

Abstract
Cells evolved robust homeostatic mechanisms to protect against oxidation or alkylation by electrophilic species. Glutathione (GSH) is the most abundant intracellular thiol, protects cellular components from oxidation and is maintained in a reduced state by glutathione reductase (GR). Nitro oleic acid (NO2-OA) is an electrophilic fatty acid formed under digestive and inflammatory conditions that both reacts with GSH and induces its synthesis upon activation of Nrf2 signaling. The effects of NO2-OA on intracellular GSH homeostasis were evaluated. In addition to upregulation of GSH biosynthesis, we observed that NO2-OA increased intracellular GSSG in an oxidative stress-independent manner. NO2-OA directly inhibited GR in vitro by covalent modification of the catalytic Cys61, with kon of (3.45 ± 0.04) × 103 M−1 s−1, koff of (4.4 ± 0.4) × 10−4 s−1, and Keq of (1.3 ± 0.1) × 10−7 M. Akin to NO2-OA, the electrophilic Nrf2 activators bardoxolone-imidazole (CDDO-Im), bardoxolone-methyl (CDDO-Me) and dimethyl fumarate (DMF) also upregulated GSH biosynthesis while promoting GSSG accumulation, but without directly inhibiting GR activity. In vitro assays in which GR was treated with increasing GSH concentrations and GSH depletion experiments in cells revealed that GR activity is finely regulated via product inhibition, an observation further supported by theoretical (kinetic modeling of cellular GSSG:GSH levels) approaches. Together, these results describe two independent mechanisms by which electrophiles modulate the GSH/GSSG couple, and provide a novel conceptual framework to interpret experimentally determined values of GSH and GSSG.
Keywords: Glutathione, Glutathione reductase, Electrophile, Nitrated fatty acid, Disulfide, Nrf2, Thiol, Oxidation-reduction (redox)
1. Introduction
Glutathione (GSH) is the most abundant intracellular thiol and is central to the detoxification of electrophiles and oxidants. Enzymatic or non-enzymatic oxidation to glutathione disulfide (GSSG) and subsequent NADPH-dependent reduction to GSH by glutathione reductase (GR) is key to maintaining thiol homeostasis. Although the pKa of the GSH Cys thiol is relatively high (8.94–9.42) [1], [2], suggesting low reactivity under cellular conditions, its central role in reduction-oxidation biology stems from highly efficient synthesis, enzyme-catalyzed transfer and recycling pathways, as well as mass-action afforded by high intracellular concentrations.
The expression of enzymes responsible for biosynthesis of GSH is under the transcriptional control of Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) [3], [4]. Nrf2 also regulates expression of the import of the GSH substrate cysteine through the cystine/glutamate antiporter [5]. Under basal conditions, Keap1, a BTB/E3 ubiquitin ligase, negatively regulates the transcription factor Nrf2 by targeting it for proteosomal degradation. Oxidation or alkylation of sentinel cysteine residues in Keap1 stabilize the Keap1/Nrf2 protein complex, sequestering Keap1 and allowing newly synthesized Nrf2 to accumulate and translocate to the nucleus to activate transcription of hundreds of cytoprotective genes [4], [6], [7]. In addition to regulating GSH biosynthesis, canonical Nrf2 targets include enzymes responsible for detoxification such as heme oxygenase 1 (HO-1) and NAD(P)H-quinone dehydrogenase 1 (NQO1) and xenobiotic exporters including multidrug resistance-associated protein (MRP) family members [8], [9], [10], [11], [12], [13]. Importantly, Keap1 is highly sensitive to alkylation by electrophiles, making pharmacological intervention a viable strategy for the manipulation of Nrf2 activity and downstream antioxidant gene expression [14].
In addition to activating Nrf2, electrophiles can react enzymatically and non-enzymatically with the nucleophilic thiolate GS−, decreasing intracellular GSH levels. GS-adducts of different electrophilic Nrf2 inducers have been described in vitro and in vivo (dimethyl fumarate (DMF) [15], [16], Bardoxolone (CDDO-Me) [17], and nitrated fatty acids (NO2-FA) [18], [19], [20]). However, the net effect of these compounds on intracellular GSH pools is not known, as activation of Nrf2 induces GSH biosynthesis.
Nitro-oleic acid (NO2-OA) is a therapeutically-promising drug and nitro-conjugated linoleic acid (NO2-cLA) is the most abundant endogenous NO2-FA. Both are electrophiles that post-translationally modify protein thiols through reversible Michael addition, exerting potent protective and anti-inflammatory effects [21], [22], [23], [24] and demonstrating efficacy in diverse disease models [23], [25], [26], [27], [28], [29]. At the molecular level, specific NO2-OA binding to Cys273/288 of Keap1 via Michael addition activates Nrf2 [30], [31], [32], [18]. These findings assign relevance to the study of NO2-OA in inflammatory cells.
In this context, this study sought to rigorously define the effects of NO2-OA on GSH biosynthesis, depletion, and cycling in murine RAW264.7 macrophages. NO2-OA exposure transiently decreased intracellular GSH concentration, followed by rapid induction of GSH biosynthesis and further GSH accumulation. Although this effect on GSH suggests a more reducing intracellular milieu, a paradoxical, oxidant-stress independent accumulation of GSSG was observed. Direct and indirect mechanisms by which electrophiles modulate GSSG cycling through GR were identified. Our findings show that GR activity is primarily determined by the intracellular level of GSH, establishing the ratio of GSH to GSSG.
2. Results
2.1. Lipid electrophiles upregulate GSH biosynthesis through Nrf2 activation in RAW264.7 macrophages
Basal intracellular GSH and GSSG measured in RAW264.7 macrophages by LC-MS/MS (Suppl. Fig. 1) yielded a mean of 56.4 ± 4.2 and 0.16 ± 0.01 nmol/mg protein for GSH and GSSG respectively and an overall ratio of GSH/GSSG of 366 ± 24 (n = 4–5 from 5 independent experiments; mean ± standard error).
After 1 h of exposure to 5 M electrophilic NO2-OA, GSH levels were reduced by 23%, returned to baseline level by 3 h and increased two-fold by 6 h (Fig. 1A, red trace). Surprisingly, the GSSG levels did not significantly change under this initial mild GSH depletion but increased 8.3-fold at 6 h (Fig. 1B, red trace). Both GSH and GSSG significantly decreased by 24 h but remained elevated compared to baseline. Notably, although GSH levels significantly increased at 6 and 24 h post-NO2-OA exposure, a pronounced decrease in GSH/GSSG ratio was observed (Fig. 1C, red trace). These effects were then compared to those of the TLR4 activator lipopolysaccharide (LPS) and the GSH synthesis inhibitor buthionine sulfoximine (BSO) to evaluate the effect of macrophage activation and GSH depletion. Lipopolysaccharide induced an initial decrease in GSH that recovered to initial levels by 6 h while GSSG increased as a function of time, reaching a maximum over 10-fold above baseline at 24 h.
Fig. 1.

Modulation of GSH and GSSG levels by NO2-OA, LPS and BSO. RAW264.7 cells were treated with 5 M NO2-OA (red trace), 200 ng/mL LPS (green), or 20 μM BSO (blue trace) and GSH (A), GSSG (B) and the GSH/GSSG ratio (C) were determined. Results are expressed as mean ± standard error of 3 replicates for each condition. Results normalized to vehicle control (black, t = 0 h) ± standard error (dashed lines) of 6 replicates. (D) Time-dependent response of Nrf2 activation by NO2-OA (5 μM) in RAW264.7 cells.
In contrast, treatment with the irreversible -glutamylcysteine synthetase inhibitor BSO resulted in a linear reduction in intracellular GSH over 24 h, with an overall loss of 55% of the GSH pool (Fig. 1A, blue trace). Interestingly, the loss in GSH resulted in a more pronounced (87%) reduction in GSSG (Fig. 1B, blue trace) suggesting that the decrease in GSH was not associated to oxidative stress.
Finally, we confirmed the Nrf2-dependent effects of NO2-OA treatment on GSH biosynthesis. The expression of the modulatory subunit of gamma-glutamyl-cysteine ligase (GCLM), which is the rate-limiting step in GSH biosynthesis, was increased at 6 h and was at its highest at 24 h. By comparison, the NO2-OA modulated target HO-1 was upregulated between 3 and 6 h but trending towards baseline levels by 24 h. Despite the presence of putative ARE sequences in its promoter sequence, GR expression was not modulated by NO2-OA treatment (Fig. 1D) [33].
2.2. NO2-OA induced upregulation of GSSG occurs independently of oxidative stress
The paradoxical increase in both intracellular GSH and GSSG in cells treated with NO2-OA motivated testing whether the increase in GSSG correlated with increased oxidative stress markers within the same time-frame (Fig. 2). To this end, changes in the relative abundances of monomeric vs. dimerized peroxiredoxins (Prdx) were evaluated. The cytosolic and mitochondrial 2-Cys peroxiredoxins, Prdx1 and 3 respectively, are highly reactive antioxidant enzymes that dimerize upon oxidation by hydrogen peroxide or peroxynitrite (k 105 M−1 s−1–107 M−1 s−1) and are biomarkers of cellular changes in peroxide levels [34], [35], [36], [37].
