

SUMMARY
Disparities in cancer care have been a long-standing challenge. We estimated the genetic ancestry of The Cancer Genome Atlas patients, and performed a pan-cancer analysis on the influence of genetic ancestry on genomic alterations. Compared with European Americans, African Americans (AA) with breast, head and neck, and endometrial cancers exhibit a higher level of chromosomal instability, while a lower level of chromosomal instability was observed in AAs with kidney cancers. The frequencies of TP53 mutations and amplification of CCNE1 were increased in AAs in the cancer types showing higher levels of chromosomal instability. We observed lower frequencies of genomic alterations affecting genes in the PI3K pathway in AA patients across cancers. Our result provides insight into genomic contribution to cancer disparities.
In Brief
By analyzing TCGA cohorts, Yuan et al. show that breast, head and neck, and endometrial cancers of African Americans (AA) have higher levels of chromosomal instability than those of European Americans whereas the frequency of genetic alternations in the PI3K pathway in AA patients is lower across cancers.
Graphical Abstract
INTRODUCTION
Cancer is a genomic disease involving multi-step changes in the genome. Disparities in cancer defined by self-identified race or ethnicity (SIRE) have been a long-standing and persistent challenge (Daly and Olopade, 2015; DeSantis et al., 2016; Powell, 2007; Torre et al., 2016; Zeng et al., 2015). Certain racial and ethnic populations in the United States experience increased incidence in total, and of aggressive disease for specific cancer. For example, compared with other racial groups, African Americans (AA) exhibit higher rates of colorectal cancer incidence and death, AA women have higher mortality rates from breast and endometrial cancer, and AA men have higher incidence and mortality rates from prostate and lung cancers. However, the genomic causes of cancer disparities are still poorly understood. Genetic ancestry reflects the history of human migration, providing background information about genetic variation that is essential for inference about the genetic association of diseases (Royal et al., 2010). Due to shared continental ancestry, individuals of the same genetic ancestry group share common genetic variants. This shared genetic background may confer similarities in cancer incidence and outcomes in populations. Recent large-scale genomic profile studies in individual cancer types, such as prostate (Huang et al., 2017; Petrovics et al., 2015; Powell et al., 2013; Wang et al., 2017), breast (Ademuyiwa et al., 2017; Huo et al., 2017; Keenan et al., 2015; Loo et al., 2011), colon (Guda et al., 2015), lung (Araujo et al., 2015; Campbell et al., 2017; Kytola et al., 2017), gastric (Schumacher et al., 2017), esophageal (Deng et al., 2017), and kidney (Krishnan et al., 2016) cancers have robustly demonstrated that genomic differences in cancers exist among distinct racial and ethnic populations. Consistently, it has been reported that the genetic background of patients may influence specific somatic alterations in cancer genomes during tumorigenesis (Carter et al., 2017). The Cancer Genome Atlas (TCGA) data resource contains multi-omic profiles and clinical annotations of large-scale samples across 33 cancer types, and therefore serves as an excellent resource for pan-cancer analyses to evaluate the relationship between genetic ancestry and genomic alterations in cancers. The TCGA data portal provides SIRE information by: (1) race categorized as White, Black, Asian, American Indian/Alaska Native, and Native Hawaiian/Other Pacific Islander; and (2) ethnicity categorized as Hispanic/Latino and non-Hispanic/Latino. However, a large percentage of patients(12.36%; n = 1,375) lack race information in TCGA. For example, in the cohort of prostate cancer, a cancer type showing significant health disparities, race information is unavailable in68.67% (342/498) of patients. Rapid advances in high-density genotyping technology allow precise and quantitative estimation of genetic ancestry in recently admixed populations (Liu et al., 2013; Price et al., 2006; Pritchard et al., 2000; Sankararaman et al., 2008). Thus, we propose to estimate the genetic ancestry of each TCGA patient through a computational analysis of genome-wide genotyping data generated by TCGA, aiming to assign each individual to an ancestral population and to make this information available through a publicly accessible resource.
RESULTS
Estimation of Global Genetic Ancestry
We integrated multiple computational algorithms to estimate the global and local genetic ancestry of each TCGA patient (n = 11,122, involving 33 cancer types from 27 primary sites, Table S1), and assigned each individual to an ancestral population. EIGENSTRAT (Price et al., 2006) was applied to infer the global genetic structure of TCGA patients. Unrelated individuals from the International HapMap Project and the America panel of the Human Genome Diversity Project were used as reference populations (Table S2). Eigenvectors were estimated using all reference populations and then projected onto TCGA patients (Figure 1A). The first three principal components explained9.35%, 4.45%, and 0.96% of genetic variance, respectively. Self-identified Blacks from TCGA (n = 922) clustered with African descendants from HapMap and distinguished themselves from other SIRE groups on the first axis (Figure 1B). The second and third axes separated self-identified Asians (n = 672) and American Indians and Alaska Natives (n = 27), respectively. Using individuals of known ancestry from HapMap and HGDP, a k-nearest neighbors (k-NN) classifier was trained and applied to categorize each patient from TCGA into one of five genetic ancestry groups (European American [EA], n = 8,951, 80.5%; AA, n = 1,019, 9.2%; East Asian American [EAA], n = 677,6.1%; Native American [NA], n = 397, 3.6%; and Others [OA], n = 78, 0.7%). Similar results were obtained when the 1000 Genomes Project and the HGDP served as the reference panels (Figure S1A). To avoid confusion, in this manuscript, we categorize genetic ancestry as EA, AA, EAA, and NA, and self-identified race from TCGA as White, Black, Asian, and American Indians, and Alaska Natives. For patients with race information in TCGA, we observed that 95.6% of categorizations based on the k-NN algorithm following principal-component analysis (PCA) were consistent with race, suggesting that race and genetic ancestry identified by our approach appear to be highly correlated across TCGA patients. We inferred genetic ancestry for all TCGA specimens with SNP array genotype data, including those with unreported SIRE (Figures 1C and S1B). Notably,59.4% and 32.8% of self-identified Hispanic/Latino individuals were assigned into NA and EA groups according to genetic ancestry, respectively. Next, given that a substantial proportion of the US population is represented by admixed populations, STRUCTURE (Pritchard et al., 2000) was used to quantitatively determine the ancestral composition for each patient. To this end, unrelated individuals from CEU/TSI (European), YRI (West African), CHB/JPT (East Asian), and the America panel from HGDP (Native American) were used as proxies for “continental” ancestral populations (Table S2). As expected, a large percentage of the TCGA specimens showed genetic diversity (Figures 1D and S2). Importantly, the global genetic ancestry estimation by STRUCTURE allowed us to treat the genetic ancestry data as a continuous variable for downstream analyses.
Figure 1. Estimation of Genetic Ancestry across TCGA.

(A) Three-dimensional visualization of genetic variation of individuals from the HapMap and HGDP reference populations (left) or self-identified Black patients of TCGA (right) on the first three principal components (PCs) calculated by EIGENSTRAT. The ellipse defines the 95% confidence interval for each genetically related group.
(B) Genetic variation on each PC stratified by reference populations and TCGA self-identified racial identity. Reference populations were selected and classified according to geographical location and genetic origin. Boxplot lines reflect lower quartile, median, and upper quartile of PC scores. Whiskers extend 1.5 times the interquartile range from the upper and lower quartiles, with points outside representing outliers.
(C) Bar plot showing the numbers of TCGA patients categorized into each of the four genetic ancestry groups (EA, NA, EAA, and AA) by EIGENSTRAT across the TCGA cohort (left) and in the prostate cancer cohort (right). SIRE information is color-coded by green (White), pink (Asian), blue (Black), orange (AI/AN), and gray (unavailable). The proportion of SIRE is also represented with a circle plot.
(D) Individual ancestry of TCGA patients inferred by STRUCTURE. Each color represents one of the ancestry reference groups. Each patient is represented by a column partitioned into different colors corresponding to the genetic ancestry composition. Patients are ordered following a hierarchical clustering by Ward’s methods on distance matrix calculated as cosine dissimilarity of genetic composition. SIRE and genetic ancestry categorization as estimated by EIGENSTRAT for each patient are shown in the same order at the bottom.
(E) Three-dimensional visualization of reference populations with three patients (TCGA-06–0167, TCGA-PE-A5DD, and TCGA-VS-A9V2) used as examples for genetic ancestry (AA, EAA, and NA, respectively).
(F) Local ancestry across SNPs on 22 autosomes inferred by LAMP for these three patients. Each patient was treated as a diploid admixed genome. The colors represent ancestral reference groups, and light gray marks genomic regions unassigned because they are missing from SNPs shared by reference populations.
(G) Comparison of the percent of West African ancestry inferred from LAMP (based on distribution of local ancestry) versus STRUCTURE. TCGA patients are grouped into bins, each of which represents an interval of 1% range. The intensity of a bin represents the number of patients in the given interval group.
(H) Global (top) and local ancestry (bottom) of two unrelated admixed AA patients. To visualize local ancestry, SNPs on 22 autosomes are ordered according to genomic location. Each color represents one of the ancestry reference groups. Same color code as in (F).
(I) Genome-wide distribution of average ancestry proportion at each ancestral segment in AA patients of TCGA. Top, average proportion of West African ancestry plotted against genomic position along the 22 autosomal chromosomes (colors indicate different chromosomes). Bottom, average contribution from the four ancestral groups. Each color represents one of the ancestry reference groups. Same color code as in (F).
See also Figures S1–S3; Tables S1 and S2.
Estimation of Local Genetic Ancestry
Although a down-sampling test demonstrated that a small number of SNPs was sufficient for estimation of global ancestry (Figures S3A and S3B), the high-density SNP data generated by TCGA provided a powerful resource to estimate local ancestry (locus-specific ancestry) for each individual patient. To estimate the genetic ancestry at a particular chromosomal locus, LAMP (Sankararaman et al., 2008) was applied to the genotype data of TCGA. Specifically, LAMP inferred genetic ancestry separately within overlapping windows using a likelihood model, allowing us to assign a label of genetic ancestral origin to each locus defined by an SNP for a given individual genome (Figures 1E and 1F). As expected, local genetic ancestry estimation by LAMP yielded estimates of the individual admixture, which were comparable with global quantitative ancestry estimates by STRUCTURE (Figure 1G). However, the local genetic ancestry structures largely differed between patients with similar global genetic ancestral composition (Figure 1H). Consistent with a previous study (Bhatia et al., 2014), we did not observe significant deviation from the genome-wide average of ancestral contributions, indicating no selection influencing ancestry after admixture. In AA patients, the average proportion of African ancestry over all genomic segments with unique ancestry status was 0.786 (SD = 0.010, Figure 1I). The average proportions of East Asian and Native American ancestry were 0.945 (SD =0.047) and 0.224 (SD = 0.024) in EAA and NA patients, respectively.
