

 A migratory fluoro-alkenylation of unactivated alkyl bromides is reported; the reaction is enabled by fluorine effects and involves an alkyl nickel chain-walking mechanism.
A migratory fluoro-alkenylation of unactivated alkyl bromides is reported; the reaction is enabled by fluorine effects and involves an alkyl nickel chain-walking mechanism.
Abstract
We describe a nickel-catalyzed highly regio- and stereoselective migratory fluoro-alkenylation of unactivated alkyl bromides. A unique catalytic cycle merging alkyl nickel chain-walking and defluorinative coupling enables the introduction of a broad array of fluoroalkenyl moieties into carbon chains. Control experiments with other halogenated alkenes demonstrated the essential role of fluorine atoms in this reaction. Notably, the reaction proceeds under mild conditions and allows for the synthesis of a variety of valuable monofluoroalkenes.
Introduction
Cross-electrophile coupling has found wide application in the construction of C–C bonds and serves as a powerful and reliable alternative to the classical nucleophile/electrophile procedures.1 In particular, C(sp2)–C(sp3) cross-coupling has been thoroughly explored using the combination of aryl/alkenyl and alkyl electrophiles, wherein β-hydride elimination is inhibited to minimize olefin by-products.2 Conversely, transition-metal-catalyzed remote functionalization exploits iterative β-hydride elimination and metal-H insertion, namely the “chain-walking” process, allowing introduction of functionality into an otherwise unreactive aliphatic position.3 Recently, significant progress in this field has been achieved by the groups of Martin,4 Zhu,5 and Yin.6 Remote arylation and carboxylation of unactivated alkyl halides have been well established (Scheme 1a). However, the reported reactions are still restricted to a relatively limited number of coupling partners, thus limiting the resulting molecular diversity of the products. A notable limitation remains: the corresponding alkenylation of unactivated alkyl halides has not been reported yet, although various alkenes are frequently found in pharmaceuticals or other functional molecules. The reason for the lack of a precedent probably lies in the potential addition of disassociated metal-H to the olefinic coupling component and even the alkene product during the chain-walking process.5,7 In this regard, the development of efficient and practical routes for metal-hydride-involving remote alkenylation has remained as a formidable challenge.
Scheme 1. Migratory functionalization of unactivated alkyl halides.

To circumvent the problem of promiscuous hydrometallation, an alkenyl coupling partner inherently inert to metal-H would be required in a successful migratory alkenylation. Recently, gem-difluoroalkenes have been demonstrated by Cao, Toste, Fu, our group, and others as efficient fluoro-alkenylating reagents to access a wide range of monofluoroalkenes using radical or ionic manifolds.8 The strong electron-withdrawing nature of the two fluorine atoms renders gem-difluoroalkenes highly reactive toward nucleophilic attack on the fluorinated sp2-carbon, and fluoro-alkenylation could be accomplished through facile β-fluoride elimination. Furthermore, we considered that the fluorine-based polarization and p–π interaction would make the fluorinated olefins less favored in the hydrometallation compared with the non-fluorinated alkene intermediates and therefore amenable in the migratory alkenylation reactions.9 It could then be envisaged that gem-difluoroalkene 2 can coordinate to thermodynamically stable benzyl nickel V (generated through oxidative addition of the C–Br bond to the Ni0 and single-electron reduction followed by chain-walking) to form intermediate VI, which undergoes regioselective migratory insertion to produce a carbo-nickelation adduct (VII).5 Subsequently, β-fluoride elimination could lead to the formation of the monofluoroalkene 3.10 Reduction of Ni–F (VIII) could then regenerate the catalytically active Ni0 species (I) (Scheme 2). However, we anticipated that a few challenges would have to be overcome to realize this strategy: (1) a nickel catalyst must be able to mediate all the elementary steps including chain-walking and defluorinative coupling; (2) the resulting monofluoroalkene product should be unreactive towards the Ni–H as well as Ni-alkyl species; (3) the migration of Ni–H along the carbon chain should be much faster than the alkenylation step to ensure good regioselectivity. In this report, we disclose such a convenient synthetic route for the migratory fluoro-alkenylation of unactivated alkyl bromides with high regio- and stereocontrol (Scheme 1b). Remarkably, by virtue of this operationally simple methodology a variety of structurally privileged monofluoroalkenes11 could be obtained from easily available materials.
