

Abstract
To address the urgent need for new agents to reduce the global occurrence and spread of AIDS, we investigated the underlying hypothesis that antagonists of the HIV-1 envelope (Env) gp120 protein and the host-cell coreceptor (CoR) protein can be covalently joined into bifunctional synergistic combinations with improved antiviral capabilities. A synthetic protocol was established to covalently combine a CCR5 small-molecule antagonist and a gp120 peptide triazole antagonist to form the bifunctional chimera. Importantly, the chimeric inhibitor preserved the specific targeting properties of the two separate chimera components and, at the same time, exhibited low to subnanomolar potencies in inhibiting cell infection by different pseudoviruses, which were substantially greater than those of a noncovalent mixture of the individual components. The results demonstrate that targeting the virus−cell interface with a single molecule can result in improved potencies and also the introduction of new phenotypes to the chimeric inhibitor, such as the irreversible inactivation of HIV-1.
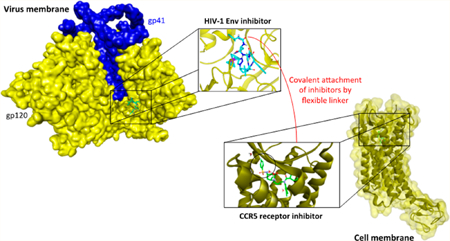
Graphical Abstract
INTRODUCTION
Inhibition of the initial entry of HIV-1 into host cells remains a compelling yet elusive means to prevent infection and the spread of the virus. HIV-1 cell infection is mediated by concerted interactions at the virus−host-cell interface between trimeric envelope-glycoprotein (Env) spikes on the virus membrane surface and two host-cell receptors, CD4 and a coreceptor that is most commonly either CCR5 or CXCR4.1 Each Env trimer consists of two noncovalently associated glycoproteins, a gp41 transmembrane protein and an external gp120 surface protein. During viral infection, the interaction between CD4 and the most exposed Env protein, gp120, causes a conformational rearrangement of the latter, leading to an increased affinity for a coreceptor interaction at an initially cryptic site in gp120. This cascade exposes structural components in gp41 necessary to promote virus- and cell-membrane lipid mixing, fusion, pore formation, and infection. Inhibitors that can potently block virus−cell interactions and cell entry would hold great promise in inhibiting initial HIV-1 infections.
Currently approved inhibitors of the entry steps are available clinically but have properties that limit their therapeutic usefulness. Maraviroc is an approved entry-inhibitor that binds to the coreceptor CCR5 and prevents binding to HIV Env gp120 and, as a consequence, prevents the full exposure of gp41 and suppresses virus−cell fusion.2,3 However, the effectiveness of this drug requires matching the administered therapeutic to the coreceptor used by the viral variants infecting each patient. In most cases, HIV infects cells after binding to the CCR5 receptor in the early stages of disease, but as the disease progresses, the virus can switch to another receptor, CXCR4. Therefore, patients need to be tested for viral tropism before commencing the CCR5-inhibition therapy. In addition, maraviroc induces a tropism switch to CXCR4-expressing cells.4 Hence, maraviroc is currently only approved for use in treatment-experienced patients.5 Enfuvirtide (T20) is a 36-residue peptide that mimics the part of the C-terminal helix in gp41 that mediates fusion. As such, it blocks the binding of the gp41 N-terminal helix to the C-terminal helix and formation of the six-helix bundle, which is the process that drives fusion between the viral and cellular membranes. Because of its high cost of production, its short half-life, and the need for subcutaneous injections, enfuvirtide is also only approved as salvage therapy in patients who have failed multiple lines of therapy. Moreover, rapid mutations in a 10-residue stretch of the gp41 N-terminal helix were observed to lead to resistance to this drug.6 Recently, it also has been shown that HIV-1 can develop resistance to fusion inhibitors and become inhibitor-dependent, because of the critical kinetics required for these inhibitors.7
Identifying new entry-targeting inhibitors, in particular those that could function in combination to increase potency and overcome viral resistance, remains an important goal of HIV-1-drug-discovery efforts. To date, delineation of such combinations has been limited mainly to combinations of coreceptor inhibitors and T20.8−11 Such combinations have demonstrated the potential for the strong synergy between CCR5 (TAK-220, SCH-C, and Aplaviroc) and CXCR4 (AMD-3100) inhibitors in assays of infection of peripheral blood mononuclear cells (PBMCs) with clinical HIV-1 isolates. Synergy between coreceptor inhibitors and Env-gp41 inhibitors has been proposed to be due to a kinetic linkage between the steps of entry that they antagonize.9−12 This work points to the potential to achieve increased potency through combinations of inhibitors targeting different components of the virus−cell interface. This could be particularly important as researchers develop new small-molecule coreceptor inhibitors that target CXCR4 in addition to CCR5, as well as new inhibitors of the Env protein complex, including the most exposed protein component, gp120.