Fig. 2.

NO2-OA does not affect oxidation of Prdx1/Prdx3. RAW264.7 macrophages were incubated for 1, 3 or 6 h with DMSO vehicle or NO2-OA (5 μM and 10 μM). Dimerization of Prdx1 (A, B) and Prdx3 (A,C) was assessed by non-reducing gel electrophoresis followed by immunoblotting and densitometry. Representative immunoblot shown; vertical lines superimposed on blot to facilitate interpretation. Densitometry is expressed as mean ± standard deviation of 6 samples from 3 independent experiments. (D) Prdx oxidation by 10 μM H2O2 occurs rapidly (within 10 min) and is neither inhibited nor promoted by NO2-OA. (E) Prdx1 and Prdx3 oxidation after macrophage activation with 200 ng/mL LPS for 0, 1, 3, or 6 h. As positive control, cells were exposed to 10 μM H2O2 for 10 min, which oxidized both Prdx1 and Prdx3.
Under basal conditions, RAW264.7 macrophages presented 35% of Prdx1 and 60% Prdx3 covalently dimerized (Fig. 2A–C). Treatment with NO2-OA had no effect on the proportion of covalent Prdx dimers compared to controls, despite the significant elevation in intracellular GSSG (Fig. 2A–C). In the setting of exacerbated oxidative stress, the peroxidatic cysteine in typical 2-Cys Prdx can be hyperoxidized to a SO2/3 via a sulfenic acid intermediate [37]. Before and after NO2-OA treatment, oxidized Prdx were detected only in the dimeric form, with absence of formation of monomeric hyperoxidized Prdx (Suppl. Fig. 2). In contrast, treatment of RAW264.7 cell with 400 M H2O2 for 10 min led to significant formation of monomeric hyperoxidized peroxiredoxins. Taken together, no evidence for steady-state oxidant exposure was observed after NO2-OA treatment, indicating that the accumulation in intracellular GSSG is independent of oxidative stress.
To evaluate whether NO2-OA affected Prdx function or interfered with Prdx dimerization, RAW264.7 cells were incubated with 10 M H2O2 in the presence or absence of 5 M NO2-OA. Fig. 2D shows that both Prdx1 and 3 oxidized fully within 20 min of exposure regardless of the presence of NO2-OA. In line with its role as a TLR4 ligand and potent pro-inflammatory mediator, LPS treatment caused significant Prdx oxidation with a similar time course as shown for GSSG formation (Fig. 2E). Exposure to high H2O2 levels consistently decreased the detection of loading control.
2.3. Inhibition of MRP1 is insufficient to increase intracellular GSSG
Cellular efflux of GSH-conjugated electrophiles and GSSG is mediated by the ATP-binding cassette (ABC) transporter class proteins including the multidrug resistance protein 1 (MRP1) [38]. Thus it is conceivable that the increases in GSSG levels induced by NO2-OA might be mediated by a direct effect on MRPs. To test this hypothesis, RAW264.7 were treated with the nonspecific MRP/organic anion transporter (OAT) inhibitor probenecid (250 M) or the MRP1-selective inhibitor MK-571 (25 M) and changes in GSSG levels determined. In contrast to NO2-OA, neither of these inhibitors was able to significantly increase intracellular GSSG (Fig. 3A–C). Although these results do not negate the possibility that NO2-OA might influence MRP function, they demonstrate that MRP inhibition is insufficient to increase the levels of GSSG.
Fig. 3.

Inhibition of MRP1 does not increase intracellular GSSG. Incubation of RAW264.7 macrophages with the specific MRP1 inhibitor MK571 (25 M) or the pan-MRP inhibitor probenecid (250 M) for 6 h did not cause accumulation of intracellular GSSG. NO2-OA was included as control. Results are expressed as mean ± standard error, n = 3. ; ; Statistical analysis by one-way ANOVA.
2.4. NO2-OA is a covalent reversible inhibitor of GR
Since NO2-OA did not induce cellular oxidative stress (Fig. 2), and appreciating that Nrf2 activation upregulates GSH biosynthesis and NADPH production in vivo [39], it was hypothesized that NO2-FA may inhibit GR, reducing GSH regeneration and increasing intracellular GSSG. The GR active site contains two critical cysteines that form an internal disulfide during the catalytic reduction-oxidation cycle of the enzyme. Both NO2-OA and NO2-cLA inhibited S. cerevisiae GR activity, with NO2-OA effecting a more pronounced inhibition (Fig. 4A). Interestingly, whereas incubation of GR with 3 M NO2-OA in the presence of NADPH resulted in a complete inhibition of GR activity, GR pre-incubation with 3 M NO2-OA in the presence of GSSG but absence of NADPH did not result in GR inhibition, which only became evident after NADPH addition (Fig. 4B).
Fig. 4.

NO2-OA is a reversible covalent inhibitor of GR. A) Inhibition of GR activity by NO2-OA, NO2-cLA, or non-electrophilic OA (5 μM each) as measured by the DTNB-reduction assay after 10 min incubation. Results are mean ± SD from 3 to 5 independent experiments. For each experiment 3.36mU GR were incubated in 145 L containing 5 M NO2-FA, OA or vehicle control for 10 min at ambient temperature. by one-way ANOVA vs. control. (B) TNB production by GR pre-incubated with vehicle control, NO2-OA + GSSG (purple) or NO2-OA + NADPH (red) for 10 min prior to addition of missing substrate. Results are summarized as mean ± SD from 3 to 5 independent experiments. (C) Reduced GR (2 nM) was incubated with 800 nM NO2-OA. At increasing times, aliquots (1.6 nM) were mixed with 0.1 mM NADPH and 1 mM GSSG, and the initial rate of absorbance decay at 340 nm and 25 °C was measured. The solid line represents the best fit to a single exponential equation. (D) kobs values at increasing concentrations of NO2-OA (200–1200 nM) were determined from kinetic traces as in (A); kobs values for the reaction between NO2-OA and GAPDH.
To further characterize the inhibition and reactivity of GR with NO2-OA, rate constants were determined using pseudo-first-order conditions (Fig. 4C). A linear dependence was observed between the pseudo-first order rate constant (kobs) and NO2-OA concentration (Fig. 4D). The forward second-order rate constant (kon) was determined from the slope of the kobs versus NO2-OA concentration plot as (3.45 ± 0.04) × 103 M−1 s−1. The non-zero y-axis intercept indicates a reversible reaction with a koff= (4.4 ± 0.4) × 10−4 s−1 and (Keq) was (1.3 ± 0.1)× 10−7 M (25 °C, pH 7.4).
In order to compare the values obtained with other abundant thiol-containing intracellular enzymes, we kinetically characterized the reaction with glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a known target for NO2-OA (Fig. 4D, inset) [18]. The values obtained were kon= 389 ± 64 M−1 s−1, koff= (3.2 ± 0.9)× 10−3 s−1 and Keq= (8 ± 4)× 10−6M (25 °C, pH 7.4) indicating that GR reacts 10 times faster with NO2-OA than GAPDH. In the case of GSH, the forward rate constant with NO2-OA is 64 ± 1 M−1 s−1 (25 °C, pH 7.4), 50 times slower than GR [40].
2.5. NO2-OA modifies Cys61 of GR
Next, we sought to evaluate whether the inhibition involved covalent binding between GR and NO2-OA using biotin-labeled NO2-OA. GR contains 5 Cys and 15 His residues, all of which could be potential nucleophilic targets of NO2-OA. Since the derivatization of NO2-OA carboxylic acid with biotin significantly impacts its size and charge, the inhibition of GR by biotin-NO2-OA was first confirmed (Suppl. Fig. 3). Incubation of purified GR with a 100-fold molar excess of biotin-NO2-OA in the absence of NADPH resulted in only low levels of adducted biotin-NO2-OA detected by non-reducing immunoblot. The adducted level of NO2-OA increased significantly in the presence of NADPH, suggesting Cys61 and Cys66 as potential targets (Fig. 5A). Digestion of GR under native conditions (non-reduced) yielded 69% coverage of the primary sequence encompassing 5/5 Cys and 11/15 His residues, and displayed the expected disulfide bond between Cys61 and Cys66 (Fig. 5D, E). When reduction was followed by alkylation with iodoacetamide, 81% sequence coverage was obtained, presenting stable carbamidomethyl-adducts at the active site cysteines (Fig. 5D, F). Likewise, incubation of GR with a 100-fold molar excess of NO2-OA followed by tryptic digestion yielded only the Cys61-Cys66 disulfide bridge without evidence of nitroalkylation of either catalytic Cys and only minor alkylation of other Cys and His (not shown). In contrast, incubation of GR with NO2-OA in the presence of NADPH permitted the identification of Cys61 as the specific nucleophilic target (84% primary sequence coverage) (Fig. 5G).
