

Abstract
Unfractionated heparin (UFH), the standard anticoagulant for cardiopulmonary bypass (CPB) surgery, carries a risk of post-operative bleeding and is potentially harmful in patients with heparin-induced thrombocytopenia-associated antibodies. To improve the activity of an alternative anticoagulant, the RNA aptamer 11F7t, we solved X-ray crystal structures of the aptamer bound to factor Xa (FXa). The finding that 11F7t does not bind the catalytic site suggested it could complement small-molecule FXa inhibitors. We demonstrate that combinations of 11F7t and catalytic-site FXa inhibitors enhance anticoagulation in purified reaction mixtures and plasma. Aptamer-drug combinations prevent clot formation as effectively as UFH in human blood circulated in an extracorporeal oxygenator circuit that mimics CPB, while avoiding side effects of UFH. An antidote can promptly neutralize the anticoagulant effects of both FXa inhibitors. Our results suggest that drugs and aptamers with shared targets can be combined to exert more specific and potent effects than either agent alone.
Unfractionated heparin (UFH) is the most potent and frequently administered intravenous anticoagulant for patients who require rapid onset systemic anticoagulation1. The most thrombogenic indication for UFH is cardiopulmonary bypass (CPB) surgery, which is performed annually on over a million patients worldwide2,3. CPB surgery triggers fulminant activation of both the intrinsic (contact-mediated) and extrinsic (tissue factor-mediated) coagulation pathways due to extracorporeal blood circulation and surgical trauma respectively4-8. UFH achieves its robust anticoagulant effect through an indirect, mechanism mediated by antithrombin (AT)2,9,10and accelerates AT-mediated irreversible inhibition of multiple procoagulant proteases including thrombin, factor (F)Xa, FIXa, FVIIa, and FXIa2,9,10. However UFH has several limitations that contribute to morbidity and mortality associated with CPB2,5,6,8,9,11-15.
The major drawbacks of UFH-facilitated CPB include that UFH cannot prevent continuous generation of thrombin during CPB, partly due to its ineffectiveness inhibiting FXa within prothrombinase and clot-bound thrombin6,8,12,16,17. Thrombin generation exacerbates complications of CPB and clotting factor consumption predisposes to post-operative bleeding8,18,19. UFH’s depletion of AT during CPB can diminish its efficacy2,5,9,20. Prolonged or repeated exposure to UFH can trigger heparin-induced thrombocytopenia (HIT), a potentially life-threating antibody-mediated thrombotic syndrome6,21. Moreover, UFH’s antidote, protamine, is associated with toxicities, and despite its routine administration for UFH reversal, post-operative bleeding remains a major adverse event after CPB6,14,15,22-25. Thus, a potent, antidote-controllable anticoagulant alternative to UFH without these limitations remains an unmet medical need.
Because UFH’s efficacy lies in its multimodal ability to enhance inhibition of thrombin and the proteinases responsible for thrombin formation26-28, an anticoagulation strategy intended to match UFH’s potency will also likely need to act at multiple steps that lead to thrombin formation. We previously identified an anticoagulant RNA aptamer, 11F7t, that binds a FXa exosite and the corresponding FX proexosite29. 11F7t inhibits prothrombinase formation by inhibiting (1) the binding of FXa to FVa, (2) FXa-catalyzed cleavage of FVIII, and (3) activation of FX by intrinsic tenase29. Through these multiple mechanisms, 11F7t achieves a substantial anticoagulant effect, although less potent than UFH’s29,30. Here, we tested whether addition of a FXa active site inhibitor might augment the anticoagulant intensity of 11F7t. Active site inhibitors of FXa, which include the small molecule drugs rivaroxaban, apixaban, or edoxaban, are already clinically approved31. We also investigate whether the combination of 11F7t plus a FXa active site inhibitor can be effectively and concomitantly neutralized by GD-FXaS195A, an inactive FXa variant that resembles an antidote for FXa inhibitors in late stage clinical trials32-35.
Results
Structure of aptamer 11F7t bound to GD-FXaS195A
We determined a high-resolution X-ray structure of 11F7t complexed with GD-FXaS195A, a FXa variant that lacks the membrane binding γ-carboxyglutamic acid (Gla) domain and contains an alanine substitution at the catalytic serine (Fig. 1a, Table S1, Fig. S1a). Molecular replacement was used to solve the structure at 2.0 Å resolution. The tertiary fold of 11F7t presents an extended molecular surface for interactions with GD-FXaS195A with ~1400 Å2 of solvent accessible surface area buried within the complex. Contacts between the aptamer and the protein are specified by 15 H-bonds with nucleotide bases in a central loop encompassing C8, A10, A21, C28-C30 and proteinase domain residues L59, R64, V88, I89, N92, R93, K236 and R240 (Fig. S1a). This broad surface includes residues implicated in heparin binding and in binding FVa based on modest changes in function upon mutagenesis36.
Figure 1: X-ray crystal structures of aptamer 11F7t bound to GD-FXaS195A both in the absence (a) and presence (b) of rivaroxaban.

The proteinase domain of GD-FXaS195A is depicted in the standard orientation. Residues that comprise the catalytic site are illustrated as red sticks, and residues that form hydrogen bonds with 11F7t are denoted as blue sticks. Bound Na+ (purple), Ca2+ (green) and rivaroxaban (yellow) are rendered as spheres.
11F7t binding does not appear to occlude the catalytic triad or access to the S1 binding pocket. In line with this observation, we were also able to crystallize and solve the structure of a ternary complex between GD-FXaS195A, 11F7t, and rivaroxaban bound to the active site of the proteinase at 2.25 Å resolution (Fig. 1b, Table S1, Fig. S1b). The proteinase-aptamer complex in the two structures are essentially identical (RMSD = 0.17 Å), as is the proteinase-rivaroxaban component within the ternary complex and a previously published structure of FXa bound to rivaroxaban (RMSD = 0.29 Å)37. Therefore, 11F7t and the active site ligand likely bind independently to FXa to separately inhibit macromolecular interactions and catalytic function. Structural evidence indicates that rivaroxaban and apixaban bind to FXa in a very similar way and both are also chemically analogous to the third FXa active site inhibitor, edoxaban, approved for clinical use31,38.
The interface between 11F7t and FXa is distant from the 165 helix of the proteinase domain, previously proposed to be essential for its binding to FVa36 and also implicated in the interaction of FVIIa and FIXa with their respective cofactors. A minimally two-part interaction between proteinase and cofactor is implied by the recent structure of the snake venom orthologs of FV bound in solution to FX from Pseudonaja textilis39. In addition to contacts between the 165 helix and the body of the FV component, an additional and extensive contact is made by a peptide from the C-terminus of the A2 domain of FV extending across a second face of the proteinase domain distant from this helix39. Superposition of the structure of GD-FXaS195A bound to 11F7t with the snake venom structure (Fig. S2, Supplementary Discussion) reveals that the aptamer occupies the same surface as the A2 polypeptide from snake venom FV. If such comparisons have merit, they imply a comparable interaction between an acidic sequence at the C terminus of the A2 domain of FVa and an extended surface encompassing the heparin binding face of the proteinase domain of FXa. Despite additional interactions between proteinase and cofactor, occlusion of this surface by 11F7t is sufficient to reduce the affinity of FXa for FVa to an undetectable level29 (Supplementary Discussion)
Potentiated inhibition of thrombin formation
To assess whether the structural findings correctly predict that inhibition by 11F7t may be potentiated if paired with an active site-directed inhibitor of FXa, we measured the effects of each anticoagulant, alone or in combination, using systems of thrombin generation of increasing complexity. We used reaction mixtures containing prothrombin, FVa, and membranes, wherein thrombin formation was initiated by FXa addition. The robust initial velocity of thrombin formation seen in the absence of inhibitors was partially inhibited by apixaban or 11F7t added individually (Fig. 2a & 2b), but the extent of inhibition was far greater (>90%) when both apixaban and 11F7t were present.
Figure 2: Inhibitory effects of 11F7t and apixaban and anticoagulant reversal.