In the current work, we tested the hypothesis that the covalent fusion of HIV-1 gp120 and coreceptor inhibitors could yield an HIV-1-cell-infection inhibitor with enhanced potency compared with the sum of the potencies of the individual components. New classes of coreceptor inhibitors have been identified that can target either CCR5 or CXCR4 with strong antiviral effects.13−17 At the same time, a class of gp120 targeting peptide triazoles (PTs) has been identified that blocks both host-cell-receptor interactions and causes irreversible virus inactivation by triggering gp120 shedding.18,19 We chose the LJC24015 (coreceptor-targeting, see Figure 2 for the structure) and UM1520 (gp120-targeting, see Figure 2 for the structure) inhibitors for the initial prototype-chimera components on the basis of the already-established antiviral efficacies of these components on their own. The conjugation points of LJC240 to UM15 were determined by examining target-binding models as well as their structure−activity relationships, which identified the structural elements of the inhibitors that were tolerant to modification. The protocol for the chimera synthesis was derived from the already-established solid-phase syntheses of the peptide triazoles themselves21−24 with follow-up sequential additions of the linker and the free-acid form of LJC240−COOH (10) before the introduction of the triazole by click conjugation. The functional properties of the resulting prototype chimera, LJC240−L4−UM15 (11), were examined by a combination of cellular, virological, and molecular assays. The results demonstrated that the covalent chimera retained the individual components’ functions and, at the same time, a synergistically enhanced antiviral potency compared with that of a noncovalent mixture of the two component inhibitors. The work provides strong support for the hypothesis that a bifunctional chimera could engage and synergistically block the infection-causing functions of host-cell coreceptors and virus envelope gp120 at the HIV-1−cell interface.
Figure 2.

Interaction maps of UM15 (left) docked within gp120 of the HIV-1 envelope (PDB code: 4NCO)25,28 and LJC24015 (right) docked onto the CCR5 structure (PDB code: 4MBS).26 The gray circles represent the solvated (solvent-exposed) moieties.
RESULTS
1. Chimera Design and Synthesis.
We designed a bifunctional molecule that is able to target both the HIV-1 envelope and the cellular coreceptor (here, CCR5). When the HIV-1 envelope encounters the cell receptors, the released gp120 V3 loop docks into the cellular-coreceptor-binding site. We envisioned that a bifunctional molecule that has gp120- and CCR5-targeting components would be able to bridge the envelope−CCR5-encounter complex. This kind of complex has not been resolved at high resolution. However, structural information is available for the individual components.25,26 Because PTs target the CD4-binding area of gp120, we measured the distance between the CD4-binding site (residue Asp368) and the tip of the released V3 loop (Figure 1, generated from PDB code 2QAD).27 This distance represents the hypothetical minimal linker length required to tether the gp120 and CCR5 inhibitors together in a way that mimics the encounter complex.
Figure 1.

Geometry of the gp120 protein showing the distance between the CD4-binding site and the released V3-loop tip (PDB code: 2QAD).27
We also examined the possible positions for chemical ligation through modeling of the inhibitors in the context of their individual protein targets (Figure 2), in order to avoid any unexpected reduction in potency. As shown in Figure 2 (left), the UM15 N-terminal (protonated) residue is in a solvent-exposed moiety. On the other hand, the fluorine atom of LJC240 (Figure 2, right) is well solvated and actually can displace water in the crystal structure of the CCR5 receptor. This docking result, combined with prior structure−function relationships in LJC240-derived CCR5 inhibitors,15 argues that structural variation replacing the fluorine atom should be tolerated.
On the basis of the predicted topology presented above (Figures 1 and 2), we envisioned a linker of approximately 59 Å would be required to tether the UM15 N-terminus to the fluoro-phenyl ring. The hydrophilicity built into the linker was predicted to help keep the two components apart and avoid any nonspecific hydrophobic interactions with either of the target proteins. We chose four units of amino-ethoxy-ethoxy acetic acid as the linker (hereafter referred to as L4) as it would provide the distance on the basis of the bond-length measurements.
