

Abstract
Hereditary angioedema (HAE) is a genetic disorder mostly caused by mutations in the C1 esterase inhibitor gene (C1INH) that results in poor control of contact pathway activation and excess bradykinin generation. Bradykinin increases vascular permeability and is ultimately responsible for the episodes of swelling characteristic of HAE. We hypothesized that the use of RNA interference (RNAi) to reduce plasma Factor XII (FXII), which initiates the contact pathway signaling cascade, would reduce contact pathway activation and prevent excessive bradykinin generation. A subcutaneously administered GalNAc-conjugated small-interfering RNA (siRNA) targeting F12 mRNA (ALN-F12) was developed, and potency was evaluated in mice, rats, and cynomolgus monkeys. The effect of FXII reduction by ALN-F12 administration was evaluated in two different vascular leakage mouse models. An ex vivo assay was developed to evaluate the correlation between human plasma FXII levels and high-molecular weight kininogen (HK) cleavage. A single subcutaneous dose of ALN-F12 led to potent, dose-dependent reduction of plasma FXII in mice, rats, and NHP. In cynomolgus monkeys, a single subcutaneous dose of ALN-F12 at 3 mg/kg resulted in >85% reduction of plasma FXII. Administration of ALN-F12 resulted in dose-dependent reduction of vascular permeability in two different mouse models of bradykinin-driven vascular leakage, demonstrating that RNAi-mediated reduction of FXII can potentially mitigate excess bradykinin stimulation. Lastly, ex vivo human plasma HK cleavage assay indicated FXII-dependent bradykinin generation. Together, these data suggest that RNAi-mediated knockdown of FXII by ALN-F12 is a potentially promising approach for the prophylactic treatment of HAE.
Keywords: hereditary angioedema, GalNAc-siRNA, Factor XII, bradykinin, vascular permeability, HK cleavage
INTRODUCTION
Hereditary angioedema (HAE) is an autosomal dominant disease characterized by recurrent episodes of potentially life-threatening angioedema, with particular focus on the skin, gastrointestinal submucosa, and upper airway (Zuraw 2010; Zuraw and Christiansen 2011; Walford and Zuraw 2014). The prevalence of HAE ranges from 1:10,000 to 1:50,000 individuals. HAE is primarily caused by deficiency or malfunction of the C1 esterase inhibitor (C1INH) (type I and II, respectively) (Bork and Davis-Lorton 2013). C1INH regulates the activation of complement and intrinsic coagulation (contact activation pathway) by inhibiting a number of plasma proteases including C1r, C1s, plasma kallikrein, FXIIa and FXIa. With respect to HAE, plasma kallikrein and FXIIa are of particular interest as they are key regulators in the kallikrein–kinin branch of the contact pathway that regulates bradykinin production. In HAE patients with C1INH deficiency, the absence of a functional “brake” leads to uncontrolled kallikrein–kinin pathway activation and the generation of excessive amounts of bradykinin, a key mediator for vascular permeability and subsequent angioedema (Carugati et al. 2001; Nussberger et al. 2002; Pappalardo et al. 2002).
FXII, also known as Hageman factor (OMIM 234000), is a protein zymogen primarily produced in the liver that sits atop the kallikrein–kinin pathway (Schmaier 2000). When it comes into contact with negatively GalNAc-siRNA charged surfaces, FXII auto-activates into active serine protease FXIIa. Although the precise trigger for FXII activation in patients with HAE is currently unknown, various in vivo activators of FXII have been identified, including heparin, misfolded protein aggregates, polyphosphates and nucleic acids (Müller et al. 2009; Björkqvist et al. 2014). Upon activation, the direct downstream targets of FXIIa include Factor XI (FXI) and plasma prekallikrein (PK). The FXIIa cleavage of FXI into FXIa drives the activation of the intrinsic coagulation pathway and gives rises to fibrin formation. Activation of PK by FXIIa results in the production of plasma kallikrein serine protease (PKa) which then cleaves HK. Cleavage of HK leads to the release of bradykinin, a nine amino acid signaling peptide that controls vascular permeability through its interaction with bradykinin-2 receptor (B2R); the remaining polypeptide, two-chain HK, is comprised of two polypeptide chains (heavy and light) linked by a disulfide bond (Zhang et al. 2000). In patients, it is the uncontrolled generation of excess bradykinin that leads to HAE attacks.
As an initiator of the bradykinin generation pathway, FXII appears to be an intriguing target for HAE treatment. In line with this therapeutic concept, the F12-46C/T polymorphism (rs1801020) results in lower plasma FXII levels and is associated with a delay in HAE disease onset and decreased need for long-term prophylaxis (Kanaji et al. 1998; Speletas et al. 2015). In addition to the two classical types of HAE with diminished C1INH function (type I and II), some HAE patients present normal C1INH functionality levels (type III HAE). Among this population, mutations in the F12 gene have been reported to lead to loss of glycosylation, which led to increased contact-mediated autoactivation of zymogen FXII, resulting in excessive activation of the kallikrein–kinin pathway (Cichon et al. 2006; Bork 2013; Björkqvist et al. 2015; de Maat et al. 2016). In the European type III HAE population, F12 mutations were identified in up to 25% of cohorts (Bork et al. 2009). Both aspects of the genetic evidence support the therapeutic concept of targeting F12 for HAE treatment.
To evaluate the effect of knocking down F12 in the alleviation of HAE, we developed ALN-F12, an F12 targeting small-interfering RNA (siRNA) conjugated with trivalent N-acetylgalactosamine (GalNAc) ligand (Nair et al. 2014; Matsuda et al. 2015) to allow hepatocyte-specific delivery after subcutaneously dosing (Fig. 1A). Currently, the GalNAc-siRNA technology has been used in multiple ongoing clinical programs (Fitzgerald et al. 2017a,b; Zimmermann et al. 2017). We hypothesized that reduction of plasma FXII levels by ALN-F12 can compensate for the defect resulting from the loss of functional C1INH and alleviate excess bradykinin generation (Fig. 1B).
