

Abstract
Aminoglycosides containing a 2-deoxystreptamine core (AGs) represent a large family of antibiotics that target the ribosome. These compounds promote miscoding, inhibit translocation, and inhibit ribosome recycling. AG binding to helix h44 of the small subunit induces rearrangement of A-site nucleotides A1492 and A1493, which promotes a key open-to-closed conformational change of the subunit and thereby increases miscoding. Mechanisms by which AGs inhibit translocation and recycling remain less clear. Structural studies have revealed a secondary AG binding site in H69 of the large subunit, and it has been proposed that interaction at this site is crucial for inhibition of translocation and recycling. Here, we analyze ribosomes with mutations targeting either or both AG binding sites. Assaying translocation, we find that ablation of the h44 site increases the IC50 values for AGs dramatically, while removal of the H69 site increases these values modestly. This suggests that the AG–h44 interaction is primarily responsible for inhibition, with H69 playing a minor role. Assaying recycling, we find that mutation of h44 has no effect on AG inhibition, consistent with a primary role for AG–H69 interaction. Collectively, these findings help clarify the roles of the two AG binding sites in mechanisms of inhibition by these compounds.
Keywords: protein synthesis, ribosome, translocation, tRNA, mRNA
INTRODUCTION
Aminoglycosides are chemically diverse antibiotics widely used to treat bacterial infections (Magnet and Blanchard 2005). Most aminoglycosides are structurally similar and contain a central 2-deoxystreptamine (2-DOS) ring. These aminoglycosides (henceforth abbreviated “AGs”) fall into two subgroups: 4,5-substituted 2-DOS compounds, exemplified by neomycin (Neo) and paromomycin (Par); and 4,6-substituted 2-DOS compounds, exemplified by gentamicin (Gen) and tobramycin (Tob). These AGs bind to ribosomal RNA (rRNA) and exert multiple effects on bacterial protein synthesis. They cause mRNA miscoding (Davies et al. 1965, 1966), inhibit mRNA-tRNA translocation (Cabañas et al. 1978; Misumi et al. 1978), and inhibit ribosome recycling (Hirokawa et al. 2002).
The primary binding site of AGs lies in helix 44 (h44) of the 16S rRNA, near the A site of the 30S subunit. Previous studies have shown that single mutations in h44 (e.g., A1408G, G1491U, or U1495A) are sufficient to confer high-level resistance to various AGs in vivo (Recht and Puglisi 2001; Hobbie et al. 2005, 2006a,b; Nessar et al. 2011; Salian et al. 2012; Perez-Fernandez et al. 2014). These mutations disrupt key interactions between helix h44 and functional groups of ring I and/or II, thereby reducing AG affinity.
AGs stabilize tRNA in the A site, and structural studies provide an explanation for this activity (Carter et al. 2000; Ogle et al. 2001, 2002, 2003; Peske et al. 2004; Walker and Fredrick 2006). Co-crystal structures have shown that Neo, Par, and Gen bind h44 in a similar way (Fig. 1; Carter et al. 2000; Ogle et al. 2001; Borovinskaya et al. 2007a). Ring I intercalates into h44, stacking on G1491 and forming hydrogen bonds with A1408. This occludes A1492 and A1493 from within h44, inducing a “flipped out” conformation of these nucleotides. Similar conformational changes of A1492 and A1493 normally occur upon codon–anticodon pairing in the A site. Thus, AG binding to h44 reduces the energy barrier of this rearrangement, which promotes A-site tRNA (A-tRNA) binding and decoding errors (Ogle et al. 2003; Satpati et al. 2014; Zhang et al. 2018). By stabilizing A-tRNA, this rearrangement also inhibits translocation, because A-tRNA cannot readily move to the P site (Peske et al. 2004; Shoji et al. 2006).
FIGURE 1.
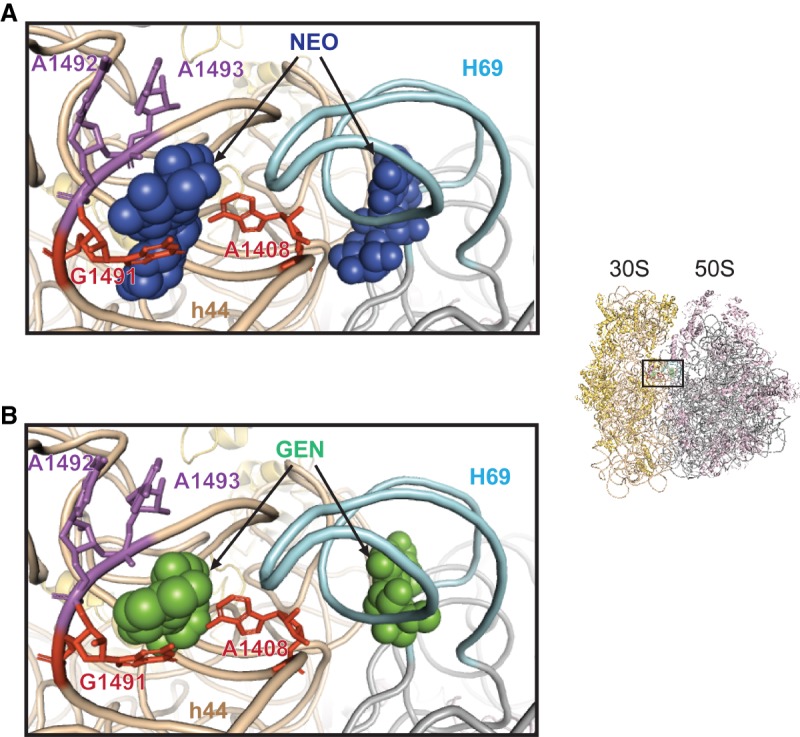
2-DOS aminoglycosides bind h44 and H69 of the ribosome in a similar way. Co-crystal structures with Neo (A) and Gen (B) are shown as examples. Thumbnail on the right shows the perspective of view. 16S rRNA is colored beige, with key A-site nt A1492 and A1492 (violet) and h44 nt A1408 and G1491 (red) highlighted. 23S rRNA is colored gray, with H69 highlighted in light cyan. Aminoglycosides (as indicated) are shown as space-filled models. Images were made using PDB entries 4V52 and 4V53.