(a-b): Thrombin formation was measured following addition of 0.2 nM FXa to reaction mixtures containing 1.4 μM prothrombin, 30 μM PCPS, 9 nM FVa, and either no anticoagulant or one of the following anticoagulant strategies: 20 nM apixaban (apix) alone, 2 μM 11F7t alone, or 20 nM apixaban plus 2 μM 11F7t in n = 2 independent experiments. Panel (a) illustrates progress curves for thrombin formation over time, and panel (b) displays the initial velocities of thrombin generation in the presence of each anticoagulation strategy. The latter results are presented after normalization as V/Vo, where Vo is the initial rate observed in the absence of an anticoagulant. (c-f): Thrombin formation was measured following addition of 2 nM FIXa to reaction mixtures containing 1.4 μM prothrombin, 30 μM PCPS, 9 nM FVa, 0.9 nM FVIII, 110 nM FX, and either no anticoagulant (n = 15) or the same anticoagulation strategies tested in the FXa-initiated system (a-b): 20 nM apixaban alone (n = 6), 2 μM 11F7t alone (n = 5), or 20 nM apixaban plus 2 μM 11F7t (n = 6). Panel (c) depicts progress curves for thrombin generation, while panels (d) and (e) respectively compare the time required for the thrombin concentration to reach 10 nM and the total thrombin concentration generated after 10 minutes in the presence of each anticoagulation strategy within the FXa- and FIXa-initiated systems. Panel (f) displays the effect of GD-FXaS195A (2 μM) or the 11F7t-specific antidote oligonucleotide AO5-2 (10 μM) on neutralization of 2 μM 11F7t plus 20 nM apixaban in the FIXa-initiated system of thrombin generation in n = 2 independent experiments. The same concentration of GD-FXaS195A did not appreciably affect thrombin formation when added in the absence of anticoagulants. In all panels, error bars indicate standard error. Statistical analysis was performed by one-way ANOVA followed by Tukey’s multiple comparisons test.
We then measured thrombin formation in reaction mixtures containing prothrombin, FX, FVa and FVIII supplemented with membranes and initiated by the addition of FIXa (Fig. 2c). In the absence of an anticoagulant, the time required for 10 nM thrombin formation following initiation with FIXa was approximately 0.5 min (Fig. 2d). While apixaban or 11F7t alone prolonged that interval significantly (~ 1 and 4 min respectively), the largest prolongation to more than 11 min was seen in the combined presence of 11F7t plus apixaban (Fig. 2d). Similarly, the combination of apixaban and 11F7t produced a striking 99% inhibition of thrombin formation 10 min after initiation (Fig. 2e).
Inhibitor neutralization
Two independent strategies have been employed to neutralize the anticoagulant effects of active site-directed inhibitors of FXa and of inhibitory aptamers targeting coagulation enzymes. The first employs a catalytically inactive decoy protein like GD-FXaS195A capable of engaging inhibitors at the active site but unable to bind membranes and FVa with high affinity32. In this case, GD-FXaS195A scavenges the circulating inhibitor, without interfering with the function of endogenous FXa, to restore normal clotting. In the second, sequence-specific complementary oligonucleotides are used to disrupt aptamer secondary structure and binding to its target40. We compared the ability of the 11F7t-specific antidote oligonucleotide, AO5-241, and GD-FXaS195A to reverse the anticoagulant effect produced by 11F7t plus apixaban in the FIXa-triggered model of thrombin generation (Fig. 2f). AO5-2 partially restored thrombin generation to levels similar to those observed in the presence of apixaban alone, in accord with its specific neutralization of aptamer-dependent inhibition. In contrast, GD-FXaS195A completely reversed the inhibitory effects of both 11F7t and apixaban, and restored thrombin generation to levels observed in the absence of an anticoagulant.
Inhibition of TF-initiated thrombin generation
The above reconstituted systems assess inhibition of thrombin formation but do not include the contribution of FX activation by the FVIIa/TF pathway. Because a viable anticoagulation strategy for CPB must inhibit both extrinsic and intrinsic hemostatic activation, we compared the ability of UFH with that of 11F7t plus a FXa active site inhibitor to limit thrombin generation triggered by adding TF to platelet rich plasma (PRP) in Calibrated Automated Thrombogram (CAT) assays42. In the absence of an anticoagulant, addition of 50 pM TF to PRP elicited a robust rise and fall in active thrombin concentration over the ensuing 60 minutes, reflecting thrombin’s formation by procoagulant proteinases and its subsequent inactivation by plasma serpins (Fig. 3a). The area under the curve, referred to as the endogenous thrombin potential (ETP)42, was 1789.0 ± 119.7 nM·min (mean ± SE). The presence of either UFH alone, 11F7t alone, or a FXa active site inhibitor (rivaroxaban, apixaban, or edoxaban) alone resulted in a concentration-dependent decrease in the ETP (Fig. S3a, S3b). UFH (5U/ml), the approximate circulating concentration during clinical CPB, reduced the ETP and thrombin generation to undetectable levels in all assays (Fig. 3a, 3b). In comparison, 2μM 11F7t alone, 2μM rivaroxaban alone, 2 μM apixaban alone and 2μM edoxaban alone only modestly, reduced the ETP to 995.5 ± 263.0 nM·min, 1251.6 ± 97.4 nM·min, 1041.5 ± 181.1 nM·min, and 1137.2 ± 169.7 nM·min respectively (Fig. 3a, 3b, S3b). Equimolar combinations of 11F7t and each FXa active site inhibitor (0.25 μM or 0.5 μM) reduced the ETP to lower levels than each inhibitor alone at the same concentrations but did not completely prevent thrombin production (Fig. S4). However, the 1μM combination of 11F7t plus either rivaroxaban, apixaban, or edoxaban completely abrogated thrombin generation, diminishing the ETP to undetectable levels similarly to 5 U/mL UFH (Fig. 3a, 3b). In addition to these inhibitory effects on the ETP, similar trends were also observed with respect to other parameters of thrombin generation evaluated by CAT analysis such as the thrombin lag time, peak thrombin concentration, and time to peak thrombin production42 (Table S2).
Figure 3: Inhibitory effects of 11F7t and a FXa catalytic site inhibitor.
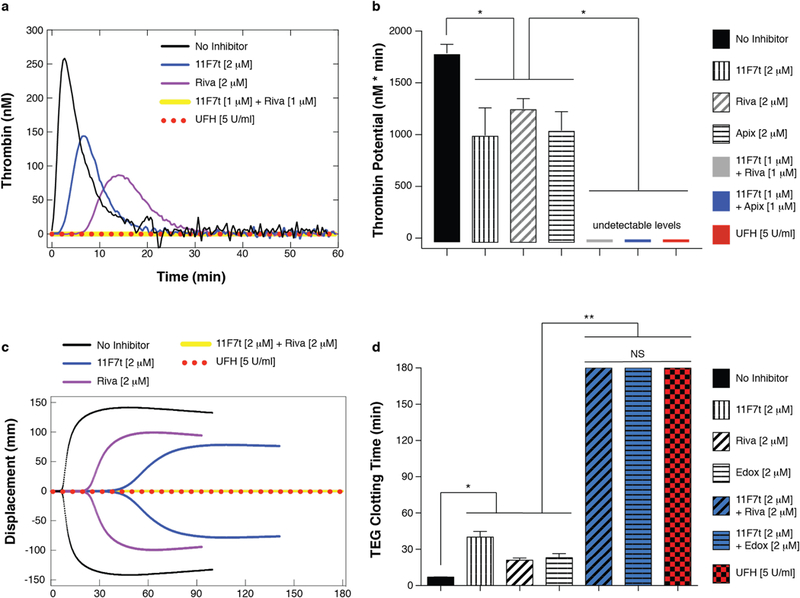
(a-b): Calibrated Automated Thrombogram (CAT) assays were performed to measure thrombin generation in platelet rich plasma (150,000 platelets/μL) following addition of 50 pM TF in the absence of an anticoagulant (n = 6 independent experiments) or in the presence of one of the following anticoagulation strategies: 2 μM 11F7t (n = 3), 2 μM rivaroxaban (riva) (n = 3), 2 μM apixaban (apix) (n = 3), 1 μM 11F7t plus 1 μM rivaroxaban (n = 3), 1 μM 11F7t plus 1 μM apixaban (n = 3), or 5 U/mL UFH (n = 4). Representative mean CAT tracings (a) and the computed endogenous thrombin potential (ETP) or area under the curve (b) for each condition are plotted. (c-d): Thromboelastography (TEG) assays were performed on human whole blood containing no anticoagulant (n = 126 independent experiments) or one of the following anticoagulation strategies: 2 μM 11F7t (n = 17), 2 μM rivaroxaban (riva) (n = 6), 2 μM edoxaban (edox) (n = 4), 2 μM 11F7t plus 2 μM rivaroxaban (n = 4), 2 μM 11F7t plus 2 μM edoxaban (n = 7), or 5 U/mL UFH (n = 4). Representative TEG tracings (c) and the time until detectable clot formation following kaolin-initiated coagulation for each condition are depicted (d), with the maximum time limit of a TEG assay being 180 minutes. In all panels, error bars indicate standard error. NS and * respectively denote “not significant” and p < 0.05 by one-way ANOVA followed by Tukey’s multiple comparisons test. Exact p-values for panels (b) and (d) are provided as follows; b(1) vs b(2): p = 0.002; b(1) vs b(3): p = 0.049; b(1) vs b(4): p = 0.004; b(2) vs b(5), b(2) vs b(6): p = 0.009; b(4) vs b(6): p = 0.0005; b(3) vs b(5), b(2) vs b(7), b(3) vs b(7), b(4) vs b(7), d(1) vs d(2), d(1) vs d(3), d(1) vs d(4), d(2) vs d(5), d(2) vs d(6), d(2) vs d(7), d(3) vs d(5), d(3) vs d(7), d(4) vs d(6), d(4) vs d(7): p < 0.0001; d(5) vs d(7), d(6) vs d(7): p > 0.9999.