We employed solid-phase synthesis to build the L4−UM15 precursor. We also synthesized a modified version of LJC240 (compound 10, Figure 3) in which the fluorine atom (Figure 2) was replaced with a carboxylic group to enable ligation with the amino group of the L4−UM15 N-terminus (Figure 4). The coupling reagents of HBTU/HOBt and DIEA were selected for the amide-coupling, which was followed by an on-resin click reaction to form the triazole component on the UM15-azido-Pro residue (Figure 4).
Figure 3.

Synthesis of the coreceptor inhibitor LJC240−COOH (10). Reagents and conditions: (a) NH2OH·HCl, NaHCO3, EtOH/H2O, rt; (b) Na, pentanol, reflux; (c) (Boc)2O, TEA, THF, rt; (d) 10% Pd/C, HCO2NH4, methanol, reflux; (e) 1-bromo-3-chloropropane, KI, TEA, DMF, rt; (f) 1-acetylpiperidine-4-carboxylic acid, SOCl2, TEA, DCM, rt; (g) 5, KI, K2CO3, MeCN, reflux; (h) HCl, methanol, rt; (i) 4-(chlorosulfonyl)benzoic acid, Na2CO3, dioxane/H2O, rt.
Figure 4.

Solid-phase synthesis of chimera 11. A Liberty blue microwave synthesizer was used for the assembly of the L4−UM15 precursor segment. The latter was joined with the LJC240 free acid (10), which was followed by ferrocenyl triazole formation using a click reaction, leading to the final chimera (11).
2. Potent Neutralization of HIV-1 Infection by the Bifunctional Chimera.
We measured the antiviral potency of the bifunctional chimera in a cell-infection-inhibition assay. Pseudotyped viruses bearing the BaL.01, JR-FL, and YU2 envelope proteins and carrying a luciferase reporter gene were incubated with the serially diluted chimera for 30 min at 37 °C and then added to human osteosarcoma (HOS) cells expressing CD4 and CCR5 (HOS.T4.R5). Following the completion of the infection-inhibition assay, the cells were lysed and monitored for luciferase activity. UM15 and LJC240−L4 (with the same L4 linker used for the chimera) served as controls for the experiment because they best represented the parental molecules that made up the chimera. LJC240−L4−UM15 (11) displayed subnanomolar to low-nanomolar inhibition of HIV-1 envelope-pseudotyped viruses (Figure 5). The chimera had improved potencies that ranged from >2-fold (YU2) to >50-fold (BaL.01) greater than the most potent parental component, LJC240−L4. We also evaluated an additional control of a 1:1 noncovalent mixture of UM15 and LJC240−L4 (against the tier-2 JRFL-pseudotyped virus), which mimicked the molar ratio between the inhibitors in the chimera, to determine whether the covalent attachment resulted in synergy and increased the potency beyond that of the combination of the individual inhibitors. Indeed, the bifunctional chimera was more potent (4 nM) than the 1:1 mixture (34 nM, Figure 5B), thus demonstrating the value of the covalent attachment for the potency compared with that of the sum of the parts.
Figure 5.

Neutralization of HIV-1 Env-pseudotyped viruses (A) Bal.01, (B) JRFL, (C) YU2, and (D) HxBc2, as examples of X4-tropic viruses, by the bifunctional chimera (red), UM15 (black), LJC240−L4 (blue), and a 1:1 mixture of UM15 and LJC240−L4 (green). ND indicates not determined.
2.1. Synergistic Effects of the Covalent Chimera.
To further define the value of the covalent attachment of UM15 to LJC240−L4 versus that of the noncovalent mixture, we performed a quantitative synergy analysis. Initially, we evaluated the noncovalent mixtures against a Bal.01 HIV-1 strain (because of the ease of virus production and assay handling) on the basis of their individual activities reported in Figure 5. Interestingly, both UM15 and LJC240−L4 showed ~7-fold improved potency in the presence of each other (Figure 6A); UM15 showed an IC50 value of 56 nM in the mixture compared with 378 nM alone (Figure 5A), and LJC240−L4 showed an IC50 value of 2.9 nM in the mixture (Figure 6A) compared with 20 nM alone (Figure 5A). The IC50 value obtained for the covalent chimera in the same assay (0.86 nM, Figure 6A) clearly showed a strong increase in potency versus that of the noncovalent mixture. These results argue for the value of the covalent joining of the two inhibitors, further confirming the difference with the 1:1 noncovalent mixture (Figure 5B). Representation of the data with an isobologram29 (Figure 6B) shows the synergy in the noncovalent mixture (1:30 molar ratio of UM15/LJC240−L4) when we plotted the 50% inhibition of the mixture (red dot in Figure 6B), compared with the individual IC50 values of the separate inhibitors (connected by the blue line in Figure 6B). Interestingly, plotting the 50% inhibition by the covalent chimera, 11, (green dot in Figure 6B) shows the even stronger synergistic effect of the latter.