FIGURE 1.

ALN-F12 for HAE prophylaxis: therapeutic hypothesis. (A) ALN-F12 silences hepatic F12 gene expression. N-acetylgalactosamine (GalNAc) conjugated siRNA duplex ALN-F12 specifically binds hepatocytes via the asialoglycoprotein receptor (ASGPR). Following calthrin-mediated endocytosis and endosomal escape, ALN-F12 is unwound and loaded into RISC (RNA-induced silencing complex) and specifically recognizes and degrades F12 mRNA, leading to a reduction of secreted Factor XII (FXII) protein levels in plasma. (B) Conversion of FXII to FXIIa initiates an enzymatic signaling cascade that results in the production of bradykinin, a peptide signal that controls vascular permeability. C1 esterase inhibitor (C1INH), which binds and inhibits FXIIa and plasma kallikrein (PKa), negatively regulates this process. In HAE patients, loss of C1INH activity leads to excess bradykinin generation, resulting in increased vascular permeability and subsequent edema. RNAi-mediated knockdown of F12 gene expression by ALN-F12 would decrease circulating FXII protein levels and therefore preventing excess bradykinin generation.
RESULTS
In vitro and in vivo screenings for ALN-F12
A set of 79 siRNAs targeting FXII bearing GalNAc-ligand were tested for in vitro activity by transfection in the human hepatoma Hep3B cell line (Supplemental Table S1A). From this set, 12 efficacious siRNAs were selected for dose-response assessment (Supplemental Table S1B). Following the identification of three potent lead sequences, two chemically modified analogs targeting the same transcript site were prepared for each lead and all six siRNAs were evaluated for in vivo activity in mice (Supplemental Table S1C). ALN-F12 was selected based on both in vitro and in vivo screenings.
To assess the potential for off-target effects, a bioinformatic search against the human transcriptome was conducted for ALN-F12. The top 31 predicted potential off-target transcripts were evaluated in Hep3B cells, and none of the predicted potential off-target transcripts were inhibited substantially by ALN-F12 (data not shown).
ALN-F12 demonstrates robust potency and durability in rodents
To evaluate the potency and stability in vivo, GalNAc conjugated siRNA duplex ALN-F12 was subcutaneously dosed into female C57BL/6 mice at 0.3 or 1.0 mg/kg. The plasma total FXII levels were measured by ELISA and normalized to the average of the PBS control group. A single dose of ALN-F12 in mice led to dose-dependent, maximum, suppression of circulating FXII of ∼51% at 0.3 mg/kg and 93% at 1 mg/kg on Day 10. FXII levels returned to baseline by ∼64 d after treatment (Fig. 2A). Similarly, a single subcutaneously dose of ALN-F12 in female Sprague–Dawley rats led to robust, dose-dependent reduction of plasma FXII (Fig. 2B). The suppression of liver F12 mRNA levels is comparable with the reduction of plasma FXII protein levels in both mice and rats (Supplemental Fig. S1).
FIGURE 2.
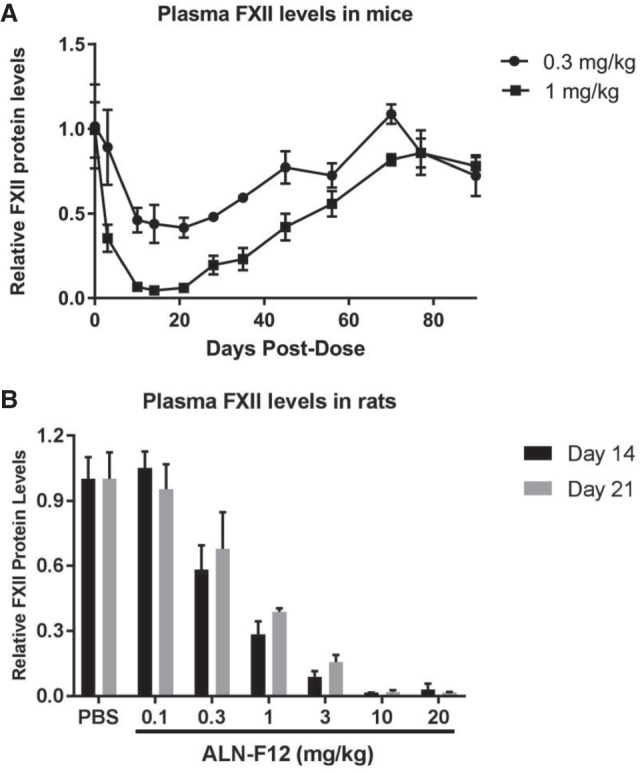
ALN-F12 dose-dependent knockdown of circulating FXII in mice and rats. (A) Female C57BL/6J mice were subcutaneously administered a single dose of ALN-F12 (n = 3 per group). Plasma FXII levels were measured by ELISA and normalized to PBS control group. (B) Female Sprague–Dawley rats were subcutaneously administered a single dose of ALN-F12 (n = 3 per group). Plasma FXII levels were measured by ELISA and normalized to the average of PBS control group. Error bars are SD.