The X-ray crystallography studies revealed that AGs bind not only h44 but also helix H69 of the 50S subunit (Fig. 1; Borovinskaya et al. 2007a). H69 contacts a number of 16S rRNA nucleotides, including nt 1406–1409 and 1494–1495 of h44, forming intersubunit bridge B2a/d (Liu and Fredrick 2016). This bridge plays an important role in translation initiation (Liu and Fredrick 2015) and needs to be disrupted during the subunit-splitting step of ribosome recycling. Co-crystal structures showed that ribosome recycling factor (RRF) displaces H69 from its normal position, disrupting B2a/d (Borovinskaya et al. 2007a). Binding of AG to the major groove of H69 reverses these effects of RRF, presumably by stabilizing bridge B2a/d. This may explain the mechanism by which AGs inhibit ribosome recycling (Borovinskaya et al. 2007a).
It has been suggested that AG–H69 interaction also plays a key role in the inhibition of translocation. In single-molecule studies, Blanchard and coworkers found that an EF-G-dependent conformational change was inhibited by Neo, and mutation A1408G of h44 had no bearing on the observed inhibition (Wang et al. 2012). At the time, they attributed to this conformational change to translocation and proposed that, by binding H69, Neo stabilizes a partially rotated conformation of the ribosome and thereby blocks translocation. However, this model is difficult to reconcile with the fact that the mutation A1408G confers high-level resistance to Neo in cell growth assays (MIC > 800 µM) (Recht and Puglisi 2001; Hobbie et al. 2006b).
To clarify the contribution of the two AG binding sites, we examined reassociated mutant ribosomes with either or both sites ablated. We find that the inhibition of translocation is primarily due to AG–h44 interaction, whereas the inhibition of ribosome recycling is independent of the h44 site.
RESULTS
Inhibition of translocation by aminoglycosides depends largely on the h44 site
To determine the contribution of h44 and H69 binding sites to AG activities, we targeted each by mutagenesis. To abrogate the h44 site, we introduced A1408G or dual A1408G/G1491U substitutions. Mutation A1408G and G1491U each confers a distinct spectrum of AG resistance (Hobbie et al. 2006a). Cells carrying both mutations grew normally and were highly resistant to multiple AGs in vivo, consistent with earlier studies (Hobbie et al. 2006b). To eliminate the H69 site, we used mutation ΔH69, which replaces the entire helix with a single nucleotide (Ali et al. 2006; Liu and Fredrick 2015). 30S subunits were purified from an E. coli Δ7 prrn strain, using conventional sucrose-gradient sedimentation (Qin et al. 2007), while aptamer-tagged 50S subunits were purified using affinity chromatography, as previously described (Youngman and Green 2005). 70S ribosomes were then formed by reassociation of control and/or mutant subunits, yielding ribosomes with no, one, or both AG binding sites mutated. Previous work has shown that rates of translocation in tagged and reassociated control ribosomes and in wild-type tight-couple ribosomes are comparable (Walker et al. 2008; Devaraj et al. 2009; Liu and Fredrick 2013).
It is well known that AGs can completely inhibit translocation (Cabañas et al. 1978; Fredrick and Noller 2003; Studer et al. 2003; Shoji et al. 2006). Accordingly, we measured the effects of various AGs on the extent of translocation by toeprinting, which tracks mRNA position in the ribosome (Hartz et al. 1989; Fredrick and Noller 2002; McGarry et al. 2005; Shoji et al. 2006). Complexes containing message m291 (Shoji et al. 2006), tRNAfMet in the P site, and N-acetyl-Phe-tRNAPhe in the A site were made and incubated with various concentrations of AG. EF-G and GTP were then added, and the extent of translocation was determined (Fig. 2). In the absence of AG, the fraction of post-translocation (POST) state ribosomes observed was ∼0.5, somewhat lower than the value expected (∼0.8) from earlier studies involving tight-couple ribosomes (Fredrick and Noller 2003; Shoji et al. 2006; Balakrishnan et al. 2014). This could be due to partial A-site occupancy, a portion of 30S complexes, and/or a portion of inactive 50S subunits. The extent of translocation decreased as a function of AG concentration. Neo strongly inhibited translocation in control ribosomes, with an IC50 value of 0.4 µM (Table 1; Fig. 2C). The dual A1408G/G1491U mutation of h44 increased the IC50 by 100-fold. The activity of Par, another 4,5-substituted 2-DOS aminoglycoside, was also substantially weakened by mutation A1408G/G1491U (Table 1; Fig. 2D). The 4,6-substituted 2-DOS AGs Gen and Tob were also investigated. Mutation A1408G/G1491U completely relieved the inhibition by Gen (Fig. 2E) and increased the IC50 of Tob by >30-fold (Table 1; Fig. 2F). We also compared the effects of the single A1408G mutation, which Wang et al. (2012) used, on Neo and Tob inhibition (Table 1; Fig. 2C,F). In both cases, the degree of resistance conferred by A1408G was similar to that conferred by the dual A1408G/G1491U mutation. These strong effects of A1408G and A1408G/G1491U indicate that AG interaction with h44 is critical for the inhibition of translocation.
FIGURE 2.