Scheme 2. Reaction design.

Results and discussion
To test our hypothesis, we selected gem-difluoroalkene 2a as an alkenylating reagent to react with 1-bromo-2-phenylethane 1a (Table 1). After careful evaluation of the reaction parameters, we found that a combination of inexpensive and bench-stable Ni(ClO4)2·6H2O as a pre-catalyst, 6,6′-dimethyl-2,2′-bipyridyl (L1) as a ligand, and Mn as a reducing agent to generate Ni0 in DMA at 25 °C gave the benzylic fluoro-alkenylation product 3a with Z-configuration in 28% NMR yield within 12 hours (entry 1).12 Pleasingly, the product was obtained with excellent regioselectivity and stereoselectivity. Furthermore, it was found that introduction of MgCl2 as an additive13 gave an improved yield (44%), thus indicating that the Lewis acid has a positive effect on the reaction yield. Encouraged by this result, extensive screening of Lewis acid additives was conducted (Table S1†), and Yb(OTf)3 led to a favorable result (70% yield). The application of YbCl3 afforded 3a in comparable yield (63%), reflecting the importance of the ytterbium cation (entry 4). On the other hand, changing the reducing agents to Zn, B2pin2 or HCOONa had a deleterious effect on the outcome of the reaction (Table S1†). Moreover, Ni(ClO4)2·6H2O proved to be the optimal catalyst after examination of various nickel salts (Table S1†). Subsequently, the ligand was further optimized. Increasing the steric profile of the substituent at the ortho position to the nitrogen in the bipyridyl scaffold (L2) hampered the reactivity of this alkenylation reaction, and no product was detected when the reaction was treated with bipyridyl (L3) as a ligand, suggesting the essential role of such substituents (entries 5 and 6). The structural analogue neocuproine (L4) also exhibited high catalytic efficiency while bathocuproine (L5) was less effective (entries 7 and 8). Slightly higher yields of 3a were obtained when the reaction was treated with fewer equivalents of Mn (entry 9). Of particular note, the process is readily scaled up, the reaction of 1a (2.5 mmol) with 2a (1.0 mmol) gave 3a in 70% yield (entry 10).
Table 1. Optimization of reaction conditions a .
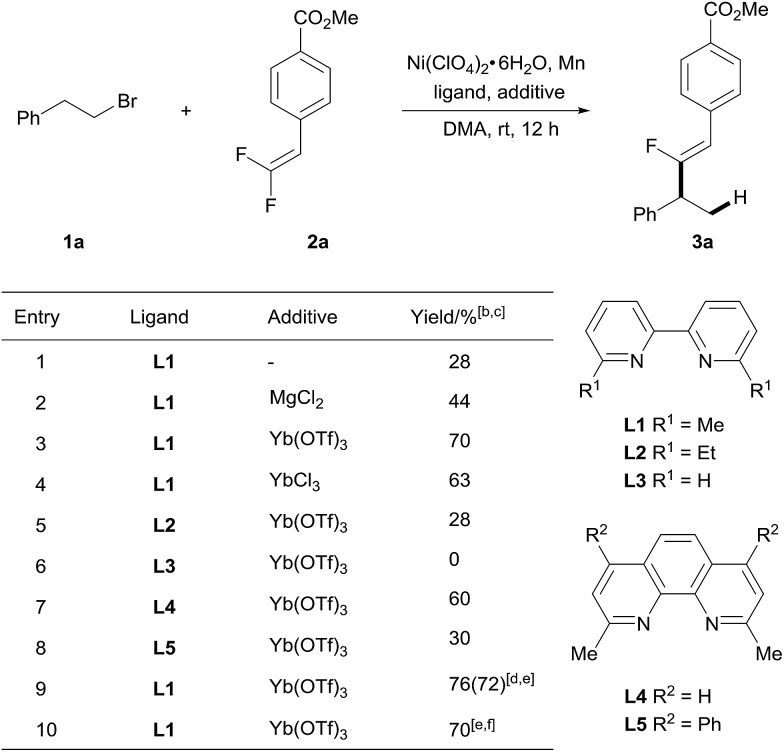
|
aUnless otherwise noted, reactions were carried out with 0.5 mmol of 1a, 0.2 mmol of 2a, 5 mol% of Ni(ClO4)2·6H2O, 6 mol% of ligand, 0.5 mmol of Mn powder and 0.1 mmol of additive in 1 mL of DMA at 25 °C for 12 h.
bDetermined by 1H NMR versus an internal standard.
cThe regioisomeric ratio was higher than 100 : 1 according to 19F NMR of the crude product.
d0.48 mmol of Mn powder was used.
eIsolated yield.
f1.0 mmol scale reaction.