RNAi-mediated FXII reduction alleviated ACE inhibitor-induced vascular leakage
It has been demonstrated previously that a vasodilator angiotensin-converting enzyme (ACE) inhibitor, such as captopril, can increase vascular permeability by blocking the breakdown of bradykinin (Migdalof et al. 1984; Han et al. 2002). To examine the effect of FXII suppression on captopril-induced vascular leakage, ALN-F12 was dosed in female C57BL/6 mice at different levels (0.1, 0.3, 1, and 3 mg/kg) 7 d before captopril induction to allow maximum knockdown. Vascular flow was traced by IV injection of Evans blue dye, a high affinity inert binder to plasma albumin. IV injection of 2.5 mg/kg of captopril led to dramatic vascular leakage within 30 min represented by decreased Evans blue dye extraction from the circulation system and increased extraction from peripheral tissue (Fig. 3A,B). Suppression of plasma FXII mitigated the captopril-induced vascular leakage in a dose-dependent manner, represented by increased Evans blue dye extraction from the circulation and decreased from the intestine. Interestingly, in this model, ∼50% F12 mRNA reduction completely prevented vascular leakage induced by captopril (Fig. 3C,D). The same studies were also performed by knocking down contact activation pathway downstream components, PK or HK to different levels using GalNAc-siRNAs specifically targeting each of them. In contrast to F12 mRNA, ∼75% silencing of PK mRNA and >90% silencing of HK mRNA was necessary to prevent captopril-induced vascular permeability (Fig. 3E,F; Supplemental Figs. S2, S3). No synergistic effect was observed by knocking down both PK and FXII (data not shown).
FIGURE 3.

Inhibition of ACE inhibitor-induced vascular permeability following a single dose subcutaneous administration of ALN-F12. Female C57BL/6J mice were subcutaneously administered a single dose of ALN-F12 at dose levels of 0, 0.1, 0.3, 1, or 3 mg/kg (n = 10 per group). Seven days after administration, animals were IV injected with the ACE inhibitor captopril (2.5 mg/kg); the vascular permeability tracer, Evans blue dye (30 mg/kg), was IV injected 15 min after captopril injection. Animals were euthanized 15 min after Evans blue dye injection. Blood (A) and intestine (B) were collected to evaluate Evans blue extravasation; livers were collected for F12 mRNA analysis (C). In panels A, B, and C, symbols denote data from individual animals, the group mean ± SD are also plotted. Statistical analysis performed using one-way ANOVA with Dunnett's post-hoc test with respect to PBS-treated animals in the captopril treatment group. (**) P < 0.01; (****) P < 0.0001. A correlation of intestinal Evans blue uptake and F12 mRNA levels (D). Panels E and F are the correlation of intestinal Evans blue extraction and PK (and HK) mRNA levels (Supplemental Figs. S2, S3).
FXII knockdown mitigated mustard oil-induced vascular leakage in C1INH knockdown mice
Clinically, both type I and type II HAE result from C1INH deficiency or dysfunction. Although C1INH-deficient mice do not develop symptomatic angioedema, they have increased baseline vascular permeability and higher sensitivity to the topical application of inflammatory stimulator mustard oil (Han et al. 2002). To generate C1INH knockdown mice, we developed a GalNAc-siRNA duplex targeting mouse C1INH (C1INH-siRNA). C1INH is mainly synthesized in the liver, thereby allowing efficacious suppression of plasma C1INH by GalNAc-conjugated C1INH-siRNA. A single SC dose of C1INH-siRNA at 10 mg/kg led to >95% decrease of liver C1INH mRNA levels (Supplemental Fig. S4B). Inflammatory stimulator, mustard oil, was topically applied to the right ear to increase vascular leakage. The left ear of each animal served as a control. Consistent with the homozygous C1INH-deficient mice, 10 mg/kg C1INH-siRNA-treated mice also displayed higher baseline vascular permeability and increased sensitivity to mustard oil stimulation (Fig. 4A). Knocking down of FXII by ALN-F12 in C1INH knockdown mice mitigated the vascular fluid leakage in a dose-dependent manner, as demonstrated by decreased Evans blue dye extraction of both untreated and mustard oil stimulated ears (Fig. 4A,B). Knocking down other components in the contact activation pathway, such as PK and HK, also mitigated the vascular leakage in this model (Supplemental Fig. S4). It is worth noting that in this model, in contrast to the ACEi-induced vascular leakage model, suppression of FXII, PK, and HK all yielded a similar outcome.
FIGURE 4.

ALN-F12-rescued mustard oil-induced vascular permeability in a C1INH knockdown mouse model. Female CD1 mice were subcutaneously dosed with a siRNA targeting C1INH (10 mg/kg) and ALN-F12 at different dose levels (n = 10 per group). Seven days post-administration, Evans blue dye (30 mg/kg) was given IV, and 5% mustard oil was topically applied to the right ear, followed 15 min later by a second application. The animals were euthanized 15 min after the second application of mustard oil. Both right (mustard oil-treated) and left (control) ears were collected for dye extraction (A). Livers were collected for mRNA measurements (B). Group mean ± SD are plotted. Statistical analysis in Panel A performed using two-way ANOVA with Tukey's post-hoc test; asterisks indicate statistical comparisons to mustard oil-treated ears in the PBS control group. Statistical analysis in Panel B performed using one-way ANOVA with Dunnett's post-hoc test; statistical comparisons to PBS-treated animals. For both analyses: (**) P < 0.01; (****) P < 0.0001.
ALN-F12 led to dose-dependent, sustainable suppression of plasma FXII in nonhuman primate (NHP)
ALN-F12 is also designed to target NHP and human F12 genes. To determine the efficacy and durability of ALN-F12 in NHP, a single dose of ALN-F12 was subcutaneously administered at 0.1, 0.3, 1, or 3 mg/kg in female cynomolgus monkeys (n = 3 per group). Plasma was collected at pre-dose (Days 5, 3, and 1) and a series of time points after dosing. Levels of circulating FXII protein were measured by plasma ELISA and normalized to the pre-dose average of each individual animal. A single SC dose of ALN-F12 led to dose-dependent suppression of circulating FXII in cynomolgus monkeys with maximal suppression of ∼21% at 0.1 mg/kg, 32% at 0.3 mg/kg, 68% at 1 mg/kg, and 83% at 3 mg/kg on Day 28 (Fig. 5A). Initial recovery of plasma FXII was observed on Day 56 for the 3 mg/kg dosage group, with FXII levels returning to baseline by ∼160 d post-dose (Fig. 5B).