Inhibition of EF-G-dependent translocation by aminoglycosides. (A) Sequence of model mRNA, m291, with the relevant codons indicated. The first nucleotide of codon 1 (defined as position +1) is shown in bold text and the Shine–Dalgarno (SD) sequence is underscored. (B) Example of a toeprinting experiment that measures AG inhibition of translocation. The pretranslocation complex (PRE) was formed by incubating m291-programmed ribosomes with tRNAfMet to bind the P site and N-acetyl-Phe-tRNAPhe to bind the A site. The complex was further incubated in the absence (lanes 1,2) or presence of AG (lanes 3–8, various concentrations), and then EF-G and GTP were added to promote translocation (lanes 2–8). The position of mRNA was then mapped in each case to quantify the fraction of ribosomes in the post-translocation (POST) state. (C,D) Shown is the extent of translocation (Fraction POST) in control and mutant ribosomes (as indicated, see KEY) plotted against the concentration of Neo (C), Par (D), Gen (E), and Tob (F). Data were fit to a modified dose response equation to obtain IC50 values (Table 1).
TABLE 1.
AG inhibition of EF-G-dependent translocation in control and mutant ribosomes
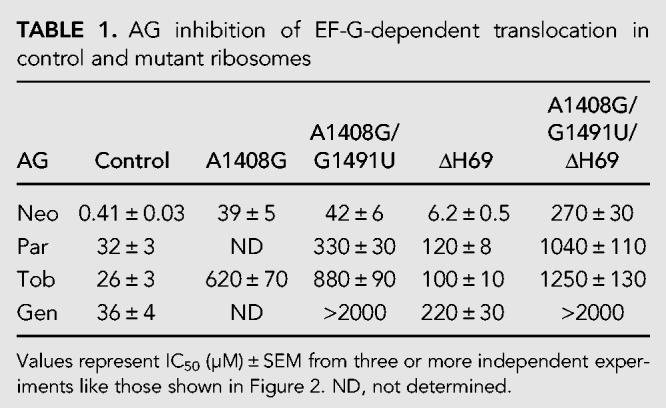
Removal of H69 had a smaller impact on AG inhibition of translocation. Mutation ΔH69 increased IC50 for Neo, Par, Gen, and Tob by about 15-, four-, four-, and sixfold, respectively (Table 1; Fig. 2C–F). We also compared the behavior of ribosomes with both h44 and H69 sites abrogated. This triple A1408G/G1491U/ΔH69 mutation conferred the highest resistance to Neo and Par (Table 1; Fig. 2C,D). Removal of H69 from A1408G/G1491U ribosomes increased the IC50 for Neo and Par by sixfold and threefold, respectively, effects similar to those of ΔH69 in ribosomes with the h44 site intact. The simplest interpretation of these data is that, for 4,5-substituted 2-DOS compounds, AG–H69 interaction contributes to inhibition through a mechanism that is largely independent of h44. In the case of the 4,6-substituted 2-DOS compound Tob, the triple mutant ribosomes were essentially no more resistant than the A1408G/G1491U ribosomes (Table 1; Fig. 2F). This differs from Neo and Par cases and suggests that the observed effect of ΔH69 on inhibition depends on Tob–h44 interaction. For Gen, the dual h44 mutation A1408G/G1491U conferred complete resistance (IC50 > 2000 µM), in the presence or absence of H69 (Table 1; Fig. 2E).
We also analyzed inhibition of translocation by Neo in control and mutant ribosomes in polymix buffer (Ehrenberg et al. 1990), conditions analogous to those of Wang et al. (2012). The results obtained (Fig. 3) were qualitatively similar to those described above (Table 1; Fig. 2C), providing further evidence that Neo–h44 interaction is primarily responsible for the inhibition of translocation.
FIGURE 3.
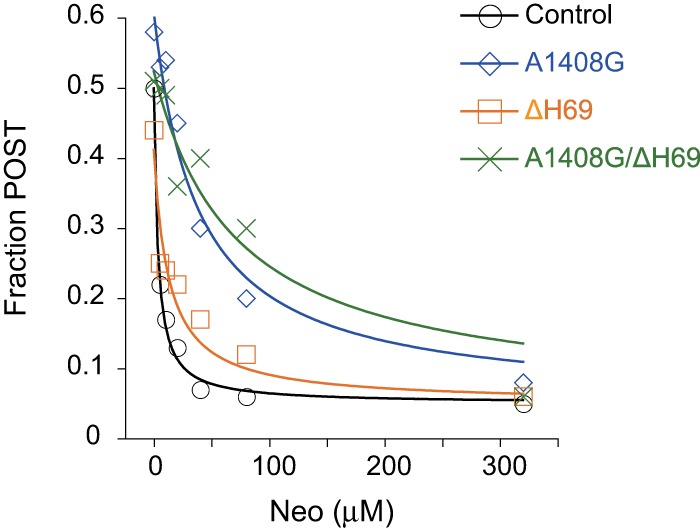
Inhibition of EF-G-dependent translocation by Neo in polymix buffer. Shown is the extent of translocation (Fraction POST) in control and mutant ribosomes (as indicated) plotted against the concentration of Neo. Data were fit to a modified dose response equation, yielding the following micromolar IC50 values (mean ± SEM, n ≥ 3): Control, 3 ± 0.4; A1408G, 45 ± 10; ΔH69, 15 ± 3; A1408G/ΔH69, 130 ± 30.