Figure 6.

(A) Synergy-assay results between UM15 and LJC240−L4 against HIV-Bal.01. The IC50 values (blue for LJC240−L4 and black for UM15) are the effective inhibitory concentrations of the individual components in the mixture. Chimera 11 (red) was also included in the experiment. (B) Isobologram representation of the UM15−LJC240−L4 combination. The blue line connects the IC50 values of the two inhibitors; the red dot is the effective concentration of the noncovalent mixture (1:30 ratio which achieved 50% inhibition), whereas the green dot is the effective concentration of the covalent chimera, 11.
3. Activity Retained by Individual Components of the Chimera.
In order to confirm that the covalent attachment of the inhibitors did not interfere with the ability of each molecule to bind to its respective target in the virus−cell interface, we performed a series of experiments to measure the magnitudes of the peptide triazole and coreceptor inhibitor functions of the bifunctional chimera.
3.1. Activity against X4-tropic HIV.
First, we performed an infection-inhibition assay with HxBc2 (Figure 5D), an X4-tropic virus, on HOS cells expressing CD4 and CXCR4 (HOS.T4.X4). By using an X4-tropic virus on X4-expressing cells, we ruled out the contribution of the coreceptor inhibitor toward the potency of the chimera. As expected, the bifunctional-chimera potency was nearly identical to that of the UM15 control (Figure 5D), thereby demonstrating that the UM15 component of the chimera retained full activity.
3.2. Full gp120 Dual Antagonism Retained by Chimera 11.
Surface-plasmon-resonance (SPR) analysis was carried out to further confirm the retention of the UM15 part’s gp120-binding efficiency. Both soluble CD4 and 17b (a gp120-targeting antibody that stabilizes the bridging sheet) were immobilized separately on sensor-chip surfaces, and gp120 was flowed across in increasing concentrations of inhibitor to assess binding. The binding of the bifunctional chimera was again equivalent to that of the UM15 control (Figure 7).
Figure 7.

Efficacy of the chimeric inhibitor for gp120-protein−ligand binding determined by SPR gp120-competition analyses. (Left) Representative sensorgrams showing dose-dependent inhibition of monomeric-YU2-gp120 binding to the receptors CD4 and 17b by LJC240−L4−UM15. (Right) Dose−response curves derived from the sensorgrams of YU2-gp120-binding inhibition by LJC240−L4−UM15 and UM15 (n = 3).
These findings confirm previous observations that N-terminal extensions on the peptide do not interfere with pharmacophore binding30,31 and also match the docking model for UM15 (Figure 2).
3.3. Irreversible Inactivation of HIV by Chimera 11 via gp120 Shedding.
Western-blot analysis was used to detect the gp120-shedding function of the peptide triazole component. As observed in the infection-inhibition (Figure 5D) and gp120-binding analyses (Figure 7), the shedding ability of UM15 was not affected by the covalent attachment to LJC240, with the chimera and UM15 both inducing gp120 shedding with similar potencies (Figure 8). This argues that the unique irreversible-virus-inactivation capacity is still retained by the chimeric inhibitor.
Figure 8.

Capacity of the bifunctional chimera, LJC240−L4−UM15, to induce gp120 shedding similarly to UM15.