FIGURE 5.

ALN-F12 dose-dependent reduction of circulating FXII in female cynomolgus monkeys. Female cynomolgus monkeys were subcutaneously administered a single dose of ALN-F12 (n = 3 per group). Plasma FXII levels were measured by ELISA and normalized to the average of individual animal's pre-dose average of Days −5, −3, and −1 (error bars = SD). Panel A represents the relative plasma FXII level on Day 28 post-dosing; Panel B depicts the relative FXII levels at all collected time points for all dosing groups.
FXII-dependent HK cleavage in human plasma
Activation of the contact pathway leads to the cleavage of full-length high-molecular weight kininogen (HK) and the release of vasodilator bradykinin. HK cleavage can be evaluated by the detection of the disappearance of full-length HK upon contact pathway activation (Reddigari and Kaplan 1988; Baroso et al. 2016; Banerji et al. 2017). Evaluating the correlation between human plasma FXII levels and the extent of HK cleavage could potentially provide guidance on the level of FXII knockdown needed for efficacy. The steady-state HK cleavage levels are ∼50% in HAE patient plasma versus ∼8.3% in plasma from healthy individuals (Banerji et al. 2017). To investigate the correlation between FXII levels and the extent of HK cleavage, we developed an ex vivo assay system using kaolin to induce contact pathway activation. To mimic the level of steady-state contact pathway activation in HAE patient plasma, normal human plasma was incubated with 0.2 µg/mL Kaolin for 10 min at 37°C. This mild contact activation condition led to ∼50% HK cleavage in normal pooled human plasma (Fig. 6, 100% FXII). By mixing the normal and congenital FXII-deficient (<1% FXII activity, nondetectable from ELISA, data not shown) human plasma, we generated a gradient concentration of FXII in the plasma mixture. Under the mild Kaolin contact activation condition that was described above, the percentage of HK cleavage was highly correlated with the levels of FXII. 15% FXII in the plasma resulted in ∼23% HK cleavage, and in FXII-deficient plasma, there was no detectable HK cleavage (Fig. 6). These data support the hypothesis that reducing plasma FXII levels can potentially suppress bradykinin generation. The same experiment was also performed for PK-deficient plasma and similar results were observed (data not shown). Consistent results were also observed in plasma samples from cynomolgus monkeys treated with ALN-F12 (Supplemental Fig. S5).
FIGURE 6.
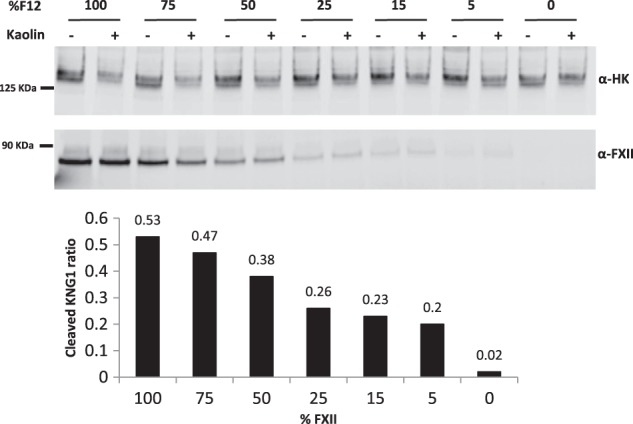
FXII dose-dependent HK cleavage in human plasma. Normal human pooled plasma was mixed with FXII-deficient (<1%) human plasma at different ratios to generate different concentration of FXII in the mixture. The plasma mixtures were activated under mild conditions (0.2 µg/mL Kaolin, 37°C, 10 min) and loaded into SDS-PAGE for western blot analysis. The anti-HK antibody detects the intact, full-length HK. The uncleaved full-length HK signal ratio of kaolin treatment to no kaolin treatment is represented in Table 1 below.
DISCUSSION
Type I and II HAE is characterized by a lack of functional C1INH that leads to the uncontrolled activation of contact pathway and excessive bradykinin generation. The contact pathway is initiated by auto-activation of FXII in contact with negatively charged surfaces, such as protein aggregates or polyphosphates. Other than fresh frozen plasma or C1-INH concentrate (Cinryze, Berinert, Haegarda) to supply functional C1INH, the other approved treatments for HAE also target the bradykinin generation pathway, including plasma kallikrein inhibitor (Kalbitor and Takhzyro) and B2R antagonist (Firazyr). There is compelling genetic evidence supporting the correlation between plasma FXII levels and HAE clinical phenotypes. The F12-46C/T polymorphism is associated with decreased plasma FXII levels at ∼50% (Kanaji et al. 1998), and F12-46C/T carriers exhibited a ∼7-yr delay on disease onset decreased demand for long-term treatment regardless of C1INH mutational status (Kanaji et al. 1998; Grunbacher et al. 2005; Bors et al. 2013; Speletas et al. 2015). Furthermore, among HAE patients with normal C1INH, also known as type III HAE, several distinct mutations in FXII have been identified. These FXII mutations, such as Thr309Lys, lead to the loss of glycosylation in FXII which results in the “gain-of-function” of FXIIa and excess activation of the bradykinin generation pathway (Cichon et al. 2006; de Maat et al. 2016).
