inflammation that characterizes IBD.
With the utilization of novel systems biology technologies, we can now begin to understand not only details about compositional changes in the gut microbiota in IBD, but increasingly also the alterations in microbiota function that accompany these. Technologies such as metagenomics, metataxomics, metatranscriptomics, metaproteomics and metabonomics are therefore allowing us a deeper understanding of the role of the microbiota in IBD. Furthermore, the integration of these systems biology technologies through advancing computational and statistical techniques are beginning to understand the microbiome interactions that both contribute to health and diseased states in IBD.
This review aims to explore how such systems biology technologies are advancing our understanding of the gut microbiota, and their potential role in delineating the aetiology, development and clinical care of IBD.
Keywords: bioinformatics, genomics, gut microbiota, inflammatory bowel diseases, interactome, metagenomics, metatranscriptomics metabonomics, proteomics
Introduction
Crohn’s disease (CD) and ulcerative colitis (UC) are relapsing and remitting chronic inflammatory conditions that fall under the umbrella term of inflammatory bowel diseases (IBD). Both disorders generally tend to become apparent in people between the age of 20 and 40 years. The prevalence of IBD in the UK is 0.5–1% with an estimated 620,000 people thought to be affected. 1 Although there are methods of inducing remission in both diseases, currently neither are curable.
IBD are multifactorial. The exact aetiology is incompletely understood, but it is thought to involve the complex interaction between a patient’s genetic predisposition, environment, gut microbiota and immune system. To date, when studying these components in isolation, each has shown changes that are associated with CD and UC, but it has not generally been possible to directly link any of these factors in isolation as a cause of IBD. This disconnect leads us to conclude that these components form intricate and complex interactions that contribute both to the initiation and maintenance of the intestinal inflammation that typifies IBD.
Specific interest in the contribution of the gut microbiota to IBD has been growing. It has been established that perturbation of the structure of the gut microbiota has been linked to intestinal inflammation. 2 Characteristic findings include a decrease in bacterial diversity and richness3–6 and a decrease in temporal stability. 7 Further-more, key changes have been identified in IBD such as a reduction in species derived from the Firmicutes phylum (such as Faecalibacterium prausnitzii 8 ) and increases in species derived from Proteobacteria (including members of the family Enterobacteriaceae9,10). Currently, it is not understood whether perturbation of the structure of the gut microbiota is the cause of, or the result of, intestinal inflammation.
Before the utilization of systems biology techniques, our knowledge regarding the gut microbiota was limited to culture-based techniques, which are labour-intensive and not high-throughput. In particular, they require specific conditions to optimize bacterial growth (e.g. an anaerobic environment and selective media for each species). Despite these limitations, however, Browne and colleagues created a workflow that demonstrated that by using a complex, broad range bacteriological medium, it was possible to archive bacteria representing 96% of the bacterial abundance at the genus level and 90% of the bacterial abundance at the species level. 11
Despite such advancements in culturing techniques, they remain limited by their inability to detect other key microbiome components, such as the virome, mycobiome and archaea. However, recent advancements in high-throughput ‘omic’ systems biology techniques, designed to detect the entire spectrum of particular components under scrutiny in biofluids or tissues, have given novel insight into the structure and function of the gut microbiota.
To begin to understand the complexity of each of these components that contribute to IBD, novel techniques have been utilized to begin to understand their function on a different level. Through the use of bioinformatics pipelines, we are increasingly able to analyze biological molecules and profile microorganisms in greater detail than ever before. The development of novel systems biology techniques - including genomics, metabonomics, transcriptomics and proteomics - presents a new frontier in attempts to understand the complex interactions and the multifactorial nature of IBD. These ‘omic’ technologies allow us direct analysis of members of the microbiota, their genes, transcripts, metabolites and proteins from biological samples, which overcomes the bias and limitation of previous culture techniques, but sometimes brings new biases and challenges.
These systems biology platforms provide us with not only details about compositional changes in IBD, but in particular give us a better comprehension of the functional alterations that may contribute to IBD. Such techniques provide exciting technology that may help us understand the underlying cause for IBD, as well as highlight predictors of disease and novel therapeutic markers.
Systems biology platform studies can be performed on both the host and the microbiota community. Specific examples of omic studies in the host are genomic studies, the mostly widely studied omic studies in IBD, which have identified up to 163 IBD-specific loci. 12 Despite this advancement in genomics, as yet their impact on disease pathways remains unclear, suggesting that genetics contribute to but do not entirely account for the development of IBD. Genomics is the study of the ‘static’ DNA of a host, while transcriptomics is the study of the dynamic expression of RNA molecules and how they may vary under different circumstances. This approach therefore allows us to study the genes that are actively expressed at any given time and circumstance. 13 Specific progress investigating transcriptomics in the host has highlighted that protein-coding and noncoding RNA such as micro RNA have a role in immune regulation in IBD.14–20 Furthermore, through host transcriptomics, potential microRNAs have been highlighted as potential biomarkers of IBD.17,21
This review will focus on the use of systems biology technologies to better understand the nature of microbial communities. We will summarize our knowledge to date regarding omics in IBD and their role in understanding the gut microbiota (also see summary in Table 1 and Figure 1).
Table 1.
Summary of changes in IBD using a systems biology approach when compared with healthy people.
| Metagenomics | • Genes associated with butanoate and propanoate metabolism genes are decreased in IBD • Decrease in genes associated with the biosynthesis of amino acids and an increase in amino acid transporters and metabolism of the sulfur containing amino acid cysteine in IBD |
| Metataxonomics | • Depletion of bacteria with anti-inflammatory effects, including Bifidobacterium, Lactobacillus, Faecalibacterium prausnitzii and other SCFA-producing bacteria • Relative expansion in pathogenic bacteria, including Proteobacteria such as adherent-invasive Escherichia coli |
| Metatranscriptomics | • Gut microbial functional potential (based on metagenomics) is often but not always proportional to metatranscriptomic profiles. • Multiple metabolic pathways were found to be differentially expressed such as methylerythritol phosphate pathway predominantly by Alistipes putredinis and dTDP-L-rhamnose biosynthesis by F. prausnitzii |
| Metaproteomics | • Depletion of a large range of microbial proteins (Crohn’s disease, especially ileal disease) • Distinct protein modules at the mucosal-luminal interface differed between healthy controls and patients with IBD |
| Metabonomics | • IBD patients have low urinary hippurate levels • IBD patients have lower levels of faecal short-chain fatty acids • Specifically, stool butyrate and acetate were reduced in Crohn’s disease |
IBD, inflammatory bowel disease; SCFA, short-chain fatty acid.