Fig. 5.

NO2-OA inhibits GR via Cys61 modification. (A) Catalytic cysteines are a main target of NO2-OA as assessed by 10 min incubation with biotinylated NO2-OA (8 M) in the presence or absence of cofactor NADPH and GSSG (n = 2). (B) Sequence of tryptic peptide encompassing the GR catalytic site ALGGTC61VNVGC66VPK with b+ and y+ fragmentation patterns and potential Cys modifications. (C) Elution profile of the native and alkylated peptides containing Cys61. (D) Tabular summary of calculated (black) and identified (blue/red) b+ and y+ ions, respectively. MS/MS spectra of the GR catalytic site peptide obtained after control, IAM and NO2-OA treated GR showing native (E), carbamidomethylated (F), and nitroalkylated (G) Cys61 peptide.
2.6. GSH noncovalently modulates GR activity
The presence of free thiols has been shown to promote the elimination of NO2-OA from preformed Michael adducts [18], [41]. To evaluate the reversibility of GR inhibition by NO2-OA, GR was pretreated with NO2-OA to achieve complete inhibition. Addition of -mercaptoethanol (BME, 5 mM) resulted in a rapid and complete recovery of GR enzymatic activity as shown by NADPH consumption (Fig. 6A). A dose-response was performed showing complete restoration of GR activity in the presence of 0.5 mM BME (Fig. 6B). Thiol-reversibility of the covalent inhibition of GR by NO2-OA was recapitulated with the endogenous thiol GSH. However, unlike BME, GSH elicited a hormetic response in which increasing concentrations (up to 2.5 mM) partially restored GR activity but higher levels resulted in a dose-dependent decline of the enzymatic activity (Fig. 6C). To test if this reduction was a result of product inhibition in vitro, GR activity was assayed as a function of [GSH](Fig. 6D). GR activity was inhibited by 50% above 10 mM GSH suggesting that this mechanism is relevant at physiologic GSH concentrations and may affect GSH equilibria inside the cell.
Fig. 6.

GR is sensitive to product inhibition in context of electrophile-induced upregulation of [GSH] (A) Representative kinetic traces showing that addition of BME (5 mM) rapidly restores GR activity and NADPH consumption of pre-inhibited GR (34mU GR + 5 M NO2-OA). (B) GR activity restoration is dependent of BME concentration. Results are reported as mean ± SD. For some points, the error bars are shorter than the height of the symbol (C) Hormetic reversal of NO2-OA inhibition of GR by GSH. GR inhibited with NO2-OA was treated with increasing concentrations of GSH and activity was assayed. (D) GSH inhibits GR activity. GR rate was measured in absence or presence of physiologically relevant concentrations of GSH and displayed dose-dependent reduction in activity suggesting product inhibition. (E) Electrophiles CDDO-Im, DMF, and MMF do not inhibit GR activity in vitro. (C–E) For each experiment 3.36mU GR was incubated in 145 L 5 M NO2-OA or vehicle control for 10 min at ambient temperature. Rates are reported as % control ± SD (C–D) or nmol/sec GSSG (E) from 3 to 5 independent experiments; for some points, the error bars are shorter than the height of the symbol. (F) CDDO-Im (100 nM) induces GCLM expression in RAW264.7 macrophages. Vertical line indicates intervening lanes cropped to approximate non-adjacent bands from one blot showing two biological replicates. Increased levels of GSH (E) and GSSG (F) in RAW264.7 macrophages following treatment with 5 μM NO2-OA (red) or 100 nM CDDO-Im (dashed) for indicated times. (I) GSH, (J) GSSG, and the ratio of GSH:GSSG (K) in RAW264.7 cells after 6 h treatment with indicated electrophilic ligands. Statistical analysis by one-way ANOVA with Sidak correction for multiple comparisons; for some groups, error is less than thickness of the error bar.
Next, we evaluated the inhibitory effects of other electrophiles known to activate Nrf2 signaling and increase GSH levels. None of the three electrophiles tested inhibited the GR activity, indicating that this effect was specific for NO2-FA and not a general characteristic of electrophilic drugs (Fig. 6E).
Based on the product inhibition observed with GSH, it was hypothesized that electrophiles which do not inhibit GR directly may nonetheless exert indirect inhibition and induce GSSG accumulation through upregulation of GSH. CDDO-Im activated Nrf2 and strongly upregulated GCLM (Fig. 6F), an effect shared by DMF (data not shown). To compare temporal effects of CDDO-Im and NO2-OA, a time-course experiment was performed evaluating GSH and GSSG 0–24 h after electrophile exposure. Changes in GSH induced by CDDO-Im were comparable to those induced by NO2-OA. Despite the lack of direct GR inhibition in vitro, CDDO-Im treatment significantly increased GSSG (Fig. 6G, H). Notably, whereas all electrophiles had similar effects in GSH levels and all increased GSSG, NO2-OA resulted in significantly larger increases in GSSG levels and concomitant decreases in the GSH: GSSG ratio (Fig. 6I–K).
The large increase in GSSG induced by NO2-OA was found to be dose-dependent with respect to [NO2-OA], indicating that NO2-OA had a direct effect on GR activity in cells (Fig. 7A–B). In contrast, the increase in intracellular GSSG in response to CDDO-Im led to 50% increase in [GSH] and 4-fold increases in GSSG (Fig. 7C–D). In a separate experiment (Fig. 7E), CDDO-Im (200 nM) was not found to increase Prdx oxidation, suggesting that product inhibition by GSH and not oxidative stress was responsible for the observed GSSG accumulation.
Fig. 7.

NO2-OA but not CDDO-Im dose-dependently increases intracellular GSSG. RAW264.7 macrophages incubated with increasing concentrations of NO2-OA (A–B) or CDDO-Im (C–D) for 6 h followed by measurement of GSH (A, C) and GSSG (B, D). Statistical analysis by one-way ANOVA with Dunnett correction for multiple corrections, comparing treatment groups to control (DMSO), n = 3–8. (E) Immunoblotting for monomeric and dimeric Prdx1 and Prdx3 after 1 and 6 h treatment with 200 nM CDDO-Im revealed no increase in cytosolic or mitochondrial Prdx oxidation.
Together these results implicate two separate and specific mechanisms by which electrophiles induce the accumulation of intracellular GSSG: covalent inhibition at catalytic Cys61 in the case of NO2-OA and product inhibition secondary to activation of Nrf2-induced biosynthesis and elevation of GSH levels for all electrophiles.
2.7. Theoretical predictions of GSH/GSSG levels with GSH inhibition of GR based on published kinetic parameters
We show that increasing total GSH pool ([GS]T; [GSH] + 2x [GSSG]) without change in oxidative environment results in an increase in [GSSG] relative to [GSH] and provide evidence suggesting that this may be a result of inhibition of GR by GSH, as reported previously for the isolated enzyme [42], [43]. It is possible to kinetically model the effects of this inhibition in cells and to quantitatively compare our experimental results with theoretical predictions. Although kinetic modeling approaches have been applied previously to the GSH/GSSG system in cells [44], [45], [46], [47], [48], [49], [50], [51], [52], [53], [54], to our knowledge the inhibition of GR by GSH has not been included. To perform this modeling, we assume a constant level of cellular H2O2 of 1 M [55] where the major hydroperoxidase is selenocysteine-containing glutathione peroxidase (GPx) [54], with regeneration of GSH exerted mainly by GR [56]. We assume regeneration of reduced GR by NADPH is more rapid than these enzymatic reactions. Scheme 1 presents an overview of the cellular metabolism of H2O2, GSH, and GSSG with GSH inhibition of GR and v1 and v2 denoting the rates of GR and GPx respectively.
Scheme 1.
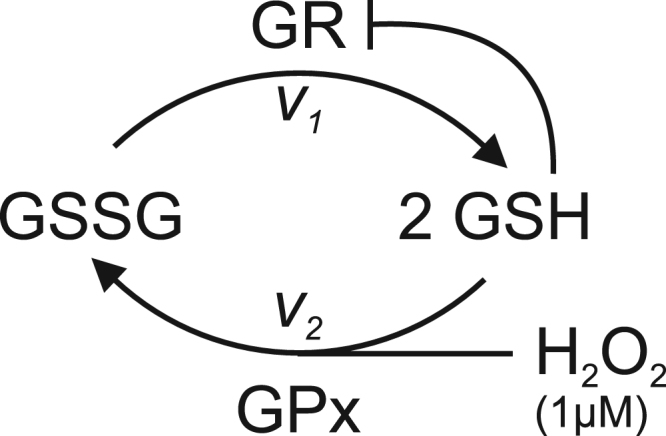
Scheme showing metabolism of H2O2, GSH, and GSSG with v1 and v2 denoting the rates of GR and GPx respectively.
