

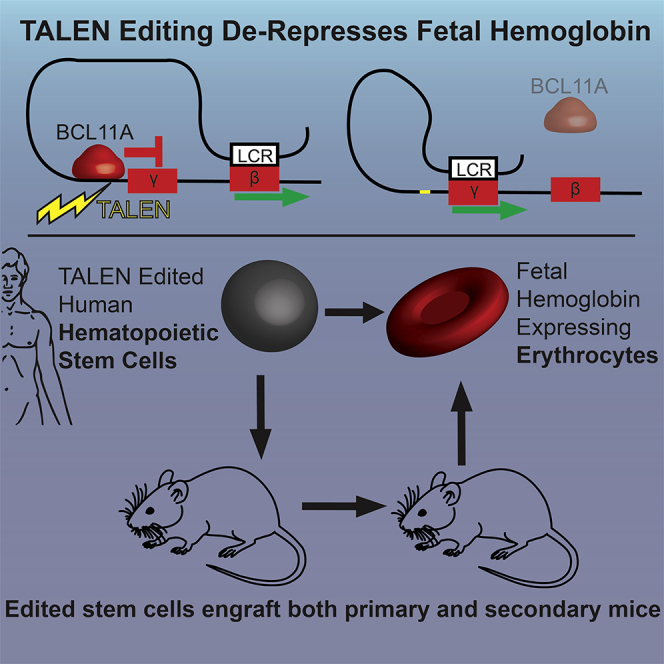
Abstract
Elements within the γ-hemoglobin promoters (HBG1 and HBG2) function to bind transcription complexes that mediate repression of fetal hemoglobin expression. Sickle cell disease (SCD) subjects with a 13-bp deletion in the HBG1 promoter exhibit a clinically favorable hereditary persistence of fetal hemoglobin (HPFH) phenotype. We developed TALENs targeting the homologous HBG promoters to de-repress fetal hemoglobin. Transfection of human CD34+ cells with TALEN mRNA resulted in indel generation in HBG1 (43%) and HBG2 (74%) including the 13-bp HPFH deletion (∼6%). Erythroid differentiation of edited cells revealed a 4.6-fold increase in γ-hemoglobin expression as detected by HPLC. Assessment of TALEN-edited CD34+ cells in vivo in a humanized mouse model demonstrated sustained presence of indels in hematopoietic cells up to 24 weeks. Indel rates remained unchanged following secondary transplantation consistent with editing of long-term repopulating stem cells (LT-HSCs). Human γ-hemoglobin expressing F cells were detected by flow cytometry approximately 50% more frequently in edited animals compared to mock. Together, these findings demonstrate that TALEN-mediated indel generation in the γ-hemoglobin promoter leads to high levels of fetal hemoglobin expression in vitro and in vivo, suggesting that this approach can provide therapeutic benefit in patients with SCD or β-thalassemia.
Keywords: TALEN, hemoglobinopathy, HBG, hemoglobin, HPFH, SCD, sickle cell disease, thalassemia, gene editing, HSC
Graphical Abstract
Introduction
Hemoglobin disorders, including sickle cell disease (SCD) and β-thalassemia, comprise the most common group of human single-gene diseases and exert a major impact on global public health. Recent WHO data suggest that 1% of all couples are at risk for having a child with a hemoglobin disorder, and population-modeling indicates that disease prevalence is on the rise.1, 2 A unifying feature of this heterogeneous group of diseases is reduced functional hemoglobin due to altered protein structure in SCD or insufficient β-hemoglobin production in thalassemia, respectively.3 Clinical management includes blood transfusion and use of hydroxyurea and pain control in SSD. Morbidity, however, remains high and life expectancy lags behind population averages.4, 5 Currently, the only curative therapy for hemoglobin disorders is allogeneic stem cell transplant, and this approach is limited by donor availability and transplant-associated medical complications.6
Hemoglobin disorder disease phenotype varies based upon both the underlying genetic mutation and genetic background. Patients who also carry gene variants or deletions that induce expression of fetal (γ or HBG) hemoglobin (HbF) can exhibit a less severe phenotype and decreased morbidity and mortality.7 A naturally occurring 13-bp deletion (d13) within the HBG1 promoter (−102 to −114) was initially reported in two siblings with the SCD mutation. Strikingly, this deletion resulted in increased expression of HbF and amelioration of SCD disease symptoms, a phenotype referred to as hereditary persistence of fetal hemoglobin (HPFH).8 HPFH represents a rare genetic condition that can effectively modulate the severity of β-hemoglobin disorders, and recent advances in the development of designer nucleases capable of mediating targeted double-stranded DNA cleavage make the artificial disruption of this site possible.9, 10, 11, 12 Consistent with this idea, recent work has demonstrated that CRISPR/Cas9-mediated disruption of the HBG1 or 2 promoter region predominantly results in the generation of a 13-bp deletion. Importantly, gene disruption also created a broad range of unique smaller indels (>40) that also induced HbF expression in primary hematopoietic stem cells in vitro.13
To directly evaluate and expand upon this potential therapeutic approach, we generated TALEN (transcription activator-like effector nucleases) pairs targeting regulatory elements within the HBG1 (Aγ) promoter proximal to the distal CCAAT box (−111 to −115). We determined whether delivery of TALEN pairs using mRNA transfection functioned to disrupt the HBG1 and HBG2 promoter in mobilized human CD34+ peripheral blood stem cells (hPBSCs). We show that TALEN delivery achieves our goal of efficient generation of double-strand breaks leading, via non-homologous end joining (NHEJ), to a variety of deletions including the naturally occurring 13-bp HPFH deletion and multiple smaller deletions in the promoter of HBG1 and HBG2. We also demonstrate that these events lead to efficient de-repression of fetal hemoglobin expression in vitro following differentiation of hPBSCs into erythroid cells. Most notably, we show that TALEN-edited hPBSCs with therapeutic edits engraft in vivo in both primary and secondary recipients in a humanized mouse model. Taken together, these findings indicate that this approach can result in sustained therapeutic editing in human long-term hematopoietic stem cells, suggesting that this strategy may be amenable to direct clinical translation.