Figure 1.

Summary of systems biology platforms.
Metagenomics and metataxonomics
The advent of next generation sequencing (NGS) in the last decade has facilitated a remarkable insight into the characteristics and functionality of the gut microbiome with unprecedented throughput and resolution. The first critical studies set the baseline by defining the diversity and composition of the gut microbiota from stool samples or colonic biopsies from healthy individuals. 22 Intriguingly, they revealed that nearly 70–80% of the bacteria inhabiting the human gut were previously unknown and were thus considered ‘unculturable’ at that time.11,23 Further studies then began to explore the differences in the gut microbiome in health and disease states while elucidating potential host–microbiome interactions and pathophysiological mechanisms.
Metataxonomics: 16S rRNA gene sequencing and analysis
Most of the culture-independent characterization of the gut microbiome in IBD has been directed towards sequencing of 16S rRNA genes, which are present in all cellular organisms. 24 This gene was chosen as it is relatively small (∼1.5 kb) and has a highly significant level of sequence conservation between bacterial species to facilitate reliable and robust alignments with sufficient variation to infer evolutionary relationships. Through barcoded primer sets that target highly conserved regions of the 16S rRNA gene, metataxonomics seeks to amplify and subsequently sequence the hypervariable regions of the gene from bacteria and archaea within a given sample. 25 The sequences are clustered into phylotypes according to their likeness to previously annotated sequences in a reference database or constructed into operational taxonomic units (OTUs) by clustering sequences based on their similarity. 26
Metataxonomics provides a highly cost effective and rapid means of defining microbial community (16S rRNA genes for bacteria and 18S rRNA genes for eukaryotes) richness and semi-quantitative relative taxonomic abundance data. 27 It also remains the primary technique for untargeted characterization of mucosally-adherent bacteria in the colon or other tissues that have a relatively low bacterial biomass. This technique is however known to be limited by the challenges associated with polymerase chain reaction (PCR)-based short read length sequencing including GC bias, sequencing errors and difficulties in assessing OTUs. 28 Furthermore the characterization of closely related species by 16S rRNA gene is limited and resolution rarely differentiates strains of the same species. However, third generation approaches are starting to open up the possibility of species and strain-level metataxonomic approaches by combining the MinIon platform and longer amplicons. 29 Unlike metagenomics, insights into the metabolic potential of a community cannot be obtained through a metataxonomic approach. Bioinformatics pipelines such as PICRUSt and Tax4Fun, however, allow predictions of the functional capability of a community based on a 16S rRNA gene dataset with significant metagenome correlation for biosamples obtained from the lower gastrointestinal (GI) tract.30,31
Metagenomics
Metagenomics or ‘shotgun metagenomics’, refers to the untargeted sequencing of whole-community DNA in an environment. 32 In a sample such as stool that consists of a complex microbial community, shotgun sequencing is primarily used to profile its taxonomic composition (down to the strain level) and directly identify functional potential. Unlike metataxonomics, rather than targeting a specific marker gene for amplification, all the extracted DNA in a given sample is sheared into small fragments, barcoded and independently sequenced. 33 The resulting DNA sequences (or reads) are either assembled or left unassembled, and aligned to databases to provide accurate quantitative taxonomic and functional characterization. Consequently, metagenomics provides the opportunity to simultaneously explore two aspects of a microbial community; exactly who is there and what they are potentially capable of doing.
Metagenomics has enabled large-scale investigations of complex microbiomes and helped understand functional differences in healthy and diseased states. It provides strain-level resolution of gut bacteria and additionally characterizes nonbacterial microbial communities such as fungi and viruses that have recently been shown to potentially play a crucial role in host health.34,35 This technique, although powerful, does have many limitations: relatively, it is significantly more expensive than 16S rRNA gene sequencing; furthermore, there are many incompletely annotated bacterial genomic sequences, and uncertainties about the accuracy and even coverage of databases. As metagenomics bioinformatics tools rely on availability of annotated genomes they are therefore affected by limitations in reference sequence databases. Moreover, lack of annotations for a large number of microbial species when profiling metabolic potential leads to a bias towards highly conserved pathways (such as housekeeping genes), even when there are significant differences in the taxonomic composition.33,36 Furthermore, the lack of host DNA depletion kits mean metagenomics is not reliable on tissues with low host–microbe biomass ratio, such as colon biopsies, where >95% of DNA sequenced is nonmicrobial.
Metataxonomic and metagenomic insights into IBD
Gut microbial taxonomic and functional profiling studies of stool and mucosal biopsies through NGS has provided us with a wealth of evidence that a dysfunctional gut microbiome plays a crucial role in the pathogenesis of IBD.10,37 There are consistent data demonstrating that patients with IBD have a decrease in the compositional diversity and stability largely due to a reduction in the phylum Firmicutes and an increase in Proteobacteria.38–40 Shifts in specific taxonomic classes have been consistently reported in IBD. Broadly, gut bacterial classification studies in IBD have identified depletions in bacteria with anti-inflammatory effects, including Bifidobacterium, Lactobacillus, Faecalibacterium prausnitzii and other short-chain fatty acid (SCFA)-producing bacteria, along with a relative expansion in pathogenic bacteria, including Proteobacteria such as adherent-invasive Escherichia coli.38,41–43 These compositional differences highlight potential mechanisms that contribute to the inflammatory mechanisms of disease in IBD. For example in several landmark studies, reduced abundances of ileal mucosal F. prausnitzii are associated with a higher risk of recurrent CD after ileocaecal resection, and recovery of F. prausnitzii after a flare in UC is associated with maintenance of clinical remission. 42 Metagenomic studies have further highlighted differences in the functional composition of the gut microbiota in IBD. 9 Genes associated with butanoate and propanoate metabolism are decreased and this change is consistent with the reductions seen in SCFA-producing Firmicutes clades from studies profiling gut bacterial taxonomy. Furthermore, a decrease in genes associated with the biosynthesis of amino acids and an increase in amino acid transporters and metabolism of the sulfur-containing amino acid cysteine is noted amongst many findings.