By using the optimized reaction conditions, the scope of this Ni-catalyzed migratory fluoro-alkenylation of unactivated alkyl bromide with gem-difluoroalkenes was evaluated. As shown in Scheme 3, the present protocol shows a remarkably broad scope with respect to the gem-difluoroalkene coupling partner. 1-Aryl-2-bromoethane was initially coupled with various aryl-gem-difluoroalkene derivatives to afford the fluoro-alkenylation products (3a–r). The method tolerated a variety of substitution patterns on the phenyl group. In general, the reaction displayed a noticeable preference for substrates bearing electron-withdrawing groups, such as cyano, trifluoromethyl, ester and ketone, which is in sharp contrast to radical-associated alkylation wherein electron-rich gem-difluoroalkenes were favored.8i For example, para-substituted phenyl-gem-difluoroalkenes were converted into the corresponding products (3a, 3d, 3g, 3j and 3k), mostly in moderate to good yields. The use of meta-substituted phenyl-gem-difluoroalkenes was also explored, which led to moderate yields of products (3b, 3e and 3h). Notably, sterically demanding starting materials that bear ortho-substituents on the phenyl ring were amenable under the optimized reaction conditions; however, the yields of the desired products were reduced (3c, 3f and 3i). Chlorine and fluorine substituents were also compatible in the present reaction (3l and 3m). Moreover, aryl-gem-difluoroalkenes with electron-donating substitution, including OMe, OTs and even the protic amide NHAc, were employed, and these transformations took place smoothly, leading to the compounds 3n–3p with moderate yields. Additionally, nitrogen-containing heterocycle derived substrates could also be converted by the catalytic system (3q and 3r). We next aimed to extend the scope of alkyl bromides. A range of β-bromoethylarenes having Me, OMe, OTBS, Cl and CF3 groups on the phenyl ring were subjected to the alkenylation reaction with an acetyl or a methoxylcarbonyl phenyl-gem-difluoroalkene (2a and 2j). The products 3s–3z were obtained in useful yields with excellent regioselectivity and stereoselectivity. It is worth mentioning that an unprotected hydroxyl group was also tolerated, despite affording the product in decreased yield. To further expand the scope of the alkyl bromides, long range alkenylation was then examined. Alkenylation occurred favorably at the benzylic position, providing the corresponding fluoro-alkenylated propane, butane and even pentane derivatives (3aa–ac). The yields decreased progressively as the carbon chain increased which might be attributed to the increased bulkiness of the benzyl–nickel intermediate. The yields of other related regioisomers were only slightly increased according to 19F NMR analysis of the crude product. These results suggest that formation of the benzyl-Ni species is faster compared to the alkenylation step. Then the more challenging secondary alkyl bromide was subjected to an alkenylation reaction with 2j, and proved to be competent in this reaction, providing the expected benzylic fluoro-alkenylation product with good regioselectivity. The relatively lower yield of 3aa from the secondary alkyl bromide (35% and 30% from (2-bromobutyl)benzene and (3-bromobutyl)benzene, respectively) than the primary one (40% from (4-bromobutyl)benzene) may indicate that the oxidative addition of the alkyl bromide to nickel is sensitive to the steric bulkiness, thus attenuating the reaction efficiency.
Scheme 3. Substrate scope. See the ESI† for experimental details. Isolated yields are indicated. The regioisomeric ratio (rr, the ratio of the benzylic fluoro-alkenylation product to the other regioisomers) was determined by 19F NMR analysis of the crude product. PMP = p-methoxyphenyl. Ts = tosyl. TBS = tert-butyldimethylsilyl.

