

Short abstract
To reveal cellular mechanisms for antinociception produced by clinically used tramadol, we investigated the effect of its metabolite O-desmethyltramadol (M1) on glutamatergic excitatory transmission in spinal dorsal horn lamina II (substantia gelatinosa; SG) neurons. The whole-cell patch-clamp technique was applied at a holding potential of −70 mV to SG neurons of an adult rat spinal cord slice with an attached dorsal root. Under the condition where a postsynaptic action of M1 was inhibited, M1 superfused for 2 min reduced the frequency of spontaneous excitatory postsynaptic current in a manner sensitive to a μ-opioid receptor antagonist CTAP; its amplitude and also a response of SG neurons to bath-applied AMPA were hardly affected. The presynaptic effect of M1 was different from that of noradrenaline or serotonin which was examined in the same neuron. M1 also reduced by almost the same extent the peak amplitudes of monosynaptic primary-afferent Aδ-fiber and C-fiber excitatory postsynaptic currents evoked by stimulating the dorsal root. These actions of M1 persisted for >10 min after its washout. These results indicate that M1 inhibits the quantal release of L-glutamate from nerve terminals by activating μ-opioid but not noradrenaline and serotonin receptors; this inhibition is comparable in extent between monosynaptic primary-afferent Aδ-fiber and C-fiber transmissions. Considering that the SG plays a pivotal role in regulating nociceptive transmission, the present findings could contribute to at least a part of the inhibitory action of tramadol on nociceptive transmission together with its hyperpolarizing effect as reported previously.
Keywords: O-Desmethyltramadol, μ-opioid receptor, glutamatergic synaptic transmission, spinal substantia gelatinosa, patch-clamp, antinociception
Introduction
Tramadol, (1RS; 2RS)-2-[(dimethylamino) methyl]-1-(3-methoxyphenyl)-cyclohexanol hydrochloride, is a clinically used drug which acts as an analgesic in the central nervous system.1 Behavioral studies have demonstrated that the administration of tramadol inhibits nociceptive responses in rats.2–5 Among cellular mechanisms for this antinociception, there are (1) μ-opioid receptor activation2,4 and (2) inhibition of the reuptake of noradrenaline and serotonin, neurotransmitters involved in the descending inhibitory pathway to the spinal dorsal horn from brainstem.4,6,7 The former mechanism has been revealed from the results of a μ-opioid receptor binding of tramadol8 and its [35S]GTP-γ-S binding stimulation.9 Consistent with the latter mechanism, Kimura et al.3 have demonstrated in in vivo microdialysis experiments that intraperitoneally applied tramadol increases the levels of noradrenaline and serotonin in the rat spinal cord.
The substantia gelatinosa (SG; lamina II) of the spinal dorsal horn plays an important role in regulating nociceptive transmission through primary-afferent Aδ-fiber and C-fiber contained in the dorsal root.10 Antinociception produced by opioids, noradrenaline, and serotonin has been attributed to their modulatory actions on synaptic transmission in SG neurons.11–14 Thus, it is possible that at least a part of the antinociception produced by tramadol is due to a modulation of synaptic transmission in SG neurons.
Tramadol is metabolized to various compounds including O-desmethyltramadol (M1) via N- and O-dimethylation in humans and animals15; M1 appears in blood immediately after a single intravenous administration of tramadol in rats.16 M1 has the highest affinity for the cloned human μ-opioid receptors among the metabolites9 and is more effective in antinociception than tramadol according to a behavioral study in rats.2 M1 has an ability to inhibit the reuptake of noradrenaline and serotonin.6,17 It is, therefore, possible that the antinociceptive action of tramadol is partly due to a modulatory action of M1 on excitatory transmission in SG neurons through the activation of μ-opioid, noradrenaline, and serotonin receptors. It has not been fully examined yet how M1 affects excitatory transmission in SG neurons. This study examined the effect of M1 on glutamatergic spontaneous excitatory transmission in SG neurons in adult rat spinal cord slices with a focus on an involvement of their receptors. Since primary-afferent Aδ-fiber and C-fiber transfer different types of nociceptive information,10 we also investigated the effect of M1 on monosynaptic excitatory transmission evoked in SG neurons by stimulating each of the fibers. These experiments were performed by using the whole-cell patch-clamp technique under the condition where a previously reported hyperpolarized effect of M118 was inhibited.
Materials and methods
This study was approved by the Animal Care and Use Committee of Saga University and was conducted in accordance with the Guiding Principles for the Care and Use of Animals in the Field of Physiological Science of the Physiological Society of Japan. All efforts were made to minimize animal suffering and the number of animals used.