Metagenomics and metataxonomic studies (in conjunction with other microbial omics) highlight multiple potential pathways through which gut microbiota in IBD contribute to immune dysregulation, gut barrier breakdown and intestinal inflammation. There are significant inter-study discrepancies largely due to multiple confounding factors, such as tissue source (stool or mucosa), disease activity, medication, diet, age and differences in both wet and dry lab techniques. On its own, this microbial compositional and functional profiling in IBD has only demonstrated disease associations and potential mechanisms.44,45 We now need well designed studies in IBD that use metagenomics or metataxonomics as one part of the jigsaw in proving causative mechanisms and predicting or ameliorating disease.
Metatranscriptomics
The recent emergence of highly parallel RNA-sequencing technologies has allowed us to gain insights into gene expression profiles of the host and microbial community. While metagenomics tells about the genomic potential of microbes in a community, metatranscriptomics informs us on the actual genetic activity within a community phenotype. The gut microbial transcriptional activity is determined by a multitude of factors such as changes in host health and disease state, immune micro-environment, diet and the microbial ecosystem. The metatranscriptome, therefore, is dynamic and contextualizes microbial functional activity to the host phenotype, and when used in conjunction with metagenomics provides a powerful understanding on the molecular mechanisms by which gut bacteria contribute to health and disease.46–48 It provides significant value in shifting our current descriptive gut microbiome knowledge towards a deeper understanding of host–microbial causal mechanisms in contributing to homeostasis and disease.
A metatranscriptome experiment involves isolation of total RNA from a tissue, such as colon biopsy or stool. This isolation can be followed by depletion of the host mRNA, for example, by using hybridization probes that take advantage of the poly-A tail on eukaryotic mRNA. In eukaryotic and prokaryotic cells, approximately 80–90% of the total RNA is comprised of ribosomal RNA (rRNA) and 15% of transfer RNA (tRNA); protein-coding mRNA constitutes only 2–5% in a sample. 49 Consequently, this makes depletion of both human and bacterial small and large rRNA an imperative step of any metatranscriptome experiment. 50 Libraries of cDNA from the rRNA-depleted mRNA are generated followed by ligation to adapters before amplification and sequencing. Bioinformatic pipelines such as HumaNN2 and SAMSA2 can be used to process the generated reads, perform quality control assessment, and undertake removal in silico of any rRNA and host transcriptome contamination. The filtered sequences are then aligned to a microbial translated protein sequence such as the UniProt database and functional databases such as KEGG or SEED.
There are several major limitations of metatranscriptomics. Tissues such as colonic biopsies consist of a significant amount of host contamination where host cells make up nearly 95% of the biomass. Such cases require deep sequencing of the total mRNA in order to obtain a representative window into the mucosally-adherent microbial gene expression profile. 46 The microbial transcriptome or translated protein databases are not comprehensive and consist of a large number of genes that are currently not yet annotated to a known function. This knowledge gap often leads to an incomplete, and to a certain extent, biased interpretation of the microbial functional profile, but is likely to change as this field evolves over time.
Metatranscriptomics in IBD
As metatranscriptomics is a relatively new technology, there is a paucity of data for gut microbial transcriptomic profiling in health or any given disease. The largest faecal metatranscriptome study in IBD was conducted as part of the Integrative Human Microbiome project (IBD multi’omics database). 51 In this study, metagenomic analysis was paired with metatranscriptomic analysis from 117 individuals (24 non-IBD healthy controls, 59 patients with CD, 34 with UC). They found that the gut microbial functional potential (based on metagenomics) is often but not always proportional to metatranscriptomic profiles. Multiple metabolic pathways were found to be differentially expressed, such as the methylerythritol phosphate pathway predominantly by Alistipes putredinis and dTDP-L-rhamnose biosynthesis by F. prausnitzii. These pathways are associated with inducing or regulating inflammation, immune response and altering interspecies interactions in the gut. This study represented the first step towards a new way of interpreting ‘dysbiosis’ in IBD by going beyond microbial compositional profiling and contextualizing altered microbial gene expression in relation to disease. The field of metatranscriptomics will continue to evolve our understanding of host–microbiome relationship in IBD in the coming years. 52
Metaproteomics
Metaproteomics involves the high-throughput characterization of the entire constituent profile of microbial proteins within a biofluid or tissue sample. A key utility of metaproteomic studies is that the identification of the protein content of a sample, coupled with insight to their interactions, abundances, and modifications, gives direct information about the true functional activity of the gut microbiota. As already discussed, this level of functional insight is not typically captured by studies focused on microbial sequencing alone. A range of different methodologies may be used for proteomic studies, including both gel-based and gel-free techniques, mass spectrometry, nuclear magnetic resonance, and microarray-based technologies.53,54
However, as for other omics technologies, there are potential limitations that must be considered when performing and interpreting proteomic studies. The proteome is vast in its scale and complexity (with proteins often interacting in networks rather than functioning singularly), which translates to high complexity in the processing and analysis of proteomic data. While the Human Proteome Project database (https://hupo.org/human-proteome-project) is available to researchers, there are no definitive reference metaproteomic databases available at present. Differences exist between proteome profiles established using alternate methodologies or after analysis in different laboratories. 55 There are disparities between the metaproteome of gut mucus, luminal content and faecal material 56
Metaproteomics in IBD
While metaproteomics as applied to IBD is a relatively novel field, there are a growing number of studies in which it has been applied. In one of the first such investigations, different stool metaproteome profiles were observed between human patients with CD in comparison with healthy individuals, with patients with CD (and particularly with those with the disease in an ileal distribution) having a particular depletion of a large range of microbial proteins. 57 A further area in which metaproteomics has been applied has been in the analysis of the mucosal-luminal interface, with the aim of better elucidating any aberrations in gut microbiota–host interactions that may contribute to the onset or activity level of IBD. A study from Li and colleagues identified distinct protein modules at the mucosal-luminal interface; these differed between healthy controls and patients with IBD, and, in the case of certain modules, differentiated UC from CD. 58 Metaproteomic analysis on the mucosal-luminal interface has also recently been reported for a paediatric IBD inception cohort 59 This demonstrated upregulation of microbial proteins related to oxidative stress responses in children with IBD compared with controls. In addition, the expression of human proteins related to oxidative antimicrobial activities was also increased in IBD cases and correlated with the identified changes in microbial functions.