3.4. Evaluation of Chimera 11 and Its Individual Components against CD4-Independent HIV.
To further validate the bifunctional approach, we evaluated chimera 11 and the individual components against an adapted HIV-1 pseudovirus, J1HX, and its CD4-independent mutant, N197S. Removal of the glycosylation site at asparagine 197 in the V1−V2 stem was found to be sufficient to enable a conformation that is more primed for CCR5 binding in the absence of CD4 and for HIV-1 entry into CD4-negative cells expressing CCR5.32 As shown in Figure 9, chimera 11 was functionally active and showed synergy against the parent non-CD4-independent J1HX WT with an IC50 >16-fold more active than that of LJC240−L4 (Figure 9A). With the CD4-independent mutant, the individual components remained active: the potency of the UM15 component did not change compared with that of the WT, arguing that the binding of UM15 to gp120 was not disrupted, whereas the LJC240−L4 component showed ~3-fold enhanced potency (Figure 9B) compared with that of the WT. In contrast with the results of the WT J1HX, chimera 11 was only slightly more active, if at all, than the LJC240−L4 component alone against the J1HX N197S virus (Figure 9B). The reduced synergistic potency of chimera 11 observed with J1HX N197S could be due to either (1) the masking of the chimera effect by the enhanced LJC240−L4 potency or (2) an altered dual Env−CCR5 encounter by the chimera components due to rearrangements in the gp120 V1−V2 region. Further investigation will be required to better define the mechanism causing reduced synergy.
Figure 9.

Evaluation of chimera 11 (red) and the individual components, UM15 (black) and LJC240−L4 (blue), against J1HX (A) and the CD4-independent J1HX mutant, N197S (B).
3.5. Coreceptor-Binding Efficiency Retained by Chimera 11.
The coreceptor-inhibitory function of the chimera was assessed, initially using a calcium-mobilization assay to determine the extent of antagonism of RANTES function (Figure 10, top panel). A comparison of LJC24015 (Figure 2) and LJC240−L4 was included to determine whether the linker would negatively impact antagonistic ability. Indeed, the added linker appeared to interfere with the potency of the molecule, as judged by an activity decrease from 1.59 nM with LJC240 to 27.44 nM with LJC240−L4. No further decrease of function was observed with LJC240−L4−UM15 (11), as judged by the IC50 value of 22.09 nM.
Figure 10.

Dose-dependent CCR5 antagonism by the bifunctional chimera and the parent small-molecule inhibitors. (Top) Dose−response curves of the extent of the inhibition of RANTES (10 nM) stimulation of calcium mobilization measured for LJC240-containing compounds. Maraviroc was included as a control. (Bottom) Chemotaxis inhibition of CEM CCR5 cells to RANTES by LJC240−L4−UM15. One million CEM CCR5 cells were seeded in the apical chambers of 8 μm transwell inserts, and chemoattraction was induced using RANTES (10 ng/mL) in the presence or absence (control, No Chemokine) of the inhibitors maraviroc (MRVC); LJC240; LJC240−L4; or the peptide triazole chimera, LJC240−L4−UM15, at the IC50 value for each compound. Data are presented as cells recovered in the basolateral chamber postincubation normalized to cells migrating to RANTES in the absence of inhibitor. Two-way ANOVA was used to compare the inhibitor-treated groups to the RANTES-only control group, and Student’s t-test was used to compare individual groups. Error bars represent means ± SEM; *p ≤ 0.05, **p ≤ 0.01. These data are representative of two independent experiments.
Antagonism of the coreceptor was confirmed using chemotaxis assays with the parent molecules, the linker-containing inhibitor, and the bifunctional chimera. As shown in Figure 10 (bottom panel), the compounds containing the coreceptor-inhibitor component at the IC50 values determined in the calcium-mobilization assay all prevented chemotaxis of CEM CCR5 cells to the RANTES chemokine in a transwell assay. The in vitro cell toxicity was measured postincubation, and the inhibitors did not lead to cell death as measured in the apical and basolateral chambers post-treatment (data not shown). These results support the finding that the bifunctional chimera retains the function of antagonizing the CCR5 coreceptor.
4. Cytotoxicity and HIV Specificity.
To assess cellular safety and HIV specificity, we evaluated the chimera (11) in a WST cytotoxicity assay as well as its activity against acute murine leukemia virus (AMLV). As shown in Figure 11, chimera 11 did not show any cytotoxicity nor any activity against AMLV at the concentration range used.
Figure 11.

Cytotoxicity evaluation of chimera 11 (left) and evaluation of chimera 11 against AMLV (right).
DISCUSSION
In this work, we engineered a covalent bifunctional chimera capable of inhibiting HIV cell infection through a combination of effects on both sides of the virus−host-cell interface. A linker designed to span the distance between the Env and the coreceptor was utilized to join the peptide triazole and the CCR5 antagonist. The first prototype bifunctional chimera generated, namely, LJC240−L4−UM15 (11), not only displays enhanced inhibitory potency compared with that of a 1:1 (mol/mol) noncovalent mixture of the parent molecules that make up the chimera, but also shows a better synergistic effect compared with that of the noncovalent mixture in a quantitative synergy analysis. This potency was made possible because each component of the chimera is able to function coordinately despite the covalent attachment to the other.