Anticoagulation of whole blood
Next, we compared the anticoagulant efficacy of 11F7t, alone and in combination with an active site directed FXa inhibitor, with that of UFH in whole blood using thromboelastography (TEG)43. This assay measures time-dependent, viscoelastic parameters of clot formation after coagulation is triggered by addition of kaolin to stimulate activation of the contact/intrinsic pathway43. In control TEG assays performed on non-anticoagulated whole blood from healthy donors, the time between kaolin addition and detectable clot formation was 7.1 ± 0.2 minutes (mean ± SE) (Fig. 3c, 3d). The presence of 5 U/ml UFH prolonged the TEG clotting time to > 180 min in each assay (Fig. 3c, 3d). When added individually, 11F7t, rivaroxaban or edoxaban each prolonged the TEG clotting time in a dose-dependent manner (Fig. S5a-S5c) but to a lesser extent than UFH. For example, 2 μM 11F7t, 2 μM rivaroxaban, or 2 μM edoxaban increased the TEG clotting time to 40.1 ± 4.7 min, 21.0 ± 1.9 min, and 23.1 ± 3.3 min respectively (Fig. 3b). Equimolar concentrations of 11F7t and either rivaroxaban or edoxaban, at 0.5 μM or 1 μM, prolonged the TEG clotting time to a significantly greater degree than each inhibitor alone at the same concentrations but not to the same extent observed with 5 U/mL UFH (Fig. S5d, S5e). However, the combined presence of 2 μM 11F7t plus either 2 μM rivaroxaban or 2 μM edoxaban each prolonged the TEG clotting time to > 180 minutes in all samples tested (Fig. 3c, 3d). In addition to the inhibitory effects on the clotting time, analogous patterns were also noted in regards to other viscoelastic properties of clot formation measured by TEG analysis such as the maximum amplitude and α angle, which reflect the clot strength and rate of formation respectively43 (Table S3). Hence, even in whole blood, the aptamer and active site-directed inhibitors of FXa yield a combined inhibitory effect that can match the anticoagulant potency achieved by UFH.
For comparison, we also similarly evaluated the effects of bivalirudin, a direct thrombin inhibitor that is occasionally employed as an alternative for CPB anticoagulation in patients with severe contraindications to UFH such as HIT2,6,44. However, bivalirudin is used very cautiously because its use during CPB surgery is associated with a notable risk of clotting in areas prone to blood pooling such as the operative field or CPB circuit2,6,44. Consistent with its weaker potency, even at supra-therapeutic concentrations as high as 36.7 μM, bivalirudin only extended the TEG clotting time to 34.3 ± 4.1 minutes (Fig. S6).
Anticoagulation during extracorporeal circulation
The anticoagulant efficacy of 11F7t plus a FXa active site inhibitor evident from the CAT and TEG studies suggested that this combination of anticoagulants might also achieve anticoagulation potent enough to facilitate CPB. To begin to assess this possibility, we tested the ability of this dual FXa anticoagulant approach to prevent clot formation in fresh human blood subjected to a 2-hour period of continuous recirculation within an ex vivo circuit that includes a membrane oxygenator30. The activated clotting time (ACT) is the assay used to assess the adequacy of anticoagulation during CPB2. The normal ACT range is 100-125 seconds, while an ACT range of 400-480 seconds in the presence of UFH is considered therapeutic for CPB2. As expected, UFH (5U/mL) increased the initial ACT to 462.3 ± 23.3 seconds (mean ± SE) (Fig. 4a) and prevented visually identifiable clot formation in the extracorporeal circuit during the 2-hour recirculation period. Post-circulation scanning electron micrograph (SEM) images of oxygenator membranes from those circuits also showed minimal fibrinous and cellular debris (Fig. 4b). The addition of protamine (0.05 mg/mL) to a post-circulation sample restored the ACT to baseline levels in all cases (Fig. 4a).
Figure 4: Anticoagulant effects during extracorporeal circulation of human blood.
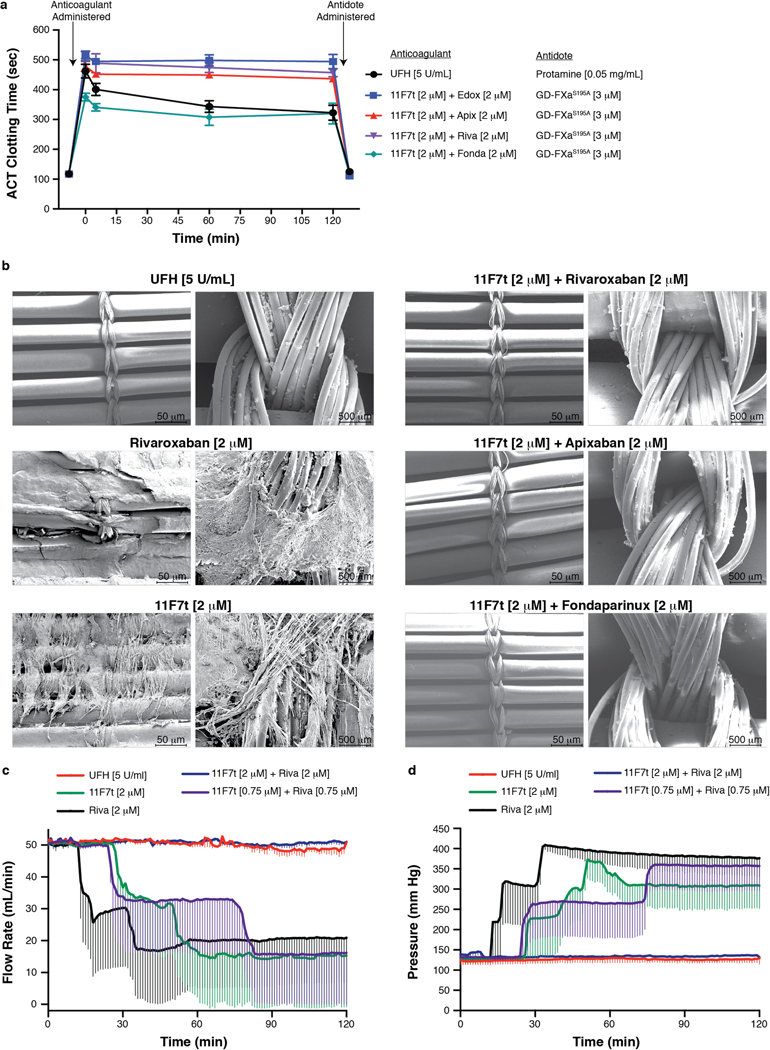
(a-b) Whole blood was anticoagulated with one of the following strategies: 5 U/mL UFH (n = 11 independent experiments), 2 μM 11F7t alone (n = 3), 2 μM rivaroxaban (riva) alone (n = 3), 2 μM 11F7t plus 2 μM rivaroxaban (n = 9), 2 μM 11F7t plus 2 μM apixaban (apix) (n = 8), 2 μM 11F7t plus 2 μM edoxaban (edox) (n = 9), or 2 μM 11F7t plus 2 μM fondaparinux (fonda) (n = 5) prior to continuous ex vivo circulation for 120 minutes. (a) Assessment of ACT levels for each of the indicated anticoagulation strategies at the following time points: (1) prior to anticoagulant addition, (2) before initiation of blood flow within the circuit (t = 0 min), (3) at 5 min, 60 min, and 120 min after circulation, and (4) after antidote addition post-circulation. Protamine (0.05 mg/ml) or GD-FXaS195A (3 μM), respectively, was given to reverse the effects of UFH or each combination of 11F7t plus a FXa catalytic site inhibitor. (b) Representative scanning electron microscopy images (100X and 1000X magnification) of oxygenator membranes obtained from ex vivo circuits after circulation of whole blood with each of the indicated anticoagulation strategies. (c-d) Blood flow rate and pressure within the circuit were monitored in real-time in a subset of experiments conducted with the indicated combinations of 11F7t and rivaroxaban (n = 3 independent experiments) in comparison to either UFH (5 U/mL) (n = 3), 11F7t (2 μM) (n = 3), or rivaroxaban (2 μM) (n = 3) alone. Error bars in panels (a), (c), and (d) represent standard error.