Development of the Cancer Genetic Ancestry Atlas
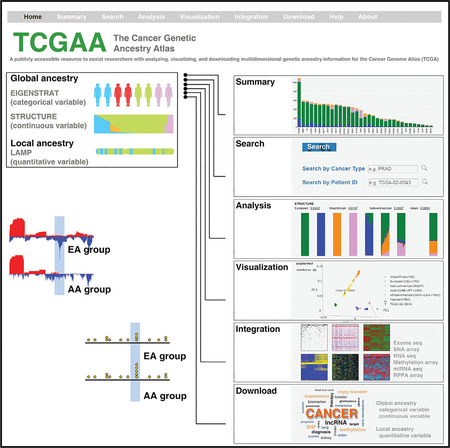
After estimating both global (as a categorical variable and a continuous quantitative variable) and local genetic ancestry for each patient of TCGA (Figure 2A), we integrated our analyses and TCGA clinical annotations and multidimensional genomic profiles (Figure 2B) to develop a comprehensive academic research database: The Cancer Genetic Ancestry Atlas (TCGAA). The TCGAA data portal provides six modules (Summary, Search, Analysis, Visualization, Integration, and Download) by integrating genetic ancestry, clinical annotations and genomic profiles of the TCGA project (Figure 2C). The patient information was also directly linked to the data portals of the Genomic Data Commons and the TCGA. The TCGAA database contains information for 11,122 cancer patients across 27 primary sites (33 cancer types, Figure 3A). Across all cancer types, 80.5% (n = 8,951) were EA, 9.2% (n = 1,019) were AA,6.1% (n = 677) were EAA, and 3.6% (n = 397) were NA. Breast cancer and liver cancer had the largest number of AA (n = 183;16.7%) and EAA (n = 163; 43.2%) patients, respectively (Figure 3B). In 16 cancer types, the number of patients with AA ancestry estimated by EIGENSTRAT is larger than 20, while eight cancer types have >20 EAA patients and seven cancer types have >20 NA patients (Figure 3B and Table S3). We also analyzed the cancer types that show evidence of racial disparities, and found that race and ethnicity were considered during the sample collection of TCGA for most of these cancer types (Figure 3B). For example, higher incidence or mortality rates from stomach, liver, and thyroid cancers are reported in Asian American patients; correspondingly, greater numbers of EAA samples were collected in the TCGA specimens for these cancer types. Taken together, although the sample sizes of racial minorities in TCGA may still not be sufficient for de novo identification of racial group-specific genomic alterations at a cancer type-specific level (Huang et al., 2017; Spratt et al., 2016), TCGA provides one of the largest sample cohorts across most common adult cancer types, with comprehensive clinical information and multidimensional genomic profiles for studies of the effects of genetic ancestry on genomic alterations. Using the same computational approaches, we also estimated global and local genetic ancestry for 1,251 cancer cell lines, derived from 29 primary sites (Table S4), whose SNP array genotyping data were retrieved from the Cancer Cell Line Encyclopedia and the Genomics of Drug Sensitivity in Cancer. For the cell lines with SIRE information in ATCC (n = 378), we observed that 94.71% of categorizations based on the k-NN algorithm following PCA analysis were consistent with SIRE, suggesting that SIRE and genetic ancestry identified by our approach appear to be highly correlated across cell lines. This information is provided to the public via the Cancer Cell Line Genetic Ancestry Atlas (CCGAA, http://52.25.87.215/CCGAA, Figure 2B).
Figure 2. The Cancer Genetic Ancestry Atlas.

(A) Summary of analysis and integration strategies for genotype data. The global and local genetic ancestry for each patient of TCGA was estimated by three algorithms (EIGNSTRAT, STRUCTURE, and LAMP). Unrelated individuals from the HapMap and HGDP projects were used as reference populations. The genetic ancestry information was integrated with genomic profiles and provided through the TCGAA data portal. N, the number of cancer types (TCGA) or reference populations (HapMap and HGDP); n, the number of individuals.
(B) Overview of TCGAA data portal. The TCGAA database contains integrated information for 11,122 primary cancer specimens across 27 primary sites (33 cancer types). The global and local genetic ancestry information for 1,251 established cancer cell lines with a detailed genetic and pharmacologic characterization is also provided via a sub-database (CCGAA).
(C) The TCGAA and CCGAA provide six modules (Summary, Search, Analysis, Visualization, Integration, and Download) by integrating genetic ancestry, clinical annotations, and genomic profiles of the TCGA project.
Figure 3. The Genetic Ancestry of TCGA Patients.

(A) Summary of genetic ancestry of TCGA patients across 33 cancer types. The size of each circle corresponds to the number of samples of a given cancer type, and the proportion of each genetic ancestry is indicated by color. A color-coded square indicates that the sample number of a given minority genetic ancestry group is larger than 20.
(B) Summary of the patient numbers of each minority genetic ancestry group. The cancer types in each minority group are ranked by the number of minority patients in the group. The cancer types that show evidence for racial disparities are labeled by a color-coded circle in each minority group.
See also Tables S3 and S4.
Pan-cancer Analysis of AA Genetic Ancestry and Genomic Alterations
Given that, across the TCGA sample cohorts, the number of tumor samples from NA populations (3.6%; n = 397) is too small for informative statistical analysis, we decided to compare the difference in genomic alterations between two genetic ancestry groups. This also makes our results comparable with the previous studies in individual cancer types, since most of them were performed in two racial populations. In this regard, we chose AA as the minority population of interest for this current study, and used EA as a reference population. We investigated the relationship between AA genetic ancestry and the genomic alterations of cancers by studying tumors from 8 primary sites (10 cancer types), each of which had more than 40 AA patients: breast invasive carcinoma (BRCA), colon adenocarcinoma (COAD), glioblastoma multiforme (GBM), head and neck squamous cell carcinoma (HNSC), kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP), lung adeno-carcinoma (LUAD), lung squamous cell carcinoma (LUSC), prostate adenocarcinoma (PRAD), and uterine Corpus Endometrial Carcinoma (UCEC). We treated the genetic ancestry information inferred by EIGENSTRAT as a categorical variable (i.e., AA versus EA), and the ancestral population allele frequency estimated by STRUCTURE as a continuous quantitative variable(i.e., percentage of AA ancestry).
AA Genetic Ancestry and Somatic Copy-Number Alterations
We deconstructed the copy-number profiles into three different levels of somatic copy-number alterations (SCNAs) (i.e., focal, arm, and chromosome levels). An overall SCNA score was calculated through modification of the methods described by Davoli et al. (2017). For each tumor, the overall SCNA score was defined as the unweighted numeric sum of the standardized SCNA scores from the above three levels. Adjusting for clinical factors, AA patients had a significantly higher overall SCNA score for three cancer types (BRCA, UCEC, and HNSC), and a significantly lower score for two kidney cancer types (KIRC and KIRP) than EA patients with a false discovery rate (FDR < 10%) (Figure 4A). There were no significant differences observed for the other five cancer types examined. Similarly, when we treated genetic ancestry as a continuous variable, an identical result was found using regression analysis (Figure S3C). Given that the SCNAs in arm/chromosome level and focal level may arise through distinct molecular mechanisms, we also analyzed them separately (Figures 4A–4D). With FDR < 10%, both focal and arm/chromosomal SCNAs differed significantly between AA and EA patients in BRCA, UCEC, and HNSC (Figure 4A). Interestingly, in KIRC, SCNAs at focal level were the dominant contributor to the difference in overall SCNAs between AA and EA patients, and no significant difference was observed at the arm/chromosomal level. The opposite pattern was observed in KIRP, where SCNAs at arm/chromosomal level were the dominant source of differences in overall SCNAs between AA and EA patients, and no significant difference was observed at the focal level. Furthermore, we estimated the whole-genome doubling (WGD) event for each tumor by the ABSOLUTE algorithm (Carter et al., 2012) based on allele-specific copy-number profiles. With FDR < 10%, BRCA was the only cancer type that demonstrated different frequencies of WGD between AA and EA patients (Figure 4E), with higher frequency among AA patients.
Figure 4. AA Genetic Ancestry and Global Somatic Copy-Number Alterations.

(A) Volcano plot of log10 (p value) against effect size (AA versus EA), representing the difference in SCNA scores between AA and EA patients across 10 cancer types. Each circle corresponds to a cancer type with size proportional to median burden of SCNA: weighted genomic instability index at overall level, weighted sum of SCNA events at the focal level or the arm/chromosomal level. Significance (y axis) and effect size (x axis) were calculated by linear regression adjusting for a clinical factors-derived propensity score. SCNA scores were rank-scaling transformed as a conservative measure to avoid results driven by outliers. Positive effect size corresponded to elevation of SCNA score in AA patients and negative values to reduction. The cancer types with significantly elevated or reduced SCNA scores in AA patients (FDR < 10%) are shown in red or blue, respectively. Cancer types with non-significant results are colored in gray.
(B) Comparison of the alteration frequency for recurrent focal SCNAs in AA versus EA patients of BRCA, UCEC, HNSC, and KIRC, respectively. Dots represent recurrent focal SCNAs (peak regions) identified by GISTIC. The y and x axes represent the alteration frequency for a peak region in AA and EA patients, respectively. The gray line indicates the null hypothesis (y = x) that AA patients are affected at an equal rate with EA patients at each peak region. A fitted line on all dots is plotted, with slope indicating the overall difference in alteration rate at peak regions. The fitted line is colored red if the slope is greater than one and blue if the slope is less than one.
(C) Histogram of Z values by logistic regression comparing alteration frequency of recurrent focal SCNAs between AA and EA patients, with clinical factors adjusted. For each cancer type, boxplot lines reflect lower quartile, median, and upper quartile of Z values. Whiskers extend 1.5 times the interquartile range from the upper and lower quartiles, with points outside representing outliers. Each point represents a recurrent focal SCNA. Boxes are colored red if the lower quartile is above zero and blue if the upper quartile is below zero.
(D) Comparison of the alteration frequency of arm-level SCNAs across the whole genome in BRCA, UCEC, HNSC, and KIRP. An arm-level value of the log2 copy-number change ratio larger than 0.25 was considered an arm copy-number alteration. For each chromosome arm in a certain cancer type, the frequency of gain (red, above horizontal line) or loss (blue, under horizontal line) was calculated and plotted separately. Alteration frequency of each chromosome arm in a given cancer type is plotted as lines or filled bars for AA or EA patients, respectively.
(E) Frequency of genome doubling stratified by genetic ancestry (AA versus EA) in each cancer type. The cancer types with significantly different odds of WGD event in AA patients (FDR < 10%) are marked with an asterisk.
See also Figure S3.
The recurrent focal SCNAs of each cancer type were estimated by GISTIC (Mermel et al., 2011), and recurrent focal SCNAs with significantly different alteration frequencies between AA and EA patients were identified at both pan-cancer and cancer type-specific levels. For pan-cancer analysis, we analyzed the recurrent focal SCNAs shared by multiple cancer types using Stouffer’s Z score (Stouffer et al., 1949). In brief, a Z score was calculated to assess whether AA and EA patients differed significantly in the frequency of a given recurrent focal SCNA in a certain cancer type, controlling for variations in genomic disruption. Z scores contributing to a same genomic region from different cancer types were then summarized into a pan-cancer meta-Z score (amplified and deleted SCNAs were considered separately). In summary, three recurrent focal SCNAs were identified with significantly different alteration frequencies between AA and EA patients at pan-cancer level after adjusting for clinical factors (with FDR < 10%, Figure 5A; Table S5). For example, the recurrent focal amplification residing within the 19q12 region had a significantly higher amplification frequency in AA patients across four cancer types (BRCA, GBM, LUSC, and UCEC) compared with EA patients (raw p value = 9.37 × 10−4, corrected p value = 0.072). The quantile-quantile plot demonstrated strong deviation of the observed p values from the null expectation for the above three recurrent focal SCNAs (Figure S3D). Since analyses of the genes in recurrent genomic alterations have led to the successful identification of cancer-driver genes, we hypothesized that certain genes located in the above three recurrent focal SCNAs may play functional roles in the generation of cancer disparities. To identify such genes, four criteria were used (Figure 5B). Using these criteria, 60 protein-coding and 115 non-coding genes were initially mapped into the above 3 recurrent focal SCNAs, and 34 genes (31 protein-coding and 3 non-coding) fulfilled the four criteria (Figures 5C and 5D; Table S6). Two examples of identified genes, CCNE1 and non-coding gene VPS9D-AS1, are shown in Figures 5E and 5F, respectively. As the mechanisms driving cancer development among cancer types may differ, we also identified recurrent focal SCNAs with significantly different alteration frequencies between AA and EA patients at a cancer type-specific level, based on separate analyses of each individual cancer type. After adjusting for clinical factors, five recurrent focal SCNAs differed significantly in frequency between AA and EA patients with FDR < 10% (Table S7). In addition, when we relaxed the significance threshold (FDR < 25% within each cancer type) to detect “near significance,” we found 16 additional hits. For example, in prostate cancer, we identified a single recurrent focal SCNA located on 10q23.31, which showed significantly lower alteration frequency in AAs compared with EAs (raw p value = 3.55 × 10−4, corrected p value = 0.21). Applying the four criteria described above, we further identified 30 protein-coding genes and 5 non-coding genes potentially contributing to disparities at a cancer-specific level (Table S8).