Metabolomics and metabonomics
Metabolomics is defined as the quantitative measurement of the dynamic multi-parametric metabolic response of living systems to pathophysiological stimuli or genetic modification. 60 Metabonomics is defined as ‘the quantitative measurement over time of the metabolic responses of an individual or population to drug treatment or other intervention’. 61 Despite often being used interchangeably, the subtle differences are that metabolomics places a greater emphasis upon metabolic profiling on a cellular or organ level, while metabonomics extends to metabolic profiling that includes the contributions of environmental influences such as diet, toxins, drugs and the gut microbiota. 62 A key importance of metabonomics is using an integrated systems biology approach which provides a way of investigating the metabolic status of an organism or ecosystem by studying ‘real’ metabolic endpoints. 62
Metabonomics can be used to predict responses to medical treatment (termed pharmacometabonomics), 63 as well in the prediction of diseased states, raising the potential for personalized medicine.64,65 This integrated technology utilizes 1H-nuclear magnetic resonance (NMR) and mass spectrometry (MS), which is split into liquid chromatography-MS (LC-MS) and gas chromatography-MS (GC-MS). There is also a growing interest in ambulatory MS techniques, such as rapid evaporative ionization MS (REIMS) and desorption MS imaging (DESI)66–68 Complex multivariate statistical models and bioinformatics are used to enable interpretation of metabolic profiling data.
Metabonomics therefore enables profiling of the unique end product or metabolites found in biofluids. This can enable longitudinal assessment of metabolic changes, metabolic changes in response to treatment and metabolic profiles in both healthy and diseased states. Metabonomic profiling can provide insights into unique fingerprints of biochemical perturbations that are characteristic of a disease process. 69 This can therefore be the basis of finding novel biomarkers.
Metabonomics in IBD
The unique advantage of metabonomics is that it can link metabolites found to specific metabolic pathways which can directly link in with the bacterial metabolic pathways, therefore advancing the interplay of the microbiota and metabolic pathways on disease aetiology. For instance, it has been shown that there are low levels of hippurate (a metabolite that is derived from the gut microbiota) in the urine of patients with IBD. This finding is of interest as hippurate levels have been shown to correlate with the presence of Clostridia in the gut. 70 Furthermore, Williams and colleagues, using NMR profiling, found that significant decreases in urinary hippurate were found in patients with IBD. 71
Serum has been another biofluid that has been analyzed in IBD.72–75 Despite differences being shown between amino acids and and molecules of the tricarboxylic acid (TCA) cycle between UC and CD74,75 as a tightly regulated biofluid, the ability to discriminate between UC and CD is reduced. It is also likely that due to homeostatic regulation and that the fact that serum is not directly in contact with the gut microbiota, this biofluid is less likely to be able to provide information on important changes in the gut microbiota through metabonomic processing.
The dysbiosis found in IBD has created a lot of interest in the metabolic profiling of faeces. Marchesi and colleagues highlighted that patients with IBD showed a lower level of faecal SCFAs when compared with healthy people through NMR profiling. 76 Specifically, they found that a depletion in SCFAs including acetate and butyrate in patients with CD when compared with healthy people. The SCFAs are mainly produced by the fermentation of complex carbohydrates via the gut bacteria. Furthermore, they found that methylamine and trimethylamine were decreased in patients with CD. In trying to link to the gut microbiota, it has been shown that these two compounds are derived from intestinal degradation of food components such as choline and carnitine by microbiota. 77 The role of SCFAs was supported by Le Gall and colleagues, who reported that butyrate and acetate were reduced in CD when compared with healthy people; they also found elevated levels of taurine in active UC when compared with healthy people and suggested that this could be high due to the gut microbiota’s role in the deconjugation of bile acids. 78 Further work on faeces was performed by Jansson and colleagues who found that patients with ileal CD had a greater abundance of Bacteroides vulgatus (BV), B. ovatus (BO) and E. coli when compared with healthy people which correlated most strongly to bile acids, including taurocholic and cholic acids, and fatty acids, including stearic and docosapentanoic acids, and concluded that there are correlations between metabolites and the bacterial microbiota but causality will need further exploration. 79
Tissue is another source of material that can be analyzed using metabonomics. It is one of the less studied. Sharma and colleagues highlighted that the metabolic profile of amino acid membrane components and lactate were similar between noninflamed IBD segments and inflamed segments. 80 Bjerrum and colleagues reported that colonic biopsies from patients with active UC had higher levels of antioxidants and a range of amino acids, but lower levels of lipid, glycerophosphocholine (GPC), myo-inositol, and betaine when compared with healthy people. Whilst both these studies suggest mechanistic pathways, neither integrated metabonomic with microbiota data.