As predicted by the partial inhibition of thrombin or clot formation in the CAT and TEG assays respectively, 11F7t or any of the active site directed inhibitors of FXa alone failed to provide satisfactory anticoagulation during recirculation. Clot accumulation was apparent within the circuit reservoir, on the oxygenator membrane, or in the circuit tubing (Fig. S7), while SEM images of oxygenator membranes from these circuits showed significant fibrin and cellular deposition (Fig. 4b). Moreover, ACT values with each of these strategies failed to exceed 320 seconds. Clot also formed when circuit blood was anticoagulated with fondaparinux9, another clinically approved FXa inhibitor that on its own was also found to be less effective than UFH (Fig. S8). In contrast, the combined presence of 2 μM 11F7t plus 2 μM rivaroxaban, apixaban, edoxaban, or fondaparinux each prevented visible clot formation in the circuit, and resulted in minimal deposition of fibrin and cellular debris on the oxygenator membranes upon SEM inspection (Fig. 4b). Moreover, consistent with their apparent anticoagulant efficacy, the aforementioned combinations of inhibitors raised the ACT to 512.4 ± 15.1 sec, 516.7 ± 12.9 sec, 478.5 ± 16.8 sec, and 375.2 ± 12.6 sec respectively, which remained elevated to a comparable level throughout the 2-hour period of continuous circulation (Fig. 4a). Furthermore, adding GD-FXaS195A (3 μM) to post-circulation blood samples from those circuits restored the ACT to baseline levels (Fig. 4a).
To further quantify the efficacy of this dual anti-FXa anticoagulant strategy in the ex vivo oxygenator model, blood pressure and flow rate within the circuit were monitored in real-time in a subset of experiments. As expected, when 5 U/mL UFH was used as the anticoagulant, no significant deviation in the initial flow rate (50 mL/min) or pressure was observed throughout the 2-hour period of circulation, reflecting the successful maintenance of circuit patency (Fig. 4c, 4d). In contrast, when 2 μM rivaroxaban or 2 μM 11F7t was used as the sole anticoagulant, a rapid decrease in flow rate and a concomitant elevation in pressure within the circuit was detected after approximately 20 or 40 minutes of circulation respectively, due to the formation of obstructing clot (Fig. 4c, 4d). However, similarly to UFH, the combination of 2 μM 11F7t plus 2 μM rivaroxaban effectively maintained circuit patency without any significant deviation in blood flow rate or pressure (Fig. 4c, 4d).. These results demonstrate that combinations of 11F7t plus a FXa catalytic site inhibitor can prevent clotting in human blood recirculated continuously for 2 hours within an ex vivo membrane oxygenator circuit as effectively as UFH, and that GD-FXaS195A can effectively neutralize the effects of both anticoagulants following circulation.
Patient morbidity associated with UFH-facilitated CPB is partly attributable to intraoperative generation of thrombin, which is commonly measured by assessing circulating levels of prothrombin fragment 1.2 (F1.2), a peptide cleaved from prothrombin during its activation45. Pre-circulation levels of F1.2 were within the normal range46 in blood samples anticoagulated with each of the evaluated strategies (0.19 ± 0.02 nM; mean ± SEM). In circuit blood anticoagulated with UFH, F1.2 levels were significantly elevated (5.91 ± 1.46 nM, p = 0.0002) following 2-hours of recirculation, indicating that substantial thrombin generation had occurred despite the absence of detectable clot formation (Fig. 5). By contrast, in blood anticoagulated with 2 μM 11F7t plus a 2 μM of a FXa active site inhibitor, mean post-circulation F1.2 levels were less than 0.5 nM and significantly (p<0.01) lower than those observed with UFH by greater than 10-fold (Fig. 5). Thus, in human blood subjected to continuous extracorporeal circulation, the combination of 11F7t plus a FXa active site directed inhibitor not only duplicated UFH’s anticoagulant effect, but was also more effective than UFH in limiting thrombin generation.
Figure 5: Thrombin generation during extracorporeal circulation of human blood.

Pre- and post-circulation plasma prothrombin fragment F1.2 levels were measured after whole blood was anticoagulated with one of the following strategies prior to continuous ex vivo recirculation for 120 minutes: 5 U/mL UFH (n = 10 independent experiments), 2 μM 11F7t alone (n = 3), 2 μM rivaroxaban (riva) alone (n = 3), 2 μM 11F7t plus 2 μM rivaroxaban (n = 9), 2 μM 11F7t plus 2 μM apixaban (apix) (n = 6), 2 μM 11F7t plus 2 μM edoxaban (edox) (n = 7), or 2 μM 11F7t plus 2 μM fondaparinux (fonda) (n = 6). Bar graphs depict the mean ± standard error. Statistical analysis was performed by one-way ANOVA followed by Tukey’s multiple comparisons test.
For comparison, in circuits anticoagulated with 2 μM 11F7t or 2 μM rivaroxaban alone, wherein extensive clot was observed, post-circulation F1.2 levels in the remaining fluid were 184.5 ± 86.7 nM and 325.4 ± 27.3 nM, more than 350-fold higher than the levels observed when the two inhibitors were paired at those same concentrations (Fig. 5). Finally, post-circulation levels of bradykinin, a pro-inflammatory product of the kallikrein-kinin system that is generated through contact pathway activation and also becomes elevated during UFH-facilitated CPB47, were similarly increased in circuits anticoagulated with UFH or each combination of 11F7t plus a FXa active site inhibitor (Fig. S9), consistent with the notion that bradykinin generation is not thrombin-dependent and that all of these anticoagulant strategies limit such activation to a similar degree.
Reduction in the risk of HIT antibody-mediated platelet activation
To assess whether administration of 11F7t to a patient with HIT antibodies would elicit pathological platelet activation, as normally occurs with UFH, we compared the ability of purified IgG from three patients with a clinical diagnosis of HIT to induce platelet aggregation in the presence of UFH or 11F7t, alone or in combination with a FXa active site inhibitor. Purified IgG (25-100 μg/mL) from all three HIT patients consistently caused UFH-dependent platelet aggregation in PRP, with a maximum light transmission (Mx%) of 79 ± 15 % (mean ± SE) in the presence of 2 U/mL UFH (Fig. S10). In contrast, HIT IgG triggered platelet activation was not observed when UFH was replaced by a range of 11F7t concentrations (0.1 to 5 μM) with or without the additional presence of either edoxaban or fondaparinux (Fig. S10).
Discussion
Our finding that multistep inhibition of coagulation is key for generating robust anticoagulation is consistent with prior studies demonstrating that aptamer-mediated inhibition of individual procoagulant proteases is not sufficient for preventing clotting during extracorporeal circulation30. For example, while we previously showed that an aptamer targeting factor IXa could successfully replace UFH in a neonatal porcine model of CPB48, evidence of gross thrombus formation was readily seen when the same aptamer was used either for CPB anticoagulation in adult pigs (unpublished data) and baboons49, or during circulation of human blood in an ex vivo oxygenator circuit identical to that used in the present study30. This example also highlights the value of such an ex vivo circulation model as a stringent predictor of the potential efficacy of an anticoagulation strategy for in vivo CPB.
In fact, prior to the present study, only a handful of other anticoagulants besides UFH have been reported to prevent clot formation in human blood subjected to similar circulation within an ex vivo oxygenator circuit50,51. Among these are certain low molecular weight heparins such as enoxaparin, the thrombin-specific inhibitor hirudin51, and the FXa-specific inhibitor tick anticoagulant peptide50. Nevertheless, with each of these anticoagulants, investigators noted that circulating markers of thrombin generation or activity were either similarly increased or elevated compared to those seen with UFH50,51. Hence, our finding that the combination of 11F7t plus a FXa active site inhibitor suppresses post-circulation F1.2 levels considerably more than UFH supports the notion that minimizing thrombin formation by inhibiting multiple upstream procoagulant steps is not only an effective approach for achieving UFH-like anticoagulation, but may also reduce the risk of common clinical complications of UFH-facilitated CPB exacerbated by thrombin’s pro-thrombotic and pro-inflammatory activities8,52. These include CPB-induced SIRS18, thromboembolism8,13,53, ischemia-reperfusion injury54-56, consumptive coagulopathy6,8, and platelet dysfunction5.
We also demonstrate that the FXa mutant GD-FXaS195A can simultaneously reverse the anticoagulant effects of 11F7t plus an active site-directed inhibitor of FXa. GD-FXaS195A is nearly identical in sequence to the recombinant FXa mutant, Andexanet Alfa, which is efficacious as a reversal agent for direct and indirect FXa inhibitors32,33,34. Thus, our findings suggest that Andexanet Alfa may have additional applicability as an antidote for 11F7t, when used either by itself or in combination dosing strategies.