We assume that cellular NADP+/NADPH ratio is relatively low, especially considering the well-established regulation of respiratory substrate availability including NADPH [57], [58]. Under these conditions, GR will operate predominantly via the ping-pong, as opposed to sequential, branch pathway [59]. At steady-state the rate of GR v1(Eq. (1)) is thus given by the Michaelis-Menten equation with noncompetitive parabolic inhibition by GSH, with Km= 88 M for the murine enzyme [59], Vmax=k2[GR]T, and values for the inhibition constants K3–6 as presented in Suppl. Table 1.
| (1) |
where
| (2) |
| (3) |
The steady-state rate of GPx v2 (Eq. (4)) is given by the rate equation for the enzyme-substitution mechanism with published rate constants and 1 M [H2O2] incorporated in the k7 term [60], as presented in Suppl. Table 1.
| (4) |
We use numerical integration of the governing rate equations to predict the relative concentrations of GSSG and GSH as the total [GS]T is increased. As time is gradually increased, enzymatic instantaneous steady-state conditions for each time point are achieved by employing enzymatic rate constants that are much faster than the rate of increase in [GS]T (reaction (10) in Suppl. Table 1). The values for K3 and K4 for noncompetitive parabolic inhibition are taken from published values for the rat liver enzyme and the values for K5, K6, and Vmax (for GR, v1) and [GPx]0 (for GPx, v2) are set to achieve optimal fit to the data (steady-state [GSSG], [GSH]) for all electrophiles except NO2-OA.
Fig. 8A presents the computed increases in [GSH] and [GSSG] with time using the rate constants shown in Suppl. Table 1. The results are identical whether [GS]T is increased by generation of GSH or GSSG (red vs. black), demonstrating rapid steady-state interconversion determined by the enzymatic mechanisms. Fig. 8B shows that steady-state is achieved very early during the calculation (within 5 s) and the inset shows that the rates of each enzymatic reaction are equal (v1 = v2) at every point throughout the calculation, a requirement for steady-state. In addition, this shows that the calculated results are identical to the values predicted by the steady-state derivation (Eq. (1), (2), (3), (4).
Fig. 8.

Theoretical predictions of GSH/GSSG couple support product inhibition of GR. (A) Computed increases in [GSH] (dashed line) and [GSSG] (solid line) with increasing [GS]T by generation of GSH (red) or GSSG (black), using the model described in Suppl. Table 1 and as described in Results. (B) Rapid establishment of steady-state between GR (black) and GPx (red). Inset: Fidelity of predictions by calculation (integrated rate expressions) and steady-state (Michaelis-Menten) theory. (C) Unbiased assessment of GSH plotted as a function of GSSG in cells treated with vehicle (red), CDDO-Im (light blue), CDDO-ME (purple), DMF (dark blue), or BSO (green) for 6 h. Each data point depicts one cell culture sample. Solid line presents results from the kinetic modeling. (D) Simulation of relative abundances of active and inactive (inhibited) GR forms GRi1-4. (E) [GSH] and [GSSG] in cells treated with pharmacologically active concentrations of NO2-OA compared to simulation model and data shown in C (yellow). (F) Relative proportions of GR inhibition by adduction (red) compared to product inhibition (black) with increasing [NO2-OA]. (G) Percent adduction by NO2-OA as a function of GSH concentration. (H) Increase of steady-state [GSSG] as a function of increasing loss of active enzyme (decrease in [GR]) with increasing [NO2-OA].
Fig. 8C shows that the model accurately predicts the experimental results for [GSSG] and [GSH] for all treatments with the exception of NO2-OA. It is clear that as the total [GS]T approaches 10 mM there is a dramatic increase in [GSSG] with only modest increases in [GSH]. To examine the mechanistic basis for this phenomenon, Fig. 8D shows the distribution of GR between different forms as [GS]T is increased. The noncompetitive parabolic binding of GSH to both GR and GR-GSSG results in conversion of active enzyme (GR) into inactive GRi forms; however, the overall rate of GSSG reduction to GSH (v1) remains relatively constant (Fig. 8B inset). This is because at [GSH] 5 mM GPx is saturated (Km = k7/k9 = 0.5 mM, Eq. (4)) and so the rate v2 is maximal and constant, equal to k7[GPx]T.
Fig. 8D shows that even under baseline conditions the enzyme is substantially inhibited; the enzyme is 65% inhibited at [GSH]= 5 mM, 90% inhibited at [GSH]= 7.5 mM and increases to 98% at 9.5 mM. This is because of the steady-state condition v1 = v2 and so v1 is also constant. Therefore, the only way to counteract loss of active enzyme is to increase substrate [GSSG] for the remaining active enzyme. This illustrates the concept that major changes in the relative amounts of GSH and GSSG can occur independently of changes in oxidative conversion of GSH into GSSG (indeed, the rate of this conversion, v2, is essentially unchanged, Fig. 8B inset).
Fig. 8E superimposes experimentally measured [GSSG] as a function of [GSH] from RAW264.7 macrophages treated with NO2-OA data over the values obtained with compounds which do not inhibit GR, as well as the computational prediction. In contrast to the other electrophiles (Fig. 8C), the extent of [GSSG] increase (due to GR inhibition) is much more dependent on the NO2-OA concentration and not on the GSH concentration. This emphasizes our conclusion that the primary mechanism for NO2-OA is by covalent adduction rather than an increase of [GS]T.
In order to test how much tolerance there is in the values for the adjusted parameters K3 and K4, Suppl. Fig. 4 shows that the model provides a reasonable fit to the data only when these values vary by no more than approximately ± 15%.
We utilized modeling to predict the quantitation of GR inhibition due to adduction by NO2-OA. From the steady-state assumption we presume increase in [GSSG] is due to decrease in the amount of active enzyme [GR]T by the less than unity factor α, as predicted from the relationship v1 = v2.
| (5) |
Thus, for each experimental result using NO2-OA, the percent inhibition due to formation of the inactive adducted enzyme (GRa) is given by 100 * (1-α). Although not shown, obtaining α by decreasing [GR]T with trial and error using the integrated rate approach provides results essentially identical to the steady-state value and also allows determination of the total enzyme present in the four inhibited forms from product inhibition ([GRi]T =[GRi1]+[GRi2]+[GRi3]+[GRi4]). Fig. 8F shows the relative percent inhibition for increasing [NO2-OA]. In all cases the enzyme is greatly inhibited, between 94% and 97%, and surprisingly the relative proportion from adduction vs. product inhibition is not determined by increasing [NO2-OA]. These proportions do not correlate with [GSSG], but as Fig. 8G shows there is a linear inverse relationship between percent adducted (GRa) and [GSH]. This in vivo prediction validates the in vitro studies (Fig. 6C) demonstrating that increasing GSH has opposing effects on the activity of adducted enzyme, decreased activity from product inhibition and increased activity from reversal of adduction. Finally, Fig. 8H shows that increasing [NO2-OA] monotonically (although nonlinearly) decreases the total active GR, even with variable proportions of adduction vs. product inhibition (Fig. 8F).
3. Discussion
GSH, with intracellular concentrations in the millimolar range and numerous enzymatic and non-enzymatic modes of reactivity, is decisively positioned to act not only as a regulator of thiol oxidation status, but also as a main target of electrophiles. The molecular pleiotropy of the GSH system is further affirmed by its ubiquity, penetrance and complexity in modulating cell signaling activities, detoxification and downstream post-translational protein modifications. The pharmacological manipulation of the GSH/GSSG couple allowed us to study its impact on temporal responses to oxidative and electrophilic stress. Cell exposure to NO2-OA for 1 h leads to a slight decrease in GSH levels, likely caused by adduct formation, which has been previously documented [18]. This initial decrease was followed by a rebound in intracellular GSH levels that peaked at 6 h. The increase in GSH was not temporally linked to GCLM protein expression, which was maximal at 24 h and under Nrf2 control, suggesting GCLM levels are not the rate limiting factor on GSH synthesis at the early time points. It is known that activation of Nrf2 may also impact import of cystine, a substrate in the synthesis of GSH [61].
Notably, the increase in GSH levels (1.5–2 fold) occurred concomitantly with an unexpected and paradoxical increase in intracellular GSSG. This result is in close agreement with prior observations which showed that both the thiol antioxidant/GSH-precursor N-acetylcysteine and GCLC/GCLM over-expression concomitantly increase GSH and GSSG levels [62]. However, the authors in this prior study concluded that the mechanism underlying GSSG accumulation was related to cellular damage induced by reductive stress. In the setting of NO2-OA-induced Nrf2 activation, analysis of Prdx 1 and 3 dimerization status did not indicate significant alterations in endogenous peroxide levels or impaired ability to respond to oxidative insults. Additionally, inhibition of MRP1 with known inhibitors MK571 and probenecid proved insufficient to elevate intracellular GSSG.
Rather, the present investigation revealed two discrete mechanisms underlying this phenomenon, both of which converge on the intuitive, yet underappreciated concept that the GSH:GSSG ratio is regulated primarily by the impact of [GSH] on the activity of GR. Specifically, our findings indicate that electrophiles modulate the GSH redox couple by direct pharmacological inhibition in the case of NO2-OA and indirect product inhibition through elevation in GSH levels (Scheme 2).