Using our GalNAc-conjugate siRNA delivery platform, we developed ALN-F12, a potent and stable GalNAc-siRNA duplex to efficiently target liver F12 mRNA and diminish plasma FXII levels. ALN-F12 has demonstrated robust potency and durability in both rodents and nonhuman primates. The durability of the pharmacodynamic effect of ALN-F12, and other GalNAc-siRNAs, is presumed to be due to the durability of the assembled RISC as well as the enhanced metabolic stability of the GalNAc-siRNA that allows for an extended duration of functional RISC loading (Foster et al. 2018). Off-target effects of ALN-F12 were mitigated through both sequence-based bioinformatics and screening approaches as well as through the use of chemical modifications to abrogate innate immune responses. These approaches have been successfully applied in prior GalNAc-siRNA compounds evaluated in human clinical trials (Zimmermann et al. 2017).
To explore the effect of ALN-F12 on in vascular permeability, we implemented a modified ACE inhibitor stimulated vascular leakage model in mice (Han et al. 2002; Bhattacharjee et al. 2013). In this model we knocked down FXII, PK, or HK at different levels using corresponding GalNAc-siRNAs, and observed the mitigation effect on vascular leakage. PK and HK silencing was evaluated to determine whether other genes in the pathway may be alternative therapeutic targets. Interestingly, ≥50% F12 mRNA knockdown fully rescued the vascular leakage to peripheral tissues, while PK and HK mRNAs needed to be reduced to higher extents (>75% and 90%, respectively) to achieve an equivalent effect. However, we did not observe the superior effect of FXII knockdown over PK or HK suppression in the mustard oil stimulation model in animals with reduced C1INH levels. The superiority of FXII reduction over PK (or HK) in the ACE inhibitor model is a unique observation and this might be due to the experimental setting and mice genetic background.
Since many aspects of HAE attacks are not well understood, such as the triggers, the attack sites and the extent of FXII activation, it is difficult to predict how much suppression of plasma FXII would be sufficient to prevent HAE attacks. To explore the level of FXII reduction that may have an impact on HAE attacks, we developed an ex vivo human plasma assay to evaluate the correlation between FXII levels and bradykinin generation. In the anti-kallikrein antibody DX-2930 Phase 1b trial, HAE attacks were significantly decreased during Day 8 to Day 50 and ∼15%–25% of plasma HK was cleaved during this period compared to a steady-state activation level of 50% in HAE plasma (Banerji et al. 2017). In our ex vivo assay, FXII levels of 15% of normal led to ∼25% HK cleavage, which is comparable to the level of HK cleavage achieved in the DX-2930 trial. These data suggest that >85% F12 knockdown may be sufficient to mitigate the frequency or severity of HAE attacks. This level of knockdown is achievable with subcutaneous administration of ALN-F12, based on both the efficacy of ALN-F12 in NHP, and the preliminary results from other Alnylam clinical programs utilizing GalNAc-siRNA conjugates (Fitzgerald et al. 2017a,b; Pasi et al. 2017; Zimmermann et al. 2017).
It is encouraging that human homozygous F12 deficiency (Hageman trait) has been described and has not been associated with increased bleeding tendency or other overt clinical phenotype. However, the safety of long-term suppression of FXII by ALN-F12 remains to be established in clinical trials. Furthermore, beyond a role in HAE, in recent years reports have suggested an important role of FXII in thrombosis and inflammation (Matafonov et al. 2014; Kenne et al. 2015), Alzheimer's disease (Zamolodchikov et al. 2016), and wound healing (Stavrou et al. 2018). Therefore, ALN-F12 mediated FXII lowering may potentially have broad clinical utility.
MATERIALS AND METHODS
Care and use of laboratory animals
Mice and rats were purchased from the Jackson Laboratory. All rodent studies were conducted at Alnylam Pharmaceuticals, an AALAS accredited animal facility, using protocols consistent with local, state and federal regulations and approved by the Institutional Animal Care and Use Committee (IACUC) at Alnylam Pharmaceuticals. Procedures using female cynomolgus monkeys were conducted by WIL Research/Charles River Laboratories, Ohio (2.5–5 yr of age; 2.3–5 kg of bodyweight; Study No. WIL-268503).
Design and synthesis of ALN-F12 and other GalNAc-siRNAs targeting mouse PK, HK, and C1INH
ALN-F12 is an investigational RNA interference (RNAi) therapeutic designed and synthesized by Alnylam Pharmaceuticals. The siRNA was designed to reduce liver production of Factor XII (FXII) by targeting a 23-nt region of F12 mRNA conserved among mouse, rat, cynomolgus monkey, and human. The drug substance of ALN-F12 is a chemically synthesized double-stranded oligonucleotide covalently linked to a ligand containing three N-acetylgalactosamine (GalNAc) residues to enable delivery to hepatocytes via binding with asialoglycoprotein receptor (ASGPR) followed by clathrin-mediated endocytosis. The other GalNAc-siRNAs designed and synthesized by Alnylam Pharmaceuticals for this publication are as follows: PK-siRNA (NM_008455), HK-siRNA (NM_001102409), and C1INH-siRNA (NM_009776). Sequences and chemistry details of GalNAc-siRNA conjugates are listed in Table 1. Quality control parameters of the GalNAc-siRNA conjugates are summarized in Supplemental Table S2. Design of siRNA duplexes used an algorithm for chemical modification to optimize metabolic stability and intrinsic RNAi activity (Foster et al. 2018). For in vivo studies, all GalNAc-siRNAs were diluted in sterile 1× PBS and subcutaneously injected into the animals at 10 µL/g body weight for mice, 5 µL/g body weight for rats and 2 µL/g body weight for monkeys.
TABLE 1.