While the direct thrombin inhibitor bivalirudin has been used as an alternative CPB anticoagulant to UFH in patients with proven or suspected HIT, bivalirudin requires continuous infusion owing to its 25-minute half-life, and its use during CPB carries a well-known risk of clot formation in blood that becomes stagnant in the operative field or in the CPB circuit 2,6,44. Although bivalirudin is approved for CPB in Canada, it has not received approval by the U.S. FDA or European Medicines Agency even for patients with HIT. At our institution, patients with known or suspected HIT requiring CPB are treated by attempting to remove HIT antibodies by blood apheresis prior to surgery, an expensive and time-consuming process, to allow for UFH-use during CPB and avoid using bivalirudin. While a prior report suggested that certain nucleic acid aptamers can bind patient-derived HIT antibodies analogously to UFH to induce immunogenic platelet activation57, we did not observe this with 11F7t or any combination of 11F7t plus an active site-directed FXa inhibitor.
Given that clinical CPB necessitates infusion of an intravenous anticoagulant, it must be noted that each of the three direct FXa active site inhibitors tested in the present study (rivaroxaban, apixaban, and edoxaban) is only approved for oral administration and require solubilization in dimethylsulfoxide (DMSO) for in vitro or ex vivo testing. The findings of this study may encourage the re-formulation of one of the above agents for IV administration. Additionally, further assessment of the combination of 11F7t and an active site-directed inhibitor of FXa, together with their antidote(s), as a CPB anticoagulation strategy requires preclinical studies in mammals large enough to undergo cardiac procedures. Since 11F7t does not effectively inhibit canine, porcine, or bovine FXa, evaluating the efficacy of this strategy necessitates studies on non-human primates.
Using nucleic acid aptamers to complement existing small molecule drugs directed against the catalytic site of the same enzyme may be a generalized approach for identifying potent combinatorial therapies in a variety of clinical contexts. This is because aptamer-based therapies typically engage their targets by burying large surface-exposed regions and blocking exosite-dependent macromolecular interactions58, in contrast to the mechanism of action of catalytic site-directed inhibitors such as small molecule drugs. For example, we recently showed in vitro that the concomitant presence of distinct RNA aptamers against the GPCR β2 adrenoceptor could selectively stabilize the binding of small molecule β2 drug agonists or antagonists respectively in an allosteric fashion59. Furthermore, our preliminary studies suggest that dual targeting of thrombin using the combination of an exosite-directed aptamer and an active site-binding small molecule can similarly lead to potentiated anticoagulation.
Online Methods
Anticoagulants.
The 11F7t FXa aptamer (5’-GAGAGCCCCAGCGAGAUAAUACUUGGCCCCGCUCUU-3’) lacking a 5’ phosphate was purchased from Nitto Denko Avecia Inc29. The pyrimidines in the sequence are modified ribonucleotides, where “C” denotes a 2’Fluorocytosine and “U” denotes a 2’Fluorouracil. 11F7t’s complementary DNA antidote AO5-2 (5’-TATTATCTCGCTGGG −3’) was synthesized by Integrated DNA Technologies, Inc41. UFH, apixaban, rivaroxaban, fondaparinux, and bivalirudin were purchased from the Duke University Hospital Pharmacy. Edoxaban was purchased from Professor Jun-ichi Nishimura, Department of Hematology and Oncology, Osaka University, Osaka, Japan.
Reagents.
Small unilamellar phospholipid vesicles (PCPS) composed of 75% (w/w) hen egg L-α-phosphatidylcholine and 25% (w/w) porcine brain L-α-phosphatidylserine (Avanti Polar Lipids) were prepared and quality-controlled as described29. The thrombin S2238 peptidyl substrate was purchased from Chromogenix. Enzygnost Prothrombin Fragment 1.2 micro-ELISA kit was purchased from Siemens. Bradykinin ELISA kit was purchased from Enzo Life Sciences.
Proteins.
Prothrombin, factor X, factor IX, and factor V were isolated from human plasma as previously described60-62. Human factor X was further depleted of trace contaminating FXa by chromatography with soy bean trypsin inhibitor-sepharose63. Factors Xa, Va, and IXaβ were prepared by proteolytic activation of factors X, V, and IX, respectively, and re-purified as described elsewhere62,63. Recombinant factor VIII was re-purified as previously described64. GD-FXaS195A was prepared by digestion of recombinant FXaS195A with chymotrypsin and purification by anion exchange chromatography as previously described35. Protein concentrations were determined using the following molecular weights and extinction coefficients (E280, mg−1•cm2): prothrombin, 72,000, 1.47 65; thrombin, 37,500, 1.89 65; FX, 56,500, 1.16 66; FXa, 45,300, 1.16 66; GD-FXaS195A, 38,000, 1.16 66; FVa, 168,000, 1.74 63,67; FIXaβ, 46,000, 1.32 68; and FVIII, 264,700, 1.22 69.
Structure of the 11F7t:GD-XaS195A Complex
Equivalent binding constants observed for the interaction of 11F7t with GD-FXaS195A or wild type FXa imply that the details revealed here adequately model the essential features of its interaction with wild type FXa29. GD-FXaS195A was dialyzed into 20 mM Hepes, 0.15 M NaCl, 5 mM Ca2+, concentrated to ~13 mg/ml by centrifugal ultrafiltration (Amicon Ultra-15, Millipore) and stored in aliquots at −80 C. A concentrated solution of 11F7t was also dialyzed into the same buffer. For crystallization trials, GD-FXaS195A was mixed with small volumes of 11F7t and rivaroxaban to achieve 2 equivalents of aptamer per mole of proteinase either in the absence or presence of 1 equivalent of rivaroxaban. Trials were conducted by vapor diffusion at room temperature using sitting drop plates (Cryschem M, Hampton Research), with 500 μl precipitant in the reservoir and by mixing 1 μl of the complexes of 11F7t:GD-FXaS195A or 11F7t:GD-FXaS195A:rivaroxaban with 1 μl precipitant solution. The final concentrations were 67 μM GD-FXaS195A and 134 μM 11F7t or 62.5 μM GD-FXaS195A, 125 μM 11F7t and 75 μM rivaroxaban in the initial drop. Crystals for either complex appeared in several conditions within 4-6 days. Those appearing with Morpheus Screen condition 2-33 (Molecular Dimensions) were cryoprotected using the same buffer containing 20% (v/v) glycerol and used for data collection. Final data sets were collected on crystals obtained in 0.1 M Bicine, 0.1 M Tris base, pH 8.5 with 0.1 M Carboxylic acids (0.1 M each of sodium formate, ammonium acetate, tribasic sodium citrate dihydrate, sodium potassium tartarate tetrahydrate, and sodium oxamate), 10% (w/v) PEG 20,000 and 20% (v/v) PEG 500 monomethyl ester. Diffraction data was collected at 100 K either at the NE-CAT 24-ID-C beamline at APS (λ= 0.9793 A) for GD-FXaS195A/11F7t with a Pilatus 6M detector or at the U. Penn X-ray facility equipped with a Rigaku MicroMax-007 HF generator (λ= 1.5418 A) and a MAR imaging plate for FXaS195A/11F7t/rivaroxaban. Diffraction data were processed with DENZO or XDS70,71. Data were minimally truncated to give I/Iσ significantly larger than 1 in the highest resolution cell. A starting structure was solved by molecular replacement using Phaser and the highest resolution published structure for GD-FXa (2Y5G)72. The aptamer and rivaroxaban were manually built into unaccounted density following molecular replacement of the polypeptide. Ca2+ and Na+ were placed in the polypeptide structure based on density, the known binding sites for these cations to the proteinase domain81. Two Mg2+ atoms were placed in the aptamer structure based on observed density and the requirement of 1 mM Mg2+ for aptamer re-annealing prior to use34. Automated refinement was performed using the Phenix suite (Ver. 1.13-2998) of programs73. The aptamer was refined using ERRASER under Rosetta included in the Phenix suite74. Manual model building and refinement was done using Coot (Ver. 0.8.8) 75. A final cycle of refinement with automated parameter selection was performed using PDB_REDO (REFMAC 5.8.0210) to provide readily reproducible fitted models and statistics76. Interacting residues on the aptamer and protein were analyzed using PISA (Ver. 1.51) 77. Final structural models were validated using Molprobity and visualized using Pymol (Ver. 2.04) 78,79.
Progress Curves for FXa-initiated Prothrombin Activation.
Thrombin generation in reaction mixtures (200 μL) containing 1.4 μM prothrombin, 9 nM FVa, 30 μM PCPS with or without varying concentrations of 11F7t and/or apixaban at 25 °C was initiated by adding 0.2 nM FXa. Activity measurements were performed in assay buffer consisting of 20mM Hepes, 150mM NaCl, 5mM CaCl2, 0.1% (w/v) polyethylene glycol 8000, pH 7.5. Prior to addition, the aptamer was diluted in assay buffer, heated to 65°C for 5 min, and cooled to ambient temperature for 3 min to re-nature the RNA. 10 μl aliquots were removed at various time points after reaction initiation and quenched by mixing with 90 μl of assay buffer lacking Ca2+ but containing 50 mM EDTA, followed by further dilution (1:10) in the same buffer. After addition of 100 μM S2238, initial rates of peptidyl substrate cleavage were calculated based on the change in absorbance (405 nm) using a Gemini kinetic plate reader (Molecular Devices). Initial rates of S2238 cleavage were determined based on the linear appearance of product as a function of time, which were then converted to rates of thrombin formation based on the linear relationship between this initial rate and known concentrations of thrombin.