Results
TALEN Design for Editing of the HBG1 and HBG2 Promoter Region
Hemoglobin switching in the post-natal period is orchestrated by key transcription factors binding the promoter elements within the β-hemoglobin gene cluster. Elements of the HBG1 (Aγ) promoter proximal to the distal CCAAT box (−111 to −115) serve as putative binding sites for transcription factors that negatively regulate γ-globin expression (Figure 1). While several transcription factors likely play roles in hemoglobin silencing, recent evidence suggests that a key player is the zinc finger protein BCL11A.8, 14, 15, 16, 17, 18 The naturally occurring 13-bp HPFH deletion is located at position −102 to −114 within the HBG1 promoter and is predicted to impact the binding of BCL11A (Figure 1).
Figure 1.

Human β Hemoglobin Locus and TALEN Design
Schematic of the human β hemoglobin locus on chromosome 11 highlighting the homologous sequences of the HBG1 and HBG2 (Aγ and Gγ) promoters and structural elements. The sequences for each promoter (shown schematically for the −80 to −140 region) are entirely homologous. The putative BCL11A binding sequence (TGACCA) is underlined in red and a red representation of the transcription factor is shown above the sequence. Green * indicates the location of published HPFH SNPs. The bracketed green line highlights the 13-bp HPFH deletion found naturally in HBG1. TALENs selected in silico and tested in vitro are shown below the promoter sequence. Blue boxes represent the repeat variable diresidues (RVDs), and their positions overlay the corresponding nucleotide bound within the conserved HBG promoter regions. Scissors represent FokI endonuclease, and the dotted line and indicated bp numbers represent the spacer length between the TALEN pairs. The distal (–111 to 115) and proximal (–84 to 88) CCAAT boxes in the HBG1 and HBG2 promoters are underlined in brown.
TALENs comprise an attractive designer nuclease platform for several reasons. First, TALENs function to introduce targeted double-stranded breaks by means of stringent, protein-based DNA sequence recognition. DNA binding is paired with endonuclease cleavage via use of a FokI nuclease fused to each of the respective DNA-binding domains requiring two unique DNA binding sites in a defined proximity leading to a low degree of off-target cutting.19 TALEN delivery as mRNA avoids the requirement for viral delivery or for protein co-transfection and may thereby avoid pre-existing adaptive immunity to viral components or to Cas9.20 Based upon this combined rationale, we used in silico prediction to design three alternative TALEN pairs to bind sequences flanking the distal CCAAT box of the HBG promoter with varied repeat variable diresidue (RVD) sequences and spacer lengths (Figure 1). Prognos off-target prediction and subsequent sequence-based screening yielded no evidence for significant cleavage (Figure S7). Screening of TALEN pairs in hPBSCs using a T7 assay showed that only the “TAL1” pair resulted in significant insertion or deletion (indel) generation in both the HBG1 and HBG2 promoters and was used in all subsequent studies (Figure S1).
Optimized Editing of HBG1 and HBG2 in Human PBSCs
We next optimized the transfection of hPBSCs with TAL1 mRNA (Figure 2A). Total cell number and viability decreased with increasing mRNA concentration with a 4-μg transfection resulting in a 35% reduction in viability and 75% decrease in cell expansion at 24 h compared to mock-transfected (0 μg) cells. Transient cold shock (16 h recovery at 30°C) led to additional cell toxicity when using mRNA concentrations >1 μg (Figure 2B). Despite initial cell losses at higher TALEN doses, overall cell viability and culture growth rates normalized in all treatment conditions between 48 and 72 h post-electroporation.
Figure 2.

Optimizing TALEN Editing Conditions
(A) Experimental timeline for TALEN transfection of hPBSCs.
(B) Cell viability (right axis) and total cell number (left axis) at 24 h post-TALEN transfection and assessed at two recovery temperatures (37°C and 30°C) versus increasing doses of TALEN mRNA (n = 6 /condition, ANOVA *p < 0.05, **p < 0.005, ***p < 0.0005).
(C) Schematic of ddPCR fall-off assay designed to detect NHEJ events at both the HBG1 and HBG2 promoters. A common set of primers (green) are used to amplify the HBG1 and HBG2 alleles. A common NHEJ ddPCR probe linked to HEX binds over the TALEN target cut site and is designed to fall off if a single base insertion or deletion is present in the binding region. Unique HBG1 and HBG2 Ref ddPCR probes linked to FAM bind to a region with three unique nucleotides allowing for editing of HBG1 and HBG2 to be assessed independently.
(D) ddPCR results for hPBSCs edited with increasing concentrations of TALEN mRNA at two recovery temperatures (data pooled from three donors).
(E) ddPCR-based assessment of NHEJ rates following transfection with 1 μg TALEN across three donors.
(F) NHEJ rates as assessed Illumina MiSeq-based amplicon sequencing; data pooled from three donors.
(G) Indel frequency in HBG1 and HBG2 according to size (bp) in alternative recovery temperatures (37°C upper bars; 30°C lower bars). Solid bars represent HBG1; open bars represent HBG2; data pooled from three donors.
Error bars represent SD.