Scheme 2.

Schematic summary of findings. Electrophiles including NO2-OA and Bardoxolone (CDDO) activate Keap1/Nrf2 signaling to up-regulate genes under Antioxidant Response Element (ARE) control, including enzymes catalyzing GSH biosynthesis. GSH reduces oxidized cellular constituents through activity of GPX and is concomitantly oxidized to GSSG. GSSG is reduced to GSH through GR. Electrophiles inhibit GR activity through product inhibition resulting from up-regulation of GSH, or through covalent inhibition at catalytic Cys61. Both GSH and GSSG participate in protein S-glutathionylation reactions, through glutathione S-transferase activity and disulfide exchange, respectively.
NO2-OA is known to modify several enzymes that contain catalytic thiol centers, such as glyceraldehyde-3-phosphate dehydrogenase [18], 5-lipoxygenase [63], and soluble epoxide hydrolase [64]. Based on the fact that GR has two catalytic cysteines that form a disulfide during enzymatic redox cycling and that these cysteines are covalently modified by GR inhibitors [65], we explored the modulation of GR activity by NO2-OA.
NO2-OA directly inhibits GR in a NADPH dependent pathway that involved nitroalkylation of Cys61 reacting with GR ten times faster than with GAPDH. While the precise extent of inhibition of GR by NO2-OA in cells was not determined in this study, increases in GSSG induced by NO2-OA displayed dose-dependence, suggesting that the in vitro inhibition of GR was recapitulated in a complex, intact biological system. The inhibition of GR by NO2-OA was rapidly reversed by the low molecular weight thiol BME. Furthermore, when the reversibility assay was repeated using different concentrations of GSH as the competing thiol, the response was bell-shaped, with concentrations of GSH up to 2.5 mM partially restoring GR activity while concentrations above 2.5 mM reducing GR activity. In this regard, macrophage incubation with other electrophiles (CDDO-Im, CDDO-Me, DMF) that did not directly inhibit GR, resulted in GSH and GSSG accumulation. The increase in GSSG elicited by CDDO-Im was found to be proportional to the increase in [GSH] but independent of electrophile concentration. This led us to propose an additional mechanism for GSSG accumulation based on product inhibition of GR.
Evidence for product inhibition of GR by GSH was first published nearly three decades ago using isolated GR preparations but these observations were never further explored or extended to complex, dynamic cellular systems [42], [43]. Of relevance, the range of concentration at which GSH inhibits GR activity is between 8 and 13 mM [42], [43]; in our experiments IC50 (10 mM) directly relates to GSH concentrations found in cells (between 6 and 9 mM). Offering support to this regulatory process, depletion of GSH with BSO resulted in 2-fold greater reduction in GSSG than in GSH (Fig. 1).
Collective unbiased expression of [GSSG] as a function of [GSH] showed a positive non-linear correlation (Fig. 8C). BSO-treated cell populations clustered in the bottom-left quadrant of the distribution suggesting de-repression of GR, while electrophile-treated populations clustered in upper-right, signifying product inhibition. Computational simulation of the steady-state GSH system using a numeric integration approach with published parameters showed that product inhibition of GR was necessary to accurately fit the model to these experimental data. The simulations are not intended to provide a complete kinetic analysis of GR, which has been performed by others. Rather, it provides an illustration of how our experimental results are compatible with (indeed, predicted by) the consequences of the inhibition of GR by GSH. The results from both experiments and simulation are incompatible with a static concept of GSH cycling, in which the relative abundances of GSH and GSSG are solely dependent upon oxidant load and where GSH depletion via oxidation is prerequisite to GSSG accumulation. Instead, the observations suggest that in the absence of oxidative stress, [GSSG] is determined by flux through GR, which is subject to product inhibition by GSH.
It is possible that the responsiveness of GR to intracellular [GSH] evolved as a mechanism to optimize allocation of reducing equivalents. In a reducing or unstressed state, this mechanism could redeploy energetically expensive reducing equivalents (NADPH) from generating unneeded GSH to other biosynthetic pathways, such as production of lipids and nucleic acids. At the same time, this would permit cells to immediately increase flux through the GSH cycle when contending with oxidative insults by bypassing the need for de novo GR expression. In aggregate, the control of GSH:GSSG couple under our conditions is shown to be independent of oxidative stress and supports the hypothesis that the kinetic couple reflects the antagonistic actions of enzymatic consumption and regeneration of GSH, as opposed to a thermodynamic state with discrete physiological correlates [55]. In contrast, the covalent inhibition of GR by NO2-OA resulted in dose-dependent increase in GSSG levels, demonstrating the importance of functional tuning of electrophile reactivity for specific targets and applications.
Electrophiles have previously been observed to modulate the expression and/or activity of enzymes involved in thiol homeostasis. In addition to biosynthesis through GCLC/GCLM and reduction by GR, the relative amounts of GSH and GSSG are impacted by use of GSH as a substrate in protein- and peroxide-reduction reactions catalyzed by glutaredoxins (GRX) and glutathione peroxidases (GPX). While previous studies have not identified GRX as a target gene of Nrf2 [6], [58], an electrophilic dithiocarbamate derivative has been previously identified to inhibit both GR [65] and GRX1 [66]. It is possible that NO2-OA may likewise inhibit GRX in addition to GR; indeed, our unpublished results suggest that mixed glutathione-protein disulfides are increased by NO2-OA. The biochemical characterization and biological sequelae of this interaction remain a topic of further study. In contrast to GRX, expression of GPX1 has been found to be dependent on Nrf2 activity and is responsive to electrophiles [58]. In addition, electrophilic pollutants from diesel exhaust and tobacco smoke have previously been found to inhibit GPX activity [67]. While expression of GPX is known to be dependent on Nrf2, in our experiments NO2-OA did not affect cell sensitivity to peroxide exposure, suggesting that activity of GPX is not impacted by this electrophile.
In conclusion, we have shown that electrophiles impact both the levels of GSH and GSSG through modulation of GSH synthesis to impact GR activity and, in the case of NO2-OA by reacting with catalytic Cys61. This has broad implications, as changes in the GSH/GSSG couple and EGSH have been widely used as a marker for oxidative stress and increased oxidative environments.
4. Experimental procedures
4.1. Materials
Primary antibodies were purchased from the following suppliers: GR (Abcam, Cambridge, MA), GCLM (Proteintech, Chicago, IL), HO-1 (Cell Signaling, Beverly, MA), GAPDH (Trevigen, Gaithersburg, MD), Histone H3 (Cell Signaling, Danvers MA) Peroxiredoxin 1 (R & D Systems, Minneapolis, MN), Peroxiredoxin 3 (R & D Systems, Minneapolis, MN), Peroxiredoxin1-4 SO2/3 (Abcam, Cambridge, MA). Secondary antibodies were purchased from Santa Cruz Biotechnologies (Dallas, TX). LPS derived from E.coli 0127:B8, purified GR from S. cerevisiae, DMF, MMF,GSH, 13C215N GSH and GSSG were from Sigma-Aldrich (St. Louis, MO). Oleic acid was obtained from Nu-Check Prep, Inc. (Elysian, MN). NO2-OA, biotin-NO2-OA and NO2-cLA were synthesized as previously reported [21]. NO2-OA was used as a mixture of 9- and 10-NO2 regio-isomers while NO2-cLA was a mixture of 9- and 12-NO2 regio-isomers. CDDO-Im and -Me were from Toronto Research Chemicals (Toronto, Canada. Solvents were LC-MS quality and purchased from Burdick and Jackson (Morristown, NJ)). Formic acid was purchased from Fisher Scientific (Pittsburgh, PA).
4.2. Enzyme activity assays
Glutathione reductase activity was determined following the initial rates of dithionitrobenzoate (DTNB) reduction using an established biochemistry method adapted from Rahman et al. [68]. Briefly, purified GR from S. cerevisiae (3mU-12mU; consistent within experiments) was incubated with vehicle or inhibitor in the presence of NADPH (150 M) or GSSG (1 mM) and reaction was started with either GSSG or NADPH respectively. Consumption of NADPH (340 = 6.22 × 103 M−1 cm−1) or production of chromogenic TNB ( 412 = 1.42 × 104 M−1 cm−1) were followed on a Agilent 8453 UV–Vis spectrophotometer.
4.3. Cell culture
RAW264.7 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA) and grown per ATCC instructions in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
4.4. Immunoblotting
Cells were washed 2x with cold PBS and lysates were prepared in 4 °C RIPA buffer with MiniTab protease inhibitor cocktail (Roche, Switzerland). For Prdx experiments, cells were incubated in alkylation buffer (40 mM HEPES, 50 mM NaCl, 1 mM EGTA, 1 mM EDTA, 100 mM NEM, pH 7.4) for 10 min at room temperature before lysing with Triton X-100 and protease inhibitor cocktail. Protein was quantified by BCA assay (Pierce/Thermo Fisher) and samples containing 15–25 g total cellular protein were loaded on 4–12% Bis-Tris gel. Proteins were resolved by reducing or non-reducing SDS-PAGE, transferred to nitrocellulose membrane, blocked with 5% non-fat dry milk in TBS-0.1% Tween 20 for 1 h. Primary and secondary antibodies were used at 1:1000 and 1:5000 dilutions unless otherwise indicated and visualized with Clarity ECL chemiluminescence kit and ChemiDoc imager (Bio-Rad, Hercules, CA).