The ability of Neo to promote spontaneous reverse translocation is eliminated by h44 mutations
It has been shown previously that Neo, Par, and Gen can similarly increase the rate of spontaneous reverse translocation (Shoji et al. 2006; Borovinskaya et al. 2008). While the basis of this activity remains unclear, it has been proposed that these AGs shift the microscopic equilibrium of tRNA binding in the transition-state complex (also known as “unlocked” state), thereby enhancing the reaction rate (Shoji et al. 2009b). To determine whether this AG activity involves h44, we measured spontaneous reverse translocation in control and mutant ribosomes (Fig. 4), as detailed previously (Shoji et al. 2006; Borovinskaya et al. 2008). E-site tRNAfMet was added to m292-programmed ribosomes containing N-acetyl-Val-tRNAVal in the P site and the fraction of ribosomes in the POST state was monitored as a function of time. In line with previous observations (Shoji et al. 2006; Borovinskaya et al. 2008), Neo increased kobs by approximately threefold in control ribosomes. In ribosomes harboring either A1408G or G1491U, the rate was virtually unaffected by Neo (Fig. 4). These data clearly indicate that h44 binding is critical for the ability of Neo to speed reverse translocation. Although there was no EF-G in these reactions, the spontaneous reaction is fundamentally related to EF-G-dependent translocation (Fredrick and Noller 2003; Shoji et al. 2006, 2009b; Fischer et al. 2010; Liu and Fredrick 2013). Hence, these results further implicate AG–h44 interaction in the mechanism of translocation inhibition.
FIGURE 4.

The ability of neomycin to stimulate spontaneous reverse translocation depends on the h44 site. Reverse translocation in control (A), A1408G (B), or G1491U (C) ribosomes was measured in the absence and presence of Neo (as indicated). The fraction of ribosomes in the POST state was plotted versus time, and the data were fit to a single exponential function to obtain the observed rate at 0, 0.5, and 3 µM Neo (respectively): Control, kobs = 0.16, 0.54, 0.60 min−1; A1408G, kobs = 0.15, 0.15, 0.15 min−1; G1491U, kobs = 0.15, 0.14, 0.20 min−1.
Inhibition of ribosome recycling by aminoglycosides
Kaji and coworkers showed that RRF/EF-G-dependent ribosome recycling could be strongly inhibited by AGs (Hirokawa et al. 2002), but the basis of this activity has remained unclear. Structural studies have shown that RRF binds the large subunit and causes H69 to swing away from the subunit interface, breaking bridge B2a/d (Borovinskaya et al. 2007a). The additional presence of Gen or Par completely suppressed this RRF-induced movement of H69. It was proposed that AG binding to H69 restricts the dynamics of the helix and stabilizes B2a/d, thereby inhibiting the subunit-splitting step of recycling (Borovinskaya et al. 2007a). Yet, single mutations in h44 confer high-level AG resistance (Recht and Puglisi 2001; Hobbie et al. 2005, 2006a,b; Nessar et al. 2011). The corresponding nucleotides (A1408, G1491, U1495, C1409, and U1406) lie close to the intersubunit bridge B2a/d, raising the alternative possibility that AG–h44 interaction is involved in recycling inhibition.
To test the role of the h44 site in recycling inhibition, we used a filter-binding method to assay recycling at various concentrations of AGs. Model post-termination complexes containing radiolabeled tRNAPhe in the P site were formed, and the tRNA dissociation rate (koff) was measured in the absence and presence of RRF, EF-G (GTP), and IF3. The factors stimulated the rate of tRNA release by an order of magnitude, an effect that required all three factors (Fig. 5A). Although quite slow, the observed rate of this reaction is comparable to rates seen previously for analogous POST complexes in which SD–ASD pairing occurs (Peske et al. 2005; Chen et al. 2017). As expected, Neo and Tob inhibited recycling of the control ribosome, with IC50 values of 1.9 ± 0.3 µM and 23 ± 3 µM, respectively (Fig. 5B,D,E). Ablation of the h44 site (via mutation A1408G or dual mutation A1408G/G1491U) had virtually no effect on recycling inhibition (Fig. 5C–E). If anything, complexes formed with the mutant ribosomes exhibited slightly smaller IC50 values (Neo IC50 = 1.5 ± 0.2, 1.3 ± 0.2 µM; Tob IC50 = 20 ± 2, 18 ± 2 µM). These data indicate that AG–h44 interaction plays no role in the inhibition of recycling, as measured in this in vitro assay. The effect of ΔH69 could not be investigated because the mutation causes recycling to become independent of RRF (Ali et al. 2006), an observation that we confirmed.
FIGURE 5.

Inhibition of ribosome recycling by AGs. (A) An in vitro assay for ribosome recycling. Dissociation of deacyl 3′-[32P]-tRNAPhe from the P site of m291-programmed ribosomes was measured in the absence or presence of various factors (as indicated). Stimulation of tRNA release required RRF, EF-G (GTP), and IF3, as reactions with single omissions showed no apparent catalysis. (B,C) Examples of experiments measuring the effect of Neo (various concentrations, as indicated) on tRNA release from control (B) or A1408G/G1491U (C) ribosomes in the presence of RRF, EF-G (GTP), and IF3. Fraction of radiolabeled tRNA bound was plotted against time, and the data were fit to a single exponential equation to obtain koff. (D,E) koff values were plotted as a function of concentration of Neo (D) and Tob (E). Data were fit to a modified dose response equation to generate IC50 values for control, A1408G, and A1408G/G1491U cases, respectively, as follows: Neo IC50 = 1.9 ± 0.3, 1.5 ± 0.2, 1.3 ± 0.2 µM; Tob IC50 = 23 ± 3, 20 ± 2, 18 ± 2 µM.