We developed a Droplet Digital Polymerase Chain Reaction (ddPCR) drop-off probe assay to estimate indel generation within both the HBG1 and HBG2 targets (Figure 2C). Editing rates at HBG1 and HBG2 increased ∼2-fold when cells were exposed to 30°C cold shock compared with 37°C culture when using 1 μg TALEN mRNA (17% ± 4.1% to 43.3% ± 9.3% and 41% ± 3.6% to 74% ± 2.4%, respectively). Dose titration of TALEN mRNA from 0.5 to 4 μg did not significantly increase indel rates with 37°C culture (HBG1 14% ± 1.8% to 20% ± 4.5%, HBG2 33% ± 3% to 41% ± 5%). In contrast, 30° cold shock was associated with maximal editing rates using 0.5–1 μg of mRNA and a progressive decline in editing rates as mRNA concentration increased above 1 μg (Figure 2D) correlated with decreased cell number and viability at higher mRNA concentrations. Importantly, high rates of gene editing using the 30°C condition were consistent across multiple independent PBSC donors (n = 3). Editing rates at both HBG promoters were also consistent across donors regardless of recovery temperature (Figure 2E) or mRNA concentration (Figure 2D). Notably, despite identical target sites at each locus, editing rates were consistently higher (∼40%–50% relative increase) at HBG2 compared HBG1 (Figures 2D and 2E).
As an independent means to assess both indel rates and sizes, we utilized next-generation sequencing (NGS). Overall, indel rates at both HBG1 and HBG2 assessed by NGS were similar, although slightly lower, than those obtained by ddPCR. Consistent with ddPCR findings, NGS demonstrated a significant increase in editing following mRNA delivery in the setting of a 30°C cold shock with indel rates increased ∼2-fold from 26% ± 3% to 52% ± 4% at the 1-μg mRNA transfection dose (p < 0.0005; Figure 2F). With respect to the spectrum of indels, the majority comprised 0- to 10-bp deletions centered around the TALEN cut site. Notably, there was also a consistent and significant increase in 13-bp deletions (3.8% at 37°C and 6.3% at 30°C) recapitulating the naturally occurring HPFH deletion (Figure 2G).8 As previously described,13 this specific deletion likely reflects the impact of micro-homology-templated DNA repair of double-stranded DNA breaks in this region. Importantly, TALEN mRNA delivery in the setting of 30°C cold shock not only resulted in increased editing but also shifted the indel spectrum to favor larger deletions with 17% versus 3.4% of all sequences exhibiting deletions of at least 5–8 bp in the cold shock compared to 37°C condition (Figure 2G). These smaller deletions, similar to the 13-bp deletion, would be predicted to limit the recruitment of transcriptional repressors, including BCL11A and others, to the −110 to −120 region containing the distal CCAAT box adjacent to the TAL1 nuclease cleavage site.
TALEN-Induced Deletions Promote Fetal Hemoglobin Expression in Differentiated Erythroid Cells
Using the experimental approach outlined in Figure 3A, we assessed the impact of TALEN-based editing on fetal hemoglobin expression in vitro. Following a shift to erythroid differentiation culture media, both mock- and TALEN-transfected hPBSCs expanded and differentiated into erythroid progeny. Differentiated cells exhibited matching profiles of CD235a expression (Figure 3B), CD71 expression, and time to visual hemoglobin detection in cell pellets (data not shown). Compared to mock-edited cells, the presence of HbF expressing F cells was significantly increased in TALEN-edited conditions (p < 0.0005). 37°C culture step resulted in a 1.6-fold increase in HbF expression (25% ± 4% versus 44% ± 4% for 0 versus 1 μg mRNA, respectively) while TALEN delivery with 30°C cold shock resulted in a 2.3-fold increase in HbF expression (25% ± 2% versus 57% ± 3% for 0 μg versus 1 μg mRNA; Figure 3C). Consistent with in vitro editing rates (as assessed by ddPCR or NGS), the highest levels of HbF were observed using 0.5–1 μg TALEN mRNA and a 30°C cold shock.
Figure 3.

TALEN Editing Drives Fetal Hemoglobin Expression in Differentiated hPBSCs
(A) Experimental timeline for erythroid differentiation and analysis following transfection.
(B) Representative flow cytometry data at day 14 comparing erythroid progeny in mock (blue) versus TALEN-edited (orange) hPBSCs and isotype control in brown. Left, CD235a+ staining; Right, HbF expression.
(C) Combined analysis of HbF expression at increasing doses of TALEN mRNA and at different recovery temperatures (30°C, blue circles; 37°C, black circles).
(D) Hemoglobin expression by HPLC following transfection with 1 μg TALEN mRNA at alternative recovery temperatures. (ANOVA *p < 0.05, **p < 0.005, ***p < 0.0005); data pooled from three donors.
Error bars represent SD.
We also utilized HPLC to directly measure HbF production. We observed a significant increase in total HbF (HBG1 and HBG2) protein levels by HPLC in cells transfected with ≥1 μg TALEN mRNA. Using 1 μg mRNA and 37°C culture, we observed a 2.4-fold increase in total HbF (26% ± 3%). In contrast, use of the 30°C cold shock resulted in a 4.6-fold increase in HbF protein expression (41% ± 8%; p < 0.005) (Figure 3D). Notably, despite the higher indel rates at HBG2 (as detected by ddPCR), the majority of the HbF protein detected by HPLC was HBG1 with a smaller contribution made by HBG2. This latter observation is consistent with association of the HPFH phenotype with deletions specifically within the HBG1 promoter and may also suggest that the larger deletions generated by dual editing of HBG1 and HBG2 may also be functionally active. Finally, HbF induction was consistently associated with a reciprocal reduction in HbA thereby maintaining an equivalent ratio of α and β hemoglobin proteins (Figure S3). These combined data demonstrate that TALEN-mediated indels in the HBG1 and HBG2 promoters result in increased numbers of F cells as well as overall fetal hemoglobin protein production. The increased indel generation seen following 30°C cold shock correlates with higher rates of fetal hemoglobin production.