Progress Curves for IXa-initiated Prothrombin Activation.
Assays were performed exactly as described above for prothrombin cleavage by prothrombinase, except that reaction mixtures (200 μL) contained 1.4 μM prothrombin, 110 nM FX, 0.9nM FVIII, 9 nM FVa, 30 μM PCPS, with or without varying concentrations of 11F7t and/or apixaban at 25 °C and thrombin generation was initiated with 2 nM FIXaβ. For inhibitor neutralization assays, varying concentrations of GD-FXaS195A or AO5-2 were added to the reaction mixture after inclusion of all other reaction components and prior to initiation with FIXaβ. The sigmoidal progress curve for thrombin formation in this system (Fig. 2c) arises from the slow rate of FXa formation catalyzed by FIXa alone, the subsequent amplification of FXa formation following assembly of intrinsic tenase resulting from activation of FVIII by FXa and/or traces of thrombin, and the explosive generation of thrombin upon assembly of prothrombinase. Given the complexity of the system, empirical measures of the time required to generate 10 nM thrombin (Fig. 2d) or the concentration of thrombin produced at 10 minutes (min) following initiation (Fig. 2e) were used to assess the inhibitory effects of apixaban and 11F7t added alone and in combination.
Blood.
For thrombin generation, thromboelastography, extracorporeal membrane oxygenator circuit, and platelet aggregation studies, blood was drawn from healthy, consenting volunteers under a Duke University Institutional Review Board approved protocol, where blood draw procedures were in accordance with institutional guidelines. Blood was anticoagulated with 3.2% sodium citrate unless otherwise noted.
Calibrated Automated Thrombography.
Citrated whole blood from healthy donors as described above and centrifuged at 180g for 20 min to isolate PRP. In Immunlon 2HB clear U-bottom 96-well plates (Thermo Labsystems), PRP (80 μL) was mixed with 10 μL of anticoagulant (either UFH or 11F7t and/or a FXa catalytic site inhibitor) and incubated at 37°C for 5 min. Prior to addition, 11F7t was diluted in Hepes-saline assay buffer (20 mM Hepes pH 7.4, 150 mM NaCl, and 2 mM CaCl2) and renatured as described above. 50 ρM TF (20 μL, Diagnostica Stago) was then added to the mixture, followed by incubation at 37°C for another 5 min. Thrombin generation was initiated by addition of 10 μL of Flu-Ca reagent (Diagnostica Stago), which contains a fluorogenic thrombin specific substrate in a calcium containing buffer, and measured for 60 minutes at 37°C with a Fluoroskan Ascent plate reader (Thermo Labsystems), per the manufacturer’s instructions. Concentrations of thrombin formed over time were quantified based on the known activity of thrombin calibrator reagent (Diagnostica Stago) towards the particular fluorogenic substrate, which was analyzed in concomitant assays per the manufacturer’s instructions. Parameters of thrombin generation, including the endogenous thrombin potential (ETP), thrombin lag time, peak thrombin concentration, and time to peak thrombin concentration, were automatically calculated by the Thrombinoscope software (Thrombinoscope BV).
Thromboelastography (TEG).
Citrated whole blood (320 μl) from healthy donors was first mixed with anticoagulant (10 μl, diluted in the Hepes-saline buffer used for the CAT studies) and kaolin (Haemonetics) (10 μl) to initiate clotting. After addition of 20 μL of 0.2M CaCl2, the mixture was immediately added to a plain disposable plastic TEG cup (Haemonetics), and the assay was run at 37°C according to the manufacturer’s instructions. Anticoagulant concentrations were calculated based on a final reaction volume of 360 μL. Clot formation was measured with a Thromboelastograph Analyzer (Haemonetics) until a stable clot was formed (i.e., a maximum amplitude was reached) or for 3 hr. The time until clot formation, the α angle, and the maximum amplitude were automatically calculated by the TEG Analytical Software version 4.2.3 (Haemonetics).
Extracorporeal Membrane Oxygenator Circuits.
The particular extracorporeal membrane oxygenator circuit model used for these studies has been described in more detail previously30. Briefly, the circuit consisted of a custom-designed 8 mL Plexiglass venous reservoir, a mechanical roller pump (MasterFlex, Cole-Parmer Instrument), and a custom-designed 4mL oxygenator that are all connected via silicone tubing (MasterFlex). The oxygenator is comprised of two Plexiglass shells that contains a disposable three-layer artificial diffusion membrane (10 cm X 10 cm X 0.1 cm) composed of hollow polypropylene fibers affixed in a crosswise fashion. Membrane oxygenation was maintained used a 95% O2/5% CO2 gas mixture. In order to provide temperature control, one of the shells contains a water jacket heat exchanger connected to a circulating water bath system (Gaymar Industries). The temperature of the circulating blood was maintained at approximately 33°C as monitored by a reusable temperature probe inserted into the reservoir. Blood flow rate was monitored using an in-line flow probe (2N806 flow probe and T208 volume flowmeter; Transonics Systems). Blood pressure was measured using an SPR-524 ultra-miniature Nylon pressure catheter (Millar) connected to a MPVS Ultra Pressure-Volume Loop System (Millar) and analyzed using LabChart 7 software (AD Instruments).
Prior to addition of blood, the circuit was primed with PBS (containing CaCl2 and MgCl2), circulated continuously at a flow rate of 50 ml/min for approximately 30 min. As described above, approximately 35 mL of blood was drawn from individual donors, of which 30 mL was immediately added to a 50 mL conical tube containing the anticoagulant(s) to be evaluated at the appropriate concentrations and mixed. Likewise, 4.5 mL of blood was anticoagulated with 3.2% sodium citrate and centrifuged at 3000 rpm for 10 min to prepare platelet-poor plasma (PPP) for ELISA analysis of baseline F1.2 levels. A small amount of leftover non-anticoagulated blood was also kept for immediate assessment of baseline Activated Clotting Time (ACT+) level with a Hemochron Jr. Signature point-of-care device (ITC Nexus Dx). After draining the PBS from the circuit, the anticoagulated blood was added and circulation was initiated (time = 0 min) at a flow rate of 50 ml/min with 95% O2/5% CO2 delivery at approximately 33°C. Blood samples (~ 200 μL) were withdrawn from the circuit at 5, 60, and 120 min after circulation was initiated for ACT+ measurement. After 120 min of circulation, as much of the circuit blood as possible was collected. 68 μL of circuit blood was mixed with the corresponding antidote (12 μL) and immediately analyzed by ACT+ assay. 4.5 mL of the withdrawn blood was further anticoagulated with 3.2% sodium citrate and centrifuged as described above for analysis of post-circulation F1.2 or bradykinin levels. The circuit was then rinsed with PBS and the oxygenator membrane was removed, while excised sections were subsequently prepared for analysis by scanning electron microscopy as described in detail previously30.
HIT Platelet Aggregation Assay
Plasma from three patients with documented clinical history of heparin-induced thrombocytopenia were obtained with informed consent under an IRB approved protocol. Polyclonal IgG was purified from plasma as previously described80. Platelet aggregation using a PAP-8E Platelet Aggregation Profiler (Bio Data Corp., Horsham, PA) with the following modifications81. Individual patient IgG (25-100 μg/mL final concentration) samples were pre-mixed with varying concentrations of anticoagulant (UFH, 11F7t, or combinations of 11F7t plus edoxaban or fondaparinux). The resulting mixture (25 μL) was added to PRP (225 μL) and platelet aggregation was analyzed for 30 min according to the manufacturer’s instructions. A positive aggregation result is defined by an increase in maximum light transmission (Mx%) of greater than 20%82.
Statistical Analysis
All experimental data were plotted and analyzed using Graphpad Prism 7. For purified reaction mixture studies of FIXa-initiated thrombin generation, differences in mean lag time and mean thrombin concentration at 10 min were analyzed by one-way ANOVA followed by Tukey’s multiple comparisons test (Fig. 2d, 2e). For comparison of FXa- and FIXa-mediated thrombin generation, analysis was performed by two-tailed student’s t-test. For CAT and TEG studies, comparison of mean ETPs (Fig. 3b) and mean clotting times (Fig. 3d) was performed by one-way ANOVA followed by Tukey’s multiple comparisons test. For analysis of anticoagulant synergy in the CAT (Fig. S4, S8) and TEG (Fig. S5, S8) studies, two-way ANOVA was performed using JMP Pro software. For extracorporeal circuit studies, differences in mean post-circulation F1.2 levels and bradykinin were analyzed by one-way ANOVA followed by Tukey’s multiple comparisons test (Fig. 5, Fig. S9). For comparison of pre- and post-circulation F1.2 levels, analysis was performed by two-tailed student’s t-test (Fig. 5). For platelet aggregation studies, differences in mean maximum light transmission were analyzed by one-way ANOVA followed by Tukey’s multiple comparisons test.