Designs and sequences of the GalNAc-siRNA conjugates

Liver RT-QPCR and plasma FXII ELISA
Mouse or rat liver tissues were harvested and snap frozen in liquid nitrogen to prevent mRNA degradation. Frozen liver tissue was ground, and tissue powder (∼20 mg) was lysed in 700 µL RLT lysis buffer provided from the RNeasy Mini Kit (QIAGEN). mRNA was extracted according to the manufacturer's instructions. The extracted RNA was eluted in 50 µL of water. 10 µL of eluted RNA (∼2 µg) was used for cDNA synthesis utilizing a High Capacity cDNA synthesis kit (Thermo Fisher). Synthesis cDNA (2 µL) was used for MultiPlex qPCR reactions. GAPDH was used as an internal control.
Plasma FXII total antigen levels were evaluated using ELISA kits from Molecular Innovations (mouse: MFXIIKT-TOT; rat: RFXIIKT-TOT; monkey: HFXIIKT-TOT) following the manufacturer's instructions. Briefly, K2EDTA-collected plasma samples were thawed on ice and diluted (mouse: 1:20,000; rat: 1:10,000; monkey: 1:2,000) with blocking buffer (3% BSA, 100 mM Tris-HCl, pH 7.4, 150 mM NaCl). 100 µL of diluted sample (or standard) was added to capture antibody-coated plate and incubated, followed by detection antibody and HRP-conjugated secondary antibody incubation, with washing steps included in between. The calculated plasma FXII concentrations for the test groups were then normalized to the average of the PBS-treated group or the corresponding individual animal's pre-dose average.
ACE inhibitor-induced vascular permeability model
Female C57BL/6J mice were subcutaneously (SC) administered a single dose of ALN-F12 at 0.1, 0.3, 1, or 3 mg/kg (n = 10 per group). Seven days after ALN-F12 administration, animals were intravenously (IV) injected with 2.5 mg/kg ACE inhibitor captopril (Sigma-Aldrich), dissolved in 1× PBS at 10 µL/g body weight. The vascular leakage inert tracer, Evans blue dye (30 mg/kg in 1× PBS), was IV injected in a tail vein 15 min after captopril injection. Animals were euthanized 15 min after Evans blue dye injection. To assess vascular permeability, 100 µL of blood, collected via cardiac puncture, was mixed with 300 µL ice-chilled acetone. After mixing thoroughly, the mixtures were centrifuged at 14,000 rpm for 5 min. Supernatant (150 µL) was transferred to a 96-well flat-bottom plate for absorbance measurements at 620 nm. A section of intestine (∼15 cm) was open longitudinally, rinsed twice in 0.9% sodium chloride, and transferred into balanced formamide (Sigma-Aldrich,) for Evans blue dye extraction. The tubes were incubated overnight at 55°C, mixed, and centrifuged at 14,000 rpm for 10 min. Supernatant (150 µL) was then transferred to a 96-well flat-bottom plate for absorbance measurements at 620 nm. Livers were collected for mRNA analysis.
Mustard oil-induced vascular permeability model in C1INH knockdown mice
The in vivo potency of the GalNAc-siRNA targeting mouse C1INH (C1INH-siRNA) was confirmed (>95% silencing of liver C1-INH mRNA, Supplemental Fig. S4B). Female CD-1 mice were given a single SC dose of a C1INH-siRNA at 10 mg/kg followed by a single dose of ALN-F12 (or PK-siRNA or HK-siRNA) at different doses within 10 min (n = 10 per group). Seven days after GalNAc-siRNAs administration, animals were IV injected in the tail vein with Evans blue dye (30 mg/kg in 1× PBS at 10 µL/g body weight). Immediately thereafter, 5% mustard oil (Sigma-Aldrich, diluted with Mineral oil) was topically applied to the right ear of each animal followed by a second mustard oil application to the same ear 15 min later. The left ear remained untreated and served as the negative control. The animals were euthanized 30 min after the injection of Evans blue dye. To evaluate vascular permeability, each ear was collected, weighed, minced and 1 μL/mg tissue balanced formamide added for dye extraction as above. Livers were collected for mRNA analysis.
Plasma HK cleavage assay
Direct measurement of bradykinin generation is burdensome due to the instability and very short half-life (<1 min) of bradykinin (Murphey et al. 2000; Marketou and Vardas 2012). Instead, the levels of bradykinin generation can be quantified by plasma western blot using antibodies detecting the full-length, heavy chain, or light chain of HK. Normal pool and congenital FXII-deficient human plasma were purchased from George King Bio-Medical (Cat. No. 0010 and 1200). Cynomolgus plasma was obtained from animals dosed with ALN-F12 (Study No. WIL268503). In a 96-well PCR plate, 18 µL Kaolin solution (Kaolin: Sigma-Aldrich, K7375; prepared in HBS buffer [20 mM HEPES, pH7.5, 140 mM NaCl]) was added into each well followed by the addition of 2 µL plasma. The plate was put on a shaking incubator at 37°C for precisely 10 min. Samples containing an equivalent of 0.5 µL of plasma were loaded on denaturing NuPAGE SDS-PAGE gels (Invitrogen) for Western blot analysis using full-length HK and FXII antibodies from Abcam (α-HK: ab79650; α-FXII: ab196670). A LiCoR Odymmey Infrared Imager (Li-COR Biosciences) was applied for quantification.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.068916.118.