Sustained Multi-lineage Engraftment of TALEN-Edited hPBSCs in a Humanized Murine Model
Having demonstrated efficient TALEN-based gene editing of hPBSC in vitro, we next assessed the capacity of edited cells to engraft in vivo. We utilized transplantation of hPBSC into the immune-deficient W41 mouse strain (Jackson Labs 026622) mouse strain to assess long-term engraftment and function of gene-edited cells (Figure 4A). Compared to the NOD-scid IL2Rgnull (NSG) animals, the W41 strain permits enhanced human hematopoietic chimerism and erythroid development in the marrow with minimal conditioning.21 Representative flow analysis of the bone marrow at week 24 demonstrates robust human engraftment (hCD45 = 52% ± 22% mock, 54% ± 16% TALEN edited) (Figure 4B). Human cells were present in all lineages, including CD19+ B cells, CD33+ myeloid cells, CD34+ lymphoid and myeloid progenitors, and in erythroid lineage cells including pro-erythroblasts (CD71+), erythroblasts (CD71+CD235a+), and reticulocytes (CD235a+) (Figure S4). There was no significant difference in the overall engraftment rates in recipients of TALEN-edited versus mock-transfected control hPBSCs (Figure 4C). To definitively assess engraftment and editing in long-term repopulating stem cells (LT-HSCs), secondary transplants were established. For these studies, we utilized bone marrow cells isolated at 24 weeks post-transplant from primary recipients in transplant 2. At 9 weeks post-secondary transplant, we observed an average human engraftment of 5%, including multiple lineages, findings consistent with previously reported secondary engraftment rates in the human xenograft W41 mouse model21 (Figure 4B).
Figure 4.

Sustained Multi-lineage Engraftment of TALEN-Edited hPBSCs in Primary and Secondary Recipient W41 Mice
(A) Experimental schema for hPBSC editing and engraftment in primary and secondary recipient W41 mice.
(B) Human cell engraftment (% hCD45+ cells) at time of sacrifice in primary and secondary recipient mice. Shapes designate individual mice within each experimental cohort.
(C) FACS analysis of human hematopoietic cell lineages in primary recipients at sacrifice (transplant 1, 16 weeks; transplant 2, 24 weeks).
(D) NHEJ rates in HBG1 and HBG2 as detected by ddPCR at time of sacrifice in primary and secondary recipients.
(E) Indel frequency in HBG1 (filled bars) and HBG2 (open bars) both pre- (upper bars) and post-transplant (lower bars) as measured by next-gen sequencing. Values are displayed as percentage of all sequence reads corresponding to a given insertion or deletion size. n = 5 animals.
(F) Comparison of percent modification (deletion) at each nucleotide pre- (black lines) and post-transplant (red lines). Closed and open circles designate HBG1 and HBG2, respectively. Location of BCL11A binding site (red line), distal CCAAT box (green line), and 13-bp HPFH deletion are indicated.
(G) Flow analysis of HbF expression in human cells (CD45+, CD235+) within the bone marrow at week 24 in transplant 2 (time of sacrifice).
(H) Flow analysis of HbF+ cells following ex vivo liquid erythroid differentiation from the same experiment. Bone marrow cells differentiated in liquid differentiation media result in a heterocellular phenotype with high background HbF expression.47 t test, *p < 0.05, **p < 0.005.
Error bars represent SD.
At the time of sacrifice in primary recipients, editing rates were approximately half of the input rate (average 50% ± 4% in input versus 23% ± 8% in engrafted cells [at 16 weeks in transplant 1], and 24% ± 2% versus 9.4% ± 4% [at 24 weeks in transplant 2; Figure 4D]). Indel distribution was similar to that observed pre-transplant with the exception of a decrease in proportion of the 13-bp deletion from 17.4% of detected indels in the HBG1 promoter to 6.9% and from 16.9% to 8% in the HBG2 promoter (Figures 4E and S5). While the overall editing rates decreased 2-fold post-transplant, the proportional editing by nucleotide did not change significantly. Most notably, the majority of deletions were located within in the BCL11A binding sequence (HBG1, 31% to 40%; HBG2, 34% to 40%) and the distal CCAAT box (HBG1, 42% to 47%; HBG2, 45% to 43%) with proportionally fewer persistent indels over the distal 13-bp HPFH deletion (HBG1, 93% to 69%; HBG2, 86% to 60%) (Figure 4F). Of note, no previous studies of globin gene editing have assessed editing rates in secondary transplants. Importantly, our findings indicate that editing rates and indel distribution are maintained following secondary transplant (9.4% ± 4% in primary and 10.4% ± 4% in secondary recipients, respectively), suggesting that the edited population contains LT-HSCs capable of secondary engraftment (Figure 6).
We also assessed fetal hemoglobin induction in transplant recipients. F cells were increased significantly at the time in the marrow of animals that received the TALEN-edited cells (32% ± 15% on average) compared to mock controls (20% ± 1% average) (p < 0.05, Welch’s t test) (Figure 4G). These cells represent human erythrocytes present in the mouse marrow space. Unsorted marrow cells were also cultured in erythroid differentiation media to assess the potential of engrafted human stem and progenitor cells to differentiate into erythroid progeny. Cultured marrow from recipients of TALEN-edited PBSCs produced human erythrocytes with more F cells (70% ± 6%) compared to bone marrow from control recipient animals (59% ± 2%) (Figure 4H). Erythroid colonies differentiated in methylcellulose expressed significantly higher levels of fetal hemoglobin if indels were present (Figure S8). Taken together, these data demonstrate that TALEN-edited hPBSCs include a population of LT-HSCs that retain indels and express higher levels of fetal hemoglobin.
Discussion
The advent of targeted gene editing has raised the possibility of modifying the genome of hematopoietic cells as a means of therapeutic fetal hemoglobin induction. Previous work has begun to define key genomic loci and transcription factors that facilitate silencing of fetal hemoglobin in the post-natal period.22, 23, 24 While the molecular mechanisms that regulate developmental hemoglobin switching are complex, the reduced morbidity and mortality following deletions that lead to de-repression of HBG1 and 2, suggest that this regulatory nexus represents an excellent target for therapeutic gene editing.