Omics data integration methods
Integrating the omics is challenging because of multiple types of data set, but the process is improving. Each of the omic data sets convey knowledge from different labels (layers) of the molecular organization; for example, gene expression is about genes that changes globally but also those that change significantly between diseases versus controls (for example IBD and non-IBD patients). Other omic data sets (such as metabonomics, lipidomics, metagenomics, and microbiome) also provide similar types of knowledge. Linking, fusing or integrating (Figure 2) those multi-omics matrices provides a holistic picture of the disease versus control patients in terms of the mechanistic understanding of the disease state. Multi-omics data usually consist of two or more matrices (for example, transcriptomics and metabonomics, metabonomics and microbiome, lipidomics and transcriptomics, metabonomics and proteomics) that share the same patients (sample numbers or same objects) but contain different biological features (variables) such as genes, metabolites, lipids or OTUs.
Figure 2.
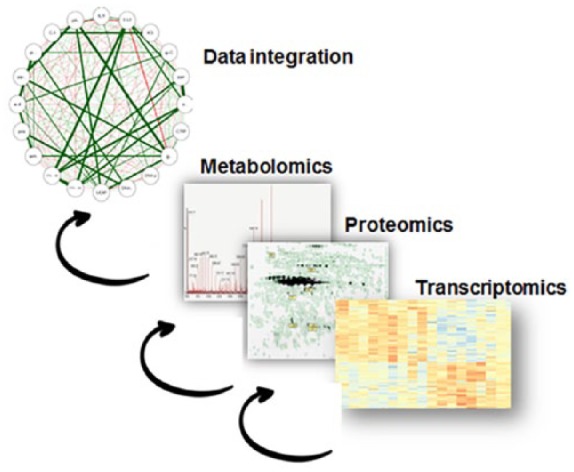
Integration of system biology platform.
Linking multiple omics data sets (e.g. transcriptomics, metabolomics, proteomics) and trying to forge mechanistic understanding of pathways.
Univariate versus multivariate methods
Different statistical approaches - specifically, univariate or multivariate analyses - are applied to understand associations (or correlations) between biological features (variables) in different omic data sets. In the literature there are many methods described starting from simple univariate to multivariate methods to determine if there are relationships between individual genes, metabolites, lipids or OTUs (microbiome). In the literature both parametric and nonparametric univariate methods have been applied to link OTUs and metabolomics data. Some examples are that where Theriot and colleagues performed univariate nonparametric correlation analysis (Spearman correlation analysis) between microbiome and metabolomics data from the mouse gut to identify relationships between features (metabolite versus taxonomic features or OTUs)81,82, and work by Mao and colleagues, 83 who used parametric correlation analysis (Pearson correlation analysis) to find associations between taxonomic features or OTUs and metabolites.82,83 A major advantage of the univariate correlation is that those methods are easy to understand as they are simple models, but they do not take into account the correlation structure of the data. Multivariate approaches, on the other hand, take into account the correlation (association) structure of the omic data sets, but suffer from high dimensionality of the data. Such high dimensionality is caused by the number of features from any type of omic data set. In the high dimensional settings, we have two issues or challenges:
(a) The number of parameters, features or variables (p) are large compared with the number of the samples, experimental units or individuals (n), in high dimensional statistics known as p>>n (large ‘p’ small ‘n’ problem) and due to that, is not possible to apply many statistical models, such as multiple linear regression.
(b) The features, variables or parameters are correlated.
To overcome the above problems, researchers have suggested some tentative solutions, for example, to create a new feature called a latent feature, using a linear combination of the original variables. Such approaches are called dimension reduction methods. Omic data sets are usually taken into account together to find out joint variations in the features. Some of the methods include partial least squares regression (PLS) 84 and two-way orthogonal partial least squares analysis (O2PLS).85,86 Aidy and colleagues 87 used O2PLS to connect transcriptomics, metabolomics and microbiome. Morgan and colleagues used linear discriminant analysis to link host transcriptomics, microbiome and clinical data. 88 Canonical correlation analysis (CCA) 89 is another type of method that maximizes the correlation structure of the two omic data sets, for example, metabolomics and proteomics. Recently, in the literature many versions of the CCA appeared; for example Sparse CCA 90 and Kernel CCA 91 tools have been developed 38 with statistical methods like PLS, O2PLS and CCA included in the packages. An example is Mixomics (R version 6.32) that aims to help draw the variable omics together to create a logical integrative story. Acharjee and colleagues, 92 developed omicsFusion, which is a web application that can perform statistical analysis based on regularization93,94 like LASSO 93 and Elastic net 94 together with univariate regression and dimension reduction methods.
The other type of integration is treating each of the omics data separately with a clinical outcome and selecting important features from them and integrating. Procedures for selecting subsets of features/variables are called variable or feature selection procedures. By doing this, it is possible to reduce the dimensionality of the data set and perhaps to get rid of some or even many noise variables (variables that have no predictive power for the response variable) in the data set.
Therefore, such type of integration is essentially integration of two labels. This integration provides us with two types of information: first, the type of the omics data, which is important for future experiments; and second, features that are important for prediction. Acharjee and colleagues 95 used a random forest approach to selecting features from metabolomics and lipidomics data and linked these with clinical outcomes in mouse data sets. Furthermore, Acharjee and colleagues 96 used a similar approach to integrate transcriptomics and metabolomics/metabonomic data in plant species.
Conclusion
This review has highlighted the potential of omics to integrate all the various variables that contribute to IBD to enable us to begin to understand their interactions within the gut microbiota. It is important to highlight that as well as integrating the known omics, future studies need to integrate the role of environmental factors that may contribute to alterations to the microbiota in IBD, including diet, medication and environmental exposure. Such integration (omics and non-omics data sets) for IBD will open up new therapeutic targets and mechanistic understanding of IBD.
Acknowledgments
The Division of Integrative Systems Medicine and Digestive Disease at Imperial College, London, UK receives financial support from the National Institute of Health Research Imperial Biomedical Research Centre based at Imperial College Healthcare NHS Trust and Imperial College London. BHM is the recipient of a UK Medical Research Council Clinical Research Training Fellowship (grant reference: MR/R000875/1).
JPS, BHM, MNQ were responsible for conception, literature review, writing and revising the manuscript and are joint first authors. AA conducted a literature review and contributed to writing and revising the manuscript. HW, TI, AH, and JRM gave critical revisions and helped revise the manuscript. All authors agreed to the final version
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributor Information
Jonathan P. Segal, Inflammatory Bowel Disease Department, St Mark’s Hospital, Harrow HA1 3UJ, UK.