Preparation of spinal cord slices
The methods used for obtaining slice preparations of the adult rat spinal cord were similar to those described elsewhere.13,18 Briefly, male rats (7–8 weeks old) were deeply anesthetized with urethane (1.2 g/kg, i.p.), and then a lumbosacral laminectomy was performed. The lumbosacral segment (L1-S3) of the spinal cord with the ventral and dorsal roots attached was removed and placed in ice-cold (1–4°C) Krebs solution preequilibrated with 95% O2 and 5% CO2; the rats were then immediately sacrificed by exsanguination. After cutting all ventral and dorsal roots near the root entry zone, except for the L4 or L5 dorsal root on one side, the arachnoid membrane was removed. The spinal cord was mounted on a microslicer (DTK-1000, Dousaka, Kyoto, Japan), and then a 650-μm-thick transverse slice with a dorsal root attached was cut. The slice was placed on a nylon mesh in the recording chamber and was completely submerged and superfused at a drip rate of 15–18 ml/min with Krebs solution equilibrated with 95% O2 and 5% CO2 at 36 ± 1°C. Flow of solution was induced by gravity through a polyethylene tube from the reservoir. The composition of Krebs solution used was (in mM); NaCl 117, KCl 3.6, CaCl2 2.5, MgCl2 1.2, NaH2PO4 1.2, NaHCO3 25, and glucose 11 (pH = 7.4).
Patch-clamp recordings from SG neurons
Blind whole-cell recordings were made from SG neurons with patch-pipettes fabricated from a thin-walled fiberglass (1.5 mm OD) using a single-stage horizontal puller (P-97/PC; Sutter Instrument, Novato, CA), as reported previously.13,18 The patch-pipettes had a tip resistance of 8–15 MΩ when filled with a solution of the following composition (in mM): Cs2SO4 110, CaCl2 0.5, MgCl2 2, EGTA 5, HEPES 5, Mg-ATP 5, tetraethylammonium (TEA) 5, and guanosine 5′-O-(2-thiodiphosphate) (GDP-β-S) 1 (pH = 7.2); where GDP-β-S and K+-channel blockers (Cs+ and TEA) were used to inhibit a postsynaptic effect of M1 through the action of G-proteins and to block an activation of K+ channels which results from the postsynaptic effect, respectively.18
Signals were gained with a patch-clamp amplifier (Axopatch 200B; Axon Instruments, Foster City, CA). Data were low-pass filtered at 5 kHz, digitized at 333 kHz with an A/D converter, stored, and analyzed with a personal computer using pCLAMP data acquisition program (Version 6.0, Axon Instruments). The program (AxoGraph 4.0, Axon Instruments) used to analyze spontaneous excitatory postsynaptic currents (EPSCs) detects spontaneous events if the difference between the baseline and a following current value exceeds a given threshold of 5 pA, and the peak is separated from an adjacent peak by an intervening valley that is deeper than 50% of the adjacent peak. A validity of whether spontaneous EPSCs (sEPSCs) were accurately detected by the program was confirmed by measuring visually individual sEPSCs in a fast time scale on a computer screen in several cases. Orthodromic stimulation of the dorsal root to elicit EPSCs was performed with a suction electrode with a constant current source of pulse at a frequency of 0.1 Hz unless otherwise mentioned. The strength of stimuli (duration: 0.1 ms) used was 1.2 times the threshold to elicit EPSCs. The conduction velocities (CVs) of primary-afferent fibers involved in the production of the EPSCs were calculated from the latency of monosynaptic EPSC from a stimulus artifact and the length of dorsal root. Under the voltage-clamp condition, the holding potential (VH) used was −70 mV at which GABAA and glycine receptor-mediated synaptic currents were invisible.12 Neurons from which recordings were made were identified as SG neurons under a binocular microscope, where the SG could be easily distinguished as a colorless band located in the superficial dorsal horn.
Application of drugs
Drugs were dissolved in Krebs solution and applied by perfusing via a three-way stopcock Krebs solution containing drugs of a known concentration without an alteration in the perfusion rate and temperature; a change in solutions in the recording chamber completed within 20 s. The drugs used in this work were (±)-M1 (M1), (±)-noradrenaline, serotonin creatine sulfate, D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH22 (CTAP; Sigma-Aldrich Chemical Co., St. Louis, MO), α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (Tocris Neuramin, Bristol, England). The concentration of M1 used was 1 mM, at which concentration M1 produced a maximal outward current in SG neurons.18 Data are presented as mean ± SEM. Statistical significance was determined as P < 0.05 using either the paired or unpaired Student’s t test. In all cases, n refers to the number of neurons studied.
Results
Whole-cell patch-clamp recordings were made from a total of 92 SG neurons. Stable recordings could be obtained from neurons in spinal cord slices maintained in vitro for more than 6 h, and recordings were made from single neurons for up to 2 h. All experiments were performed at least 10 min later after the establishment of whole-cell configuration; this time was enough for GDP-β-S and K+-channel blockers in patch-pipette solutions to diffuse into SG neurons. M1 (1 mM) hardly changed holding currents at −70 mV.