Consistent with this idea, we demonstrate that TALEN editing of the HBG1 and HBG2 promoter in hPBSCs efficiently drives fetal hemoglobin induction in vitro and in vivo. The double-stranded DNA breaks induced by TALEN cleavage resolve primarily into small deletions near the TALEN cut site. In vitro, a significant proportion of the resultant deletions also recapitulate the naturally occurring 13-bp HPFH deletion as described in patients with SCD.8 Generation of this canonical deletion likely occurs due to a high degree of microhomology within surrounding sequences that increase the likelihood of DNA repair events that resolve with this specific sequence.13 d13 HPFH subjects express fetal hemoglobin at levels that abrogate the sickle phenotype.8 Notably, the majority of TALEN-based edits generated using the targeting approach described in this study, including the majority of edits that are retained in vivo, result in deletions that are smaller than the canonical 13-bp deletion. However, similar to the 13-bp deletion, based upon their location, these deletions are predicted to similarly disrupt the binding sites for key repressive elements including BCL11A and the DRED and COUP-TFII complexes. Our combined findings showing sustained editing and sustained fetal hemoglobin induction in vivo following transplantation of TALEN-edited PBSC strongly support this idea.
As part of our study, we assessed the impact of transient cold shock following TALEN mRNA transfection. Our findings demonstrate that this approach leads a near doubling of indel generation at both the HBG1 and HBG2 target sites as well as an increase in large intergenic deletions. Similar use of cold shock for zinc finger nuclease (ZFN)-based gene editing has been previously reported, and this increase in efficiency likely reflects delayed mRNA and protein turnover and/or molecular chaperone induction.25 NGS of TALEN target sites revealed that, in addition to increased indel generation, the size of resulting indels was also significantly increased in the setting of transient cold shock. The increased indel frequency and size yielded an overall increase in total HbF expression in vitro of as high as 41%, levels well above a clinically relevant threshold of 10%–20%.7
Interestingly, we noted a preferential expression of HBG1 protein by HPLC despite higher editing rates detected by ddPCR in the HBG2 promoter. A likely explanation for this finding is generation of a 4.9-kb intergenic deletion following cleavage at both TALEN sites, an event that is resolved as a fusion gene that expresses HBG1 protein using the HBG2 promoter. The ddPCR drop-off assay used in our study was not designed to detect this large intergenic deletion. Thus, results from this ddPCR assay likely underreport the true rate of editing at the HBG1 locus while accurately reflecting editing rates at HBG2. To directly assess for the presence of this predicted larger deletion, we utilized a modified ddPCR assay (Figure S2). Using this assay, we demonstrate that the editing rate differences detected at HBG1 versus HBG2 are largely accounted for by the prevalence of this large deletion event. While we have not directly assessed the full impact of this deletion in our study, the relatively high occurrence rate and lack of obvious impact on cellular viability or phenotype, suggest such events are well tolerated in TALEN-edited PBSCs and unlikely to be detrimental.
A critical finding in this study is the demonstration that TALEN-edited hPBSCs are capable of sustained primary and secondary engraftment in a humanized mouse model system. We believe this is the first time this has been demonstrated in the field of hemoglobin gene editing. Following primary transplantation, we observed a 2-fold reduction in indel rates. This decline is consistent with previous reports assessing engraftment of gene-edited human HSCs and likely reflects a combination of events, including higher editing rates in rapidly dividing committed progenitors that are lost over time in vivo and, possibly, decreased engraftment of LT-HSCs that undergo and repair double-strand breaks.26, 27 Importantly, despite this initial decline in indel frequency, we demonstrate maintenance of clinically relevant indel rates in both primary and secondary transplant recipients, findings consistent with having edited an LT-HSC population. As noted above, post-transplant indel profiles identified by NGS revealed partial loss of the d13 HPFH mutation with retention of smaller indels. The skewing of retained edits may reflect differences in the activity of specific DNA repair pathways in various hPBSC progenitors, implying that long-term engrafted cells exhibit limited activity of micro-homology-directed repair and primarily generate indels by the NHEJ pathway.28, 29 Analysis of retained deletions at each nucleotide (Figure 4F) revealed that the majority of deletions occur in the BCL11A and DRED/COUP-TFII repressor-binding motifs.17 The resultant phenotype of increased F cells despite the relative decrease of the d13 HPFH deletion suggests that smaller deletions disrupting these putative negative repressor binding sites are sufficient for fetal hemoglobin expression.
Therapeutic gene editing has the potential to become a curative option for patients with hemoglobinopathies. Clinical trials involving the lentiviral delivery of an anti-sickling βA(T87Q)-globin gene are well underway.30 Other clinical trials in the recruitment phase aim to induce fetal hemoglobin expression by targeted disruption or small hairpin RNA (shRNA) knockdown of the transcription factor BCL11A.31, 32, 33, 34 One potential drawback to this approach is that BCL11A plays an important role in other hematopoietic cell types including B lymphocytes and is also expressed in non-hematopoietic cells.35, 36, 37 While clinical trials based on this approach will utilize editing strategies that preferentially impact erythroid cells, this approach will require careful monitoring to assess potential non-erythroid events.38 TALEN-targeted disruption of the γ globin promoter yields at least equivalent to if not higher HbF expression than that seen when ZFNs are used to disrupt the erythroid enhancer of BCL11A.27 Recent data suggest that the 13-bp HPFH deletion overlaps the major BCL11A binding site within the HBG1 and/or HBG2 promoter (in addition to other putative repressive elements).17 Thus, targeting the multiple transcription factor binding sites described here and in previous studies using either ex vivo39 or in vivo13 CRISPR/Cas9-mediated editing may specifically ablate the activity of BCL11A within this locus, thereby limiting the impact of edits more strictly to the erythroid lineage. This target site may also be amenable to megaTAL-based nuclease editing.40 While it remains to be determined which of these alternative nuclease platforms and delivery approaches will reach clinical application, our demonstration of sustained editing in LT-HSCs in secondary recipient humanized mice support the potential for clinical translation.