Figure 5. AA Genetic Ancestry and Focal Copy-Number Alterations.

(A) Three recurrent focal SCNAs with significantly different alteration frequencies between AA and EA patients were identified by a pan-cancer meta-analysis across 10 cancer types. The top dot plots show the significance (y axis) of the meta-analysis. Dots represent recurrent focal SCNAs (peak regions) identified in at least two cancer types, ordered by genomic location. The red or blue dots represent the recurrent focal SCNAs identified to be altered at significantly different rates in AA patients compared with EA patients (with FDR < 10%) by a pan-cancer meta-analysis across ten cancer types (red represent amplification and blue represent deletions, respectively). The bottom heatmaps show schematic boundaries of peak regions identified by GISTIC in each cancer type. Cancer types are clustered by similarity of independent significance upon analysis on the cancer-specific level by controlled permutation test. Significance for each recurrent focal SCNA on the cancer-specific level is colored with intensity (a higher-intensity color represents a more significant difference; orange represents higher alteration rate in AA patients and green represents lower alteration rate in AA patients, respectively).
(B) Simplified workflow of the computational approaches used to identify recurrent focal SCNAs with significantly different alteration frequencies between AA and EA patients (top), and genes potentially contributing to disparity through SCNAs (bottom).
(C) Diagram shows the number of candidate genes during the stepwise filtering depicted in (B).
(D) Word cloud of the genes potentially contributing to disparity through SCNAs identified by pan-cancer analysis. The size of the font indicates the significance (p value on a negative log scale) of differential expression between AAs and EAs after adjusting for clinical factors. Gray indicates the function of the gene in cancer is unknown and the intensity of red color indicates prediction score of gene function in cancer.
(E and F) Violin plots showing the cancer type-adjusted RNA expression levels of CCNE1 (E) and VPS9D1-AS1 (F) across given cancer types, with samples grouped based on gene copy number (left) or genetic ancestry (right). The central line within each violin represents the median value. Correlations between RNA expression and predicted gene copy numbers (left) were calculated by meta-analysis. Tests for differential expression between AA and EA tumors (right) were calculated by meta-analysis adjusting for clinical factors.
See also Figure S3; Tables S5–S8.
AA Genetic Ancestry and Somatic Mutations
We analyzed the difference in mutation burden between AA and EA patients. Defining tumors of high mutation burden as those in the upper quartile, we found that there was no significant difference in the frequency of tumors with high mutation burden after adjusting for clinical factors. Next, we compared the mutational process activity (Alexandrov et al., 2013) between AA and EA patients adjusting for clinical factors, and no statistically significant differences were observed (Table S9). We generated a list of recurrently mutated genes for each cancer type by using a combination of four resources (Table S10). The recurrently mutated genes with significantly different alteration frequencies between AA and EA patients were identified at both pan-cancer and cancer type-specific levels, controlling for clinical factors. For pan-cancer meta-analysis, we chose 30 recurrently mutated genes whose mutation frequencies were higher than 5% across the 10 cancer types analyzed in this study (Figure 6A). At a pan-cancer level, we found that TP53 mutations were significantly enriched in AA patients compared with EA patients (raw p value =1.02 × 10−3, corrected p value = 3.05 × 10−2, Figure 6A). In addition, when we relaxed the significance threshold (FDR < 25%) to detect mutations with near significance, we found four additional hits that were less frequently mutated in AA: CREBBP, ARID1A, PIK3CA, and PTEN (Figure 6A). Finally, we repeated this analysis within each cancer type utilizing cancer type-specific lists of recurrently mutated genes (Table S10). At a cancer type-specific level, five genes were identified with FDR < 10% (Figure 6B and Table S11). Consistent with the pan-cancer analysis, differential mutation frequencies of TP53 and PIK3CA were observed in more than one cancer type (Figure 6B). AA patients had significantly higher TP53 mutation frequency in BRCA. COAD fell just below the threshold of significance but also demonstrated the trend of TP53 mutation enrichment in AAs (odds ratio [OR] =1.82, p = 0.043). Significant enrichment of PIK3CA mutations in EA patients was observed in BRCA. HNSC fell just below the significance threshold, but also followed the trend of fewer PIK3CA mutations in AAs (OR = 0.29, p = 0.048). Taken together, at both pan-cancer and cancer type-specific levels, TP53 showed significantly higher mutation frequency in AA patients compared with EA patients, while the genes in the phosphatidylinositol 3-kinase (PI3K) pathways appeared to be less frequently mutated in AA patients.
Figure 6. AA Genetic Ancestry and Somatic Mutation.

(A) Summary of pan-cancer meta-analysis on recurrently mutated genes between AA and EA patients across 10 cancer types. The top bar plot shows the significance (y axis) of the meta-analysis for each recurrently mutated gene. Red and blue bars represent the genes whose mutation frequencies are significantly higher and lower in AAs compared with EA, respectively. The middle dot plot shows independent differences in mutation frequency in each cancer type. The intensity of color corresponds to effect size of AA ancestry compared with EAs (red and blue indicate higher and lower frequencies in AA, respectively). The size corresponds to overall mutation frequency of a given gene in a specific cancer type. Cancer types are ordered by similarity between statistical measures (Z score based) observed at the individual cancer type level and at the pan-cancer level. The bottom bar plot shows the mutation frequency of the recurrently mutated genes across 10 cancer types.
(B) Summary of the cancer type-specific analysis on recurrently mutated genes between AA and EA patients in ten cancer types. The volcano plot of −log10 (p value) against effect size (AA versus EA) represents the difference in mutation frequency between AA and EA patients for a given cancer type after adjusting for clinical factors. Each circle corresponds to a gene tested in a specific cancer type with size proportional to overall mutation frequency. Red and blue circles represent the genes whose mutation frequencies are significantly higher and lower in AAs compared with EA, respectively.
Integrated Analysis of Genomic Alterations in Patients with AA Genetic Ancestry
Across the ten cancer types analyzed, we observed that, compared with EA, AA patients showed a significantly higher level of chromosomal instability in BRCA, UCEC, and HNSC, which was associated with increased frequencies of TP53 mutations as well as CCNE1 amplification (Figure 7A). In contrast, in kidney cancers, AA patients had a significantly lower level of chromosomal instability. Supporting this observation, RNA sequencing profile analysis from the corresponding cancer specimens of TCGA showed that an mRNA expression signature of chromosomal instability (CIN70 signature developed by Carter et al., 2006) had a consistent pattern of expression across the ten cancer types between AA and EA patients. In AA patients, the CIN70 signature was overexpressed in BRCA, UCEC, and HNSC, and was decreased in KIRP and KIRC (Figure 7A). In contrast to this varied pattern of chromosomal instability, AA patients showed a uniform decrease in alterations in the PI3K pathway relative to EA patients (Figure 7B); i.e., significantly lower mutation frequencies of PIK3CA (BRCA), PIK3R1 (UCEC), and PTEN (UCEC), as well as significantly lower levels of PTEN copy-number loss (PRAD) in AA patients. Consistent with this observation, analysis of reverse-phase protein array data from TCGA showed that the PI3K pathway activity (PI3K score developed by Zhang et al. (2017)) was globally decreased in AA compared with EA patients (Figure 7B).
Figure 7. Integrated Analysis of Genomic Alterations on Patients with AA Genetic Ancestry.

(A) Chromosomal instability and associated genes in AA patients. The upper dot plot shows the overall SCNA score and genomic alterations (TP53 mutation and CCNE1 amplification) for each cancer type. The intensity of circles represents the relative difference between AA and EA patients in the individual cancer type, with the size proportional to the significance of the association. A circle with an outline indicates a statistically significant difference (FDR < 0.1). Red, increased in AA; blue, decreased in AA. The heatmap at the bottom shows normalized significance levels (−log[p value]) for the association between AA ancestry and the expression of 70 genes correlated with chromosomal instability (CIN70 signature). For all statistical tests, clinical factors were considered.
(B) PI3K activity and associated genes in AA patients. The dot plot at the top shows the PI3K score and genomic alterations (PIK3CA, PIK3R1, and PTEN mutations; PTEN deletion) for each cancer type. The intensity of circle represents the relative difference between AA and EA patients in the individual cancer type, with the size proportional to the significance of the association. A circle with an outline indicates a statistically significant difference (FDR < 0.1). Red, increased in AA; blue, decreased in AA. The heatmap at the bottom shows normalized significance levels (−log[p value]) for the association between AA ancestry and expression of proteins (reverse-phase protein array [RPPA]) correlated with PI3K activity. For all statistical tests, clinical factors were considered.
DISCUSSION
We estimated genetic ancestry for patients in TCGA—a large genetically admixed cohort of cancer patients with multiple genomic profiles and comprehensive clinical annotations. By using integrated computational algorithms, we inferred the genetic ancestry of TCGA patients at global and local levels, designated genetic ancestry groups based on genotyping data, and developed the TCGAA data portal to make these data widely available to the research community. This resource provides a tool to help elucidate the genetic contribution to cancer disparities. Given that 33 cancer types from 27 primary anatomic sites were analyzed by a standardized genomic-profiling protocol, TCGAA significantly improves our ability to perform pan-cancer analyses for genomic mechanisms of cancer disparities across multiple cancer types. However, the absolute number of samples from racial minorities in each given cancer type is still relatively small, which limits the ability to detect racial group-specific genomic alterations in specific cancer types (Huang et al., 2017; Spratt et al., 2016). In addition, as information about the socioeconomic status of TCGA patients is unavailable, we were unable to adjust for this important variable when comparing racial differences in genomic alterations. Therefore, additional efforts are still urgently needed to identify large samples of underrepresented patients with comprehensive clinical information such as socioeconomic status to better understand the genomic basis for disparities across all racial/ethnic groups.
Leveraging the resources of TCGA, which profiles multiple cancer types by a unified genomic platform and standardized pipeline, we performed a pan-cancer analysis to investigate the relationship between AA genetic ancestry and genomic alterations in cancers. We observed that AA patients with BRCA, HNSC, or UCEC exhibit a higher level of chromosomal instability compared with EA patients, while a lower level of chromosomal instability was observed in AA patients with KIRP or KIRC. Interestingly, the frequencies of TP53 mutations and amplification of CCNE1 were significantly higher in AA patients in the cancer types that show higher levels of chromosomal instability. Tumor-igenesis is a multi-step process in which normal cells accumulate acquired genomic alterations. These alterations in cancer genomes dominantly and intrinsically influence the transcriptional phenotypes of cancers, such as gene expression signatures (e.g., PAM50 signature in breast cancer). TP53 somatic mutations are one of the well-demonstrated early driving genomic alterations during tumor development. As a transcription factor, both loss-of-function and gain-of-function mutations in the TP53 gene will have large effects on the transcriptome, such as the expression of ER and ER-regulated/associated genes in breast cancer (Angeloni et al., 2004; Borresen-Dale, 2003; Troester et al., 2006). TP53 mutant breast tumors are enriched for the triple-negative (basal-like) transcriptional pheno-type, providing a potential explanation for higher prevalence of the basal-like subtype (PAM50) in AA patients with breast cancer. In addition, we found that genomic alterations of the genes in the PI3K pathway were less frequent in AA patients compared with EA across most cancer types. These pan-cancer findings are consistent with previous results from studies based on a single cancer type. For example, recent studies on breast cancer from the TCGA cohort (Ademuyiwa et al., 2017; Huo et al., 2017; Keenan et al., 2015) demonstrated that AAs had more TP53 mutations and fewer PIK3CA mutations. These observations not only further our understanding of the contributions of genetic ancestry to cancer disparities, but may also inform personalized treatment of cancer patients from racial/ethnic minority groups.