REFERENCES
- Banerji A, Busse P, Shennak M, Lumry W, Davis-Lorton M, Wedner HJ, Jacobs J, Baker J, Bernstein JA, Lockey R, et al. 2017. Inhibiting plasma kallikrein for hereditary angioedema prophylaxis. N Engl J Med 376: 717–728. 10.1056/NEJMoa1605767 [DOI] [PubMed] [Google Scholar]
- Baroso R, Sellier P, Defendi F, Charignon D, Ghannam A, Habib M, Drouet C, Favier B. 2016. Kininogen cleavage assay: diagnostic assistance for kinin-mediated angioedema conditions. PLoS One 11: e0163958 10.1371/journal.pone.0163958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee G, Revenko AS, Crosby JR, May C, Gao D, Zhao C, Monia BP, MacLeod AR. 2013. Inhibition of vascular permeability by antisense-mediated inhibition of plasma kallikrein and coagulation factor 12. Nucleic Acid Ther 23: 175–187. 10.1089/nat.2013.0417 [DOI] [PubMed] [Google Scholar]
- Björkqvist J, Nickel KF, Stavrou E, Renné T. 2014. In vivo activation and functions of the protease factor XII. Thromb Haemost 112: 868–875. 10.1160/th14-04-0311 [DOI] [PubMed] [Google Scholar]
- Björkqvist J, de Maat S, Lewandrowski U, Di Gennaro A, Oschatz C, Schönig K, Nöthen MM, Drouet C, Braley H, Nolte MW, et al. 2015. Defective glycosylation of coagulation factor XII underlies hereditary angioedema type III. J Clin Invest 125: 3132–3146. 10.1172/JCI77139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork K. 2013. Hereditary angioedema with normal C1 inhibitor. Immunol Allergy Clin North Am 33: 457–470. 10.1016/j.iac.2013.07.002 [DOI] [PubMed] [Google Scholar]
- Bork K, Davis-Lorton M. 2013. Overview of hereditary angioedema caused by C1-inhibitor deficiency: assessment and clinical management. Eur Ann Allergy Clin Immunol 45: 7–16. [PubMed] [Google Scholar]
- Bork K, Wulff K, Hardt J, Witzke G, Staubach P. 2009. Hereditary angioedema caused by missense mutations in the factor XII gene: clinical features, trigger factors, and therapy. J Allergy Clin Immunol 124: 129–134. 10.1016/j.jaci.2009.03.038 [DOI] [PubMed] [Google Scholar]
- Bors A, Csuka D, Varga L, Farkas H, Tordai A, Füst G, Szilagyi A. 2013. Less severe clinical manifestations in patients with hereditary angioedema with missense C1INH gene mutations. J Allergy Clin Immunol 131: 1708–1711. 10.1016/j.jaci.2012.11.015 [DOI] [PubMed] [Google Scholar]
- Carugati A, Pappalardo E, Zingale LC, Cicardi M. 2001. C1-inhibitor deficiency and angioedema. Mol Immunol 38: 161–173. 10.1016/S0161-5890(01)00040-2 [DOI] [PubMed] [Google Scholar]
- Cichon S, Martin L, Hennies HC, Müller F, Van Driessche K, Karpushova A, Stevens W, Colombo R, Renné T, Drouet C, et al. 2006. Increased activity of coagulation factor XII (Hageman factor) causes hereditary angioedema type III. Am J Hum Genet 79: 1098–1104. 10.1086/509899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Maat S, Björkqvist J, Suffritti C, Wiesenekker CP, Nagtegaal W, Koekman A, van Dooremalen S, Pasterkamp G, de Groot PG, Cicardi M, et al. 2016. Plasmin is a natural trigger for bradykinin production in patients with hereditary angioedema with factor XII mutations. J Allergy Clin Immunol 138: 1414–1423.e9. 10.1016/j.jaci.2016.02.021 [DOI] [PubMed] [Google Scholar]
- Fitzgerald K, Kallend D, Simon A. 2017a. A highly durable RNAi therapeutic inhibitor of PCSK9. N Engl J Med 376: e38 10.1056/NEJMc1703361 [DOI] [PubMed] [Google Scholar]
- Fitzgerald K, White S, Borodovsky A, Bettencourt BR, Strahs A, Clausen V, Wijngaard P, Horton JD, Taubel J, Brooks A, et al. 2017b. A highly durable RNAi therapeutic inhibitor of PCSK9. N Engl J Med 376: 41–51. 10.1056/NEJMoa1609243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DJ, Brown CR, Shaikh S, Trapp C, Schlegel MK, Qian K, Sehgal A, Rajeev KG, Jadhav V, Manoharan M, et al. 2018. Advanced siRNA designs further improve in vivo performance of GalNAc-siRNA conjugates. Mol Ther 26: 708–717. 10.1016/j.ymthe.2017.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunbacher G, Marx-Neuhold E, Pilger E, Koppel H, Renner W. 2005. The functional -4C>T polymorphism of the coagulation factor XII gene is not associated with deep venous thrombosis. J Thromb Haemost 3: 2815–2817. 10.1111/j.1538-7836.2005.01651.x [DOI] [PubMed] [Google Scholar]
- Han ED, MacFarlane RC, Mulligan AN, Scafidi J, Davis AE III. 2002. Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest 109: 1057–1063. 10.1172/JCI200214211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaji T, Okamura T, Osaki K, Kuroiwa M, Shimoda K, Hamasaki N, Niho Y. 1998. A common genetic polymorphism (46 C to T substitution) in the 5′-untranslated region of the coagulation factor XII gene is associated with low translation efficiency and decrease in plasma factor XII level. Blood 91: 2010–2014. [PubMed] [Google Scholar]
- Kenne E, Nickel KF, Long AT, Fuchs TA, Stavrou EX, Stahl FR, Renné T. 2015. Factor XII: a novel target for safe prevention of thrombosis and inflammation. J Intern Med 278: 571–585. 10.1111/joim.12430 [DOI] [PubMed] [Google Scholar]