In most clinical gene-editing applications, designer nucleases are used to facilitate either NHEJ- or homology-directed repair (HDR) gene modification. In applications that utilize HDR, NHEJ events that occur in parallel are typically undesirable. In contrast, while not described here, targeting the γ-hemoglobin promoter offers a unique advantage that clinical benefit may be achieved concurrently via both NHEJ-induced indel generation and introduction of repair templates via HDR. In parallel with the NHEJ-based induction of fetal hemoglobin described here, a range of HDR templates might offer potential additional clinical benefits, including use of HDR to introduce defined deletions (including the 13-bp HPFH deletion or BCL11A binding-motif disruption) or, alternatively, larger gene cassettes that induce expression of β-hemoglobin or permit post-editing selection of gene-modified progenitors. Current studies using the approach described here including assessment in non-human primate models and using HDR templates will help fully define the clinical validity of editing the γ promoter and may provide additional therapeutic options for the treatment of hemoglobinopathies.
Materials and Methods
TALEN Generation and Production
The TALE-NT Targeter 2.0 (https://tale-nt.cac.cornell.edu/) was used to query the human HBG1 promoter (Homo sapiens, alternate assembly CHM1_1.1, 5270034-5270208), Miller architecture and Streubel guidelines.41, 42, 43, 44 Three TALEN pairs were selected for production (Table S1). Off-target site prediction was carried out using the PROGNOS bioinformatics tool.45 TALENs were assembled by Golden Gate cloning and inserted into a novel linear plasmid pEVL backbone to encode a 300-bp poly(A) tail.44, 46 Constructs were screened and validated by Sanger sequencing. mRNA production was carried out using the CELLSCRIPT T7 mScript kit following manufacturer’s protocol (CAP-1 architecture). Tail length was validated by Lonza flash gel following manufacturer’s protocol. mRNA was eluted in nuclease-free water, and concentration was confirmed by Nanodrop spectrophotometer. Plasmid data is published on GenBank (GenBank: MH_990331 and MH_990332).
ddPCR
The NHEJ assay measured editing at HBG loci using a reference FAM probe specific to HBG1 or HBG2 and a NHEJ-HEX probe specific to the unedited TALEN cut sites. Primers and probes are listed in Table S2. Editing was measured using 50 ng of genomic DNA (gDNA) with either an HBG1 or HBG2 FAM probe along with the NHEJ-HEX probe (1× assay, 900-nM primers and 250-nM probe) using ddPCR supermix for probes (no deoxyuridine triphosphate [dUTP]). Droplets were generated with the QX200 Droplet Generator (Bio-Rad) and amplified. Fluorescence was measured using the QX200 Droplet reader (Bio-Rad) and analyzed using Quantasoft. Editing rates were calculated as the relative frequency (%) of FAM+ and HEX− events compared to all FAM+ events.
Transplantation
1 × 106 CD34+ hPBSCs (control and TALEN transfected) were injected by tail vein into W41 mice (Jackson Labs Strain 026622, NBSGW21) following minimal radiation (150 cGy) 18 h post-transfection. Bone marrow was harvested and analyzed 16–24 weeks post-transplant. Secondary W41 mice were transplanted with 50% of the marrow cells harvested from the primary recipient mice. Secondary mice were sacrificed 9 weeks post-transplant. gDNA was isolated for analysis by QIAGEN DNeasy Blood and Tissue kit (#69506) per manufacturer’s protocol. Animal experiments were conducted in accordance with institutional guidelines. All animal experiments were carried out following IACUC review and approval at both the Seattle Children’s Research Institute (SCRI) and Fred Hutchinson Cancer Research Center (FHCRC) and conformed to all institutional regulatory standards.
Author Contributions
C.T.L. designed research, performed research, analyzed and interpreted data, and wrote the manuscript; S.P. designed research, interpreted data, and participated in writing of the manuscript; M.B. performed research and analyzed data; C.N. performed research; D.A.F. performed research; O.N. performed research; O.H. designed research; J.G.Y. performed research; C.L. performed research; K.J. designed research; I.B. designed research; H.-P.K. designed research; A.M.S. designed research and analyzed and interpreted data; D.J.R. designed research, analyzed data, and wrote the manuscript.
Conflicts of Interest
A.M.S. in an employee of Casebia Therapeutics. O.N., C.L., and J.G.Y. are employees of bluebird bio.
Acknowledgments
We thank Brandon Hadland for advice and mentorship, Brian Gordon for technical support and ddPCR expertise, and Samantha Lotti for technical support. This work was supported by the Seattle Children’s Research Institute, Program for Cell and Gene Therapy; the NIH (under award numbers T32CA009351, 1R01HL136135-02, and 4K12HL087165-10); bluebird bio, Inc; and the Children’s Guild Association Endowed Chair in Pediatric Immunology (D.J.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Supplemental Information includes Supplemental Materials and Methods, seven figures, and two tables and can be found with this article online at https://doi.org/10.1016/j.omtm.2018.12.008.