DISCUSSION
Our findings help clarify the roles of two AG binding sites in the inhibition of protein synthesis. We demonstrate that AG interaction with the h44 site is primarily responsible for inhibition of translocation. This is consistent with the fact that single mutations in the h44 are sufficient to confer high-level resistance to AGs in vivo (Recht and Puglisi 2001; Hobbie et al. 2005, 2006a,b; Nessar et al. 2011) and challenges an earlier conclusion of Blanchard and coworkers (Wang et al. 2012). Monitoring a fluorescence change in single-molecule studies (Munro et al. 2010), they reported no difference in AG-inhibition between control and A1408G ribosomes and proposed that AG–H69 interaction was key to translocation inhibition (Wang et al. 2012). Translocation is a complex process involving multiple conformational changes of the ribosome, tRNAs, and EF-G. Our suspicion is that their readout—an EF-G-dependent increase in Cy3-tRNA fluorescence intensity—corresponds to another event, related to translocation but distinct from codon–anticodon movement. Indeed, more recent work from the Blanchard laboratory, in which the motions of translocation were followed from multiple perspectives, suggests that codon–anticodon movement with respect to the 30S subunit occurs at a later stage in the process (Wasserman et al. 2016).
The ability of AGs to inhibit translocation is reduced to some degree by removal of H69. The simplest interpretation of these data is that AG–H69 interaction does contribute, albeit modestly, to translocation inhibition. Deletion ΔH69 similarly increases the IC50 for Neo and Par, regardless of whether the primary h44 site is mutated. Such cumulative effects of ΔH69 and A1408G/G1491U suggest that for example Par-H69 and Par-h44 interactions act in largely independent ways, each contributing to translocation inhibition. In contrast, ΔH69 fails to increase the IC50 for Tob when the primary h44 site is ablated. This indicates a difference in activity between the 4,6-linked AG Tob and the 4,5-linked AGs Neo and Par, regarding H69. Interestingly, it has been shown that Neo alters the dynamics of the ribosome, stabilizing a semi-rotated conformation (Wang et al. 2012). This activity, attributed to H69 binding, appears to be specific to the 4,5-linked AGs. Based on these collective observations, we propose that binding of 4,5-linked AGs to H69 stabilizes an intermediate of the process (Wang et al. 2012; Wasserman et al. 2016), which contributes to inhibition.
Another possibility is that the effects of ΔH69 are indirect, and AG–H69 interaction plays no role in translocation inhibition. Deletion ΔH69 removes the secondary AG binding site, of course, but also disrupts bridge B2a/d, which could weaken AG–h44 interaction and/or its inhibitory effects. Although we introduced two AG resistance mutations in an effort to eliminate the h44 site, low-affinity AG binding to h44 may still occur in A1408G/G1491U ribosomes. If so, ΔH69 could indirectly weaken AG–h44 interaction in both control and A1408G/G1491U ribosomes, thereby increasing the IC50 in multiple contexts as observed. Additional experiments will be needed to distinguish whether AG–H69 interaction plays a minor role or no role in the inhibition of translocation.
Based on structural studies, it has been proposed that AG binding to H69 is responsible for the inhibition of recycling (Borovinskaya et al. 2007a). Our data are consistent with this model, as recycling inhibition appears to be completely independent of h44. Yet, as mentioned above, single nucleotide substitutions in h44 are sufficient to confer high-level AG resistance in vivo. This implies that AG inhibition of recycling either (i) occurs with little physiological consequence or (ii) is countered somehow in the cell. The latter scenario seems more likely, given that RRF is essential for cell viability (Janosi et al. 1994, 1998; Baba et al. 2006).
MATERIALS AND METHODS
Subunit purification and reassociation
Mutations A1408G and G1491T were introduced into plasmid p278MS2 (Youngman and Green 2005), using the QuikChange (Stratagene) method. Plasmid p278MS2 and its derivatives (carrying nonlethal mutations) were moved into E. coli Δ7 prrn strain SQZ10, replacing the resident plasmid. From these strains, 30S subunits (control or mutant) were purified, using conventional sucrose-gradient sedimentation as previously described (Qin et al. 2007). Mutation ΔH69 has been described previously (Ali et al. 2006), and aptamer-tagged 50S subunits (control and mutant) were purified using affinity chromatography as previously described (Youngman and Green 2005; Liu and Fredrick 2013, 2015).
Reassociated 70S ribosomes were formed from purified subunits (Shoji et al. 2009a; Liu and Fredrick 2013). 30S subunits (control or mutant) were heat activated in the presence of 20 mM Mg2+ at 42°C for 20 min and then mixed with an equal amount of 50S subunits (control or mutant) and further incubated at 37°C for 20 min.
Factors, tRNAs, and other reagents
EF-G-His6, IF3-His6, and RRF were purified as previously described (MacDougall et al. 1997; Dallas and Noller 2001; Fredrick and Noller 2002). Purified tRNAfMet, tRNAPhe, and tRNAVal were purchased from Chemical Block, charged, and acetylated as previously described (Walker and Fredrick 2008). Messenger RNA was made by in vitro transcription and gel purified (Fredrick and Noller 2002). AGs were purchased from Sigma-Aldrich or Fisher Scientific.
Translocation assays
EF-G-dependent translocation was measured by toeprinting (Shoji et al. 2006; Fosso et al. 2015). Briefly, pretranslocation (PRE) complexes (0.5 µM; final concentrations indicated throughout) containing deacylated tRNAfMet in the P site and N-acetyl-Phe-tRNAPhe in the A site were formed at 37°C in buffer A (10 mM Tris-HCl pH 8.0, 10 mM MgCl2, 65 mM NH4Cl, 1 mM DTT). AG (variable concentration) was added, followed 10 min later by EF-G (1 µM) and GTP (0.3 mM). Under single-turnover conditions like these, the rate of translocation is too fast to measure manually (Pan et al. 2007; Walker et al. 2008; Liu and Fredrick 2013; Holtkamp et al. 2014). After 5 min, an end point chosen merely for practical reasons, complexes were subjected to primer extension analysis. Toeprints were quantified and the extent of translocation determined, as described previously (Shoji et al. 2006). The fraction of ribosomes in the POST state (F) were plotted against the AG concentration (A), using KaleidaGraph (Synergy Software), and the data were fit to the modified dose response equation
where F0 represents F prior to EF-G and GTP addition (set to 0.05 based on control experiments), Fmax represents the maximal F value, B represents IC50, and C represents the Hill coefficient. Initial data fitting showed that the Hill coefficient fell between 0.8 and 1.2, and hence C was set to 1.0 to ensure uniform comparisons.