Benjamin H. Mullish, Division of Integrative Systems Medicine and Digestive Disease, Department of Surgery and Cancer, Faculty of Medicine, Imperial College London, UK.
Mohammed Nabil Quraishi, Institute of Immunology and Immunotherapy, University of Birmingham, Department of Gastroenterology, University Hospital, Birmingham, UK.
Animesh Acharjee, College of Medical and Dental Sciences, Institute of Cancer and Genomic Sciences, Centre for Computational Biology, University of Birmingham, Birmingham, UK; Institute of Translational Medicine, University Hospitals Birmingham NHS Foundation Trust, Birmingham, UK; NIHR Surgical Reconstruction and Microbiology Research Centre, Birmingham, UK.
Horace R. T. Williams, Division of Integrative Systems Medicine and Digestive Disease, Department of Surgery and Cancer, Faculty of Medicine, Imperial College London, UK
Tariq Iqbal, Institute of Immunology and Immunotherapy, University of Birmingham, Department of Gastroenterology, University Hospital, Birmingham, UK.
Ailsa L. Hart, Inflammatory Bowel Disease Department, St Mark’s Hospital, Harrow, UK Department of Surgery and Cancer, Division of Integrative Systems Medicine and Digestive Disease, Faculty of Medicine, Imperial College, London, UK.
Julian R. Marchesi, Department of Surgery and Cancer, Division of Integrative Systems Medicine and Digestive Disease, Faculty of Medicine, Imperial College, London, UK School of Biosciences, Cardiff University, Cardiff, UK.
References
- 1. Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012; 142: 46–54.e42. [DOI] [PubMed] [Google Scholar]
- 2. Rutgeerts P, Goboes K, Peeters M, et al. Effect of faecal stream diversion on recurrence of Crohn’s disease in the neoterminal ileum. Lancet 1991; 338: 771–774. [DOI] [PubMed] [Google Scholar]
- 3. Frank DN, St Amand AL, Feldman RA, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 2007; 104: 13780–13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Manichanh C. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006; 55: 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Willing BP, Dicksved J, Halfvarson J, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 2010; 139: 1844–1854.e1. [DOI] [PubMed] [Google Scholar]
- 6. Tong M, Li X, Wegener Parfrey L, et al. A modular organization of the human intestinal mucosal microbiota and its association with inflammatory bowel disease. PLoS One 2013; 8: e80702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scanlan PD, Shanahan F, O’Mahony C, et al. Culture-independent analyses of temporal variation of the dominant fecal microbiota and targeted bacterial subgroups in crohn’s disease. J Clin Microbiol 2006; 44: 3980–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sokol H, Seksik P, Furet JP, et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis 2009; 15: 1183–1189. [DOI] [PubMed] [Google Scholar]
- 9. Morgan XC, Tickle TL, Sokol H, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 2012; 13: R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marchesi JR, Adams DH, Fava F, et al. The gut microbiota and host health: a new clinical frontier. Gut 2016; 65: 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Browne HP, Forster SC, Anonye BO, et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 2016; 533: 543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thompson AI, Lees CW. Genetics of ulcerative colitis. Inflamm Bowel Dis 2011; 17: 831–848. [DOI] [PubMed] [Google Scholar]
- 13. Zhang W, Li F, Nie L. Integrating multiple “omics” analysis for microbial biology: application and methodologies. Microbiology 2010; 156: 287–301. [DOI] [PubMed] [Google Scholar]
- 14. Iborra M, Bernuzzi F, Invernizzi P, et al. MicroRNAs in autoimmunity and inflammatory bowel disease: Crucial regulators in immune response. Autoimmun Rev 2012; 11: 305–314. [DOI] [PubMed] [Google Scholar]
- 15. Granlund A, van B, Flatberg A, Østvik AE, et al. Whole genome gene expression meta-analysis of inflammatory bowel disease colon mucosa demonstrates lack of major differences between Crohn’s disease and ulcerative colitis. PLoS One 2013; 8: e56818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coskun M, Bjerrum JT, Seidelin JB, et al. MicroRNAs in inflammatory bowel disease - pathogenesis, diagnostics and therapeutics. World J Gastroenterol 2012; 18: 4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin J, Welker NC, Zhao Z, et al. Novel specific microRNA biomarkers in idiopathic inflammatory bowel disease unrelated to disease activity. Mod Pathol 2014; 27: 602–608. [DOI] [PubMed] [Google Scholar]
- 18. Duttagupta R, DiRienzo S, Jiang R, et al. Genome-wide maps of circulating miRNA biomarkers for ulcerative colitis. PLoS One 2012; 7: e31241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haberman Y, Tickle TL, Dexheimer PJ, et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest 2014; 124: 3617–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brest P, Lapaquette P, Souidi M, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet 2011; 43: 242–245. [DOI] [PubMed] [Google Scholar]
- 21. de Bruyn M, Machiels K, Vandooren J, et al. Infliximab restores the dysfunctional matrix remodeling protein and growth factor gene expression in patients with inflammatory bowel disease. Inflamm Bowel Dis 2014; 20: 339–352. [DOI] [PubMed] [Google Scholar]
- 22. Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464: 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Walker AW, Duncan SH, Louis P, et al. Phylogeny, culturing, and metagenomics of the human gut microbiota. Trends Microbiol 2014; 22: 267–274. [DOI] [PubMed] [Google Scholar]