Effect of M1 on spontaneous excitatory transmission in SG neurons
In all SG neurons tested, sEPSCs were downward at −70 mV and were blocked by CNQX (10 μM; n = 4), as reported previously,13,19 indicating the activation of non-N-methyl-D-aspartate (non-NMDA) receptors. In all SG neurons examined (n = 31), the occurrence of the sEPSCs was depressed by M1 (1 mM) superfused for 2 min and this action persisted after washout of M1, as shown in Figure 1(a). When quantitatively estimated in some neurons, sEPSC frequency was 68 ± 5% (P < 0.05) of that before the application of M1 (control; 18.6 ± 1.5 Hz, n = 15) around 4 min after the onset of its superfusion. This action was not accompanied by a significant change in sEPSC amplitude [87 ± 7% (P > 0.05) of control (12.4 ± 0.9 pA, n = 15)], although this amplitude had a tendency to be reduced. Figure 1(b) demonstrates averages of the time courses of M1-induced changes in sEPSC frequency and amplitude that were measured from seven neurons. Such a reduction in sEPSC frequency persisted for at least more than 20 min after washout of M1 (n = 5). Consistent with insignificant change in sEPSC amplitude, M1 (1 mM) hardly affected the peak amplitude of the response of SG neurons to AMPA (5 μM) (85 ± 6% (P > 0.05) of control (42.3 ± 11.4 pA, n = 3)), as seen in Figure 1(c).
Figure 1.
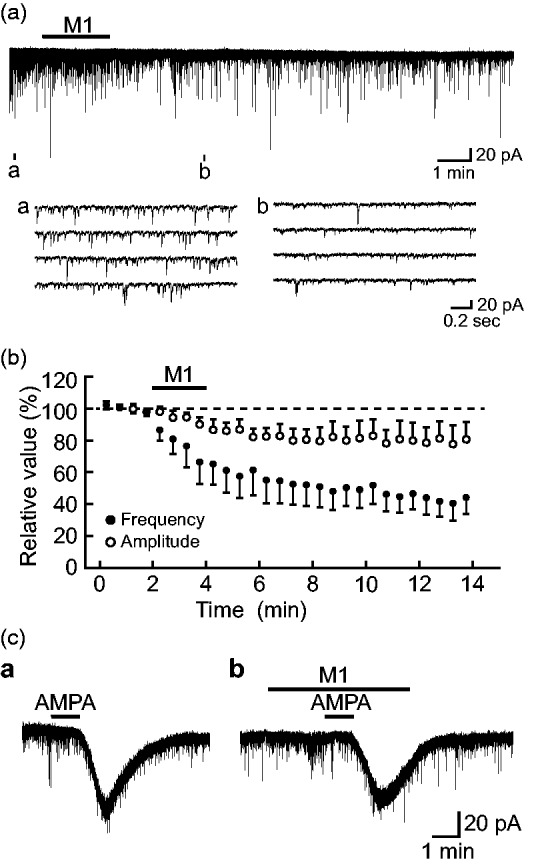
O-Desmethyltramadol (M1; 1 mM) presynaptically inhibits glutamatergic spontaneous excitatory transmission in rat substantia gelatinosa (SG) neurons. (a) Chart recording of spontaneous excitatory postsynaptic currents (EPSCs) in the absence and presence of M1. In this and subsequent figures, the duration of drug superfusion is shown by a horizontal bar above the chart recording; four consecutive traces of spontaneous EPSCs (sEPSCs) for a period indicated by a short bar designated as (a) to (d) below the chart recording are shown in an expanded scale in time. (b) Averages of the frequency (closed circles) and amplitude (open circles) of sEPSCs under the action of M1, relative to those before its application (control: frequency, 12.4 ± 0.9 Hz; amplitude, 20.3 ± 2.7 pA; n = 7), which are plotted against time. Each point with vertical bars indicates the mean values and SEM, calculated from sEPSCs measured for 30 s. (c) Current responses induced in a SG neuron by superfusing AMPA (5 μM) in the absence (a) and presence (b) of M1. Holding potential (VH) = −70 mV. AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid.