Supplementary Material
Acknowledgements:
This work was supported by NIH grants HL-74124 (S.K.), HL-125422 (S.K.), HL-065222 (B.A.S), F30 HL-127977 (R.G.), and T32 GM-007171 (R.G.). The NE-CAT 24-ID-C beamline is funded by NIH grant GM103403, the Pilatus 6M detector is funded by a NIH-ORIP HEI grant (RR029205). This research used resources of the Advanced Photon Source operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We would also like to acknowledge Dougald Monroe, Maureane Hoffman, and George Pitoc for useful discussion.
Footnotes
Competing Financial Interests Statement:
Duke University has applied for a patent on this dual anticoagulant strategy.
Data Availability
Crystallographic data have been deposited in the Protein Data Bank under accession codes 5VOE (11F7t:dG-XaS195A) and 5VOF (11F7t:dG-XaS195A:Rivaroxaban).
References:
- 1.Mureebe L Direct thrombin inhibitors: alternatives to heparin. Vascular 15, 372–375, doi: 10.2310/6670.2007.00057 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Sniecinski RM & Levy JH Anticoagulation management associated with extracorporeal circulation. Best Pract Res Clin Anaesthesiol 29, 189–202, doi: 10.1016/j.bpa.2015.03.005 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Punjabi PP & Taylor KM The science and practice of cardiopulmonary bypass: From cross circulation to ECMO and SIRS. Glob Cardiol Sci Pract 2013, 249–260, doi: 10.5339/gcsp.2013.32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boisclair MD et al. Mechanisms of thrombin generation during surgery and cardiopulmonary bypass.[see comment]. Blood 82, 3350–3357 (1993). [PubMed] [Google Scholar]
- 5.Sniecinski RM & Chandler WL Activation of the hemostatic system during cardiopulmonary bypass. Anesth Analg 113, 1319–1333, doi: 10.1213/ANE.0b013e3182354b7e (2011). [DOI] [PubMed] [Google Scholar]
- 6.Yavari M & Becker RC Anticoagulant therapy during cardiopulmonary bypass. J Thromb Thrombolysis 26, 218–228, doi: 10.1007/s11239-008-0280-4 (2008). [DOI] [PubMed] [Google Scholar]
- 7.De Somer F et al. Tissue factor as the main activator of the coagulation system during cardiopulmonary bypass. J Thorac Cardiovasc Surg 123, 951–958 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Edmunds LH & Colman RW Thrombin During Cardiopulmonary Bypass. The Annals of Thoracic Surgery 82, 2315–2322 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Garcia DA, Baglin TP, Weitz JI, Samama MM & American College of Chest, P. Parenteral anticoagulants: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 141, e24S–43S, doi: 10.1378/chest.11-2291 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frederiksen JW Cardiopulmonary bypass in humans: bypassing unfractionated heparin. Ann Thorac Surg 70, 1434–1443 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Edmunds L Inflammatory response to cardiopulmonary bypass. Ann Thorac Surg 66, S12–16; discussion S25-18 (1998). [DOI] [PubMed] [Google Scholar]
- 12.Merry AF Focus on thrombin: alternative anticoagulants. Semin Cardiothorac Vasc Anesth 11, 256–260, doi: 10.1177/1089253207311154 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Raymond PD & Marsh NA Alterations to haemostasis following cardiopulmonary bypass and the relationship of these changes to neurocognitive morbidity. Blood Coagul Fibrinolysis 12, 601–618 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Ranucci M Hemostatic and thrombotic issues in cardiac surgery. Semin Thromb Hemost 41, 84–90, doi: 10.1055/s-0034-1398383 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Paparella D, Brister SJ & Buchanan MR Coagulation disorders of cardiopulmonary bypass: a review. Intensive Care Med 30, 1873–1881, doi: 10.1007/s00134-004-2388-0 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Weitz JI, Hudoba M, Massel D, Maraganore J & Hirsh J Clot-bound thrombin is protected from inhibition by heparin-antithrombin III but is susceptible to inactivation by antithrombin III-independent inhibitors. J Clin Invest 86, 385–391 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brufatto N, Ward A & Nesheim ME Factor Xa is highly protected from antithrombin-fondaparinux and antithrombin-enoxaparin when incorporated into the prothrombinase complex. Journal of Thrombosis & Haemostasis 1, 1258–1263 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Levy JH & Tanaka KA Inflammatory response to cardiopulmonary bypass. Ann Thorac Surg 75, S715–720 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Bosch YP et al. Measurement of thrombin generation intra-operatively and its association with bleeding tendency after cardiac surgery. Thromb Res 133, 488–494, doi: 10.1016/j.thromres.2013.12.017 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Ranucci M et al. Postoperative antithrombin levels and outcome in cardiac operations. Crit Care Med 33, 355–360 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Arepally GM & Ortel TL Heparin-induced thrombocytopenia. Annual review of medicine 61, 77–90, doi: 10.1146/annurev.med.042808.171814 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nybo M & Madsen JS Serious anaphylactic reactions due to protamine sulfate: a systematic literature review. Basic Clin Pharmacol Toxicol 103, 192–196, doi: 10.1111/j.1742-7843.2008.00274.x (2008). [DOI] [PubMed] [Google Scholar]
- 23.Hobbhahn J et al. Heparin reversal by protamine in humans--complement, prostaglandins, blood cells, and hemodynamics. J Appl Physiol 71, 1415–1421 (1991). [DOI] [PubMed] [Google Scholar]
- 24.Hartmann M et al. Effects of cardiac surgery on hemostasis. Transfus Med Rev 20, 230–241, doi: 10.1016/j.tmrv.2006.03.003 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Woodman RC & Harker LA Bleeding complications associated with cardiopulmonary bypass. Blood 76, 1680–1697 (1990). [PubMed] [Google Scholar]
- 26.Ofosu FA et al. Unfractionated heparin inhibits thrombin-catalysed amplification reactions of coagulation more efficiently than those catalysed by factor Xa. Biochemical Journal 257, 143–150 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beguin S, Lindhout T & Hemker HC The mode of action of heparin in plasma. Thrombosis & Haemostasis 60, 457–462 (1988). [PubMed] [Google Scholar]
- 28.Pieters J & Lindhout T The limited importance of factor Xa inhibition to the anticoagulant property of heparin in thromboplastin-activated plasma. Blood 72, 2048–2052 (1988). [PubMed] [Google Scholar]
- 29.Buddai SK et al. An anticoagulant RNA aptamer that inhibits proteinase-cofactor interactions within prothrombinase. Journal of Biological Chemistry 285, 5212–5223 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bompiani KM et al. Probing the coagulation pathway with aptamers identifies combinations that synergistically inhibit blood clot formation. Chem Biol 21, 935–944, doi: 10.1016/j.chembiol.2014.05.016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeh CH, Fredenburgh JC & Weitz JI Oral direct factor Xa inhibitors. Circ Res 111, 1069–1078, doi: 10.1161/CIRCRESAHA.112.276741 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Lu G et al. A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nature medicine 19, 446–451, doi: 10.1038/nm.3102 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Siegal DM et al. Andexanet Alfa for the Reversal of Factor Xa Inhibitor Activity. The New England journal of medicine 373, 2413–2424, doi: 10.1056/NEJMoa1510991 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Connolly SJ et al. Andexanet Alfa for Acute Major Bleeding Associated with Factor Xa Inhibitors. The New England journal of medicine 375, 1131–1141, doi: 10.1056/NEJMoa1607887 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thalji NK et al. A rapid pro-hemostatic approach to overcome direct oral anticoagulants. Nature medicine 22, 924–932, doi: 10.1038/nm.4149 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Rezaie AR Identification of Basic Residues in the Heparin-binding Exosite of Factor Xa Critical for Heparin and Factor Va Binding. Journal of Biological Chemistry 275, 3320–3327, doi: 10.1074/jbc.275.5.3320 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Roehrig S et al. Discovery of the novel antithrombotic agent 5-chloro-N-({(5S)-2-oxo-3- [4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene- 2-carboxamide (BAY 59-7939): an oral, direct factor Xa inhibitor. J Med Chem 48, 5900–5908, doi: 10.1021/jm050101d (2005). [DOI] [PubMed] [Google Scholar]