In a pan-cancer meta-analysis, three recurrent focal SCNAs were identified with significantly different alteration frequencies between AA and EA patients. We hypothesized that certain genes located in these three recurrent focal SCNAs may play functional roles in cancer disparities, and that identification and characterization of such genes may provide molecular insight into the understanding of cancer disparities. Supporting this hypothesis, CCNE1, located within the recurrent focal amplification SCNA locus on 19q12, showed significantly different alteration frequencies between AA and EA patients, and was identified as a potential contributor to cancer disparities. The correlation between chromosomal instability and the amplification of CCNE1 further supports the idea that it may play biological roles in cancer disparities. Finally, five recurrently mutated genes at a pan-cancer level and six at a cancer type-specific level were identified with significantly different alteration frequencies between AA and EA patients. Genomic instability, epigenetic regulation and PI3K were the major molecular pathways implicated by the 39 genes found to be altered (either through SCNA or mutation) with significantly different frequency between AA and EA patients at a pan-cancer level. Notably, many genes with unknown functions were identified in our study; therefore, further characterization of the biological functions of these genes is urgently needed.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lin Zhang M.D. (linzhang@upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In this research, we used data collected by the Cancer Genome Atlas (TCGA) Research Network. Under the direction of the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI), TCGA collected both tumor and non-tumor bio-specimens from more than 10,000 human samples with informed consent under that authorization of local Institutional Review Boards (https://cancergenome.nih.gov/abouttcga/policies/informedconsent). These steps ensured that patients were exposed to no unnecessary risks and that the resulting research is legal, ethical, and well designed.
METHOD DETAILS
Data Collection and Processing
SNP Array Data
TCGA Affymetrix Human SNP Array 6.0 raw data in CEL format (n=11,122, across 33 cancer types; Table S1) were downloaded from the Cancer Genomic Cloud of the TCGA project (http://www.cancergenomicscloud.org/). The probe-level signal intensities of CEL files were combined, calibrated, and normalized for each cancer type separately using Affymetrix Power Tools version 1.18.2 (https://www.affymetrix.com/support/developer/powertools/changelog/index.html). Birdseed (Korn et al., 2008) version 2 was used for genotype-calling of SNP arrays. Segmented copy number profiles from Affymetrix Human SNP Array 6.0 (n=5,580, across 10 cancer types) were downloaded from the Cancer Genomic Cloud of the TCGA project. For each patient, a pair of segmentation files of tumor and matched control (if available) was selected for somatic copy number alteration analysis. If multiple aliquot barcodes existed for one patient, one single pair of tumor/matched control sample was kept following the rules: (1) sample type: for tumor tissues, primary > recurrent > metastatic (Sample Type code: 01 > 02 > 06); for normal control tissues, blood > solid (Sample Type code: 10 > 11); (2) molecular type of analyte for analysis: prefer D analytes (native DNA) over G, W, or X (whole-genome amplified); (3) order of sample portions: higher portion numbers were selected; and (4) order of plate: higher plate numbers were selected.
Whole-exome Sequencing Data
Mutation annotation files (MAFs) across 10 cancer types of TCGA, which were generated by the MuTect2 pipeline, were downloaded through the Genomic Data Commons (GDC) Data Transfer Tool (gdc-client_v1.2.0_Ubuntu14.04_x64) from the GDC Data Portal (https://portal.gdc.cancer.gov/). If multiple aliquot barcodes existed for one patient, one single pair of tumor/matched control sample was kept following the rules: (1) sample type: for tumor tissues, primary > recurrent > metastatic (Sample Type code: 01 > 02 > 06); for normal control tissues, blood > solid (Sample Type code: 10 > 11); (2) molecular type of analyte for analysis: prefer D analytes (native DNA) over G, W, or X (whole-genome amplified); (3) order of sample portions: higher portion numbers were selected; and (4) order of plate: higher plate numbers were selected. We excluded all mutations that were not labelled with “PASS” on the “FILTER” column. Additionally, mutations were considered as low-confidence calls and also excluded if there were: 1) less than 20 reads covering the variant site in tumor sample (t_depth < 20); or 2) less than 4 reads supporting the variant allele in tumor sample (t_alt_count < 4); or 3) more than one read supporting the variant allele in normal sample (n_alt_count > 1).
Recurrently Mutated Gene (RMG) List
To define the recurrently mutated genes (RMGs), gene lists from the following four complementary resources were combined: 1) Mut-Sig2CV analysis from the GDAC Firehose standard analysis pipeline (http://gdac.broadinstitute.org) (accessed January 28, 2016); 2) Cancer genes database by Iorio et al. (Iorio et al., 2016), which is a combination analysis of MutSigCV, OncodriveFM, and Oncodrive-CLUST; 3) Mutational driver gene database from Intogen (Rubio-Perez et al., 2015) (http://www.intogen.org/downloads), which is a database containing information on genes obtained by a method designed to detect complementary signals of positive selection in the pattern of somatic mutations; and 4) RMGs identified from the MuSiC algorithm by Kandoth et al. (Kandoth et al., 2013). For a given cancer type, if an RMG was identified by more than one of the above methods, it was considered a “confident” RMG. For KIRP, RMGs defined by the only resource GDAC Firehose (MutSig2CV) were adopted. Collectively, we identified a total of 173 RMGs for the 10 cancer types included in this study (Table S10).
RNA-sequencing Data
Gene-level RNA expression from TCGA RNA-seq profiles was downloaded through the Genomic Data Commons (GDC) Data Transfer Tool (gdc-client_v1.2.0_Ubuntu14.04_x64) from GDC Data Portal (https://portal.gdc.cancer.gov/). In the GDC RNA-seq analysis pipeline, reads were aligned to the GRCh38 reference genome, and then gene level expression was measured from HT-Seq raw read count using GENCODE v22 for gene annotation. Subsequently, RNA-Seq expression level read counts were normalized using FPKM (Fragments per Kilobase of transcript per Million mapped reads). A gene expression matrix was created for each cancer type. For expression analysis of tumor samples, one single aliquot barcode for each patient was kept following the rules: (1) sample type: primary > recurrent > metastatic (Sample Type code: 01 > 02 > 06); (2) molecular type of analyte for analysis: prefer R analytes (RNA) over T (Total RNA); (3) order of sample portions: higher portion numbers were selected; and (4) order of plate: higher plate numbers were selected. To maintain an expression value of zero for untranscribed genes, the FPKM data was log2 scaled after adding a small constant, i.e. exp = log2(FPKM + 1).
Reverse Phase Protein Array (RPPA) Data
The RPPA data (level 4 data) were downloaded from The Cancer Protein Atlas website (http://tcpaportal.org/tcpa/download.html). For each protein in a given cancer type, the RPPA data were median-centered and normalized across all samples to yield a relative protein level. The proteins chosen to estimate the PI3-kinase score were selected based on the method by Zhang et al. (Zhang et al., 2017). The PI3-kinase score was defined as the sum of normalized protein levels of selected proteins involved in the PI3-kinase pathway (Zhang et al., 2017), including AKT (S473 and T308 features), GSK3 (S9 and S21/S9 features), PRAS40, and phospho-TSC2.
Clinical Data
Clinical and biospecimen annotation files in XML format (n=11,160, across 33 cancer types) were downloaded by the GDC Data Transfer Tool (gdc-client_v1.2.0_Ubuntu14.04_x64) from the GDC Data Portal (https://portal.gdc.cancer.gov/) on July 26, 2017. R package XML was used to access clinical information and convert it to tabular text.
Health Disparities for Each Cancer Type
Cancer health disparities refer to differences in the cancer health status of different racial/ethnic groups in the US: i.e. some racial/ethnic groups have higher incidence and/or mortality rates of certain cancers compared to others. The racial/ethnic group-specific incidence and mortality rates for each cancer type were retrieved from two related cancer statistics reports. 1) The United States Cancer Statistics (USCS) Incidence and Mortality Web-based Report, which presents the official federal statistics on cancer incidence from registries and cancer mortality statistics (1999–2014) produced by the Centers for Disease Control and Prevention (CDC) and the National Cancer Institute (NCI) (https://nccd.cdc.gov/uscs/cancersbyraceandethnicity.aspx#Footnotes). 2) Cancer Statistics for racial/ethnic minority groups (African American, Asian Americans, Native Hawaiians, and Pacific Islanders) in 2016 (De-Santis et al., 2016; Siegel et al., 2016; Torre et al., 2016), reported by the American Cancer Society. A PubMed-based literature search was also performed to confirm the disparities of individual cancer types.
Genotype Data for Reference Populations
Genotype files of the International HapMap project (Phase III) were downloaded from the Hapmap ftp site (ftp://ftp.ncbi.nlm.nih.gov/hapmap/genotypes/2010–05_phaseIII/hapmap_format/polymorphic/). Of the 1,397 individuals from 11 populations in phase III data of HapMap, known relationships were described when the data were released (ftp://ftp.ncbi.nlm.nih.gov/hapmap/genotypes/2010–05_phaseIII/relationships_w_pops_041510.txt). Furthermore, Pemberton et al. (Pemberton et al., 2010) identified additional previously unidentified pairs with close relatives through evaluation by both the software package RELPAIR and allele sharing. Based on this information, we selected 1,117 unrelated individuals as reference populations from HapMap (Table S2). The populations used in our study included: ASW (African ancestry in Southwest USA, n=52), CEU (Utah residents with Northern and Western European ancestry from the CEPH collection, n=112), CHB (Han Chinese in Beijing, China, n=137), CHD (Chinese in Metropolitan Denver, Colorado, n=106), GIH (Gujarati Indians in Houston, Texas, n=97), JPT (Japanese in Tokyo, Japan, n=113), LWK (Luhya in Webuye, Kenya, n=99), MXL (Mexican ancestry in Los Angeles, California, n=54), MKK (Maasai in Kinyawa, Kenya, n=105), TSI (Toscani in Italia, n=102), and YRI (Yoruba in Ibadan, Nigeria, n=140).
Because there are no reference samples for Native Americans in the HapMap reference panel, samples from the American population from the Human Genome Diversity Project (HGDP) were chosen as a reference for a population of Native American ancestry. Genotype files of HGDP were downloaded from http://www.hagsc.org/hgdp/files.html. Of the 1,043 individuals successfully geno-typed in HGDP, 940 were from the recommended unrelated subset (H952), defined by Rosenberg (Rosenberg, 2006). 64 individuals from 5 populations (7 Colombians, 14 Karitiana, 21 Maya, 14 Pima and 8 Surui) were pooled as a reference population of Native American ancestry. Mitochondrial markers were removed since it has been reported that there are very limited associations between mitochondrial and autosomal population structures (Biffi et al., 2010).