The synthetic utility of the fluoro-alkenylation product was exemplified by further transformations of 3a (Scheme 4). Hydrogenation of the fluoroalkene moiety contained within 3a was carried out (H2, Pd/C), yielding 4a. An epoxidation reaction using 3a also proceeded well to give fluoroepoxide 4b with high yield (88%). In addition, dibromination of the C C double bond with bromine was executed and the addition product 4c was formed uneventfully. Moreover, treatment of 3a with a base to eliminate HF furnished the synthetically useful trisubstituted allene 4d in 70% yield. This sequence allows gem-difluoroalkenes to serve as a vinylidene source, and thereby an expedient migratory vinylidenation was achieved. Finally, we found that 3a could be converted into 1,2-diketone 4e by oxidation.
Scheme 4. Synthetic applications.

To gain preliminary insight into the unique fluorine effects14 that enable the migratory fluoro-alkenylation reaction, several control experiments were then performed. First, we carried out the alkenylation reaction with a series of halogenated alkenes (Scheme 5a–c). As shown in Scheme 5a, gem-chloroalkene 5a and gem-dibromoalkene 5b did not lead to the desired halo-alkenylation product. Second, we examined monohaloalkenes under the standard conditions. Not surprisingly, in these experiments no alkenylation product was detected again upon the consumption of the starting materials 7a–c (Scheme 5b).15 The comparison with various halogenated alkenes highlights the prominent role of the two fluorine atoms which entail the unique reactivity in migratory fluoro-alkenylation. Regarding the C(sp2)–C(sp3) bond formation, an alternative pathway involving oxidative addition of the C(sp2)–F bond to the benzyl–Ni followed by reductive elimination is also possible.8c,e However, the 1-bromo-1-fluoroalkene 9 failed to produce the fluoro-alkenylation product which could tentatively rule out this mechanism (Scheme 5c).16 The use of deuterium labelled alkyl bromide D2-1a gave rise to the deuterium-shift product D2-3a exclusively, which strongly supports idea that a process involving β-hydride elimination and reinsertion is operative in the present transformation (Scheme 5d). Note that no significant further hydrogen/deuterium scrambling was found in D2-3a, revealing the thermodynamic preference of the benzylic-Ni intermediate in the migration process, which intrinsically dictates the regioselectivity of this transformation. Given the Ni-catalyzed chain-walking process does not require a Lewis acid additive,4–6 it is reasonable to attribute the role of Yb(OTf)3 in activating gem-difluoroalkene towards the nucleophilic addition17 or facilitating the reduction of Ni–F species.18 To obtain additional insight into the influence of Yb(OTf)3, reactions with a stoichiometric amount of Ni(ClO4)2·6H2O/L1 were carried out (see the ESI† for details). In the reaction with Yb(OTf)3, the desired product (3a) was attained in 55% yield which is much higher than the ytterbium-free reaction (16%), implying that the Lewis acid should take effect in the nucleophilic addition step rather than the Ni–F reduction. To evaluate the ability of Ni–H to recognize the non-fluorinated alkenes and gem-difluoroalkenes, styrene 10 was subjected to the fluoro-alkenylation reaction with 1-bromopropane as the hydride source (Scheme 5e).5,19 With the same catalytic system, the desired product 3a was formed in 40% NMR yield. This result clearly demonstrates the significant impact of fluorine substituents on the alkene moiety, which differentiate the two kinds of alkenes towards the Ni–H species as proposed in Scheme 1b.
Scheme 5. Control experiments (Ar = 4-MeOCOC6H4).