The observed synergistic function of the two inhibitory components of the CoRI−PT chimera argues that both components can function at the same time. This in turn implies spatial proximity of the gp120- and coreceptor-binding sites upon the virus−cell encounter. Several hypotheses can be surmised to explain this synergy, based on the modes of action of the chimera components. Because PTs inhibit binding to the coreceptor site likely via affecting the release of the V3 loop,33 we envision that as the virus approaches the cell in the presence of the chimera, the PT part of the chimera can dock onto the virion Env, inhibiting CD4 attachment and altering the V3-loop−CCR5-epitope-release mechanism. However, the chimera has another exposed scaffold, which is the CCR5 blocker. This CCR5 blocker brings the Env−chimera complex closer to the cell to dock onto the CCR5 protein. The net result of this mechanism is the locking of the virus−cell complex in an arrested configuration, leading to the blocking of the infection and later on causing gp120 shedding by the PT-domain function. In other words, the chimera CCR5-inhibitor−L4 part can act as a false V3 loop, bringing the virus closer to the cell not for initiating infection but rather to be blocked. On the other hand, the entropic advantage of the tethered hybrid over the two separate components might be one cause of the synergistic effect.34 When the chimera binds to one site (protein) at the cell−virus interface, much of the entropy cost of binding to the second site (protein) has already been paid. Thus, the free energy of binding for the chimera is not the sum of the free energies of the two components because of entropy, leading to a synergistic potency. Importantly, the shedding function of the chimera was demonstrated in this work. Nonetheless, the sequence of bifunctional-chimera-binding events at the cell−virus interface remains speculative at present.
Although other covalent chimeras have been designed that target HIV-1 infection,35−40 the prototype chimera LJC240−L4−UM15 (11) is unique in the added value of the irreversible inactivation of the virus. Previously, chimeric inhibitors have been reported that target (1) two sites on the HIV-1 Env protein alone,36,38,39 (2) CD4 and Env gp41,35 and (3) Env gp41 and a coreceptor.37,40 The latter two findings have demonstrated the feasibility of the bifunctional engagement of both virus Env and host-cell receptors. Because the CCR5 and CXCR4 coreceptors can heterodimerize with each other,41 the CCR5-targeting CoRI−PT chimera reported in the current work has the additional potential to bind to CCR5 in the heterodimer and neutralize the X4-tropic virus by Env inactivation through the PT domain (which is active against both X4- and R5-viruses) before the CXCR4 encounter.
Bifunctional chimeras of the CoRI−PT type have an important advantage in avoiding virus resistance by combining components that can function synergistically or separately against evolving viruses and hence challenging the virus to combine escapes to two different inhibitory functions coordinately. Analogously with most if not all virus-protein-targeting antagonists, mutational escape of the Env-binding PT component can occur.31 Further, even though CoRIs target a host-cell protein rather than a virus protein, HIV-1 resistance mutations to coreceptor antagonists have been observed to occur by the formation of binding sites on the virus Env protein that can bind the antagonist-bound form of the coreceptor and in this way infect cells in the presence of CoRIs.42 We envision that, because the CCR5 antagonist in the LJC240−L4−UM15 chimera (11) is attached to a relatively large peptide and linker moiety, it would be difficult for the virus to bind the chimera-armed coreceptor because of the increased spatial blockade. However, even if the virus could overcome this blockade, the added challenge of escaping the PT component of the chimera would improve the ability of the chimera to avoid overall inhibitor escape.
The results of the current work open up both future chimera-design opportunities and the potential for expanded functional utility. The chimeric inhibitor presented in this work (11) serves as a prototype for advancing more druglike chimeric inhibitors. The recent discovery of macrocyclic PTs (cPTs),19,43 combined with existing44 and newly discovered CXCR4-targeting small molecules,45 provides the potential to develop druglike and protease-resistant CoRI−cPT variants that target both CCR5 and CXCR4 cellular infections with improved bioavailability. Increased understanding of the structural mechanisms of action of the chimeras will be helpful to guide future chimera design. As the latter is progressing, the relatively small sizes of such chimeras will make it feasible to investigate whether these could inhibit not only virus−cell infections but also cell-to-cell transmission.