We took SNPs shared by HapMap and HGDP (Native American panel), flipped strands for HGDP SNP genotypes if alleles showed discordance with HapMap, and re-formatted the data to the HapMap standard. To this end, we collected a total of 655,113 SNPs which were shared by HapMap and HGDP (Native American panel). Then, we applied a set of quality control criteria. First, 195,643 SNPs with call rates across the 1,181 reference samples < 95% or minor allele frequency (MAF) < 1% were filtered out. Then an exact test of Hardy-Weinberg equilibrium (HWE) was performed by the HWExactMat() function of R package HardyWeinberg (Graffelman, 2015) for the reference populations separately (11 populations from HapMap and 5 populations from HGDP). To be excluded on the basis of Hardy-Weinberg disequilibrium, a SNP had to have at least four copies of the minor allele in the population in which it was being assessed and possess at least one of the following properties: (1) an exact test p value < 10−5 in at least one population; 2) an exact test p value < 10−3 in at least two populations. This excluded an additional 4,578 SNPs. In all, 454,892 SNPs remained as effective markers for reference populations.
Genotype Data for TCGA Specimens
After combining all genotype data from TCGA, a total of 22,072 non-redundant samples for 33 cancer types were obtained (if multiple aliquot barcodes existed for one patient, one single file was chosen following the pre-defined criteria for tumor sample and matched normal sample respectively, see also section ‘SNP Array Data’). Of the 909,622 SNPs genotyped on Affymetrix Human SNP Array 6.0, 133,322 SNPs overlapped with the processed genotype data of the two reference population resources (HapMap and HGDP [Native American panel]). Quality control processes were applied to the overlapping SNPs on TCGA: 1) SNP call rate across all TCGA was > 95% and 2) MAF was > 1%. This yielded 103,991 SNPs effective and useful in both reference populations and the TCGA cohort. SNPs on sex chromosomes were further removed as the different variance for males complicates proper theoretical treatment (Reinhold and Engqvist, 2013). Finally, a total of 100,611 SNPs were kept for analysis, all of which were bi-allelic SNPs. Based on this set of overlapping SNPs, we combined the genotype data of TCGA and the reference populations as input data for the three independent downstream genetic ancestry analysis pipelines (EIGENSTRAT, STRUCTURE and LAMP). TCGA genotypes were flipped if necessary. Input files were prepared according to software requirements.
Genetic Ancestry Assessment
Genetic Ancestry Assessment by EIGENSTRAT
EIGENSTRAT (Price et al., 2006) is a method to study human diversity based on Principal Component Analysis (PCA), which reduces the information contained in SNP frequencies to components that capture most genetic variability. The EIGENSOFT package (EIGENSTRAT algorithms included) version 6.1.4 was downloaded from GitHub (https://github.com/DReichLab/EIG). The smartpca program was applied to run PCA on the combined genotype data of TCGA and the reference populations. All 16 reference populations (11 from HapMap and 5 from HGDP, Table S2) were used to compute eigenvectors (by supplying a poplist file using the parameter -w). Given that EIGENSTRAT results are not sensitive to the number (K) of axes of variation used (Price et al., 2006), we adopted the default value K=10 for running the smartpca program. The program then outputs the positions of each individual (either from TCGA or from reference populations) on the top ten axes of variation into a file with the extension .evec. This allows visualization of the population structure as well as estimation of relative distance between individuals or populations. To categorize the TCGA patients into genetic ancestry groups, we attempted to train a k-nearest neighbor (k-NN) classifier. We first grouped the 16 reference populations into 7 populations according to continental distribution and migration history: West Africans (YRI), European (CEU and TSI), East Asian (CHB, CHD and JPT), Native American (Pima, Maya, Colombians, Karitiana and Surui), South Asian (GIH), African American (ASW, LWK and MKK), and Mexican (MXL). Before we built a k-NN classifier on the 7 grouped populations, we performed a grid search with a nested leave-one-out validation to determine the optimal combination of two parameters: the number of eigenvectors (n) to be used to calculate distance between individuals and the number of neighbors (k) to take votes from. With candidate values from 3 to 10 for n and k, we found that all possible combinations achieved near perfect performance on reference populations (accuracy > 99%). However, the combination of k=6 and n=7 was the most robust when applying the trained classifier to TCGA patients (priority of sample type was blood > solid normal > tumor). Finally adopting this set of parameters for model training and prediction with k-NN, we observed a great consistency (95.6%) of genetic categorization with self-reported race from TCGA clinical annotation.
Genetic Ancestry Assessment by EIGENSTRAT Using Different Numbers of SNPs
To determine the effect of number of SNPs used on global ancestry estimation, we repeated our approach using subsampled SNP sets ranging from 500 to 50,000 SNPs. In each cycle, a specified number of SNPs was randomly selected from the total of 100,611 SNPs. To train a k-nearest neighbor (k-NN) classifier to categorize the TCGA patients into genetic ancestry groups, we re-defined the two parameters: the number of eigenvectors (n) to be used to calculate distance between individuals and the number of neighbors (k) to take votes from. A grid search with a nested leave-one-out validation was performed to determine the optimal combination of the two parameters (n and k). The final combination was determined as the one observed to be the most robust when applying the trained classifier to TCGA patients (priority of sample type is blood > solid normal > tumor). Overall, the number of patients classified into the four genetic ancestry groups (EA, AA, EAA and NA) remained stable, suggesting that a small number of SNPs is sufficient to assess global ancestry (Figures S3A and S3B).
Genetic Ancestry Assessment by EIGENSTRAT Using Alternative Reference Cohort
Since the 1000 Genomes Project could also serve as reference populations, we applied EIGENSTRAT to estimate genetic ancestry for all patients of TCGA using the 1000 Genomes Project as an alternative reference cohort. To use the phase 3 data of the 1000 Genomes Project as a reference panel, the VCF (Variant Call Format) files of the 1000 Genomes were downloaded from ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/. It has been reported that the American populations of the 1000 Genomes Project were admixed populations (Gravel et al., 2013) (i.e., CLM [Colombians from Medellin, Colombia], MXL [Mexican Ancestry from Los Angeles USA], PEL [Peruvians from Lima, Peru], PUR [Puerto Ricans from Puerto Rico]). Therefore, we chose the American population from the Human Genome Diversity Project (HGDP) as the reference for a population of Native American ancestry. Of the 84,805,772 variants characterized by the 1000 Genomes Project, 640,638 were shared with the HGDP (Native American panel). The genotypes of samples from the 1000 Genomes Project were formatted (i.e. 0 for AA, 1 for AB, 2 for BB and 9 for missing) before pooling with those of samples from the HGDP (Native American panel). Then, we applied a set of quality control criteria: first, a set of 4,592 SNPs whose call rate across the reference samples was < 95% were excluded. A further set of 1,550 SNPs whose minor allele frequency (MAF) was < 1% were filtered out. Then an exact test of Hardy-Weinberg equilibrium (HWE) was performed by the HWExactMat function of R package HardyWeinberg (Graffelman, 2015) for the reference populations separately. To be excluded on the basis of Hardy-Weinberg disequilibrium, a SNP had to have at least four copies of the minor allele in the population in which it was being assessed and possess at least one of the following properties: (1) an exact test p value < 10−5 in at least one population; 2) an exact test p value < 10−3 in at least two populations. This excluded an additional 841 SNPs. In all, 633,655 SNPs remained as effective markers for the alternative reference populations (the 1000 Genomes Project and HGDP [Native American panel]).
Of the 909,622 SNPs genotyped on Affymetrix Human SNP Array 6.0 for the TCGA cohort, 181,946 SNPs overlapped with the processed genotype data of the alternative reference populations (the 1000 Genomes Project and HGDP [Native American panel]). Quality control processes were applied to the overlapping SNPs on TCGA: 1) SNP call rate across all TCGA was > 95% and 2) MAF was > 1%. This yielded 150,128 SNPs effective and useful in both reference populations and the TCGA cohort. All of the 150,128 SNPs were located on autosomal chromosomes. TCGA genotypes were flipped if necessary. Based on this set of overlapping SNPs, we combined the genotype data of TCGA and the alternative reference populations as input data for the EIGENSTRAT algorithm.
For k-nearest neighbor (k-NN) classifier training to categorize the TCGA patients into genetic ancestry groups, we re-defined the two parameters: the number of eigenvectors (n) to be used to calculate distance between individuals and the number of neighbors (k) to take votes from. A grid search with a nested leave-one-out validation was performed to determine the optimal combination of the two parameters (n and k). The combination of k=9 and n=6 was observed to be the most robust when applying the trained classifier to TCGA patients (priority of sample type was blood > solid normal > tumor). Finally adopting this set of parameters for model training and prediction with k-NN, we observed a high consistency (99.6%) of genetic categorization with that reported using HapMap and HGDP as the reference cohort (Figure S1A).
Genetic Ancestry Assessment by STRUCTURE
STRUCTURE (Pritchard et al., 2000) is an algorithm using multi-locus genotype data to infer population structure utilizing a model-based clustering method. The STRUCTURE algorithm (version 2.3.4) was downloaded from http://web.stanford.edu/group/pritchardlab/structure.html. The USEPOPINFO model was applied in our analysis. Briefly, we chose individuals from the following reference populations as continental ancestors: Northern Europeans from Utah (CEU, n=112) and Tuscans from Italy (TSI, n=102) represented European ancestry; West Africans (YRI, n=140) represented African ancestry; East Asians (CHB, n=137 and JPT, n=113) represented East Asian ancestry, and Native Americans (Colombians, n=7, Karitiana, n=14, Maya, n=21, Pima, n=14 and Surui, n=8) represented Native American ancestry. Using the STRUCTURE algorithm, we calculated the proportion of an individual’s genome that originates from the assumed ancestral population. To facilitate STRCTURE efficiency and reproducibility, the STRUCTURE analysis procedure was repeated 10 times on 10 separate sets of 3,000 randomly chosen SNPs, and the final ancestry was the average of the 10 estimates. In the input file (genotype file), the ancestral reference samples were labeled as 1 in the POPFLAG column. In contrast, the other samples were labeled as 0. NUMLOCI was set to 3000 by the -L option (the number of SNPs used in each run of genetic ancestry estimation). A maximum of 4 ancestries (European, African, East Asian, and Native American) were assumed for any individual (MAXPOPS was set to 4 by the -K option). For each ancestry estimation run, we performed 10,000 iterations (BURNIN=10,000). Both USEPOPINFO and PFROMPOPFLAGONLY were set to 1.
Genetic Ancestry Assessment by LAMP
The LAMP (Sankararaman et al., 2008) algorithm (Release 2.5; http://lamp.icsi.berkeley.edu/lamp/) was used to estimate ancestries at each SNP locus for TCGA patients. To determine the local ancestry for TCGA patients, the priority of sample type was blood > solid normal > tumor. LAMP-ANC was used in our analysis. Individuals from the following reference populations were used to derive prior knowledge of population-specific allele frequencies: CEU (n=112) and TSI (n=102) represented European ancestry; YRI (n=140) represented African ancestry; CHB (n=137) and JPT (n=113) represented East Asian ancestry; HGDP (Colombians, n=7; Karitiana, n=14; Maya, n=21; Pima, n=14; Surui, n=8) represented Native American ancestry. Starting estimates of a (the mixture proportion for each population) were designated according to STRUCTURE results. Local ancestry at SNPs across 22 autosomes inferred by LAMP for each individual were averaged to yield estimated proportions of global ancestry, which were then compared with estimates by STRUCTURE. Given that local ancestry is highly correlated between neighboring SNPs due to admixture linkage disequilibrium (Tang et al., 2010), we determined chromosomal segments with unique ancestry status (or ancestry blocks). Briefly, adjacent SNPs having identical ancestry status in >95% of patients (AAs, EAAs and NAs separately) were considered within the same ancestry block. We finally determined 638, 81, and 3,976 genomic segments with independent ancestry status based on 1,019 AAs, 677 EAAs, and 397 NAs, respectively. The average proportion of West African ancestry over all AAs in TCGA and all ancestry blocks determined was 0.786 (SD=0.010 across all ancestry blocks). Similarly, the average proportions of East Asian ancestry over EAAs, and Native American ancestry over NAs were 0.945 (SD=0.047) and 0.224 (SD=0.024), respectively. Estimation using local ancestry at SNPs yielded similar results. Visualization of average ancestry contributions against genomic position of ancestry blocks revealed an even distribution of local ancestry across genomes.