Conclusions
In conclusion, a general, Ni-catalyzed migratory fluoro-alkenylation of unactivated alkyl bromides with gem-difluoroalkenes has been developed, providing ready access to diversely functionalized monofluoroalkenes, which are valuable molecules in biological and materials science. More importantly, this work extends the boundaries of the highly attractive field of remote functionalization of unactivated alkyl electrophiles since it represents the first instance in which an alkene coupling partner has been used in a NiH-mediated process. It is also noteworthy that this C(sp2)–C(sp3) bond-forming reaction proceeds with excellent regio- and stereoselectivity under nonbasic conditions at room temperature, and an array of potentially reactive functional groups are tolerated.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The work was supported by the “Thousand Talents Plan” Youth Program, the “Jiangsu Specially-Appointed Professor Plan”, the Natural Science Foundation of Jiangsu Province (BK20170984 and BK20170992), the Start-up Grant from Nanjing Tech University and the SICAM Fellowship from Jiangsu National Synergetic Innovation Center for Advanced Materials.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04162h
References
- (a) Everson D. A., Weix D. J. J. Org. Chem. 2014;79:4793. doi: 10.1021/jo500507s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Knappke C. E., Grupe S., Gartner D., Corpet M., Gosmini C., Jacobi von Wangelin A. Chem.–Eur. J. 2014;20:6828. doi: 10.1002/chem.201402302. [DOI] [PubMed] [Google Scholar]; (c) Weix D. J. Acc. Chem. Res. 2015;48:1767. doi: 10.1021/acs.accounts.5b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Moragas T., Correa A., Martin R. Chem.–Eur. J. 2014;20:8242. doi: 10.1002/chem.201402509. [DOI] [PubMed] [Google Scholar]; (b) Lucas E. L., Jarvo E. R. Nat. Rev. Chem. 2017;1:65. [Google Scholar]
- For reviews, see: ; (a) Franzoni I., Mazet C. Org. Biomol. Chem. 2014;12:233. doi: 10.1039/c3ob42050g. [DOI] [PubMed] [Google Scholar]; (b) Vasseur A., Bruffaerts J., Marek I. Nat. Chem. 2016;8:209. doi: 10.1038/nchem.2445. [DOI] [PubMed] [Google Scholar]; (c) Sommer H., Juliá-Hernández F., Martin R., Marek I., ACS Cent. Sci., 2018, 4 , 153 , ; for selected examples, see: . [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ilies L., Chen Q., Zeng X., Nakamura E. J. Am. Chem. Soc. 2011;133:5221. doi: 10.1021/ja200645w. [DOI] [PubMed] [Google Scholar]; (e) Werner E. W., Mei T.-S., Burckle A. J., Sigman M. S. Science. 2012;338:1455. doi: 10.1126/science.1229208. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Mei T.-S., Werner E. W., Burckle A. J., Sigman M. S. J. Am. Chem. Soc. 2013;135:6830. doi: 10.1021/ja402916z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Mei T.-S., Patel H. H., Sigman M. S. Nature. 2014;508:340. doi: 10.1038/nature13231. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Larionov E., Lin L., Guénée L., Mazet C. J. Am. Chem. Soc. 2014;136:16882. doi: 10.1021/ja508736u. [DOI] [PubMed] [Google Scholar]; (i) Patel H. H., Sigman M. S. J. Am. Chem. Soc. 2015;137:3462. doi: 10.1021/ja5130836. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Lin L., Romano C., Mazet C. J. Am. Chem. Soc. 2016;138:10344. doi: 10.1021/jacs.6b06390. [DOI] [PubMed] [Google Scholar]; (k) Aspin S., Goutierre A.-S., Larini P., Jazzar R., Baudoin O. Angew. Chem., Int. Ed. 2012;51:10808. doi: 10.1002/anie.201206237. [DOI] [PubMed] [Google Scholar]; (l) Dupuy S., Zhang K.