Supplemental Information
References
- 1.Modell B., Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 2008;86:480–487. doi: 10.2471/BLT.06.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piel F.B., Hay S.I., Gupta S., Weatherall D.J., Williams T.N. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10:e1001484. doi: 10.1371/journal.pmed.1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodman M.A., Malik P. The potential of gene therapy approaches for the treatment of hemoglobinopathies: achievements and challenges. Ther. Adv. Hematol. 2016;7:302–315. doi: 10.1177/2040620716653729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yawn B.P., Buchanan G.R., Afenyi-Annan A.N., Ballas S.K., Hassell K.L., James A.H., Jordan L., Lanzkron S.M., Lottenberg R., Savage W.J. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312:1033–1048. doi: 10.1001/jama.2014.10517. [DOI] [PubMed] [Google Scholar]
- 5.Origa R. Beta-thalassemia. In: GeneReviews M.P., editor. University of Seattle, Washington; 1993. [Google Scholar]
- 6.Bernaudin F., Pondarré C., Galambrun C., Thuret I. Allogeneic/Matched Related Transplantation for β-Thalassemia and Sickle Cell Anemia. Adv. Exp. Med. Biol. 2017;1013:89–122. doi: 10.1007/978-1-4939-7299-9_4. [DOI] [PubMed] [Google Scholar]
- 7.Platt O.S., Brambilla D.J., Rosse W.F., Milner P.F., Castro O., Steinberg M.H., Klug P.P. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N. Engl. J. Med. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 8.Gilman J.G., Mishima N., Wen X.J., Stoming T.A., Lobel J., Huisman T.H. Distal CCAAT box deletion in the A gamma globin gene of two black adolescents with elevated fetal A gamma globin. Nucleic Acids Res. 1988;16:10635–10642. doi: 10.1093/nar/16.22.10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LaFountaine J.S., Fathe K., Smyth H.D.C. Delivery and therapeutic applications of gene editing technologies ZFNs, TALENs, and CRISPR/Cas9. Int. J. Pharm. 2015;494:180–194. doi: 10.1016/j.ijpharm.2015.08.029. [DOI] [PubMed] [Google Scholar]
- 10.Cottle R.N., Lee C.M., Bao G. Treating hemoglobinopathies using gene-correction approaches: promises and challenges. Hum. Genet. 2016;135:993–1010. doi: 10.1007/s00439-016-1696-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Canver M.C., Orkin S.H. Customizing the genome as therapy for the β-hemoglobinopathies. Blood. 2016;127:2536–2545. doi: 10.1182/blood-2016-01-678128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavazzana M., Antoniani C., Miccio A. Gene Therapy for β-Hemoglobinopathies. Mol. Ther. 2017;25:1142–1154. doi: 10.1016/j.ymthe.2017.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Traxler E.A., Yao Y., Wang Y.-D., Woodard K.J., Kurita R., Nakamura Y., Hughes J.R., Hardison R.C., Blobel G.A., Li C., Weiss M.J. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med. 2016;22:987–990. doi: 10.1038/nm.4170. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu X., Wang Y., Pi W., Liu H., Wickrema A., Tuan D. NF-Y recruits both transcription activator and repressor to modulate tissue- and developmental stage-specific expression of human γ-globin gene. PLoS ONE. 2012;7:e47175. doi: 10.1371/journal.pone.0047175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanabe O., Katsuoka F., Campbell A.D., Song W., Yamamoto M., Tanimoto K., Engel J.D. An embryonic/fetal β-type globin gene repressor contains a nuclear receptor TR2/TR4 heterodimer. EMBO J. 2002;21:3434–3442. doi: 10.1093/emboj/cdf340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masuda T., Wang X., Maeda M., Canver M.C., Sher F., Funnell A.P., Fisher C., Suciu M., Martyn G.E., Norton L.J. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351:285–289. doi: 10.1126/science.aad3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu N., Hargreaves V.V., Zhu Q., Kurland J.V., Hong J., Kim W., Sher F., Macias-Trevino C., Rogers J.M., Kurita R. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell. 2018;173:430–442.e17. doi: 10.1016/j.cell.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martyn G.E., Wienert B., Yang L., Shah M., Norton L.J., Burdach J., Kurita R., Nakamura Y., Pearson R.C.M., Funnell A.P.W. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet. 2018;50:498–503. doi: 10.1038/s41588-018-0085-0. [DOI] [PubMed] [Google Scholar]
- 19.Joung J.K., Sander J.D. TALENs: a widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charlesworth C.T., Deshpande P.S., Dever D.P., Dejene B., Gomez-Ospina N., Mantri S., Pavel-Dinu M., Camarena J., Weinberg K.I., Porteus M.H. Identification of Pre-Existing Adaptive Immunity to Cas9 Proteins in Humans. bioRxiv. 2018;4 [Google Scholar]
- 21.McIntosh B.E., Brown M.E., Duffin B.M., Maufort J.P., Vereide D.T., Slukvin I.I., Thomson J.A. Nonirradiated NOD,B6.SCID Il2rγ-/- Kit(W41/W41) (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Reports. 2015;4:171–180. doi: 10.1016/j.stemcr.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vinjamur D.S., Bauer D.E., Orkin S.H. Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Br. J. Haematol. 2018;180:630–643. doi: 10.1111/bjh.15038. [DOI] [PubMed] [Google Scholar]
- 23.Smith E.C., Orkin S.H. Hemoglobin genetics: recent contributions of GWAS and gene editing. Hum. Mol. Genet. 2016;25(R2):R99–R105. doi: 10.1093/hmg/ddw170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antoniani C., Meneghini V., Lattanzi A., Felix T., Romano O., Magrin E., Weber L., Pavani G., El Hoss S., Kurita R. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood. 2018;131:1960–1973. doi: 10.1182/blood-2017-10-811505. [DOI] [PubMed] [Google Scholar]
- 25.Doyon Y., Choi V.M., Xia D.F., Vo T.D., Gregory P.D., Holmes M.C. Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat. Methods. 2010;7:459–460. doi: 10.1038/nmeth.1456. [DOI] [PubMed] [Google Scholar]