4.5. Cellular GSH and GSSG determination
The method for simultaneous determination of GSH and GSSG was adapted from a previously published report [69]. Briefly, RAW264.7 macrophages were seeded in 24-well plates at a density of 300,000 cells/well and were cultured overnight prior to treatments. Media was aspirated and cells were washed 2x with sterile PBS and incubated with PBS containing 25 mM N-ethylmaleimide (NEM) for 15 min at 37 °C. The PBS/NEM solution was aspirated and 150 L derivatizing solution (25 mM NEM, 40 mM HEPES/50 mM NaCl/1 mM EDTA; 2 M 13C215N GSH; 2 M 13C415N2 GSSG) was added to each well and incubated for 15 min at room temperature. Cells were detached by scraping, followed by 3 cycles of sonication in a 4 °C water bath for 30 s each. The lysate was cleared by centrifugation at 21,000 g for 10 min at 4 °C. Protein content was assessed using the BCA assay. Proteins were precipitated by addition of 9 volumes of ethanol (200 proof), cooled to −80 °C overnight, centrifuged (21,000 g, 4 °C, 10 min) and the supernatant dried under N2 stream at room temperature. Samples were reconstituted in dH2O and 20 L were injected for analysis by LC-MS/MS.
4.6. Reversed phase HPLC tandem mass spectrometry
A Shimadzu HPLC (Columbia, MD) coupled to a Thermo Scientific CTC HTS PAL autosampler (Waltham, MA) and an AB Sciex (Framingham, MA) 5000 triple quadrupole mass spectrometer were used for the quantification of GSH adducts and GSSG. Samples (20 μl) were resolved on a Phenomenex C18 column ( mm, 5 μm particle size) using the following solvent system A) aqueous 0.1% formic acid and B) 0.1% formic acid in acetonitrile at a flow rate of 600 l/min. Chromatographic conditions were as follows: 1% solvent B for 0.1 min, followed by a linear gradient to 20.3% solvent B at 6 min, to then switch to 100% solvent B for 2 min and re-equilibration to return to the initial condition (1% solvent B) for 4 min. The following settings for the mass spectrometer were used: Source temperature 550 °C; ionization spray voltage 5500 V; CAD 4.0 arbitrary units; Curtain gas 40 arbitrary units; GS1 45 arbitrary units; GS2 50 arbitrary units; EP 5.00 V; CXP 10.00 V. Multiple reaction monitoring was performed with 65 ms dwell time and a declustering potential of 60 V using the following transitions in positive ion mode: GS-NEM (433.0/304.0, CE 38), 13C215N GS-NEM (436.0/307.0, CE 38), GSSG (613.2/355.2, CE 30), 13C415N2 GSSG (619.2/361.2, CE 30).
4.7. LC-MS/MS detection and analysis of GR post-translational modifications
Purified GR (S. cerevisiae; 10 μg) was incubated in the presence or absence of GSSG (2 mM), NADPH (3 mM), NO2-OA (8 μM) for 30 min in 50 mM phosphate buffer, pH 7.4 and the reaction stopped by addition of iodoacetamide (10 mM final concentration, 10 min at room temperature). After alkylation, GR was digested using MS grade modified trypsin (trypsin/GR ratio of 1:50) for 16 h at 37 °C. The peptide digest was analyzed by LC-MS/MS for post-translational modifications using a Thermo Surveyor Plus HPLC coupled to an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). Peptides (3 μg on column) were loaded onto a Phenomenex C18 ( mm, 5 µm particle size; Torrance, CA) reverse-phase column resolved using a linear gradient of solvent A (0.1% formic acid in HPLC grade water) and solvent B (0.1% formic acid in acetonitrile) at a 650 μl/min flow rate. Chromatographic conditions were as follows: 5% solvent B for 2 min, followed by a linear gradient to 60% solvent B for 18 min, to then switch to 100% solvent B for 5 min and re-equilibration to return to the initial condition (5% solvent B) for 5 min. MS analysis was carried out in the positive ion mode with source parameters optimized for the detection of peptides containing nitroalkylated Cys. Instrument settings were as follows: source voltage, 3.75 kV; capillary temperature, 200 °C; source heater temperature, 450 °C; sheath gas flow and auxiliary gas flow, 25 arbitrary units; sweep gas flow, 20 arbitrary units; collision energy, 35 eV. MS/MS spectra was acquired using data-dependent acquisition in which one full MS spectrum was followed by MS/MS spectra of the top five ions. Peptide analysis was performed using Proteome Discoverer 2.0 (Thermo Fisher Scientific). MS/MS spectra (b and y ions) of detected modified peptides (presenting a 327.2410 atomic mass unit shift corresponding to NO2-OA) were manually validated by comparing their fragmentation pattern with the native carbamidomethylated peptides.
4.8. Kinetic modeling of cellular GSH and GSSG
Steady-state conditions were assumed for both GR and GPx. For each enzyme, validation of the steady-state assumption was accomplished by numerical integration (Berkeley-Madonna www.berkeleymadonna.com using the Rosenbrock stiff algorithm) of the elementary rate equations for each mechanism. The rate constants were chosen to reflect published experimental values; for GPx k7, k9, and [H2O2] reported in [55], for GR K3 and K4 reported [42] and Km in [59]. The parameters adjusted to optimize fit were Vmax, K5, and K6 for GR and GPx]Tfor GPx. Further detail is provided in Supplemental figures.
4.9. Statistical analysis
Statistical analyses were performed using GraphPad Prism (La Jolla, CA. USA) version 7.01 by Student's t-test, one-way or two-way analysis of variance (ANOVA) and multiple comparison testing as indicated in figure legends. P values less than 0.05 were considered statistically significant.
Acknowledgments
We would like to thank Dr. Beatriz Álvarez for helpful discussions.
This work was supported by National Institutes of Health (United States) grants R01-HL64937, R01-HL132550, and P01-HL103455 (B.A.F.), K01-HL133331 and the Vascular Medicine Institute, the Hemophilia Center of Western Pennsylvania, and the Institute for Transfusion Medicine (DAV), 5F30DK108391 (SJ), R01-GM125944 and R01-DK112854 (FJS) and American Heart Association (United States) 17GRN33660955 (FJS) and fellowships from Universidad de la República CSIC (Uruguay) (LT), and Academy of Finland (Finland) 285468 (EK).
Conflict of interest
BAF, DAV and FJS have financial interest in Complexa Inc. All other authors declare that they have no conflicts of interest with the contents of this article.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.11.008.