-F., Goutierre A.-S., Baudoin O. Angew. Chem., Int. Ed. 2016;55:14793. doi: 10.1002/anie.201608535. [DOI] [PubMed] [Google Scholar]; (m) Borah A. J., Shi Z. J. Am. Chem. Soc. 2018;140:6062. doi: 10.1021/jacs.8b03560. [DOI] [PubMed] [Google Scholar]; (n) He Y., Cai Y., Zhu S. J. Am. Chem. Soc. 2017;139:1061. doi: 10.1021/jacs.6b11962. [DOI] [PubMed] [Google Scholar]; (o) Xiao J., He Y., Ye F., Zhu S. Chem. 2018;4:1645. [Google Scholar]; (p) Zhou F., Zhu J., Zhang Y., Zhu S. Angew. Chem., Int. Ed. 2018;57:4058. doi: 10.1002/anie.201712731. [DOI] [PubMed] [Google Scholar]
- (a) Juliá-Hernández F., Moragas T., Cornella J., Martin R. Nature. 2017;545:84. doi: 10.1038/nature22316. [DOI] [PubMed] [Google Scholar]; (b) Gaydou M., Moragas T., Juliá-Hernández F., Martin R. J. Am. Chem. Soc. 2017;139:12161. doi: 10.1021/jacs.7b07637. [DOI] [PubMed] [Google Scholar]
- Chen F., Chen K., Zhang Y., He Y., Wang Y.-M., Zhu S. J. Am. Chem. Soc. 2017;139:13929. doi: 10.1021/jacs.7b08064. [DOI] [PubMed] [Google Scholar]
- (a) Peng L., Li Y., Li Y., Wang W., Pang H., Yin G. ACS Catal. 2018;8:310. [Google Scholar]; (b) Peng L., Li Z., Yin G. Org. Lett. 2018;20:1880. doi: 10.1021/acs.orglett.8b00413. [DOI] [PubMed] [Google Scholar]
- (a) Pappas I., Treacy S., Chirik P. J. ACS Catal. 2016;6:4105. [Google Scholar]; (b) Léonard N. G., Chirik P. J. ACS Catal. 2018;8:342. [Google Scholar]
- (a) Amii H., Uneyama K. Chem. Rev. 2009;109:2119. doi: 10.1021/cr800388c. [DOI] [PubMed] [Google Scholar]; (b) Zhang X., Cao S. Tetrahedron Lett. 2017;58:375. doi: 10.1016/j.tetlet.2017.04.096. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dai W., Xiao J., Jin G., Wu J., Cao S. J. Org. Chem. 2014;79:10537. doi: 10.1021/jo5022234. [DOI] [PubMed] [Google Scholar]; (d) Tian P., Feng C., Loh T.-P. Nat. Commun. 2015;6:7472. doi: 10.1038/ncomms8472. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Xiong Y., Huang T., Ji X., Wu J., Cao S. Org. Biomol. Chem. 2015;13:7389. doi: 10.1039/c5ob01016k. [DOI] [PubMed] [Google Scholar]; (f) Thornbury R. T., Toste F. D. Angew. Chem., Int. Ed. 2016;55:11629. doi: 10.1002/anie.201605651. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Dai W., Shi H., Zhao X., Cao S. Org. Lett. 2016;18:4284. doi: 10.1021/acs.orglett.6b02026. [DOI] [PubMed] [Google Scholar]; (h) Cai S.-H., Ye L., Wang D.-X., Wang Y.-Q., Lai L.-J., Zhu C., Feng C., Loh T.-P. Chem. Commun. 2017;53:8731. doi: 10.1039/c7cc04131d. [DOI] [PubMed] [Google Scholar]; (i) Lu X., Wang Y., Zhang B., Pi J.-J., Wang X.-X., Gong T.-J., Xiao B., Fu Y. J. Am. Chem. Soc. 2017;139:12632. doi: 10.1021/jacs.7b06469. [DOI] [PubMed] [Google Scholar]; (j) Yu L., Tang M.-L., Si C.-M., Meng Z., Liang Y., Han J., Sun X. Org. Lett. 2018;20:4579. doi: 10.1021/acs.orglett.8b01866. [DOI] [PubMed] [Google Scholar]; (k) Yang L., Ji W.-W., Lin E., Li J.-L., Fan W.-X., Li Q., Wang H. Org. Lett. 2018;20:1924. doi: 10.1021/acs.orglett.8b00471. [DOI] [PubMed] [Google Scholar]
- (a) Guo S., Yang P., Zhou J. Chem. Commun. 2015;51:12115. doi: 10.1039/c5cc01632k. [DOI] [PubMed] [Google Scholar]; (b) Ahrens T., Teltewskoi M., Ahrens M., Braun T., Laubenstein R. Dalton Trans. 2016;45:17495. doi: 10.1039/c6dt03027k. [DOI] [PubMed] [Google Scholar]