- 38.de Candia M, Lopopolo G & Altomare C Novel factor Xa inhibitors: a patent review. Expert Opin Ther Pat 19, 1535–1580, doi: 10.1517/13543770903270532 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Lechtenberg BC et al. Crystal structure of the prothrombinase complex from the venom of Pseudonaja textilis. Blood 122, 2777–2783, doi: 10.1182/blood-2013-06-511733 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nimjee SM, White RR, Becker RC & Sullenger BA Aptamers as Therapeutics. Annu Rev Pharmacol Toxicol 57, 61–79, doi: 10.1146/annurev-pharmtox-010716-104558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soule EE, Bompiani KM, Woodruff RS & Sullenger BA Targeting Two Coagulation Cascade Proteases with a Bivalent Aptamer Yields a Potent and Antidote-Controllable Anticoagulant. Nucleic Acid Ther 26, 1–9, doi: 10.1089/nat.2015.0565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hemker HC et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb 33, 4–15, doi:71636 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Reikvam H et al. Thrombelastography. Transfus Apher Sci 40, 119–123, doi: 10.1016/j.transci.2009.01.019 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Rehfeldt KH & Barbara DW Cardiopulmonary Bypass Without Heparin. Semin Cardiothorac Vasc Anesth 20, 40–51, doi: 10.1177/1089253215573326 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Knudsen L et al. Monitoring thrombin generation with prothrombin fragment 1.2 assay during cardiopulmonary bypass surgery. Thromb Res 84, 45–54 (1996). [DOI] [PubMed] [Google Scholar]
- 46.Ota S et al. Elevated levels of prothrombin fragment 1 + 2 indicate high risk of thrombosis. Clin Appl Thromb Hemost 14, 279–285, doi: 10.1177/1076029607309176 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Campbell DJ, Dixon B, Kladis A, Kemme M & Santamaria JD Activation of the kallikrein-kinin system by cardiopulmonary bypass in humans. Am J Physiol Regul Integr Comp Physiol 281, R1059–1070, doi: 10.1152/ajpregu.2001.281.4.R1059 (2001). [DOI] [PubMed] [Google Scholar]
- 48.Nimjee SM et al. A Novel Antidote-Controlled Anticoagulant Reduces Thrombin Generation and Inflammation and Improves Cardiac Function in Cardiopulmonary Bypass Surgery. Mol Ther 14, 408–415 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Bel A et al. Inhibition of factor IXa by the pegnivacogin system during cardiopulmonary bypass: a potential substitute for heparin. A study in baboons. European journal of cardio-thoracic surgery : official journal of the European Association for Cardio-thoracic Surgery 49, 682–689, doi: 10.1093/ejcts/ezv159 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Gikakis N et al. Effect of factor Xa inhibitors on thrombin formation and complement and neutrophil activation during in vitro extracorporeal circulation. Circulation 94, II341–346 (1996). [PubMed] [Google Scholar]
- 51.Bernabei A et al. Recombinant desulfatohirudin as a substitute for heparin during cardiopulmonary bypass. J Thorac Cardiovasc Surg 108, 381–382 (1994). [PubMed] [Google Scholar]
- 52.Brister SJ, Ofosu FA & Buchanan MR Thrombin generation during cardiac surgery: is heparin the ideal anticoagulant? Thrombosis & Haemostasis 70, 259–262 (1993). [PubMed] [Google Scholar]
- 53.Welsby IJ et al. Effect of combined anticoagulation using heparin and bivalirudin on the hemostatic and inflammatory responses to cardiopulmonary bypass in the rat. Anesthesiology 106, 295–301 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Raivio P, Lassila R & Petaja J Thrombin in myocardial ischemia-reperfusion during cardiac surgery. Ann Thorac Surg 88, 318–325, doi: 10.1016/j.athoracsur.2008.12.097 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Chong AJ et al. Tissue factor and thrombin mediate myocardial ischemia-reperfusion injury. Ann Thorac Surg 75, S649–655 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Farivar AS et al. Crosstalk between thrombosis and inflammation in lung reperfusion injury. Ann Thorac Surg 81, 1061–1067 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Jaax ME et al. Complex formation with nucleic acids and aptamers alters the antigenic properties of platelet factor 4. Blood 122, 272–281, doi: 10.1182/blood-2013-01-478966 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Long SB et al. Crystal structure of an RNA aptamer bound to thrombin. Rna 14, 2504–2512 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kahsai AW et al. Conformationally selective RNA aptamers allosterically modulate the beta2-adrenoceptor. Nat Chem Biol 12, 709–716, doi: 10.1038/nchembio.2126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
References for Online Methods
- 60.Baugh RJ & Krishnaswamy S Role of the activation peptide domain in human factor X activation by the extrinsic Xase complex. J Biol Chem 271, 16126–16134 (1996). [DOI] [PubMed] [Google Scholar]
- 61.Orcutt SJ, Pietropaolo C & Krishnaswamy S Extended interactions with prothrombinase enforce affinity and specificity for its macromolecular substrate. J Biol Chem 277, 46191–46196, doi: 10.1074/jbc.M208677200 (2002). [DOI] [PubMed] [Google Scholar]
- 62.Lu G, Broze GJ Jr. & Krishnaswamy S Formation of factors IXa and Xa by the extrinsic pathway: differential regulation by tissue factor pathway inhibitor and antithrombin III. Journal of Biological Chemistry 279, 17241–17249 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Buddai SK, Toulokhonova L, Bergum PW, Vlasuk GP & Krishnaswamy S Nematode anticoagulant protein c2 reveals a site on factor Xa that is important for macromolecular substrate binding to human prothrombinase. J Biol Chem 277, 26689–26698, doi: 10.1074/jbc.M202507200 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Fay PJ, Haidaris PJ & Smudzin TM Human factor VIIIa subunit structure. Reconstruction of factor VIIIa from the isolated A1/A3-C1-C2 dimer and A2 subunit. J Biol Chem 266, 8957–8962 (1991). [PubMed] [Google Scholar]
- 65.Mann KG, Elion J, Butkowski RJ, Downing M & Nesheim ME Prothrombin. Methods Enzymol 80 Pt C, 286–302 (1981). [DOI] [PubMed] [Google Scholar]
- 66.Di Scipio RG, Hermodson MA & Davie EW Activation of human factor X (Stuart factor) by a protease from Russell’s viper venom. Biochemistry 16, 5253–5260 (1977). [DOI] [PubMed] [Google Scholar]
- 67.Luckow EA, Lyons DA, Ridgeway TM, Esmon CT & Laue TM Interaction of clotting factor V heavy chain with prothrombin and prethrombin 1 and role of activated protein C in regulating this interaction: analysis by analytical ultracentrifugation. Biochemistry 28, 2348–2354 (1989). [DOI] [PubMed] [Google Scholar]
- 68.Di Scipio RG, Kurachi K & Davie EW Activation of human factor IX (Christmas factor). J Clin Invest 61, 1528–1538, doi: 10.1172/JCI109073 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cao W, Krishnaswamy S, Camire RM, Lenting PJ & Zheng XL Factor VIII accelerates proteolytic cleavage of von Willebrand factor by ADAMTS13. Proc Natl Acad Sci U S A 105, 7416–7421, doi: 10.1073/pnas.0801735105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kabsch W Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132, doi: 10.1107/S0907444909047337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Otwinowski Z & Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 72.Salonen LM et al. Molecular recognition at the active site of factor Xa: cation-pi interactions, stacking on planar peptide surfaces, and replacement of structural water. Chemistry 18, 213–222, doi: 10.1002/chem.201102571 (2012). [DOI] [PubMed] [Google Scholar]
- 73.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chou FC, Sripakdeevong P, Dibrov SM, Hermann T & Das R Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat Methods 10, 74–76, doi: 10.1038/nmeth.2262 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501, doi: 10.1107/S0907444910007493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Joosten RP, Joosten K, Murshudov GN & Perrakis A PDB_REDO: constructive validation, more than just looking for errors. Acta Crystallogr D Biol Crystallogr 68, 484–496, doi: 10.1107/S0907444911054515 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Krissinel E & Henrick K Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774–797, doi: 10.1016/j.jmb.2007.05.022 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21, doi: 10.1107/S0907444909042073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.8 (2015). [Google Scholar]
- 80.Rauova L et al. Role of platelet surface PF4 antigenic complexes in heparin-induced thrombocytopenia pathogenesis: diagnostic and therapeutic implications. Blood 107, 2346–2353, doi: 10.1182/blood-2005-08-3122 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cai Z et al. Atomic description of the immune complex involved in heparin-induced thrombocytopenia. Nat Commun 6, 8277, doi: 10.1038/ncomms9277 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pouplard C et al. Decision analysis for use of platelet aggregation test, carbon 14-serotonin release assay, and heparin-platelet factor 4 enzyme-linked immunosorbent assay for diagnosis of heparin-induced thrombocytopenia. Am J Clin Pathol 111, 700–706 (1999). [DOI] [PubMed] [Google Scholar]
- 83.Krishnaswamy S Exosite-driven substrate specificity and function in coagulation. Journal of Thrombosis & Haemostasis 3, 54–67 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.