Figure 2(a) demonstrates how a μ-opioid receptor antagonist CTAP (1 μM) affects the effect of M1 (1 mM) on spontaneous excitatory transmission in SG neurons. Superfusing M1 with pretreatment with CTAP for 2 min had no effect on sEPSC frequency [96 ± 6% (P > 0.05) of control (12.4 ± 1.8 Hz, n = 7) around 4 min after the beginning of M1 superfusion]. Since the antinociceptive effect of M1 is possibly due to not only μ-opioid receptor activation but also inhibition of the reuptake of monoamines such as noradrenaline and serotonin,3,4,6,7,17 we next examined the effects of M1 (1 mM) on spontaneous excitatory transmission in single SG neurons where noradrenaline (50 μM) or serotonin (40 μM) was tested. As seen in Figure 2(b), noradrenaline did not affect sEPSC frequency,13 whereas this frequency was reduced by M1. This was confirmed in four neurons; sEPSC frequencies around 4 min after the onset of noradrenaline and M1 superfusion were, respectively, 96 ± 6% (n = 4; P > 0.05) and 71 ± 8% (n = 4; P < 0.05) of control (9.4 ± 2.5 Hz). The effect of serotonin on sEPSC frequency was also different from M1’s one (Figure 2(c)). sEPSC frequencies around 4 min and 6 min after the onset of serotonin application were, respectively, 82 ± 6% (n = 8; P > 0.05) and 525 ± 101% (n = 8; P < 0.05) of control (13.8 ± 1.9 Hz), as reported previously.11 On the other hand, M1 reduced sEPSC frequency around 4 min and 6 min after the beginning of its superfusion; they were, respectively, 68 ± 7% (n = 8; P < 0.05) and 48 ± 6% (n = 8; P < 0.05) of control.
Figure 2.
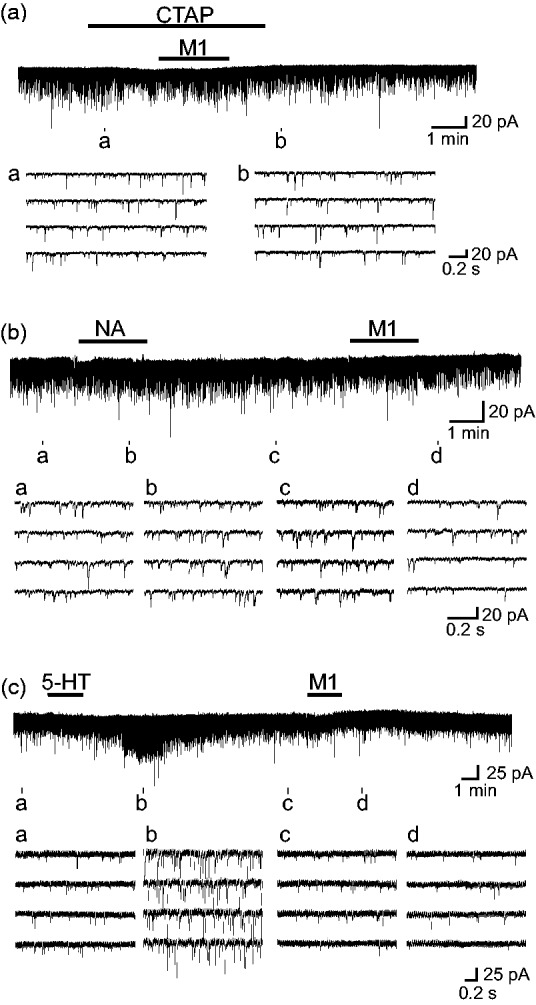
The sEPSC frequency reduction produced by M1 (1 mM) is due to μ-opioid receptor activation but not inhibition of the reuptake of noradrenaline (NA) or serotonin (5-HT) in the SG. (a) Recordings of sEPSCs in the absence and presence of M1 together with a μ-opioid receptor antagonist CTAP (1 μM). (b) and (c) Recordings of sEPSCs in the absence and presence of NA (50 μM; b) or 5-HT (40 μM; c) in a neuron where the effect of M1 on sEPSC frequency was examined. VH = −70 mV. CTAP: D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2.
Effects of M1 on monosynaptically evoked Aδ-fiber and C-fiber excitatory transmissions in SG neurons
Stimulating the dorsal root with a strength of more than 26 μA (sufficient to recruit Aδ-fibers) elicited in some neurons monosynaptic EPSCs which displayed no failure and no change in latency when examined at 20 Hz. CV values estimated from the latency of EPSC averaged to be 7.5 ± 1.0 m/s (5.1–12.0 m/s; n = 8); this was within the range of those of Aδ-fibers, as reported previously.13,19,20 Monosynaptic Aδ-fiber EPSCs evoked at 0.1 Hz had a mean amplitude of 278 ± 96 pA (range: 59–902 pA; n = 8). On the other hand, stimuli with a strength larger than 270 μA (enough to activate C-fibers) evoked in some neurons monosynaptic EPSCs which had no failures albeit there was a variety in the latency when stimulated at 1 Hz. The monosynaptic EPSCs had an average CV of 0.53 ± 0.05 m/s (0.4–0.8 m/s; n = 8), values comparable to those of C-fibers.13,19,20 Monosynaptic C-fiber EPSCs evoked at 0.1 Hz had a mean amplitude of 180 ± 95 pA (range: 38–345 pA; n = 8). Some SG neurons exhibited both monosynaptic Aδ-fiber and C-fiber EPSCs. These Aδ-fiber and C-fiber EPSCs were largely inhibited in peak amplitude by CNQX (10 μM; n = 4), indicating the activation of non-NMDA receptors.12
As seen in Figure 3(a) and (b), each of monosynaptic Aδ-fiber and C-fiber EPSCs was reduced in peak amplitude by M1 (1 mM). Figure 3(c) demonstrates the time courses of changes in the peak amplitudes of Aδ-fiber and C-fiber EPSCs, relative to control, following M1 (1 mM) superfusion. These inhibitory actions of M1 did not disappear at least 15 min after its washout, as seen in sEPSC frequency reduction produced by M1. When examined in many neurons, Aδ-fiber and C-fiber EPSC peak amplitudes were, respectively, 51 ± 7% (P < 0.05; n = 8) and 57 ± 7% (P < 0.05; n = 8) of control (278 ± 96 pA and 180 ± 95 pA, respectively) around 4 min after the beginning of M1 superfusion. These percentage values were not significantly different from each other (P > 0.05; Figure 3(d)). No difference in extent between Aδ-fiber and C-fiber EPSC peak amplitude reductions produced by M1 was observed in a single neuron where both monosynaptic Aδ-fiber and C-fiber EPSCs were observed; their extents were 47% and 40%, respectively (Figure 3(e)).