- 24. Clarridge JE, III. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev 2004; 17: 840–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci 2011; 108(Suppl. 1): 4516–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mysara M, Njima M, Leys N, et al. From reads to operational taxonomic units: an ensemble processing pipeline for MiSeq amplicon sequencing data. Gigascience 2017; 6: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carlos N, Tang YW, Pei Z. Pearls and pitfalls of genomics-based microbiome analysis. Emerg Microbes Infect 2012; 1: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Janda JM, Abbott SL. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clin Microbiol 2007; 45: 2761–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kerkhof LJ, Dillon KP, Häggblom MM, et al. Profiling bacterial communities by MinION sequencing of ribosomal operons. Microbiome 2017; 5: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aßhauer KP, Wemheuer B, Daniel R, et al. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015; 31: 2882–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Langille MGI, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013; 31: 814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Escobar-Zepeda A, Vera-Ponce de, León A, Sanchez-Flores A. The road to metagenomics: from microbiology to DNA sequencing technologies and bioinformatics. Front Genet 2015; 6: 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quince C, Walker AW, Simpson JT, et al. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 2017; 35: 833–844. [DOI] [PubMed] [Google Scholar]
- 34. Oulas A, Pavloudi C, Polymenakou P, et al. Metagenomics: tools and insights for analyzing next-generation sequencing data derived from biodiversity studies. Bioinform Biol Insights 2015; 9: 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gilbert JA, Dupont CL. Microbial metagenomics: beyond the genome. Ann Rev Mar Sci 2011; 3: 347–371. [DOI] [PubMed] [Google Scholar]
- 36. Huttenhower C, Gevers D, Knight R, et al. Structure, function and diversity of the healthy human microbiome. Nature 2012; 486: 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel diseases: current status and the future ahead. Gastroenterology 2014; 146: 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matsuoka K, Kanai T. The gut microbiota and inflammatory bowel disease. Semin Immunopathol 2015; 37: 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491: 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Manichanh C, Borruel N, Casellas F, et al. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol 2012; 9: 599–608. [DOI] [PubMed] [Google Scholar]
- 41. Loh G, Blaut M. Role of commensal gut bacteria in inflammatory bowel diseases. Gut Microbes 2012; 3: 544–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci 2008; 105: 16731–16736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Prorok-Hamon M, Friswell MK, Alswied A, et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut 2014; 63: 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ni J, Wu GD, Albenberg L, et al. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol 2017; 14: 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Olesen SW, Alm EJ. Dysbiosis is not an answer. Nature Microbiology 2016; 1: 16228. [DOI] [PubMed] [Google Scholar]
- 46. Bashiardes S, Zilberman-Schapira G, Elinav E. Use of metatranscriptomics in microbiome research. Bioinform Biol Insights 2016; 10: 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gosalbes MJ, Durbán A, Pignatelli M, et al. Metatranscriptomic approach to analyze the functional human gut microbiota. PLoS One 2011; 6: e17447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lavelle A, Sokol H. Gut microbiota: beyond metagenomics, metatranscriptomics illuminates microbiome functionality in IBD. Nat Rev Gastroenterol Hepatol 2018; 15: 193. [DOI] [PubMed] [Google Scholar]
- 49. Petrova OE, Garcia-Alcalde F, Zampaloni C, et al. Comparative evaluation of rRNA depletion procedures for the improved analysis of bacterial biofilm and mixed pathogen culture transcriptomes. Sci Rep 2017; 7: 41114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Reck M, Tomasch J, Deng Z, et al. Stool metatranscriptomics: a technical guideline for mRNA stabilisation and isolation. BMC Genomics 2015; 16: 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schirmer M, Franzosa EA, Lloyd-Price J, et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat Microbiol 2018; 3: 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bikel S, Valdez-Lara A, Cornejo-Granados F, et al. Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: towards a systems-level understanding of human microbiome. Comput Struct Biotechnol J 2015; 13: 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jin P, Wang K, Huang C, et al. Mining the fecal proteome: from biomarkers to personalised medicine. Expert Rev Proteomics 2017; 14: 445–459. [DOI] [PubMed] [Google Scholar]
- 54. Zhang X, Chen W, Ning Z, et al. Deep metaproteomics approach for the study of human microbiomes. Anal Chem 2017; 89: 9407–9415. [DOI] [PubMed] [Google Scholar]
- 55. Kim MS, Pinto SM, Getnet D, et al. A draft map of the human proteome. Nature 2014; 509: 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lichtman JS, Alsentzer E, Jaffe M, et al. The effect of microbial colonization on the host proteome varies by gastrointestinal location. ISME J 2016; 10: 1170–11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Erickson AR, Cantarel BL, Lamendella R, et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of crohn’s disease. PLoS One 2012; 7: e49138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li X, LeBlanc J, Elashoff D, et al. Microgeographic proteomic networks of the human colonic mucosa and their association with inflammatory bowel disease. Cell Mol Gastroenterol Hepatol 2016; 2: 567–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang X, Deeke SA, Ning Z, et al. Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat Commun 2018; 9: 2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yan S, Nagle DG, Zhou Y, et al. Application of systems biology in the research of TCM formulae. Syst Biol its Appl TCM Formulas Res 2018; 31–67. [Google Scholar]
- 61. Holmes E, Wilson ID, Nicholson JK. Metabolic phenotyping in health and disease. Cell 2008; 134: 714–717. [DOI] [PubMed] [Google Scholar]
- 62. Fernie AR, Trethewey RN, Krotzky AJ, et al. Metabolite profiling: from diagnostics to systems biology. Nat Rev Mol Cell Biol 2004; 5: 763–769. [DOI] [PubMed] [Google Scholar]
- 63. Andrew Clayton T, Lindon JC, Cloarec O, et al. Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature 2006; 440: 1073–1077. [DOI] [PubMed] [Google Scholar]
- 64. Everett JR, Loo RL, Pullen FS. Pharmacometabonomics and personalized medicine. Ann Clin Biochem 2013; 50: 523–545. [DOI] [PubMed] [Google Scholar]