- Marketou ME, Vardas PE. 2012. Bradykinin in the treatment of arterial hypertension: friend or foe? Hellenic J Cardiol 53: 91–94. [PubMed] [Google Scholar]
- Matafonov A, Leung PY, Gailani AE, Grach SL, Puy C, Cheng Q, Sun MF, McCarty OJ, Tucker EI, Kataoka H, et al. 2014. Factor XII inhibition reduces thrombus formation in a primate thrombosis model. Blood 123: 1739–1746. 10.1182/blood-2013-04-499111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Keiser K, Nair JK, Charisse K, Manoharan RM, Kretschmer P, Peng CG, V Kel'in A, Kandasamy P, Willoughby JL, et al. 2015. siRNA conjugates carrying sequentially assembled trivalent N-acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem Biol 10: 1181–1187. 10.1021/cb501028c [DOI] [PubMed] [Google Scholar]
- Migdalof BH, Antonaccio MJ, McKinstry DN, Singhvi SM, Lan SJ, Egli P, Kripalani KJ. 1984. Captopril: pharmacology, metabolism and disposition. Drug Metab Rev 15: 841–869. 10.3109/03602538409041080 [DOI] [PubMed] [Google Scholar]
- Müller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, Schmidbauer S, Gahl WA, Morrissey JH, Renné T. 2009. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell 139: 1143–1156. 10.1016/j.cell.2009.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphey LJ, Hachey DL, Oates JA, Morrow JD, Brown NJ. 2000. Metabolism of bradykinin in vivo in humans: identification of BK1-5 as a stable plasma peptide metabolite. J Pharmacol Exp Ther 294: 263–269. [PubMed] [Google Scholar]
- Nair JK, Willoughby JL, Chan A, Charisse K, Alam MR, Wang Q, Hoekstra M, Kandasamy P, Kel'in AV, Milstein S, et al. 2014. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc 136: 16958–16961. 10.1021/ja505986a [DOI] [PubMed] [Google Scholar]
- Nussberger J, Cugno M, Cicardi M. 2002. Bradykinin-mediated angioedema. N Engl J Med 347: 621–622. 10.1056/NEJM200208223470820 [DOI] [PubMed] [Google Scholar]
- Pappalardo E, Zingale LC, Terlizzi A, Zanichelli A, Folcioni A, Cicardi M. 2002. Mechanisms of C1-inhibitor deficiency. Immunobiology 205: 542–551. 10.1078/0171-2985-00153 [DOI] [PubMed] [Google Scholar]
- Pasi KJ, Rangarajan S, Georgiev P, Mant T, Creagh MD, Lissitchkov T, Bevan D, Austin S, Hay CR, Hegemann I, et al. 2017. Targeting of antithrombin in hemophilia A or B with RNAi therapy. N Engl J Med 377: 819–828. 10.1056/NEJMoa1616569 [DOI] [PubMed] [Google Scholar]
- Reddigari S, Kaplan AP. 1988. Cleavage of human high-molecular weight kininogen by purified kallikreins and upon contact activation of plasma. Blood 71: 1334–1340. [PubMed] [Google Scholar]
- Schmaier AH. 2000. Plasma kallikrein/kinin system: a revised hypothesis for its activation and its physiologic contributions. Curr Opin Hematol 7: 261–265. 10.1097/00062752-200009000-00001 [DOI] [PubMed] [Google Scholar]
- Speletas M, Szilágyi A, Csuka D, Koutsostathis N, Psarros F, Moldovan D, Magerl M, Kompoti M, Varga L, Maurer M, et al. 2015. F12-46C/T polymorphism as modifier of the clinical phenotype of hereditary angioedema. Allergy 70: 1661–1664. 10.1111/all.12714 [DOI] [PubMed] [Google Scholar]
- Stavrou EX, Fang C, Bane KL, Long AT, Naudin C, Kucukal E, Gandhi A, Brett-Morris A, Mumaw MM, Izadmehr S, et al. 2018. Factor XII and uPAR upregulate neutrophil functions to influence wound healing. J Clin Invest 128: 944–959. 10.1172/JCI92880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walford HH, Zuraw BL. 2014. Current update on cellular and molecular mechanisms of hereditary angioedema. Ann Allergy Asthma Immunol 112: 413–418. 10.1016/j.anai.2013.12.023 [DOI] [PubMed] [Google Scholar]
- Zamolodchikov D, Renné T, Strickland S. 2016. The Alzheimer's disease peptide β-amyloid promotes thrombin generation through activation of coagulation factor XII. J Thromb Haemost 14: 995–1007. 10.1111/jth.13209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JC, Claffey K, Sakthivel R, Darzynkiewicz Z, Shaw DE, Leal J, Wang YC, Lu FM, McCrae KR. 2000. Two-chain high molecular weight kininogen induces endothelial cell apoptosis and inhibits angiogenesis: partial activity within domain 5. FASEB J 14: 2589–2600. 10.1096/fj.99-1025com [DOI] [PubMed] [Google Scholar]
- Zimmermann TS, Karsten V, Chan A, Chiesa J, Boyce M, Bettencourt BR, Hutabarat R, Nochur S, Vaishnaw A, Gollob J. 2017. Clinical proof of concept for a novel hepatocyte-targeting GalNAc-siRNA conjugate. Mol Ther 25: 71–78. 10.1016/j.ymthe.2016.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuraw BL. 2010. Summary and conclusions: new perspectives in hereditary angioedema: molecular mechanisms and therapeutic choices: a CME symposium presented at the World Allergy Congress, Buenos Aires, Argentina (December 2009). World Allergy Organ J 3: S39–S40. 10.1186/1939-4551-3-S3-S39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuraw BL, Christiansen SC. 2011. Pathophysiology of hereditary angioedema. Am J Rhinol Allergy 25: 373–378. 10.2500/ajra.2011.25.3661 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.