Figure 3.

M1 (1 mM) reduces the peak amplitudes of monosynaptic Αδ-fiber and C-fiber EPSCs evoked in SG neurons by stimulating the dorsal root with a comparable time course and extent. (a) and (b) Aδ-fiber (a) and C-fiber EPSCs (b), respectively, in the absence (left panel) and presence of M1 (right panel; 4 min after the onset of its application). Note that Aδ-fiber and C-fiber EPSC peak amplitudes were reduced by M1 by a similar extent. (c) Time courses of changes in Aδ-fiber EPSC and C-fiber EPSC peak amplitudes, relative to control, under the action of M1. Each of them was obtained from a different neuron. Each point is an average of the peak amplitudes of six consecutive EPSCs. (d) The peak amplitudes of Aδ-fiber and C-fiber EPSC, relative to control, around 4 min after the beginning of M1 superfusion. Vertical lines accompanied by bars indicate SEM; statistical significance between data shown by bars is indicated by a horizontal line; n.s.: not significant. The number of neurons examined is shown in parentheses. (e) Monosynaptic Aδ-fiber and C-fiber EPSCs in the absence (left panel) and presence of M1 (right panel; 4 min after the onset of its application) in a single neuron. Each record in (a), (b), and (e) (stimulus strength: 30, 500, and 260 μA, respectively) shows an average of six traces of EPSCs evoked at a frequency of 0.1 Hz. VH = −70 mV. EPSC: excitatory postsynaptic current.
Discussion
This study demonstrated that bath-applied M1 (1 mM) irreversibly reduces the frequency but not amplitude of sEPSC in SG neurons, albeit its amplitude has a tendency to be reduced. This irreversibility was also seen in the production of an outward current by M1 in SG neurons.18 Consistent with this change in sEPSC amplitude, a response of SG neurons to AMPA also had a tendency to be inhibited by M1. The change in sEPSC amplitude and AMPA response appeared to be unlikely due to a rundown of non-NMDA receptors in the recording neurons, because sEPSC amplitude remained to be constant 60 min after the whole-cell configuration19 under the same condition as that in this study. Such a postsynaptic action of M1 may be due to its inhibitory effect on non-NMDA receptors, albeit insignificantly, because M1 is reported to inhibit various types of neurotransmitter receptor-coupled ion channel.21 Similar sEPSC frequency and amplitude changes produced by M1 in rat SG neurons have been demonstrated by using the in vivo patch-clamp technique.5
The sEPSC frequency decrease was observed under the condition where a postsynaptic hyperpolarizing effect of M1 was inhibited in a recording SG neuron by using GDP-β-S- and K+-channel blockers-containing patch-pipette solution. It is possible that bath-applied M1 hyperpolarizes membranes of neurons, which are synaptically connected to the recording neuron, resulting in a decrease in the spontaneous release of L-glutamate from terminals of the neurons and thus in sEPSC frequency decrease. This possibility is, however, unlikely, because sEPSCs are unaffected in amplitude and frequency by the voltage-gated Na+-channel blocker tetrodotoxin and therefore are not mediated by neuronal activities.19,22,23
M1 inhibited the reuptake of noradrenaline and serotonin in rat frontal cortex or hypothalamic synaptosomes.6,17 Bloms-Funke et al.24 have demonstrated in in vivo microdialysis experiments that tramadol increases extracellular levels of noradrenaline and serotonin in the rat ventral hippocampus. Since noradrenergic and serotonergic descending pathways from the brainstem such as the locus coeruleus and raphe complex are involved in an inhibition of nociceptive transmission in the spinal dorsal horn,10,14 it is possible that an inhibition by M1 of the reuptake of noradrenaline and serotonin increases their concentrations in the spinal dorsal horn, resulting in a modulation of spontaneous excitatory transmission in SG neurons. In support of this idea, a selective serotonin reuptake inhibitor fluvoxamine modulated spontaneous excitatory transmission in superficial dorsal horn neurons of mouse spinal cord slices.25 Antinociception produced by the intrathecal administration of tramadol has been attributed to an activation of α2-adrenoceptors in the rat spinal cord.26 In this study, in SG neurons where M1 decreased sEPSC frequency, noradrenaline did not change sEPSC frequency and serotonin increased sEPSC frequency, as reported previously in SG neurons.11,13 This observation that M1 affects sEPSC frequency in a manner different from noradrenaline and serotonin indicates that the presynaptic effect of M1 is not mediated by noradrenaline and serotonin, as different from the ideas proposed previously.3,4,6,7,17 In fact, M1 was much less active in inhibiting the reuptake of serotonin than tramadol in rat frontal cortex synaptosomes17 and in rat dorsal raphe nucleus slices.27 We previously reported that M1-induced outward current is not mediated by noradrenaline.18 To ascertain more reliably our idea that M1 activity is not mediated by serotonin, further experiments using serotonin-related drugs will be required.