Spontaneous reverse translocation was measured as detailed previously (Shoji et al. 2006; Borovinskaya et al. 2007b, 2008; Liu and Fredrick 2013). Briefly, m292-programmed ribosomes (0.5 µM) containing N-acetyl-Val-tRNAVal in the P site were incubated in polymix buffer (5 mM potassium phosphate pH 7.3, 95 mM KCl, 5 mM NH4Cl, 0.5 mM CaCl2, 5 mM magnesium acetate, 8 mM putrescine, 1 mM spermidine, 1 mM DTT) with various concentrations of Neo at 37°C for 10 min. Reverse translocation was initiated by adding deacylated tRNAfMet (8 µM) at time t = 0. Aliquots were removed at various time points and subjected to primer extension analysis. Fraction POST was quantified and plotted as a function of time. Data were fit to a single exponential function to obtain the observed rate and amplitude of the reaction.
Ribosome recycling assay
Ribosome recycling was measured using a double-membrane filtration method (Fahlman and Uhlenbeck 2004). Typically, ∼50 nM [3′-32P]-tRNAPhe, m291 (1.5 µM) and ribosomes (1 µM) were incubated at 37°C in buffer A for 20 min to bind the P site. At t = 0, a portion of the reaction (2 µL) was diluted 100-fold in buffer containing excess unlabeled tRNAPhe (0.2 µM) and GTP (0.3 mM), with or without EF-G (0.3 µM), RRF (0.3 µM), IF3 (0.3 µM), and AG (variable concentration) as specified. Aliquots (20 µL) were then filtered through a bilayer of nitrocellulose (NitroBind, GE Healthcare) and positively charged nylon (Hybond-N+) membranes (GE Healthcare) at various time points. Ribosome-bound and free tRNA were captured by the top and bottom membranes, respectively (Fahlman and Uhlenbeck 2004; Shoji et al. 2009a). Membranes were immediately washed with 150 µL of buffer, dried, and exposed to a phosphor screen (GE Healthcare). The fraction of tRNA bound was quantified and plotted against time. Dissociation rate (koff) was determined by fitting the data to a single exponential function. Then, koff was plotted as a function of AG concentration, and data were fit to a dose response equation analogous to the one described above, yielding the IC50 value.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (grant no. GM072528).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.068460.118.
REFERENCES
- Ali IK, Lancaster L, Feinberg J, Joseph S, Noller HF. 2006. Deletion of a conserved, central ribosomal intersubunit RNA bridge. Mol Cell 23: 865–874. 10.1016/j.molcel.2006.08.011 [DOI] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2: 2006.0008 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan R, Oman K, Shoji S, Bundschuh R, Fredrick K. 2014. The conserved GTPase LepA contributes mainly to translation initiation in Escherichia coli. Nucleic Acids Res 42: 13370–13383. 10.1093/nar/gku1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovinskaya MA, Pai RD, Zhang W, Schuwirth BS, Holton JM, Hirokawa G, Kaji H, Kaji A, Cate JH. 2007a. Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat Struct Mol Biol 14: 727–732. 10.1038/nsmb1271 [DOI] [PubMed] [Google Scholar]
- Borovinskaya MA, Shoji S, Holton JM, Fredrick K, Cate JHD. 2007b. A steric block in translation caused by the antibiotic spectinomycin. ACS Chem Biol 2: 545–552. 10.1021/cb700100n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovinskaya MA, Shoji S, Fredrick K, Cate JH. 2008. Structural basis for hygromycin B inhibition of protein biosynthesis. RNA 14: 1590–1599. 10.1261/rna.1076908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabañas MJ, Vázquez D, Modolell J. 1978. Inhibition of ribosomal translocation by aminoglycoside antibiotics. Biochem Biophys Res Commun 83: 991–997. 10.1016/0006-291X(78)91493-6 [DOI] [PubMed] [Google Scholar]
- Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. 2000. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 407: 340–348. 10.1038/35030019 [DOI] [PubMed] [Google Scholar]
- Chen Y, Kaji A, Kaji H, Cooperman BS. 2017. The kinetic mechanism of bacterial ribosome recycling. Nucleic Acids Res 45: 10168–10177. 10.1093/nar/gkx694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas A, Noller HF. 2001. Interaction of translation initiation factor 3 with the 30S ribosomal subunit. Mol Cell 8: 855–864. 10.1016/S1097-2765(01)00356-2 [DOI] [PubMed] [Google Scholar]
- Davies J, Gorini L, Davis BD. 1965. Misreading of RNA codewords induced by aminoglycoside antibiotics. Mol Pharmacol 1: 93–106. [PubMed] [Google Scholar]
- Davies J, Jones DS, Khorana HG. 1966. A further study of misreading of codons induced by streptomycin and neomycin using ribopolynucleotides containing two nucleotides in alternating sequence as templates. J Mol Biol 18: 48–57. 10.1016/S0022-2836(66)80075-X [DOI] [PubMed] [Google Scholar]
- Devaraj A, Shoji S, Holbrook ED, Fredrick K. 2009. A role for the 30S subunit E site in maintenance of the translational reading frame. RNA 15: 255–265. 10.1261/rna.1320109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenberg M, Bilgin N, Kurland CG. 1990. Design and use of a fast and accurate in vitro translation system. In Ribosomes and protein synthesis—a practical approach (ed. Spedding G), pp. 101–129. IRL Press, Oxford, UK. [Google Scholar]