- For review, see: ; (a) Ahrens T., Kohlmann J., Ahrens M., Braun T., Chem. Rev., 2015, 115 , 931 , ; for Ni-catalyzed β-F elimination, see: . [DOI] [PubMed] [Google Scholar]; (b) Ichitsuka T., Fujita T., Arita T., Ichikawa J. Angew. Chem., Int. Ed. 2014;53:7564. doi: 10.1002/anie.201402695. [DOI] [PubMed] [Google Scholar]; (c) Watabe Y., Kanazawa K., Fujita T., Ichikawa J. Synthesis. 2017;49:3569. [Google Scholar]; (d) Ichitsuka T., Fujita T., Ichikawa J. ACS Catal. 2015;5:5947. [Google Scholar]; (e) Ohashi M., Ueda Y., Ogoshi S. Angew. Chem., Int. Ed. 2017;56:2435. doi: 10.1002/anie.201610047. [DOI] [PubMed] [Google Scholar]; (f) Lan Y., Yang F., Wang C. ACS Catal. 2018;8:9245. [Google Scholar]
- (a) Bégué J.-P. and Bonnet-Delpon D., Bioorganic and Medicinal Chemistry of Fluorine, Wiley, Hoboken, 2008. [Google Scholar]; (b) Landelle G., Bergeron M., Turcotte-Savard M.-O., Paquin J.-F. Chem. Soc. Rev. 2011;40:2867. doi: 10.1039/c0cs00201a. [DOI] [PubMed] [Google Scholar]; (c) Malo-Forest B., Landelle G., Roy J.-A., Lacroix J., Gaudreault R. C., Paquin J.-F. Bioorg. Med. Chem. Lett. 2013;23:1712. doi: 10.1016/j.bmcl.2013.01.057. [DOI] [PubMed] [Google Scholar]; (d) Osada S., Sano S., Ueyama M., Chuman Y., Kodama H., Sakaguchi K. Bioorg. Med. Chem. 2010;18:605. doi: 10.1016/j.bmc.2009.12.005. [DOI] [PubMed] [Google Scholar]
- The Z-configuration of 3a was determined by comparing the NMR data with structurally analogous compounds reported in the literature. Larnaud F., Malassis J., Pfund E., Linclau B., Lequeux T., Org. Lett., 2013, 15 , 2450 . [DOI] [PubMed] [Google Scholar]
- (a) León T., Correa A., Martin R. J. Am. Chem. Soc. 2013;135:1221. doi: 10.1021/ja311045f. [DOI] [PubMed] [Google Scholar]; (b) Sayyed F. B., Sakaki S. Chem. Commun. 2014;50:13026. doi: 10.1039/c4cc04962d. [DOI] [PubMed] [Google Scholar]
- (a) Orsi D. L., Altman R. A. Chem. Commun. 2017;53:7168. doi: 10.1039/c7cc02341c. [DOI] [PubMed] [Google Scholar]; (b) Volla C. M. R., Das A., Atodiresei I., Rueping M. Chem. Commun. 2014;50:7889. doi: 10.1039/c4cc03229b. [DOI] [PubMed] [Google Scholar]; (c) Crich D., Vinogradova O. J. Am. Chem. Soc. 2007;129:11756. doi: 10.1021/ja0730258. [DOI] [PubMed] [Google Scholar]; (d) Yu J.-S., Liu Y.-L., Tang J., Wang X., Zhou J. Angew. Chem., Int. Ed. 2014;53:9512. doi: 10.1002/anie.201404432. [DOI] [PubMed] [Google Scholar]
- (a) Liu J., Ren Q., Zhang X., Gong H. Angew. Chem., Int. Ed. 2016;55:15544. doi: 10.1002/anie.201607959. [DOI] [PubMed] [Google Scholar]; (b) Hofstra J. L., Cherney A. H., Ordner C. M., Reisman S. E. J. Am. Chem. Soc. 2018;140:139. doi: 10.1021/jacs.7b11707. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cherney A. H., Reisman S. E. J. Am. Chem. Soc. 2014;136:14365. doi: 10.1021/ja508067c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Considering the reaction exhibits complete Z-selectivity, Ni-catalyzed β-F elimination through intermediate VII is most likely the pathway for the monofluoroalkene formation as suggested by ref. 8d, f and i.
- (a) Deacon G. B., Forsyth C. M., Junk P. C., Wang J. Chem.–Eur. J. 2009;15:3082. doi: 10.1002/chem.200802294. [DOI] [PubMed] [Google Scholar]; (b) Träff A. M., Janjetovic M., Ta L., Hilmersson G. Angew. Chem., Int. Ed. 2013;52:12073. doi: 10.1002/anie.201306104. [DOI] [PubMed] [Google Scholar]
- Tobisu M., Xu T., Shimasaki T., Chatani N. J. Am. Chem. Soc. 2011;133:19505. doi: 10.1021/ja207759e. [DOI] [PubMed] [Google Scholar]
- Wang X., Nakajima M., Serrano E., Martin R. J. Am. Chem. Soc. 2016;138:15531. doi: 10.1021/jacs.6b10351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.