The sEPSC frequency decrease produced by M1 in this study disappeared in the presence of a μ-opioid receptor antagonist CTAP, indicating an involvement of μ-opioid receptors. This result is consistent with the observations that μ-opioid receptor activation in the SG results in a decrease in the spontaneous release of L-glutamate from nerve terminals12,22 and that μ-opioid receptors exist in superficial layers of the spinal cord, particularly the SG in rats.28–30 M1-induced sEPSC frequency decrease in rat SG neurons in vivo was abolished in the presence of a nonselective opioid-receptor antagonist naloxone.5
M1 reduced not only sEPSC frequency but also the peak amplitudes of monosynaptic primary-afferent Aδ-fiber and C-fiber EPSCs evoked in SG neurons by stimulating the dorsal root. This EPSC amplitude reduction was irreversible, as seen in the sEPSC frequency reduction produced by M1. Since M1 hardly affects sEPSC amplitude, the EPSC amplitude reduction is mainly presynaptic in origin. This result was the same as that of a μ-opioid receptor agonist [D-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin (DAMGO12). The Aδ fiber is myelinated and thus transfers fast, sharp, and more-localized pain, while the C fiber is unmyelinated and thus transfers slow, dull, and less-localized pain.10 The EPSC peak amplitude reduction produced by M1 was almost comparable in extent between monosynaptic Aδ-fiber and C-fiber transmissions, as seen in the action of adenosine in SG neurons.20 On the other hand, monosynaptic Aδ-fiber and C-fiber excitatory transmissions were depressed by nociceptin,31 noradrenaline,13 and galanin23 in a different manner from each other. Noradrenaline and galanin inhibited more effectively Aδ-fiber than C-fiber transmission, while C-fiber transmission was more sensitive to nociceptin than Aδ-fiber one. M1 is suggested to suppress fast- and slow-conducting pain transmissions with a comparable efficacy. Although M1 exhibits a high affinity to μ-opioid receptors,9 this result is different from that of DAMGO. DAMGO reduced monosynaptic C-fiber evoked EPSC peak amplitude more effectively than monosynaptic Aδ-fiber one.32 Minami et al.33 have demonstrated by using a Ca2+-activated Cl− channel current assay method that M1 and DAMGO activate μ-opioid receptors in Xenopus oocytes expressing cloned human μ-opioid receptors with a less efficacy of M1 than DAMGO. A Cl− current produced by M1 appeared to be somewhat different from that of DAMGO. M1 is known to act on various types of voltage-gated and neurotransmitter receptor-coupled ion channels, and thus to affect not only μ-opioid but also other G protein-coupled receptors21 located in primary-afferent central terminals. Therefore, M1 may exhibit an action different from that of DAMGO. The difference in the reduction of evoked EPSC amplitude between M1 and DAMGO remains to be examined. This study revealed for the first time that M1 inhibits primary-afferent Aδ-fiber and C-fiber excitatory transmissions in SG neurons.
The concentration (1 mM) of M1 used in this study was much higher than 2 μM, a concentration estimated to be clinically relevant.5 Although M1 at a low concentration such as 2 μM was reported to decrease sEPSC frequency in rat SG neurons in vivo,5 this action appeared to be reversible, as different from our observation. The irreversibility of the action of M1 at the high concentration used in this study may be due to a slow rate of diffusion of M1 out of spinal cord slices.