Appendix A. Supplementary data
References
- 1.Portillo-Ledesma S., Sardi F., Manta B., Tourn M.V., Clippe A., Knoops B., Alvarez B., Coitiño E.L., Ferrer-Sueta G. Deconstructing the catalytic efficiency of peroxiredoxin-5 peroxidatic cysteine. Biochemistry. 2014;53:6113–6125. doi: 10.1021/bi500389m. [DOI] [PubMed] [Google Scholar]
- 2.Tajc S.G., Tolbert B.S., Basavappa R., Miller B.L. Direct determination of thiol pKa by isothermal titration microcalorimetry. J. Am. Chem. Soc. 2004;126:10508–10509. doi: 10.1021/ja047929u. [DOI] [PubMed] [Google Scholar]
- 3.Griffith O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999;27:922–935. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- 4.Itoh K., Wakabayashi N., Katoh Y., Ishii T., Igarashi K., Engel J.D., Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewerenz J., Hewett S.J., Huang Y., Lambros M., Gout P.W., Kalivas P.W., Massie A., Smolders I., Methner A., Pergande M., Smith S.B., Ganapathy V., Maher P. The cystine/glutamate antiporter system xc - in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013;18:522–555. doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chorley B.N., Campbell M.R., Wang X., Karaca M., Sambandan D., Bangura F., Xue P., Pi J., Kleeberger S.R., Bell D.A. Identification of novel NRF2-regulated genes by ChiP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012;40:7416–7429. doi: 10.1093/nar/gks409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen T., Sherratt P.J., Nioi P., Yang C.S., Pickett C.B. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J. Biol. Chem. 2005;280:32485–32492. doi: 10.1074/jbc.M503074200. [DOI] [PubMed] [Google Scholar]
- 8.Alam J., Stewart D., Touchard C., Boinapally S., Choi A.M.K., Cook J.L. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999;274:26071–26078. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 9.Ishii T., Itoh K., Takahashi S., Sato H., Yanagawa T., Katoh Y., Bannai S., Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi A., Suzuki H., Itoh K., Yamamoto M., Sugiyama Y. Transcription factor Nrf2 is required for the constitutive and inducible expression of multidrug resistance-associated protein 1 in mouse embryo fibroblasts. Biochem. Biophys. Res. Commun. 2003;310:824–829. doi: 10.1016/j.bbrc.2003.09.086. [DOI] [PubMed] [Google Scholar]
- 11.Maher J.M., Dieter M.Z., Aleksunes L.M., Slitt A.L., Guo G., Tanaka Y., Scheffer G.L., Chan J.Y., Manautou J.E., Chen Y., Dalton T.P., Yamamoto M., Klaassen C.D. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology. 2007;46:1597–1610. doi: 10.1002/hep.21831. [DOI] [PubMed] [Google Scholar]
- 12.Jeyapaul J., Jaiswal A.K. Nrf2 and c-Jun regulation of antioxidant response element (ARE)-mediated expression and induction of γ-glutamylcysteine synthetase heavy subunit gene. Biochem. Pharmacol. 2000;59:1433–1439. doi: 10.1016/s0006-2952(00)00256-2. [DOI] [PubMed] [Google Scholar]
- 13.Favreau L.V., Pickett C.B. Transcriptional regulation of the rat NAD(P)H: quinone reductase gene. Identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. J. Biol. Chem. 1991;266:4556–4561. [PubMed] [Google Scholar]
- 14.Itoh K., Tong K.I., Yamamoto M. Molecular mechanism activating nrf2-keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004;36:1208–1213. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt T.J., Ak M., Mrowietz U. Reactivity of dimethyl fumarate and methylhydrogen fumarate towards glutathione and N-acetyl-l-cysteine-Preparation of S-substituted thiosuccinic acid esters. Bioorg. Med. Chem. 2007;15:333–342. doi: 10.1016/j.bmc.2006.09.053. [DOI] [PubMed] [Google Scholar]
- 16.Dibbert S., Clement B., Skak-Nielsen T., Mrowietz U., Rostami-Yazdi M. Detection of fumarate-glutathione adducts in the portal vein blood of rats: evidence for rapid dimethylfumarate metabolism. Arch. Dermatol. Res. 2013;305:447–451. doi: 10.1007/s00403-013-1332-y. [DOI] [PubMed] [Google Scholar]
- 17.Samudio I., Konopleva M., Hail N., Shi Y.X., McQueen T., Hsu T., Evans R., Honda T., Gribble G.W., Sporn M., Gilbert H.F., Safe S., Andreeff M. 2-Cyano-3,12-dioxooleana-1,9-dien-28-imidazolide (CDDO-Im) directly targets mitochondrial glutathione to induce apoptosis in pancreatic cancer. J. Biol. Chem. 2005;280:36273–36282. doi: 10.1074/jbc.M507518200. [DOI] [PubMed] [Google Scholar]
- 18.Batthyany C., Schopfer F.J., Baker P.R.S., Durán R., Baker L.M.S., Huang Y., Cerveñansky C., Branchaud B.P., Freeman B.A., Duran R., Baker L.M.S., Huang Y., Cervenansky C., Branchaud B.P., Freeman B.A. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J. Biol. Chem. 2006;281:20450–20463. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker L.M.S., Baker P.R.S., Golin-Bisello F., Schopfer F.J., Fink M., Woodcock S.R., Branchaud B.P., Radi R., Freeman B.A. Nitro-fatty acid reaction with glutathione and cysteine. Kinetic analysis of thiol alkylation by a Michael addition reaction. J. Biol. Chem. 2007;282:31085–31093. doi: 10.1074/jbc.M704085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodcock C.-S.C., Huang Y., Woodcock S.R., Salvatore S.R., Singh B., Golin-Bisello F., Davidson N.E., Neumann C., Freeman B.A., Wendell S.G. Nitro-fatty acid inhibition of triple negative breast cancer cell viability, migration, invasion and tumor growth. J. Biol. Chem. 2017;293:1120–1137. doi: 10.1074/jbc.M117.814368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui T., Schopfer F.J., Zhang J., Chen K., Ichikawa T., Baker P.R.S., Batthyany C., Chacko B.K., Feng X., Patel R.P., Agarwal A., Freeman B.A., Chen Y.E. Nitrated fatty acids: endogenous anti-inflammatory signaling mediators. J. Biol. Chem. 2006;281:35686–35698. doi: 10.1074/jbc.M603357200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang J., Lee K.E., Lim J.-Y., Park S.I. Nitrated fatty acids prevent TNFalpha-stimulated inflammatory and atherogenic responses in endothelial cells. Biochem. Biophys. Res. Commun. 2009;387:633–640. doi: 10.1016/j.bbrc.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 23.Villacorta L., Chang L., Salvatore S.R., Ichikawa T., Zhang J., Petrovic-Djergovic D., Jia L., Carlsen H., Schopfer F.J., Freeman B.A., Chen Y.E. Electrophilic nitro-fatty acids inhibit vascular inflammation by disrupting LPS-dependent TLR4 signalling in lipid rafts. Cardiovasc. Res. 2013;98:116–124. doi: 10.1093/cvr/cvt002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reddy A.T., Lakshmi S.P., Kleinhenz J.M., Sutliff R.L., Hart C.M., Reddy R.C. Endothelial cell peroxisome proliferator-activated receptor γ reduces endotoxemic pulmonary inflammation and injury. J. Immunol. 2012;189:5411–5420. doi: 10.4049/jimmunol.1201487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudolph V., Rudolph T.K., Schopfer F.J., Bonacci G., Woodcock S.R., Cole M.P., Baker P.R.S., Ramani R., Freeman B.A. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovasc. Res. 2010;85:155–166. doi: 10.1093/cvr/cvp275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu H., Jia Z.Z., Soodvilai S., Guan G., Wang M.-H., Dong Z., Symons J.D., Yang T. Nitro-oleic acid protects the mouse kidney from ischemia and reperfusion injury. Am. J. Physiol. - Ren. Physiol. 2008;295:942–949. doi: 10.1152/ajprenal.90236.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J., Villacorta L., Chang L., Fan Z., Hamblin M., Zhu T., Chen C.S., Cole M.P., Schopfer F.J., Deng C.X., Garcia-Barrio M.T., Feng Y.-H.H., Freeman B.A., Chen Y.E. Nitro-oleic acid inhibits angiotensin II-induced hypertension. Circ. Res. 2010;107:540–548. doi: 10.1161/CIRCRESAHA.110.218404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reddy A.T., Lakshmi S.P., Dornadula S., Pinni S., Rampa D.R., Reddy R.C. The nitrated fatty acid 10-nitro-oleate attenuates allergic airway disease. J. Immunol. 2013;191:2053–2063. doi: 10.4049/jimmunol.1300730. [DOI] [PubMed] [Google Scholar]
- 29.Wang H., Sun J., Jia Z., Yang T., Xu L., Zhao B., Yu K., Wang R. Nitrooleic acid attenuates lipid metabolic disorders and liver steatosis in DOCA-salt hypertensive mice. PPAR Res. 2015;2015:480348. doi: 10.1155/2015/480348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kansanen E., Jyrkkänen H.-K., Volger O.L., Leinonen H., Kivelä A.M., Häkkinen S.-K., Woodcock S.R., Schopfer F.J., Horrevoets A.J., Ylä-Herttuala S., Freeman B.A., Levonen A.-L. Nrf2-dependent and -independent responses to nitro-fatty acids in human endothelial cells: identification of heat shock response as the major pathway activated by nitro-oleic acid. J. Biol. Chem. 2009;284:33233–33241. doi: 10.1074/jbc.M109.064873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levonen A.L., Hill B.G., Kansanen E., Zhang J., Darley-Usmar V.M. Redox regulation of antioxidants, autophagy, and the response to stress: implications for electrophile therapeutics. Free Radic. Biol. Med. 2014;71:196–207. doi: 10.1016/j.freeradbiomed.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uruno A., Motohashi H. The Keap1-Nrf2 system as an in vivo sensor for electrophiles. Nitric Oxide - Biol. Chem. 2011;25:153–160. doi: 10.1016/j.niox.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 33.Iskusnykh I.Y., Popova T.N., Agarkov A.A., Pinheiro De Carvalho M.A., Rjevskiy S.G. Expression of glutathione peroxidase and glutathione reductase and level of free radical processes under toxic hepatitis in rats. J. Toxicol. 2013;2013:8706–8728. doi: 10.1155/2013/870628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dubuisson M., Vander Stricht D., Clippe A., Etienne F., Nauser T., Kissner R., Koppenol W.H., Rees J.-F., Knoops B. Human peroxiredoxin 5 is a peroxynitrite reductase. FEBS Lett. 2004;571:161–165. doi: 10.1016/j.febslet.2004.06.080. [DOI] [PubMed] [Google Scholar]