- Fahlman RP, Uhlenbeck OC. 2004. Contribution of the esterified amino acid to the binding of aminoacylated tRNAs to the ribosomal P- and A-sites. Biochemistry 43: 7575–7583. 10.1021/bi0495836 [DOI] [PubMed] [Google Scholar]
- Fischer N, Konevega AL, Wintermeyer W, Rodnina MV, Stark H. 2010. Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. Nature 466: 329–333. 10.1038/nature09206 [DOI] [PubMed] [Google Scholar]
- Fosso MY, Zhu H, Green KD, Garneau-Tsodikova S, Fredrick K. 2015. Tobramycin variants with enhanced ribosome-targeting activity. Chembiochem 16: 1565–1570. 10.1002/cbic.201500256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredrick K, Noller HF. 2002. Accurate translocation of mRNA by the ribosome requires a peptidyl group or its analog on the tRNA moving into the 30S P site. Mol Cell 9: 1125–1131. 10.1016/S1097-2765(02)00523-3 [DOI] [PubMed] [Google Scholar]
- Fredrick K, Noller HF. 2003. Catalysis of ribosomal translocation by sparsomycin. Science 300: 1159–1162. 10.1126/science.1084571 [DOI] [PubMed] [Google Scholar]
- Hartz D, McPheeters DS, Gold L. 1989. Selection of the initiator tRNA by Escherichia coli initiation factors. Genes Dev 3: 1899–1912. 10.1101/gad.3.12a.1899 [DOI] [PubMed] [Google Scholar]
- Hirokawa G, Kiel MC, Muto A, Selmer M, Raj VS, Liljas A, Igarashi K, Kaji H, Kaji A. 2002. Post-termination complex disassembly by ribosome recycling factor, a functional tRNA mimic. EMBO J 21: 2272–2281. 10.1093/emboj/21.9.2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbie SN, Pfister P, Brüll C, Westhof E, Böttger EC. 2005. Analysis of the contribution of individual substituents in 4,6-aminoglycoside-ribosome interaction. Antimicrob Agents Chemother 49: 5112–5118. 10.1128/AAC.49.12.5112-5118.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbie SN, Bruell C, Kalapala S, Akshay S, Schmidt S, Pfister P, Böttger EC. 2006a. A genetic model to investigate drug-target interactions at the ribosomal decoding site. Biochimie 88: 1033–1043. 10.1016/j.biochi.2006.04.008 [DOI] [PubMed] [Google Scholar]
- Hobbie SN, Pfister P, Bruell C, Sander P, Francois B, Westhof E, Böttger EC. 2006b. Binding of neomycin-class aminoglycoside antibiotics to mutant ribosomes with alterations in the A site of 16S rRNA. Antimicrob Agents Chemother 50: 1489–1496. 10.1128/AAC.50.4.1489-1496.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtkamp W, Cunha CE, Peske F, Konevega AL, Wintermeyer W, Rodnina MV. 2014. GTP hydrolysis by EF-G synchronizes tRNA movement on small and large ribosomal subunits. EMBO J 33: 1073–1085. 10.1002/embj.201387465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janosi L, Shimizu I, Kaji A. 1994. Ribosome recycling factor (ribosome releasing factor) is essential for bacterial growth. Proc Natl Acad Sci 91: 4249–4253. 10.1073/pnas.91.10.4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janosi L, Mottagui-Tabar S, Isaksson LA, Sekine Y, Ohtsubo E, Zhang S, Goon S, Nelken S, Shuda M, Kaji A. 1998. Evidence for in vivo ribosome recycling, the fourth step in protein biosynthesis. EMBO J 17: 1141–1151. 10.1093/emboj/17.4.1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Fredrick K. 2013. Contribution of intersubunit bridges to the energy barrier of ribosomal translocation. Nucleic Acids Res 41: 565–574. 10.1093/nar/gks1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Fredrick K. 2015. Roles of helix H69 of 23S rRNA in translation initiation. Proc Natl Acad Sci 112: 11559–11564. 10.1073/pnas.1507703112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Fredrick K. 2016. Intersubunit bridges of the bacterial ribosome. J Mol Biol 428: 2146–2164. 10.1016/j.jmb.2016.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougall J, Holst-Hansen P, Mortensen KK, Freistroffer DV, Pavlov MY, Ehrenberg M, Buckingham RH. 1997. Purification of active Escherichia coli ribosome recycling factor (RRF) from an osmo-regulated expression system. Biochimie 79: 243–246. 10.1016/S0300-9084(97)83511-0 [DOI] [PubMed] [Google Scholar]
- Magnet S, Blanchard JS. 2005. Molecular insights into aminoglycoside action and resistance. Chem Rev 105: 477–498. 10.1021/cr0301088 [DOI] [PubMed] [Google Scholar]
- McGarry KG, Walker SE, Wang H, Fredrick K. 2005. Destabilization of the P site codon-anticodon helix results from movement of tRNA into the P/E hybrid state within the ribosome. Mol Cell 20: 613–622. 10.1016/j.molcel.2005.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misumi M, Nishimura T, Komai T, Tanaka N. 1978. Interaction of kanamycin and related antibiotics with the large subunit of ribosomes and the inhibition of translocation. Biochem Biophys Res Commun 84: 358–365. 10.1016/0006-291X(78)90178-X [DOI] [PubMed] [Google Scholar]