Summary
M1 inhibited the release of L-glutamate onto SG neurons from nerve terminals by activating μ-opioid receptors; this inhibition was comparable in extent between monosynaptic primary-afferent Aδ-fiber and C-fiber transmissions. This presynaptic inhibitory action of M1 could contribute to at least a part of the inhibitory action of tramadol on pain transmission together with its hyperpolarizing effect as reported previously.18
Authors’ Note
AK, TF, TN, and L-HP are currently affiliated with Koga Clinic, Fukuoka Dental University, Kansai University of Health Sciences, and Jilin University, respectively.
Author Contributions
AK, TF, L-HP, TN, and EK designed research; AK, TF, L-HP, and TN performed experiments; AK, TF, L-HP, and TN analyzed data; AK, TF, L-HP, TN, and EK wrote the paper. All authors read, edited, and approved the final manuscript.
Declaration of Conflicting Interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by Grants-in-aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan (KAKENHI: 15K08673).
References
- 1.Klotz U. Tramadol—the impact of its pharmacokinetic and pharmacodynamic properties on the clinical management of pain. Arzneimittelforschung 2003; 53: 681–687. [DOI] [PubMed] [Google Scholar]
- 2.Hennies H-H, Friderichs E, Schneider J. Receptor binding, analgesic and antitussive potency of tramadol and other selected opioids. Arzneimittelforschung 1988; 38: 877–880. [PubMed] [Google Scholar]
- 3.Kimura M, Obata H, Saito S. Antihypersensitivity effects of tramadol hydrochloride in a rat model of postoperative pain. Anesth Analg 2012; 115: 443–449. [DOI] [PubMed] [Google Scholar]
- 4.Raffa RB, Friderichs E, Reimann W, Shank RP, Codd EE, Vaught JL. . Opioid and nonopioid components independently contribute to the mechanism of action of tramadol, an ‘atypical’ opioid analgesic. J Pharmacol Exp Ther 1992; 260: 275–285. [PubMed] [Google Scholar]
- 5.Yamasaki H, Funai Y, Funao T, Mori T, Nishikawa K. Effects of tramadol on substantia gelatinosa neurons in the rat spinal cord: an in vivo patch-clamp analysis. PLoS One 2015; 10: e0125147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Driessen B, Reimann W, Giertz H. Effects of the central analgesic tramadol on the uptake and release of noradrenaline and dopamine in vitro. Br J Pharmacol 1993; 108: 806–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reimann W, Hennies H-H. Inhibition of spinal noradrenaline uptake in rats by the centrally acting analgesic tramadol. Biochem Pharmacol 1994; 47: 2289–2293. [DOI] [PubMed] [Google Scholar]
- 8.Frink MC, Hennies H-H, Englberger W, Haurand M, Wilffert B. Influence of tramadol on neurotransmitter systems of the rat brain. Arzneimittelforschung 1996; 46: 1029–1036. [PubMed] [Google Scholar]
- 9.Gillen C, Haurand M, Kobelt DJ, Wnendt S. Affinity, potency and efficacy of tramadol and its metabolites at the cloned human μ-opioid receptor. Naunyn-Schmiedeberg’s Arch Pharmacol 2000; 362: 116–121. [DOI] [PubMed] [Google Scholar]
- 10. Willis WD, Jr, Coggeshall RE. Sensory mechanisms of the spinal cord. 2nd ed New York, NY: Plenum Press, 1991, pp. 94–115. [Google Scholar]
- 11.Ito A, Kumamoto E, Takeda M, Takeda M, Shibata K, Sagai H, Yoshimura M. Mechanisms for ovariectomy-induced hyperalgesia and its relief by calcitonin: participation of 5-HT1A-like receptor on C-afferent terminals in substantia gelatinosa of the rat spinal cord. J Neurosci 2000; 20: 6302–6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohno T, Kumamoto E, Higashi H, Shimoji K, Yoshimura M. Actions of opioids on excitatory and inhibitory transmission in substantia gelatinosa of adult rat spinal cord. J Physiol 1999; 518: 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawasaki Y, Kumamoto E, Furue H, Yoshimura M. α 2 Adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology 2003; 98: 682–689. [DOI] [PubMed] [Google Scholar]
- 14.Fürst S. Transmitters involved in antinociception in the spinal cord. Brain Res Bull 1999; 48: 129–141. [DOI] [PubMed] [Google Scholar]
- 15.Lintz W, Erlaçin S, Frankus E, Uragg H. Metabolismus von Tramadol bei Mensch und Tier. Arzneimittelforschung 1981; 31: 1932–1943. [PubMed] [Google Scholar]