CONCLUSIONS
In summary, we have designed and produced a fully synthetic bifunctional chimeric inhibitor that can target both sides of the HIV−cell interface. One component blocks the viral envelope, and the second component blocks the cellular coreceptor CCR5. The chimera showed synergistic potency, compared with that of the noncovalent mixture of the individual components. This result demonstrates the ability to make potent, fully synthetic HIV-1 inhibitors by simultaneously targeting the virus Env and its cellular chemokine receptors.
EXPERIMENTAL SECTION
Docking of LJC240 onto the CCR5 Receptor.
The recently solved crystal structure of CCR5 bound to the inhibitor maraviroc was used (PDB code: 4MBS).26 The PDB file was downloaded from the Protein Data Bank and prepared with the protein-preparation wizard in Maestro 9.9 (Schrödinger Suite 2015; Schrödinger, LLC). Before docking LJC240, the docking procedures were validated; the cocrystallized maraviroc was docked onto the receptor structure using the Glide-XP docking protocol, and it showed a similar pose to that of the crystallographically solved bound state (heavy atoms RMSD = 0.31 Å, data not shown) and had a calculated receptor−maraviroc-interaction-energy value of ca. −33.2 kcal/mol as calculated using Szybki 1.8.0.2 (OpenEye Scientific Software).46 LJC240 was built and prepared using the builder and LigPrep tools in Maestro 9.9.47 The inhibitor LJC240 was then docked onto the CCR5 receptor using the same Glide-XP protocol. This yielded a stable low-energy pose (Figure 2) with a calculated receptor−ligand-interaction energy of ca. −27.1 kcal/mol (calculated using Szybki 1.8.0.2, OpenEye Scientific Software).46 The docking showed the fluorophenyl moiety of the inhibitor to be the most-solvent-exposed part, with the least-important interaction. This was in agreement with the results of the SAR studies that changes in this part of the molecule can be tolerated.
Docking of UM15 onto the gp120 Protein.
Peptide UM15 was built and docked onto gp120 chain A of the SOSIP Env trimer (PDB code: 4NCO,25 structure as previously described)48 using the InducedFit docking protocol.49 The N-terminus of UM15 was shown to be well solvated (Figure 2) and could be used as an extension point for the chimera synthesis.
Chemical Synthesis.
General Information.
The starting materials were obtained from commercial sources, such as Adamas-Beta, Sigma-Aldrich, J&K, TCI, and Chem-Impex, and were used without further purification. Unless otherwise specified, all reactions were carried out in oven-dried glassware with magnetic stirring. All reagents were weighed and handled in air at room temperature. Column chromatography was performed on silica gel (200−300 mesh). The column output was monitored by TLC on silica gel (100−200 mesh) precoated on glass plates (15 × 50 mm), and spots were visualized by UV light at 254 nm. All new compounds were characterized by 1H NMR, 13C NMR, and low- or high-resolution mass spectroscopy. NMR spectra were recorded on a Bruker AVANCE III 400 NMR spectrometer. Chemical shifts for the proton-magnetic-resonance spectra (1H NMR) were quoted in parts per million (ppm) and referenced to the signals of residual chloroform (7.26 ppm) or methanol (3.30 ppm). All 13C NMR spectra were reported in ppm relative to deuterochloroform (77.23 ppm) or methanol (49.00 ppm). The following abbreviations were used to describe peak-splitting patterns when appropriate: br, broad; s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; and dd, doublet of doublets. Coupling constants, J, were reported in hertz units (Hz). Mass spectra were recorded using an ESI ion source unless stated otherwise. All melting points were measured using a BÜCHI 510 melting-point apparatus. The yields in this paper refer to the isolated yields of compounds estimated to be ≥95% pure as determined by 1H NMR. Unless otherwise specified, the purities of all new compounds were determined by analytical HPLC on an Agilent Technologies 1260 Infinity with an Agilent ZORBAX 3.5 μM SB-phenyl column (4.6 × 75 mm) in one solvent system (solvent A was 0.02% TFA in water, and solvent B was 0.02% TFA in acetonitrile). The flow rate was 1.5 mL/min, the UV detector was at 210 or 254 nm, and the gradient was 5−90% B in 12 min and then 90% B for 3 min.