- Munro JB, Wasserman MR, Altman RB, Wang L, Blanchard SC. 2010. Correlated conformational events in EF-G and the ribosome regulate translocation. Nat Struct Mol Biol 17: 1470–1477. 10.1038/nsmb.1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nessar R, Reyrat JM, Murray A, Gicquel B. 2011. Genetic analysis of new 16S rRNA mutations conferring aminoglycoside resistance in Mycobacterium abscessus. J Antimicrob Chemother 66: 1719–1724. 10.1093/jac/dkr209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogle JM, Brodersen DE, Clemons WM Jr, Tarry MJ, Carter AP, Ramakrishnan V. 2001. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 292: 897–902. 10.1126/science.1060612 [DOI] [PubMed] [Google Scholar]
- Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. 2002. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111: 721–732. 10.1016/S0092-8674(02)01086-3 [DOI] [PubMed] [Google Scholar]
- Ogle JM, Carter AP, Ramakrishnan V. 2003. Insights into the decoding mechanism from recent ribosome structures. Trends Biochem Sci 28: 259–266. 10.1016/S0968-0004(03)00066-5 [DOI] [PubMed] [Google Scholar]
- Pan D, Kirillov SV, Cooperman BS. 2007. Kinetically competent intermediates in the translocation step of protein synthesis. Mol Cell 25: 519–529. 10.1016/j.molcel.2007.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Fernandez D, Shcherbakov D, Matt T, Leong NC, Kudyba I, Duscha S, Boukari H, Patak R, Dubbaka SR, Lang K, et al. 2014. 4′-O-substitutions determine selectivity of aminoglycoside antibiotics. Nat Commun 5: 3112 10.1038/ncomms4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peske F, Savelsbergh A, Katunin VI, Rodnina MV, Wintermeyer W. 2004. Conformational changes of the small ribosomal subunit during elongation factor G-dependent tRNA–mRNA translocation. J Mol Biol 343: 1183–1194. 10.1016/j.jmb.2004.08.097 [DOI] [PubMed] [Google Scholar]
- Peske F, Rodnina MV, Wintermeyer W. 2005. Sequence of steps in ribosome recycling as defined by kinetic analysis. Mol Cell 18: 403–412. 10.1016/j.molcel.2005.04.009 [DOI] [PubMed] [Google Scholar]
- Qin D, Abdi NM, Fredrick K. 2007. Characterization of 16S rRNA mutations that decrease the fidelity of translation initiation. RNA 13: 2348–2355. 10.1261/rna.715307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recht MI, Puglisi JD. 2001. Aminoglycoside resistance with homogeneous and heterogeneous populations of antibiotic-resistant ribosomes. Antimicrob Agents Chemother 45: 2414–2419. 10.1128/AAC.45.9.2414-2419.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salian S, Matt T, Akbergenov R, Harish S, Meyer M, Duscha S, Shcherbakov D, Bernet BB, Vasella A, Westhof E, et al. 2012. Structure-activity relationships among the kanamycin aminoglycosides: role of ring I hydroxyl and amino groups. Antimicrob Agents Chemother 56: 6104–6108. 10.1128/AAC.01326-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpati P, Sund J, Aqvist J. 2014. Structure-based energetics of mRNA decoding on the ribosome. Biochemistry 53: 1714–1722. 10.1021/bi5000355 [DOI] [PubMed] [Google Scholar]
- Shoji S, Walker SE, Fredrick K. 2006. Reverse translocation of tRNA in the ribosome. Mol Cell 24: 931–942. 10.1016/j.molcel.2006.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji S, Abdi NM, Bundschuh R, Fredrick K. 2009a. Contribution of ribosomal residues to P-site tRNA binding. Nucleic Acids Res 37: 4033–4042. 10.1093/nar/gkp296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji S, Walker SE, Fredrick K. 2009b. Ribosomal translocation: one step closer to the molecular mechanism. ACS Chem Biol 4: 93–107. 10.1021/cb8002946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer SM, Feinberg JS, Joseph S. 2003. Rapid kinetic analysis of EF-G-dependent mRNA translocation in the ribosome. J Mol Biol 327: 369–381. 10.1016/S0022-2836(03)00146-3 [DOI] [PubMed] [Google Scholar]
- Walker SE, Fredrick K. 2006. Recognition and positioning of mRNA in the ribosome by tRNAs with expanded anticodons. J Mol Biol 360: 599–609. 10.1016/j.jmb.2006.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SE, Fredrick K. 2008. Preparation and evaluation of acylated tRNAs. Methods 44: 81–86. 10.1016/j.ymeth.2007.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SE, Shoji S, Pan D, Cooperman BS, Fredrick K. 2008. Role of hybrid tRNA-binding states in ribosomal translocation. Proc Natl Acad Sci 105: 9192–9197. 10.1073/pnas.0710146105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Pulk A, Wasserman MR, Feldman MB, Altman RB, Cate JH, Blanchard SC. 2012. Allosteric control of the ribosome by small-molecule antibiotics. Nat Struct Mol Biol 19: 957–963. 10.1038/nsmb.2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman MR, Alejo JL, Altman RB, Blanchard SC. 2016. Multiperspective smFRET reveals rate-determining late intermediates of ribosomal translocation. Nat Struct Mol Biol 23: 333–341. 10.1038/nsmb.3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman EM, Green R. 2005. Affinity purification of in vivo-assembled ribosomes for in vitro biochemical analysis. Methods 36: 305–312. 10.1016/j.ymeth.2005.04.007 [DOI] [PubMed] [Google Scholar]
- Zhang J, Pavlov MY, Ehrenberg M. 2018. Accuracy of genetic code translation and its orthogonal corruption by aminoglycosides and Mg2+ ions. Nucleic Acids Res 46: 1362–1374. 10.1093/nar/gkx1256 [DOI] [PMC free article] [PubMed] [Google Scholar]