- 16.Parasrampuria R, Vuppugalla R, Elliott K, Mehvar R. Route-dependent stereoselective pharmacokinetics of tramadol and its active O-demethylated metabolite in rats. Chirality 2007; 19: 190–196. [DOI] [PubMed] [Google Scholar]
- 17.Driessen B, Reimann W. Interaction of the central analgesic, tramadol, with the uptake and release of 5-hydroxytryptamine in the rat brain in vitro. Br J Pharmacol 1992; 105: 147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koga A, Fujita T, Totoki T, Kumamoto E. Tramadol produces outward currents by activating μ-opioid receptors in adult rat substantia gelatinosa neurones. Br J Pharmacol 2005; 145: 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang C-Y, Fujita T, Kumamoto E. Synaptic modulation and inward current produced by oxytocin in substantia gelatinosa neurons of adult rat spinal cord slices. J Neurophysiol 2014; 111: 991–1007. [DOI] [PubMed] [Google Scholar]
- 20.Lao L-J, Kawasaki Y, Yang K, Fujita T, Kumamoto E. Modulation by adenosine of Αδ and C primary-afferent glutamatergic transmission in adult rat substantia gelatinosa neurons. Neuroscience 2004; 125: 221–231. [DOI] [PubMed] [Google Scholar]
- 21.Minami K, Ogata J, Uezono Y. What is the main mechanism of tramadol? Naunyn Schmiedeberg's Arch Pharmacol 2015; 388: 999–1007. [DOI] [PubMed] [Google Scholar]
- 22.Fujita T, Kumamoto E. Inhibition by endomorphin-1 and endomorphin-2 of excitatory transmission in adult rat substantia gelatinosa neurons. Neuroscience 2006; 139: 1095–1105. [DOI] [PubMed] [Google Scholar]
- 23.Yue H-Y, Fujita T, Kumamoto E. Biphasic modulation by galanin of excitatory synaptic transmission in substantia gelatinosa neurons of adult rat spinal cord slices. J Neurophysiol 2011; 105: 2337–2349. [DOI] [PubMed] [Google Scholar]
- 24.Bloms-Funke P, Dremencov E, Cremers TIFH, Tzschentke TM. Tramadol increases extracellular levels of serotonin and noradrenaline as measured by in vivo microdialysis in the ventral hippocampus of freely-moving rats. Neurosci Lett 2011; 490: 191–195. [DOI] [PubMed] [Google Scholar]
- 25.Tomoyose O, Kodama D, Ono H, Tanabe M. Presynaptic inhibitory effects of fluvoxamine, a selective serotonin reuptake inhibitor, on nociceptive excitatory synaptic transmission in spinal superficial dorsal horn neurons of adult mice. J Pharmacol Sci 2014; 126: 136–145. [DOI] [PubMed] [Google Scholar]
- 26.Li C, Chen S-Q, Chen B-X, Huang W-Q, Liu K-X. The antinociceptive effect of intrathecal tramadol in rats: the role of alpha 2-adrenoceptors in the spinal cord. J Anesth 2012; 26: 230–235. [DOI] [PubMed] [Google Scholar]
- 27.Bamigbade TA, Davidson C, Langford RM, Stamford JA. Actions of tramadol, its enantiomers and principal metabolite, O-desmethyltramadol, on serotonin (5-HT) efflux and uptake in the rat dorsal raphe nucleus. Br J Anaesth 1997; 79: 352–356. [DOI] [PubMed] [Google Scholar]
- 28.Besse D, Lombard MC, Zajac JM, Roques BP, Besson JM. Pre- and postsynaptic distribution of μ, δ and κ opioid receptors in the superficial layers of the cervical dorsal horn of the rat spinal cord. Brain Res 1990; 521: 15–22. [DOI] [PubMed] [Google Scholar]
- 29.Rahman W, Dashwood MR, Fitzgerald M, Aynsley-Green A, Dickenson AH. Postnatal development of multiple opioid receptors in the spinal cord and development of spinal morphine analgesia. Develop Brain Res 1998; 108: 239–254. [DOI] [PubMed] [Google Scholar]
- 30.Stevens CW, Lacey CB, Miller KE, Elde RP, Seybold VS. Biochemical characterization and regional quantification of μ, δ and κ opioid binding sites in rat spinal cord. Brain Res 1991; 550: 77–85. [DOI] [PubMed] [Google Scholar]
- 31.Luo C, Kumamoto E, Furue H, Chen J, Yoshimura M. Nociceptin inhibits excitatory but not inhibitory transmission to substantia gelatinosa neurones of adult rat spinal cord. Neuroscience 2002; 109: 349–358. [DOI] [PubMed] [Google Scholar]
- 32.Ikoma M, Kohno T, Baba H. Differential presynaptic effects of opioid agonists on Aδ- and C-afferent glutamatergic transmission to the spinal dorsal horn. Anesthesiology 2007; 107: 807–812. [DOI] [PubMed] [Google Scholar]
- 33.Minami K, Sudo Y, Miyano K, Murphy RS, Uezono Y. µ-Opioid receptor activation by tramadol and O-desmethyltramadol (M1). J Anesth 2015; 29: 475–479. [DOI] [PubMed] [Google Scholar]