Association of AA Ancestry with Genomic Features
Adjustment for Clinical Factors
For all analyses comparing genetic characteristics between AA and EA patients, clinical factors were taken into account. Clinical factors considered included age at diagnosis, gender, pathologic stage, neoplasm histologic grade, smoking habits and alcohol consumption. Age (mean, 61.9 years; range, 10–90 years) was treated as a dichotomous variable (according to the median value specific for each cancer type). Gender was treated as a categorical variable (female, 52.0%; male, 48.0%). Pathologic stage was treated as an ordinal categorical variable (stage I, 22.4%; stage II, 22.1%; stage III 14.5%; stage IV 8.7%). Neoplasm histologic grade was also treated as an ordinal categorical variable (G1/low grade, 3.2%; G2/intermediate grade, 11.9%; G3.G4/high grade, 13.1%). Pack-years of cigarette smoking was used as a continuous variable (mean, 45.9 pack-years; range, 0.02–300 pack-years) while history of cigarette smoking was treated as a categorical variable (nonsmokers, 7.5%; smokers, 18.8%). Alcohol consumption was treated as a categorical variable (NO, 3.0%; YES, 6.3%). In order to maintain the sample size, only factors with less than 10% missing data were considered for each cancer type.
For each cancer type, the difference between AAs and EAs with respect to confounding clinical factors was estimated by propensity score (Rosenbaum and Rubin, 1983) which was later supplied as a covariate to regression models testing the association between AA ancestry status (AA as 1 while EA as 0) and genetic characteristics (e.g., overall SCNA scores, alteration status of somatic events and expression levels). To be specific, a multivariable logistic regression model was fit to regress AA ancestry status (AA as 1 while EA as 0) on all confounding clinical factors. The multivariable logistic regression analysis produced an empirically-derived formula (by weighting confounding clinical factors), which could best discriminate between the two racial groups. Applying this formula to the observed values of confounding clinical factors yielded a propensity score for each patient. Propensity scores range from 0 to 1 and reflect the likelihood of being AA given all observed clinical characteristics. Differences in propensity scores between AAs and EAs indicate imbalance in these clinical factors. Propensity scores were calculated in the context of each molecular platform separately.
SCNA Score Analysis
The GISTIC 2.0 algorithm (Mermel et al., 2011) (ftp://ftp.broadinstitute.org/pub/GISTIC2.0/) was applied to SCNA analysis. The copy number profiles of TCGA were deconstructed into three different levels of SCNAs (i.e. focal, arm, and chromosome levels). For each tumor, GISTIC 2.0 defined a combination of focal and broad (>70% of a chromosomal arm) events. Broad events were further divided into arm events and chromosomal events. Among the broad events, all cases where both arms of a chromosome had the same copy number change (in value and sign) were considered as chromosome SCNA events, while all the others were considered as arm SCNA events. Each event was then classified into one of the following according to both the sign and amplitude of its log2 copy number
in which c is the actual change in copy number for each event provided by GISTIC 2.0 and C defines the thresholded/weighted copy number change. Adopting a modification of methods described by Davoli et al. (Davoli et al., 2017), SCNA scores were calculated for each tumor at focal, arm and chromosome levels separately as the sum of thresholded/weighted copy number changes of all events, considering amplification and deletion equally.
Rank-based normalization was then performed for SCNA scores at focal, arm and chromosome levels separately for each cancer type:
Here we denote as the normalized counterparts of Sfocal, Sarm, Schrom while rfocal, rarm, rchrom is the rank of each patient in the cancer type to which he/she belongs, and n is the total number of patients in the cancer type. Normalized SCNA scores at focal, arm and chromosome levels were summed to represent an overall SCNA score:
Clinical factors were taken into account when comparing the differences in the overall SCNA score between AA and EA patients. SCNA scores were rank-scaling transformed as a conservative measure to avoid results driven by outliers. Effect size was defined as the coefficient of the regression model, representing the expected change in SCNA score percentile, given AA ancestry status (AA as 1 while EA as 0). The Benjamini and Hochberg (BH) procedure (Benjamini and Hochberg, 1995) was applied to control for false discovery rate (FDR) control. When comparing SCNA scores at different levels, arm-level and chromosomal-level were considered together.
Weighted Genome Instability Index (wGII) Analysis
To assess chromosomal instability, a weighted Genome Instability Index (wGII) was calculated based on segmentation files (Burrell et al., 2013). We first determined the ploidy of each tumor as the median copy number accounting for the length of segments. For each chromosome, GII was calculated as the fraction of the genome presenting aberrant copy numbers (differing more than 0.3) relative to the baseline ploidy. The wGII of the tumor was calculated as the mean fraction aberration across all 22 chromosomes (so that large chromosomes would not have greater effect on the score than small chromosomes).
Recurrent Focal SCNA Analysis
The recurrent focal SCNAs (peak regions) of each cancer type were identified by GISTIC 2.0 (Mermel et al., 2011). Significant peak regions were identified with q value < 0.25. Tumors which had more than 2,000 segments were excluded from our analysis. The confidence level used to calculate the region containing a driver was set to 0.95 (by the –conf option). The total number of recurrent focal SCNAs (called by GISTIC 2.0) varied among cancer types, with UCEC having the most (105 recurrent focal SCNAs) and kidney cancers having the fewest (27 and 29 recurrent focal SCNAs for KIRC and KIRP, respectively). An average number of 65 was observed for the ten cancer types included in our analysis. We carefully examined the location of each peak region. Peaks located entirely in centromeres and telomeres (within 1 Mb) were filtered out due to the poor coverage across such regions by probes of Affymetrix SNP6.0. Similar criteria were also applied by other published studies related to copy number analysis. A total of 57 peaks called by GISTIC2.0 were removed in this way, leaving an average of ~60 peaks subjected to subsequent analysis for each individual cancer type. Of all recurrent focal SCNAs (called by GISTIC 2.0), some were shared across many cancer types, while others were restricted to particular cancer types.
The frequency of focal SCNA events at each peak region was compared between AAs and EAs at both pan-cancer and cancer-specific levels, adjusting for clinical factors. At the cancer-specific level, in order to control the overall level of genomic disruption when comparing the alteration frequencies for each SCNA event between AA and EA patients, we performed a controlled permutation test in which both the fractions of the genome affected by each of the amplifications and deletions in each sample (column-wise) and the alteration frequency of each recurrent focal SCNA (row-wise) were maintained in the permuted data. We first constructed a binary matrix denoting the recurrent focal SCNA profile across all tumors for a specific cancer type, where n is the number of tumors, m is the total number of recurrent focal SCNAs and the (i,j)th element of the matrix X, Xij is determined following:
in which cij is the actual change in copy number for jth recurrent focal SCNA in ith tumor. We then permutated the matrix 10,000 times by the function permatswap() in the R package vegan. During the permutation, genomic disruption level of each tumor (row-wise sum of the matrix) as well as each recurrent focal SCNA (column-wise sum of the matrix) were maintained by supplying the parameters: method=“quasiswap”, fixedmar=“both”, shuffle=“both” and mtype=“prab”. For each permutated recurrent focal SCNA profile, a test statistic for the association with AA patients relative to EA patients for each recurrent focal SCNA was generated by a logistic regression model, which aimed to regress AA ancestry status (AA as 1, while EA as 0) using the binary alteration status as the independent variable with the clinical factor-derived propensity score as a covariate. Subsequently, the significance (Z score and raw p value) of the association with AA patients relative to EA patients for each recurrent focal SCNA was generated by examining the position of observed test statistic among the distribution of all statistics generated from the permutated recurrent focal SCNA profiles. Finally, the Benjamini and Hochberg (BH) procedure (Benjamini and Hochberg, 1995) was applied to the set of raw p values to control the false discovery rate (FDR).
At the pan-cancer level, the whole genome was divided into tiles by peak boundaries of all cancer type-specific focal events (amplification and deletion events were treated separately). Neighboring tiles within peak regions shared by at least two cancer types were merged for cross-cancer meta-analysis. Since Beroukhim et al. reported that focal SCNAs occur with a median length of 1.8 Mb (Beroukhim et al., 2010), we broadly considered peak regions from different cancer types with less than 50 kb distance as “overlapping”. In total, we obtained 158 recurrent focal SCNAs shared by different cancer types (65 for amplification, 93 for deletion) for meta-analysis. For a recurrent focal SCNAs under meta-analysis, a meta-Z score for the association with genetic ancestry was summarized from Z scores generated on individual cancer types, following Stouffer’s method (Stouffer et al., 1949): in which Zi is the Z score in cancer type i and Z is the meta-Z score. wi, the weight for cancer type i, was proportional to the squre root of event frequency.
Whole Genome Doubling (WGD) Analysis
The HAPSEG (Carter et al., 2011) algorithm (http://archive.broadinstitute.org/cancer/cga/hapseg) and ABSOLUTE (Carter et al., 2012) algorithm (http://archive.broadinstitute.org/cancer/cga/absolute) were used to calculate the purity, ploidy, and absolute DNA copy numbers of TCGA samples. First, HAPSEG was applied to the genotypes and segmented copy number profiles generated from Affymetrix SNP6 data to estimate homologue-specific copy ratios (HSCRs) for each tumor. The statistical algorithm BEAGLE (https://faculty.washington.edu/browning/beagle/beagle.html) was included for genotype imputation, with reference haplotype panel information taken from populations inferred by EIGENSTRAT (“CH” and “YOR” as pop code for EAAs and AAs respectively, otherwise “CEPH”). A paired normal sample was used if available on the Affymetrix SNP6.0 platform. A probabilistic criterion using Bayesian model comparison was applied to merge adjacent segments in order to deal with spurious breakpoints (Carter et al., 2011). The minimum segment size, outlier probability, and distance threshold for merging segments were set to 5, 0.001, and 1e-10, respectively. The output of HAPSEG was then passed through ABSOLUTE with the following parameters: sigma.p=0, max.sigma.h=0.02, min.ploidy=0.95, max.ploidy=10, max.as.seg.count=1500, max.non.clonal=0, max.neg.genome=0, platform=“SNP_6.0”, and copy_num_type=“allelic”. The output of ABSOLUTE then provided the absolute allele-specific copy number of local DNA segments, estimates of purity and ploidy, and inference of whole genome doubling status in the tumor. When comparing the frequencies of WGD between AA and EA patients, to adjust for potential confounding effects introduced by the clinical factors, a propensity score was supplied as a covariate to a regression model which aimed to regress WGD status (0 for genomes with no WGD, 1 for genomes with one WGD event and 2 for genomes with more than one WGD event) using AA ancestry status (AA as 1 while EA as0) as the independent variable. The Benjamini and Hochberg (BH) procedure (Benjamini and Hochberg, 1995) was applied to control for false discovery rate (FDR).