- 65. Everett JR. Pharmacometabonomics in humans: a new tool for personalized medicine. Pharmacogenomics 2015; 16: 737–754. [DOI] [PubMed] [Google Scholar]
- 66. Cameron SJS, Bolt F, Perdones-Montero A, et al. Rapid evaporative ionisation mass spectrometry (REIMS) provides accurate direct from culture species identification within the genus Candida. Sci Rep 2016; 6: 36788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Phelps DL, Balog J, Gildea LF, et al. The surgical intelligent knife distinguishes normal, borderline and malignant gynaecological tissues using rapid evaporative ionisation mass spectrometry (REIMS). Br J Cancer 2018; 118: 1349–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Claude E, Jones EA, Pringle SD. DESI mass spectrometry imaging (MSI). New York, NY: Humana Press, 2017, pp.65–75. [DOI] [PubMed] [Google Scholar]
- 69. Bundy JG, Lenz EM, Bailey NJ, et al. Metabonomic assessment of toxicity of 4-fluoroaniline, 3,5-difluoroaniline and 2-fluoro-4-methylaniline to the earthworm Eisenia veneta (Rosa): identification of new endogenous biomarkers. Environ Toxicol Chem 2002; 21: 1966–1972. [PubMed] [Google Scholar]
- 70. Li M, Wang B, Zhang M, et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci 2008; 105: 2117–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Williams HRT, Cox IJ, Walker DG, et al. Characterization of inflammatory bowel disease with urinary metabolic profiling. Am J Gastroenterol 2009; 104: 1435–1444. [DOI] [PubMed] [Google Scholar]
- 72. Schicho R, Shaykhutdinov R, Ngo J, et al. Quantitative metabolomic profiling of serum, plasma, and urine by H NMR spectroscopy discriminates between patients with inflammatory bowel disease and healthy individuals. J. Proteome Res 2012: 11; 3344–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Williams HRT, Willsmore JD, Cox IJ, et al. Serum metabolic profiling in inflammatory bowel disease. Dig Dis Sci 2012; 57: 2157–2165. [DOI] [PubMed] [Google Scholar]
- 74. Hisamatsu T, Okamoto S, Hashimoto M, et al. Novel, objective, multivariate biomarkers composed of plasma amino acid profiles for the diagnosis and assessment of inflammatory bowel disease. PLoS One 2012; 7: e31131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ooi M, Nishiumi S, Yoshie T, et al. GC/MS-based profiling of amino acids and TCA cycle-related molecules in ulcerative colitis. Inflamm Res 2011; 60: 831–840. [DOI] [PubMed] [Google Scholar]
- 76. Julian RM, Elaine H, Fatima K, et al. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J Proteome Res 2007; 6: 546–551. [DOI] [PubMed] [Google Scholar]
- 77. Yang F, Mao Y, Zhang X, et al. LC-MS/MS method for the determination of melamine in rat plasma: toxicokinetic study in Sprague-Dawley rats. J Sep Sci 2009; 32: 2974–2978. [DOI] [PubMed] [Google Scholar]
- 78. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res 2006; 47: 241–259. [DOI] [PubMed] [Google Scholar]
- 79. Jansson J, Willing B, Lucio M, et al. Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS One 2009; 4: e6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sharma U, Singh RR, Ahuja V, et al. Similarity in the metabolic profile in macroscopically involved and un-involved colonic mucosa in patients with inflammatory bowel disease: an in vitro proton (1H) MR spectroscopy study. Magn Reson Imaging 2010; 28: 1022–1029. [DOI] [PubMed] [Google Scholar]
- 81. Theriot CM, Koenigsknecht MJ, Carlson PE, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 2014; 5: 3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chong J, Xia J, Chong J, et al. Computational approaches for integrative analysis of the metabolome and microbiome. Metabolites 2017; 7: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mao SY, Huo WJ, Zhu WY. Microbiome-metabolome analysis reveals unhealthy alterations in the composition and metabolism of ruminal microbiota with increasing dietary grain in a goat model. Environ Microbiol 2016; 18: 525–541. [DOI] [PubMed] [Google Scholar]
- 84. Trygg J, Wold S. O2-PLS, a two-block (X-Y) latent variable regression (LVR) method with an integral OSC filter. J Chemom 2003; 17: 53–64. [Google Scholar]
- 85. Bylesjö M, Eriksson D, Kusano M, et al. Data integration in plant biology: the O2PLS method for combined modeling of transcript and metabolite data. Plant J 2007; 52: 1181–1191. [DOI] [PubMed] [Google Scholar]
- 86. Bouhaddani el S, Houwing-Duistermaat J, Salo P, et al. Evaluation of O2PLS in omics data integration. BMC Bioinformatics 2016; 17: S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. El Aidy S, Van den Abbeele P, Van de Wiele T, et al. Intestinal colonization: how key microbial players become established in this dynamic process. BioEssays 2013; 35. [DOI] [PubMed] [Google Scholar]
- 88. Morgan XC, Kabakchiev B, Waldron L, et al. Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease. Genome Biol 2015; 16: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hotelling H. Relations between two sets of variates. Biometrika 1936; 28: 321. [Google Scholar]
- 90. Lin D, Zhang J, Li J, et al. Group sparse canonical correlation analysis for genomic data integration. BMC Bioinformatics 2013; 14: 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yamanishi Y, Vert JP, Kanehisa M. Protein network inference from multiple genomic data: a supervised approach. Bioinformatics 2004; 20(Suppl. 1): i363–i370. [DOI] [PubMed] [Google Scholar]
- 92. Acharjee A, Ament Z, West JA, et al. Integration of metabolomics, lipidomics and clinical data using a machine learning method. BMC Bioinformatics 2016; 17(Suppl. 15):440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tibshiranit R. Regression shrinkage and selection via the lasso. J R Statist Soc B 1996; 58: 267–288. [Google Scholar]
- 94. Zou H, Hastie T. Regularization and variable selection via the elastic net. J R Statist Soc B 2005; 67: 301–320. [Google Scholar]
- 95. Acharjee A, Kloosterman B, Visser RGF, et al. Integration of multi-omics data for prediction of phenotypic traits using random forest. BMC Bioinformatics 2016; 17: 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Acharjee A, Kloosterman B, de Vos RCH, et al. Data integration and network reconstruction with ∼omics data using random forest regression in potato. Anal Chim Acta 2011; 705: 56–63. [DOI] [PubMed] [Google Scholar]