- 26.Peterson C.W., Wang J., Norman K.K., Norgaard Z.K., Humbert O., Tse C.K., Yan J.J., Trimble R.G., Shivak D.A., Rebar E.J. Long-term multilineage engraftment of autologous genome-edited hematopoietic stem cells in nonhuman primates. Blood. 2016;127:2416–2426. doi: 10.1182/blood-2015-09-672337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang K.-H., Smith S.E., Sullivan T., Chen K., Zhou Q., West J.A., Liu M., Liu Y., Vieira B.F., Sun C. Long-Term Engraftment and Fetal Globin Induction upon BCL11A Gene Editing in Bone-Marrow-Derived CD34+ Hematopoietic Stem and Progenitor Cells. Mol. Ther. Methods Clin. Dev. 2017;4:137–148. doi: 10.1016/j.omtm.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Negritto M.C. Repairing Double Strand DNA Breaks. Nature Education. 2010;3:26. [Google Scholar]
- 29.Kostyrko K., Bosshard S., Urban Z., Mermod N. A role for homologous recombination proteins in cell cycle regulation. Cell Cycle. 2015;14:2853–2861. doi: 10.1080/15384101.2015.1049784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Negre O., Eggimann A.-V., Beuzard Y., Ribeil J.A., Bourget P., Borwornpinyo S., Hongeng S., Hacein-Bey S., Cavazzana M., Leboulch P., Payen E. Gene Therapy of the β-Hemoglobinopathies by Lentiviral Transfer of the β(A(T87Q))-Globin Gene. Hum. Gene Ther. 2016;27:148–165. doi: 10.1089/hum.2016.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cross R. (2017). CRISPR gene editing is coming to the clinic. Chemical & Engineering News. https://cen.acs.org/articles/95/web/2017/12/CRISPR-gene-editing-coming-clinic.html31.other31
- 32.Sangamo Therapeutics, Inc. (2017). Sangamo and Bioverativ Announce FDA Acceptance of IND Application for ST-400—a Gene-Edited Cell Therapy Candidate—to Treat Beta-Thalassemia. https://investor.sangamo.com/news-releases/news-release-details/sangamo-and-bioverativ-announce-fda-acceptance-ind-application32.other32
- 33.Williams, D. (2017). Gene Transfer for Sickle Cell Disease. ClinicalTrials.gov33. https://clinicaltrials.gov/ct2/show/NCT0328265633.web33
- 34.Brendel C., Guda S., Renella R., Bauer D.E., Canver M.C., Kim Y.J., Heeney M.M., Klatt D., Fogel J., Milsom M.D. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J. Clin. Invest. 2016;126:3868–3878. doi: 10.1172/JCI87885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khaled W.T., Choon Lee S., Stingl J., Chen X., Raza Ali H., Rueda O.M., Hadi F., Wang J., Yu Y., Chin S.F. BCL11A is a triple-negative breast cancer gene with critical functions in stem and progenitor cells. Nat. Commun. 2015;6:5987. doi: 10.1038/ncomms6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dias C., Estruch S.B., Graham S.A., McRae J., Sawiak S.J., Hurst J.A., Joss S.K., Holder S.E., Morton J.E., Turner C., DDD Study BCL11A Haploinsufficiency Causes an Intellectual Disability Syndrome and Dysregulates Transcription. Am. J. Hum. Genet. 2016;99:253–274. doi: 10.1016/j.ajhg.2016.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luc S., Huang J., McEldoon J.L., Somuncular E., Li D., Rhodes C., Mamoor S., Hou S., Xu J., Orkin S.H. Bcl11a Deficiency Leads to Hematopoietic Stem Cell Defects with an Aging-like Phenotype. Cell Rep. 2016;16:3181–3194. doi: 10.1016/j.celrep.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bauer D.E., Kamran S.C., Lessard S., Xu J., Fujiwara Y., Lin C., Shao Z., Canver M.C., Smith E.C., Pinello L. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richter M., Saydaminova K., Yumul R., Krishnan R., Liu J., Nagy E.E., Singh M., Izsvák Z., Cattaneo R., Uckert W. In vivo transduction of primitive mobilized hematopoietic stem cells after intravenous injection of integrating adenovirus vectors. Blood. 2016;128:2206–2217. doi: 10.1182/blood-2016-04-711580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boissel S., Jarjour J., Astrakhan A., Adey A., Gouble A., Duchateau P., Shendure J., Stoddard B.L., Certo M.T., Baker D., Scharenberg A.M. megaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. Nucleic Acids Res. 2014;42:2591–2601. doi: 10.1093/nar/gkt1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller J.C., Tan S., Qiao G., Barlow K.A., Wang J., Xia D.F., Meng X., Paschon D.E., Leung E., Hinkley S.J. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 42.Streubel J., Blücher C., Landgraf A., Boch J. TAL effector RVD specificities and efficiencies. Nat. Biotechnol. 2012;30:593–595. doi: 10.1038/nbt.2304. [DOI] [PubMed] [Google Scholar]
- 43.Doyle E.L., Booher N.J., Standage D.S., Voytas D.F., Brendel V.P., Vandyk J.K., Bogdanove A.J. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 2012;40(Web Server issue, W1):W117–W122. doi: 10.1093/nar/gks608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cermak T., Doyle E.L., Christian M., Wang L., Zhang Y., Schmidt C., Baller J.A., Somia N.V., Bogdanove A.J., Voytas D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fine E.J., Cradick T.J., Zhao C.L., Lin Y., Bao G. An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Res. 2014;42:e42. doi: 10.1093/nar/gkt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grier A.E., Burleigh S., Sahni J., Clough C.A., Cardot V., Choe D.C., Krutein M.C., Rawlings D.J., Jensen M.C., Scharenberg A.M., Jacoby K. pEVL: A Linear Plasmid for Generating mRNA IVT Templates With Extended Encoded Poly(A) Sequences. Mol. Ther. Nucleic Acids. 2016;5:e306. doi: 10.1038/mtna.2016.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wojda U., Noel P., Miller J.L. Fetal and adult hemoglobin production during adult erythropoiesis: coordinate expression correlates with cell proliferation. Blood. 2002;99:3005–3013. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.