Correlation Analysis between Copy Number and mRNA Expression
To identify the genes whose mRNA expression levels were positively correlated with their SCNAs, the putative gene-level copy number was estimated by the GISTIC algorithm. Pearson correlation coefficients between the predicted DNA copy number (log ratio) and mRNA expression level (log scaled) of each gene were calculated by R software. When assessing the cross-cancer association between copy number and expression, a modified version of the Schmidt-Hunter method of meta-analysis was applied (Field, 2001). A weighted average of correlation coefficients calculated on each cancer type was used to measure the cross-cancer effect size: in which ri was calculated correlation coefficient for cancer type i, and wi was the weight, proportional to frequency of tumors with expression values above zero in cancer type i. The significance of the mean effect size is obtained by calculating a Z score by dividing the mean by its standard error: , in which and , where k is the number of cancer types involved in meta-analysis. The Z score generated could be converted to a p value. Finally, correction for multiple-hypothesis testing was performed using the Benjamini–Hochberg method.
Somatic Mutation Burden Analysis
Mutation burden was measured as the total number of somatic mutations present in a tumor specimen. We defined tumors of high mutation burden as those in the upper quartile for each cancer type. A logistic regression model was then applied to regress AA status (AA as 1 while EA as 0) using the binary mutation burden status (tumors of high mutation burden as 1, otherwise as 0) with the clinical factor-derived propensity score as a covariate. The Benjamini and Hochberg (BH) procedure (Benjamini and Hochberg, 1995) was applied to control the false discovery rate (FDR).
Somatic Mutation Signature Analysis
Single nucleotide variants (SNVs) of somatic mutations generated by the MuTect2 pipeline were obtained from mutation annotation files (MAFs), which were downloaded through the Genomic Data Commons (GDC) Data Transfer Tool (gdc-client_v1.2.0_Ubuntu14.04_x64) from the GDC Data Portal (https://portal.gdc.cancer.gov/). All SNVs of somatic mutations that passed quality control were used for mutational signature analysis. Each mutation fell into one of the six classes of base substitution (C>A, C>G, C>T, T>A, T>C, and T>G). At the same time, the sequence context in which a mutation occurred (the bases immediately 5’ and 3’ to the mutated base, one of the 16 possible conditions) was also considered. Thus the mutational spectra of a tumor could be summarized as a vector of length 96, in which each element represented the mutation count at one of the 96 mutated trinucleotides. A set of consensus mutational signatures identified from de novo extraction across 10,250 samples was published by the Wellcome Trust Sanger Institute (WTSI) Mutational Signature Framework (Alexandrov et al., 2013). Using the set of signatures, which they defined as prevalent in specific types of tumors, deconstructSigs (Rosenthal et al., 2016) applied a multiple linear regression model with a restriction that any coefficient must not be negative to most accurately reconstruct the observed mutational spectra. The weights of signatures were normalized between 0 and 1, making the sum of normalized weights equal to 1. By default in deconstructSigs, any signature contribution with a weight less than 0.06 is discarded. The mutational spectra of the original and reconstructed samples were compared using cosine similarity. Any signature that did not improve the cosine similarity by more than 0.02 was removed, and the sample was reanalyzed with the remaining signatures.
In the ten cancer types for analysis, we identified nine robust mutational signatures with sufficient contributions (more than 5% of the overall mutation tri-nucleotide profile). Age-associated signatures (signatures 1 and 5) were identified in all ten cancer types. However, signature 5 contributed significantly more than signature 1 to kidney and lung cancers. A significantly increased prevalence of signatures linked with activation of APOBEC cytosine deaminases (signatures 2 and 13) was observed in BRCA, HNSC and lung cancers, consistent with previous reports of APOBEC mutational process as a potential driving force across a range of cancer types. In terms of exogenous mutational processes, we found that signature 4, associated with smoking-induced mutations, was significantly prevalent in lung cancers and HNSC. Signature 6, associated with defective DNA mismatch repair, was enriched in COAD and UCEC, two cancer types where the hypermutator phenotype has been reported. Among the ten cancer types included for analysis, signature 3, associated with failure of DNA double-strand break-repair by homologous recombination, was predominantly identified in BRCA.
When we compared the difference in the contributions of each mutational signature among AA and EA patients, a regression model which aimed to regress the contribution of each mutational signature using AA ancestry status (AA as 1 while EA as 0) as the independent variable was applied. The clinical factor-derived propensity score was supplied as a covariate. As the dependent variable, the contribution of each mutational signature to a specific tumor took a value in the standard unit interval (0, 1), thus the beta regression model proposed by Ferrari and Cribari-Neto (Ferrari and Cribari-Neto, 2004) was used. Since the presence of extreme values (0 and 1), the following transformation (Smithson and Verkuilen, 2006) was applied to observed contribution percentages: , where n is the sample size of a given cancer type.
Signatures known to be associated with certain etiologies were considered together. Specifically, signatures 1 and 5 showed a correlation between the number of mutations and age of diagnosis (Alexandrov et al., 2015), and were considered together as an age-related signature. Additionally, signatures 2 and 13 exhibited predominantly C > T and C > G mutations at TpC sites and were associated with the activity of the APOBEC cytidine deaminases. Simultaneously, signatures with negligible (average less than 0.05) or dominant (average more than 0.95) contributions for a specific cancer type were skipped by the regression model. The Benjamini and Hochberg (BH) procedure (Benjamini and Hochberg, 1995) was applied to control the false discovery rate (FDR).
Recurrently Mutated Gene Analysis
Only the mutations located in gene bodies were included in the recurrently mutated gene analysis: i.e. the mutations located in intergenic regions, introns, 5’UTRs, 5’Flanks, 3’UTRs, and 3’Flanks were eliminated. Mutations in individual patient samples were aggregated to gene-level. We calculated the mutation frequency of a gene by dividing the number of patients harboring mutations in the gene body by the size of the cohort. For meta-analysis at the pan-cancer level, RMGs were pooled from individual cancer types. Thirty RMGs with overall frequency across cancer types greater than 5% were kept for analysis. For each recurrently mutated gene, z value for the association between AA ancestry and mutation frequency generated by the regression model on individual cancer types were summarized using Stouffer’s method (Stouffer et al., 1949): , in which Zi is the z value in cancer type i and Z is the meta-Z score. wi, the weight for cancer type i was proportional to the square P root of mutation frequency. The meta-Z score was then converted to a p value. At the level of individual cancer types, a list of cancer type-specific RMGs was used. Genes with mutation frequency below 5% were excluded for a given cancer type. To adjust for potential confounding effects by clinical factors, a propensity score was supplied as a covariate to a logistic regression model which aimed to regress AA ancestry status (AA as 1 while EA as 0) using the binary alteration status as the independent variable. The Benjamini and Hochberg (BH) procedure (Benjamini and Hochberg, 1995) was applied to control the false discovery rate (FDR).
Quantile-Quantile (Q-Q) Plot
We used Q-Q plots to examine the distribution of p values at both pan-cancer and individual cancer-type levels for the analyses of recurrent focal SCNAs and recurrently mutated genes. Under the null hypothesis, a uniform distribution of p values within the interval (0, 1) would be expected. Q-Q plots were generated by the function stat_qq() of R’s package ggplot2. Moreover, we calculated inflation factors using the R/Bioconductor package BACON (van Iterson et al., 2016) to assess whether inflation was introduced by the analysis.
Gene Expression Analysis
Differential gene-expression analysis on individual cancer types was performed adjusting for potential confounding effects by clinical factors. RNA expression level was rank-scaling transformed as a conservative measure to avoid results driven by outliers. A propensity score derived from clinical factors was supplied as a covariate to a logistic regression model which aimed to regress AA ancestry status (AA as 1 while EA as 0) using RNA expression level as the independent variable. z values obtained from individual cancer types were then summarized using Stouffer’s method (Stouffer et al., 1949): , in which Zi is the z value in cancer type i and Z is the meta-Z score. wi, the weight for cancer type i, was proportional to the fraction of samples with expression value above zero for the gene. The meta-Z score was then converted to a p value. The Benjamini and Hochberg (BH) procedure (Benjamini and Hochberg, 1995) was applied to control the false discovery rate (FDR). For visualization of gene expression across different cancer types, a linear model was used to estimate expression adjusting for cancer type.
Gene Expression Signature Analysis
Two independent methods were used to compare gene expression signatures of chromosomal instability (CIN70) (Carter et al., 2006) between AA and EA patients. First, an output score was defined by quantification of the composite expression of the signature in each sample. To this end, the expression of each gene included in the signature was scaled across each cancer type to mean 0 and standard deviation 1. The output score was then computed as the average of the scaled expression value of all genes for the signature and compared between AA and EA patients using a linear regression model with the clinical factor-derived propensity score as a covariate. The Benjamini and Hochberg (BH) procedure (Benjamini and Hochberg, 1995) was applied to control the false discovery rate (FDR). In addition, we ranked all genes according to the z value obtained from differential gene-expression analysis on AA versus EA, adjusting for clinical factors. Enrichment analysis was then performed for the signature using the R package fGSEA (https://bioconductor.org/packages/release/bioc/html/fgsea.html).
DATA AND SOFTWARE AVAILABILITY
Genetic ancestry assessment of each TCGA patient (n = 11,122) has been made available via the Cancer Genetic Ancestry Atlas (TCGAA, http://52.25.87.215/TCGAA).
Supplementary Material
KEY RESOURCES TABLE
Highlights.
The genetic ancestry of TCGA patients was estimated at global and local levels
The Cancer Genetic Ancestry Atlas, a publicly accessible resource, was developed
The frequencies of TP53 mutations and CCNE1 amplification were higher among AAs
The frequencies of the alterations in the PI3K pathway were lower among AAs
Significance.
The TCGA patient cohort is ethnically diverse, therefore providing a unique resource to understand the genomic basis ofcancer disparities across multiple cancer types. However, a large percentage of patients lacked self-identified race orethnicity (SIRE) information in TCGA. We integrated multiple computational algorithms to estimate the global and local genetic ancestry of each TCGA patient (n = 11,122, involving 33 cancer types from 27 primary sites) using genome-wide genotyping data, and assigned each individual to an ancestral population. We have made this information available in theCancer Genetic Ancestry Atlas(TCGAA, http://52.25.87.215/TCGAA),a publicly accessible resource, to assist researchers with analyzing, visualizing, and downloading multidimensional genetic ancestry information for each patient in TCGA.