- 35.Ogusucu R., Rettori D., Munhoz D.C., Soares Netto L.E., Augusto O. Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: rate constants by competitive kinetics. Free Radic. Biol. Med. 2007;42:326–334. doi: 10.1016/j.freeradbiomed.2006.10.042. [DOI] [PubMed] [Google Scholar]
- 36.Cox A.G., Peskin A.V., Paton L.N., Winterbourn C.C., Hampton M.B. Redox potential and peroxide reactivity of human peroxiredoxin 3. Biochemistry. 2009;48:6495–6501. doi: 10.1021/bi900558g. [DOI] [PubMed] [Google Scholar]
- 37.Peskin A.V., Dickerhof N., Poynton R.A., Paton L.N., Pace P.E., Hampton M.B., Winterbourn C.C. Hyperoxidation of peroxiredoxins 2 and 3: rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J. Biol. Chem. 2013;288:14170–14177. doi: 10.1074/jbc.M113.460881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cole S.P.C. Multidrug resistance protein 1 (MRP1, ABCC1), a multitasking ATP-binding cassette (ABC) transporter. J. Biol. Chem. 2014;289:30880–30888. doi: 10.1074/jbc.R114.609248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu K.C., Cui J.Y., Klaassen C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011;123:590–600. doi: 10.1093/toxsci/kfr183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turell L., Vitturi D.A., Coitiño E.L., Lebrato L., Möller M.N., Sagasti C., Salvatore S.R., Woodcock S.R., Alvarez B., Schopfer F.J. The chemical basis of thiol addition to nitro-conjugated linoleic acid, a protective cell-signaling lipid. J. Biol. Chem. 2017;292:1145–1159. doi: 10.1074/jbc.M116.756288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schopfer F.J., Batthyany C., Baker P.R., Bonacci G., Cole M.P., Rudolph V., Groeger A.L., Rudolph T.K., Nadtochiy S., Brookes P.S., Freeman B.A. Detection and quantification of protein adduction by electrophilic fatty acids: mitochondrial generation of fatty acid nitroalkene derivatives. Free Radic. Biol. Med. 2009;46:1250–1259. doi: 10.1016/j.freeradbiomed.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chung P.M., Cappel R.E., Gilbert H.F. Inhibition of glutathione disulfide reductase by glutathione. Arch. Biochem. Biophys. 1991;288:48–53. doi: 10.1016/0003-9861(91)90163-d. [DOI] [PubMed] [Google Scholar]
- 43.Sexton D.J., Mutus B. Glutathione reductases from a variety of sources are inhibited by physiological levels of glutathione. Comp. Biochem. Physiol. Part B: Biochem. 1992;103:897–901. doi: 10.1016/0305-0491(92)90211-9. [DOI] [PubMed] [Google Scholar]
- 44.Antunes F., Han D., Cadenas E. Relative contributions of heart mitochondria glutathione peroxidase and catalase to H2O2 detoxification in in vivo conditions. Free Radic. Biol. Med. 2002;33:1260–1267. doi: 10.1016/s0891-5849(02)01016-x. [DOI] [PubMed] [Google Scholar]
- 45.Cortassa S., Aon M.A., Winslow R.L., O’Rourke B. A mitochondrial oscillator dependent on reactive oxygen species. Biophys. J. 2004;87:2060–2073. doi: 10.1529/biophysj.104.041749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kranner I., Birtić S., Anderson K.M., Pritchard H.W. Glutathione half-cell reduction potential: a universal stress marker and modulator of programmed cell death? Free Radic. Biol. Med. 2006;40:2155–2165. doi: 10.1016/j.freeradbiomed.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 47.Kemp M., Go Y.-M.Y., Jones D.D.P. Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic. Biol. Med. 2008;44:921–937. doi: 10.1016/j.freeradbiomed.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toppo S., Flohé L., Ursini F., Vanin S., Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: variations of a basic scheme. Biochim. Biophys. Acta - Gen. Subj. 2009;1790:1486–1500. doi: 10.1016/j.bbagen.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 49.Adimora N.J., Jones D.P., Kemp M.L. A model of redox kinetics implicates the thiol proteome in cellular hydrogen peroxide responses. Antioxid. Redox Signal. 2010;13:731–743. doi: 10.1089/ars.2009.2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aon M.A., Stanley B.A., Sivakumaran V., Kembro J.M., O'Rourke B., Paolocci N., Cortassa S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study. J. Gen. Physiol. 2012;139:479–491. doi: 10.1085/jgp.201210772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brito P.M., Antunes F. Estimation of kinetic parameters related to biochemical interactions between hydrogen peroxide and signal transduction proteins. Front. Chem. 2014;2:82. doi: 10.3389/fchem.2014.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Altntaş A., Davidsen K., Garde C., Mortensen U.H., Brasen J.C., Sams T., Workman C.T. High-resolution kinetics and modeling of hydrogen peroxide degradation in live cells. Free Radic. Biol. Med. 2016;101:143–153. doi: 10.1016/j.freeradbiomed.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 53.Antunes F., Brito P.M. Quantitative biology of hydrogen peroxide signaling. Redox Biol. 2017;13:1–7. doi: 10.1016/j.redox.2017.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deponte M. The incomplete glutathione puzzle: just guessing at numbers and figures? Antioxid. Redox Signal. 2017;27:1130–1161. doi: 10.1089/ars.2017.7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flohé L. The fairytale of the GSSG/GSH redox potential. Biochim. Biophys. Acta. 2013;1830:3139–3142. doi: 10.1016/j.bbagen.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 56.Kojer K., Bien M., Gangel H., Morgan B., Dick T.P., Riemer J. Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J. 2012;31:3169–3182. doi: 10.1038/emboj.2012.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holmstrom K.M., Baird L., Zhang Y., Hargreaves I., Chalasani A., Land J.M., Stanyer L., Yamamoto M., Dinkova-Kostova A.T., Abramov A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open. 2013;2:761–770. doi: 10.1242/bio.20134853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thimmulappa R.K., Mai K.H., Srisuma S., Kensler T.W., Yamamoto M., Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 59.Pannala V., Bazil J., Camara A., Dash R. A biophysically based mathematical model for the catalytic mechanism of glutathione reductase. Free Radic. Biol. Med. 2013;65:1385–1397. doi: 10.1016/j.freeradbiomed.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Flohé L., Toppo S., Cozza G., Ursini F. A comparison of thiol peroxidase mechanisms. Antioxid. Redox Signal. 2011;15:763–780. doi: 10.1089/ars.2010.3397. [DOI] [PubMed] [Google Scholar]
- 61.Hayes J.D., Dinkova-Kostova A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 62.Zhang H., Limphong P., Pieper J., Liu Q., Rodesch C.K., Christians E., Benjamin I.J. Glutathione-dependent reductive stress triggers mitochondrial oxidation and cytotoxicity. FASEB J. 2012;26:1442–1451. doi: 10.1096/fj.11-199869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Awwad K., Steinbrink S.D., Frömel T., Lill N., Isaak J., Häfner A.-K., Roos J., Hofmann B., Heide H., Geisslinger G., Steinhilber D., Freeman B.A., Maier T.J., Fleming I. Electrophilic fatty acid species inhibit 5-lipoxygenase and attenuate sepsis-induced pulmonary inflammation. Antioxid. Redox Signal. 2014;20:2667–2680. doi: 10.1089/ars.2013.5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Charles R.L., Rudyk O., Prysyazhna O., Kamynina A., Yang J., Morisseau C., Hammock B.D., Freeman B.A., Eaton P. Protection from hypertension in mice by the Mediterranean diet is mediated by nitro fatty acid inhibition of soluble epoxide hydrolase. Proc. Natl. Acad. Sci. USA. 2014;111:8167–8172. doi: 10.1073/pnas.1402965111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seefeldt T., Zhao Y., Chen W., Raza A.S., Carlson L., Herman J., Stoebner A., Hanson S., Foll R., Guan X. Characterization of a novel dithiocarbamate glutathione reductase inhibitor and its use as a tool to modulate intracellular glutathione. J. Biol. Chem. 2009;284:2729–2737. doi: 10.1074/jbc.M802683200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sadhu S.S., Callegari E., Zhao Y., Guan X., Seefeldt T. Evaluation of a dithiocarbamate derivative as an inhibitor of human glutaredoxin-1. J. Enzym. Inhib. Med. Chem. 2013;28:456–462. doi: 10.3109/14756366.2011.649267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Staimer N., Nguyen T.B., Nizkorodov S.A., Delfino R.J. Glutathione peroxidase inhibitory assay for electrophilic pollutants in diesel exhaust and tobacco smoke. Anal. Bioanal. Chem. 2012;403:431–441. doi: 10.1007/s00216-012-5823-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rahman I., Kode A., Biswas S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2007;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 69.Harwood D.T., Kettle A.J., Brennan S., Winterbourn C.C. Simultaneous determination of reduced glutathione, glutathione disulphide and glutathione sulphonamide in cells and physiological fluids by isotope dilution liquid chromatography-tandem mass spectrometry. J. Chromatogr. B. 2009;877:3393–3399. doi: 10.1016/j.jchromb.2009.04.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.