ACKNOWLEDGMENTS
We thank the TCGA project team. This work was supported, in whole or in part, by the Basser Center for BRCA (to L.Z.), the Ludwig Institute for Cancer Research (to C.V.D.), the US NIH (R01CA142776 to L.Z., R01CA190415 toL.Z., P50CA083638 to L.Z., P50CA174523 to L.Z., R01CA057341 to C.V.D., U01CA184374 to T.R.R., P50CA083639 to A.K.S., P50CA098258 to A.K.S., R01CA163377 to R.Z., R01CA202919 to R.Z., R01NS094533 to Y.F., and T32CA009001 to K.H.K.), the Breast Cancer Alliance (to L.Z. and C.V.D.), the Frank McGraw Memorial Chair in Cancer Research (to A.K.S.), the American Cancer Society Research Professor Award (to A.K.S.), the Marsha Rivkin Center for Ovarian Cancer Research (to L.Z.), the Harry Fields Professorship (toL.Z.), the Kaleidoscope of Hope Ovarian Cancer Foundation (to L.Z.), the Ovarian Cancer Research Fund (to X.H.), and the Foundation for Women’s Cancer (to X.H. and Youyou Zhang).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes 3 figures and 11 tables and can be found with this article online at https://doi.org/10.1016/j.ccell.2018.08.019.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ademuyiwa FO, Tao Y, Luo J, Weilbaecher K, and Ma CX (2017). Differences in the mutational landscape of triple-negative breast cancer in African Americans and Caucasians. Breast Cancer Res. Treat 161, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, and Stratton MR (2015). Clock-like mutational processes in human somatic cells. Nat. Genet 47, 1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeloni SV, Martin MB, Garcia-Morales P, Castro-Galache MD, Ferragut JA, and Saceda M (2004). Regulation of estrogen receptor-alpha expression by the tumor suppressor gene p53 in MCF-7 cells. J. Endocrinol 180, 497–504. [DOI] [PubMed] [Google Scholar]
- Araujo LH, Timmers C, Bell EH, Shilo K, Lammers PE, Zhao W, Natarajan TG, Miller CJ, Zhang J, Yilmaz AS, et al. (2015). Genomic characterization of non-small-cell lung cancer in African Americans by targeted massively parallel sequencing. J. Clin. Oncol 33, 1966–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol 57, 289–300. [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia G, Tandon A, Patterson N, Aldrich MC, Ambrosone CB, Amos C, Bandera EV, Berndt SI, Bernstein L, Blot WJ, et al. (2014). Genome-wide scan of 29,141 African Americans finds no evidence of directional selection since admixture. Am. J. Hum. Genet 95, 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A, Anderson CD, Nalls MA, Rahman R, Sonni A, Cortellini L, Rost NS, Matarin M, Hernandez DG, Plourde A, et al. (2010). Principal-component analysis for assessment of population stratification in mitochondrial medical genetics. Am. J. Hum. Genet 86, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borresen-Dale AL (2003). TP53 and breast cancer. Hum. Mutat 21, 292–300. [DOI] [PubMed] [Google Scholar]
- Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, et al. (2013). Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JD, Lathan C, Sholl L, Ducar M, Vega M, Sunkavalli A, Lin L, Hanna M, Schubert L, Thorner A, et al. (2017). Comparison of prevalence and types of mutations in lung cancers among black and white populations. JAMA Oncol. 3, 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter H, Marty R, Hofree M, Gross AM, Jensen J, Fisch KM, Wu X, DeBoever C, Van Nostrand EL, et al. (2017). Interaction landscape of inherited polymorphisms with somatic events in cancer. Cancer Discov. 7, 410–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Eklund AC, Kohane IS, Harris LN, and Szallasi Z (2006). A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet 38, 1043–1048. [DOI] [PubMed] [Google Scholar]
- Carter SL, Meyerson M, and Getz G (2011). Accurate estimation of homo-logue-specific DNA concentration-ratios in cancer samples allows long-range haplotyping. Nat. Preced http://hdl.handle.net/10101/npre.2011.6494.1. [Google Scholar]
- Daly B, and Olopade OI (2015). A perfect storm: how tumor biology, genomics, and health care delivery patterns collide to create a racial survival disparity in breast cancer and proposed interventions for change. CA Cancer J. Clin 65, 221–238. [DOI] [PubMed] [Google Scholar]
- Davoli T, Uno H, Wooten EC, and Elledge SJ (2017). Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 355, 10.1126/science.aaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Chen H, Zhou D, Zhang J, Chen Y, Liu Q, Ai D, Zhu H, Chu L, Ren W, et al. (2017). Comparative genomic analysis of esophageal squa mous cell carcinoma between Asian and Caucasian patient populations. Nat. Commun 8, 1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis CE, Siegel RL, Sauer AG, Miller KD, Fedewa SA, Alcaraz KI, and Jemal A (2016). Cancer statistics for African Americans, 2016: progress and opportunities in reducing racial disparities. CA Cancer J. Clin 66, 290–308. [DOI] [PubMed] [Google Scholar]
- Ferrari S, and Cribari-Neto F (2004). Beta regression for modelling rates and proportions. J. Appl. Stat 31, 799–815. [Google Scholar]
- Field AP (2001). Meta-analysis of correlation coefficients: a Monte Carlo comparison of fixed- and random-effects methods. Psychol. Methods 6, 161–180. [DOI] [PubMed] [Google Scholar]
- Graffelman J (2015). Exploring diallelic genetic markers: the HardyWeinberg package. J. Stat. Softw 64, 1–23. [Google Scholar]
- Gravel S, Zakharia F, Moreno-Estrada A, Byrnes JK, Muzzio M, Rodriguez-Flores JL, Kenny EE, Gignoux CR, Maples BK, Guiblet W, et al. (2013). Reconstructing Native American migrations from whole-genome and whole-exome data. PLoS Genet. 9, e1004023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guda K, Veigl ML, Varadan V, Nosrati A, Ravi L, Lutterbaugh J, Beard L, Willson JK, Sedwick WD, Wang ZJ, et al. (2015). Novel recurrently mutated genes in African American colon cancers. Proc. Natl. Acad. Sci. USA 112, 1149–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FW, Mosquera JM, Garofalo A, Oh C, Baco M, Amin-Mansour A, Rabasha B, Bahl S, Mullane SA, Robinson BD, et al. (2017). Exome sequencing of African-American prostate cancer reveals loss-of-function ERF mutations. Cancer Discov. 7, 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo D, Hu H, Rhie SK, Gamazon ER, Cherniack AD, Liu J, Yoshimatsu TF, Pitt JJ, Hoadley KA, Troester M, et al. (2017). Comparison of breast cancer molecular features and survival by African and European ancestry in the cancer Genome Atlas. JAMA Oncol. 3, 1654–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Goncalves E, Barthorpe S, Lightfoot H, et al. (2016). A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan T, Moy B, Mroz EA, Ross K, Niemierko A, Rocco JW, Isakoff S, Ellisen LW, and Bardia A (2015). Comparison of the genomic landscape between primary breast cancer in African American versus white women and the association of racial differences with tumor recurrence. J. Clin. Oncol 33, 3621–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, et al. (2008). Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet 40, 1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan B, Rose TL, Kardos J, Milowsky MI, and Kim WY (2016). Intrinsic genomic differences between African American and white patients with clear cell renal cell carcinoma. JAMA Oncol. 10.1001/jamaoncol.2016.0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kytola V, Topaloglu U, Miller LD, Bitting RL, Goodman MM, D Agostino RB Jr., Desnoyers RJ, Albright C, Yacoub G, Qasem SA, et al. (2017). Mutational landscapes of smoking-related cancers in Caucasians and African Americans: precision oncology perspectives at Wake Forest Baptist Comprehensive Cancer Center. Theranostics 7, 2914–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Nyunoya T, Leng S, Belinsky SA, Tesfaigzi Y, and Bruse S (2013). Softwares and methods for estimating genetic ancestry in human populations. Hum. Genomics 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo LW, Wang Y, Flynn EM, Lund MJ, Bowles EJ, Buist DS, Liff JM, Flagg EW, Coates RJ, Eley JW, et al. (2011). Genome-wide copy number alterations in subtypes of invasive breast cancers in young white and African American women. Breast Cancer Res. Treat 127, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton TJ, Wang C, Li JZ, and Rosenberg NA (2010). Inference of unexpected genetic relatedness among individuals in HapMap Phase III. Am.J. Hum. Genet 87, 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovics G, Li H, Stumpel T, Tan SH, Young D, Katta S, Li Q, Ying K, Klocke B, Ravindranath L, et al. (2015). A novel genomic alteration of LSAMP associates with aggressive prostate cancer in African American men. EBioMedicine 2, 1957–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell IJ (2007). Epidemiology and pathophysiology of prostate cancer in African-American men. J. Urol 177, 444–449. [DOI] [PubMed] [Google Scholar]
- Powell IJ, Dyson G, Land S, Ruterbusch J, Bock CH, Lenk S, Herawi M, Everson R, Giroux CN, Schwartz AG, and Bollig-Fischer A (2013). Genes associated with prostate cancer are differentially expressed in African American and European American men. Cancer Epidemiol. Biomarkers Prev. 22, 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, and Reich D (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet 38, 904–909. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, and Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold K, and Engqvist L (2013). The variability is in the sex chromosomes. Evolution 67, 3662–3668. [DOI] [PubMed] [Google Scholar]
- Rosenbaum PR, and Rubin DB (1983). The central role of the propensity score in observational studies for causal effects. Biometrika 70, 41–55. [Google Scholar]
- Rosenberg NA (2006). Standardized subsets of the HGDP-CEPH Human Genome Diversity Cell Line Panel, accounting for atypical and duplicated samples and pairs of close relatives. Ann. Hum. Genet 70, 841–847. [DOI] [PubMed] [Google Scholar]
- Rosenthal R, McGranahan N, Herrero J, Taylor BS, and Swanton C (2016). DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royal CD, Novembre J, Fullerton SM, Goldstein DB, Long JC, Bamshad MJ, and Clark AG (2010). Inferring genetic ancestry: opportunities, challenges, and implications. Am. J. Hum. Genet 86, 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Perez C, Tamborero D, Schroeder MP, Antolin AA, Deu-Pons J, Perez-Llamas C, Mestres J, Gonzalez-Perez A, and Lopez-Bigas N (2015). In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell 27, 382–396. [DOI] [PubMed] [Google Scholar]
- Sankararaman S, Sridhar S, Kimmel G, and Halperin E (2008). Estimating local ancestry in admixed populations. Am. J. Hum. Genet 82, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher SE, Shim BY, Corso G, Ryu MH, Kang YK, Roviello F, Saksena G, Peng S, Shivdasani RA, Bass AJ, and Beroukhim R (2017). Somatic copy number alterations in gastric adenocarcinomas among Asian and Western patients. PLoS One 12, e0176045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2016). Cancer statistics, 2016. CA Cancer J. Clin 66, 7–30. [DOI] [PubMed] [Google Scholar]
- Smithson M, and Verkuilen J (2006). A better lemon squeezer? Maximum-likelihood regression with beta-distributed dependent variables. Psychol. Methods 11, 54–71. [DOI] [PubMed] [Google Scholar]
- Spratt DE, Chan T, Waldron L, Speers C, Feng FY, Ogunwobi OO, and Osborne JR (2016). Racial/ethnic disparities in genomic sequencing. JAMA Oncol. 2, 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouffer SA, Suchman EA, DeVinney LC, Star SA, and Williams RMJ (1949). The American Soldier, Vol. 1, Adjustment during Army Life (Princeton University Press; ). [Google Scholar]
- Tang H, Siegmund DO, Johnson NA, Romieu I, and London SJ (2010). Joint testing of genotype and ancestry association in admixed families. Genet. Epidemiol 34, 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre LA, Sauer AM, Chen MS Jr., Kagawa-Singer M, Jemal A, and Siegel RL (2016). Cancer statistics for Asian Americans, native Hawaiians, and Pacific Islanders, 2016: converging incidence in males and females. CA Cancer J. Clin 66, 182–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troester MA, Herschkowitz JI, Oh DS, He X, Hoadley KA, Barbier CS, and Perou CM (2006). Gene expression patterns associated with p53 status in breast cancer. BMC Cancer 6, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Iterson M, van Zwet EW, Slagboom PE, and Heijmans BT (2016). Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. bioRxiv. 10.1186/s13059-016-1131-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang BD, Ceniccola K, Hwang S, Andrawis R, Horvath A, Freedman JA, Olender J, Knapp S, Ching T, Garmire L, et al. (2017). Alternative splicing promotes tumour aggressiveness and drug resistance in African American prostate cancer. Nat. Commun 8, 15921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C, Wen W, Morgans AK, Pao W, Shu XO, and Zheng W (2015). Disparities by race, age, and sex in the improvement of survival for major cancers: results from the national cancer institute surveillance, epidemiology, and end results (SEER) program in the United States, 1990 to 2010. JAMA Oncol. 1, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kwok-Shing Ng P, Kucherlapati M, Chen F, Liu Y, Tsang YH, de Velasco G, Jeong KJ, Akbani R, Hadjipanayis A, et al. (2017). A Pan-cancer proteogenomic Atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell 31, 820–832 e823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genetic ancestry assessment of each TCGA patient (n = 11,122) has been made available via the Cancer Genetic Ancestry Atlas (TCGAA, http://52.25.87.215/TCGAA).