Synthesis of 8-Benzyl-8-aza-bicyclo[3.2.1]octan-3-one Oxime (2).50
A mixture of 8-benzyl-8-aza-bicyclo[3.2.1]octan-3-one (5.00 g, 23.2 mmol), hydroxylamine hydrochloride (1.78 g, 25.6 mmol), and NaHCO3 (2.54 g, 30.2 mmol) was stirred in EtOH/H2O (60 mL; 1:1, v/v) for 18 h. The reaction mixture was filtered, and the residue was washed with water and dried to afford compound 2 as a white solid (3.24 g, 61% yield, mp 118−120 °C). 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 7.2 Hz, 2H), 7.38−7.32 (m, 2H), 7.32−7.29 (m, 1H), 3.70 (s, 2H), 3.43−3.34 (m, 2H), 3.02 (d, J = 15.5 Hz, 1H), 2.66−2.59 (m, 1H), 2.31−2.23 (m, 1H), 2.18−2.05 (m, 1H), 2.10−2.00 (m, 2H), 1.66 (t, J = 7.9 Hz, 1H), 1.56 (t, J = 7.6 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 157.0, 139.3, 128.6, 128.3, 127.0, 58.5, 57.8, 55.6, 37.3, 31.3, 27.6, 26.7. HRMS (ESI) calcd for C14H19N2O [M + H]+: 231.1492, found 231.1489.
Synthesis of exo-8-Benzyl-8-aza-bicyclo[3.2.1]oct-3-ylamine (3).51
A solution of compound 2 (3.00 g, 13.0 mmol) in pentanol (50 mL) was heated under reflux. Sodium (3.59 g, 156.3 mmol) was added portionwise over 2.5 h. The reaction was then heated under reflux for 2 h further and then cooled in an ice bath. Water was added until no more hydrogen gas was evolved. The mixture was acidified using 6 N aqueous hydrochloric acid, and the phases were separated. The organic layer was extracted with 6 N aqueous hydrochloric acid, the combined aqueous extracts were basified to pH 12 with sodium hydroxide pellets, and the aqueous solution was extracted with ethyl acetate. The combined organic solutions were dried with MgSO4, filtered, and evaporated under reduced pressure to afford compound 3 as a colorless oil (1.91 g, 68% yield). 1H NMR (400 MHz, CDCl3) δ 7.45−7.36 (m, 2H), 7.30−7.25 (m, 2H), 7.26−7.19 (m, 1H), 3.63 (s, 2H), 3.38−3.23 (m, 3H), 2.09−1.97 (m, 2H), 1.96−1.80 (m, 4H), 1.67−1.53 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 138.2, 128.4, 127.9, 126.7, 57.5, 54.5, 43.6, 35.7, 26.1. HRMS (ESI) calcd for C14H21N2 [M + H]+: 217.1699, found 217.1704.
Synthesis of exo-(8-Benzyl-8-aza-bicyclo[3.2.1]oct-3-yl)-carbamic Acid tert-Butyl Ester (4).52
A mixture of compound 3 (3.00 g, 13.9 mmol), Boc2O (3.33 g, 15.3 mmol), and TEA (3.9 mL, 27.7 mmol) was stirred in THF (52 mL) for 18 h. The reaction mixture was evaporated under reduced pressure. The residue was purified by recrystallization, affording compound 4 as a white solid (4.19 g, 95% yield, mp 174−175 °C). 1H NMR (400 MHz, CDCl3) δ 7.38 (d, J = 7.3 Hz, 2H), 7.32 (t, J = 7.4 Hz, 2H), 7.26– 7.23 (m, 1H), 3.56 (s, 2H), 3.31−3.14 (m, 2H), 2.05−2.00 (m, 2H), 1.84−1.78 (m, 2H), 1.73−1.69 (m, 2H), 1.58−1.50 (m, 3H), 1.43 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 155.3, 140.0, 128.5, 128.2, 126.8, 79.2, 58.7, 56.1, 42.8, 38.7, 28.4, 26.5. HRMS (ESI) calcd for C19H29N2O2 [M + H]+: 317.2224, found 317.2224.
Synthesis of exo-(8-Aza-bicyclo[3.2.1]oct-3-yl)-carbamic Acid tert-Butyl Ester (5).53
To a solution of compound 4 (2.73 g, 8.6 mmol) in methanol (40 mL) were added 10% Pd/C (273 mg, ca. 50% water) and HCO2NH4 (3.81 g, 60.4 mmol), and the above mixture was heated under reflux for 15 min. After it cooled to room temperature, the reaction mixture was evaporated under reduced pressure. Purification by flash chromatography on silica gel (10:1 DCM/methanol) afforded compound 5 as a white solid (1.43 g, 73% yield, mp 271−272 °C). 1H NMR (400 MHz, CDCl3) δ 4.80−4.76 (m, 1H), 4.08−4.02 (m, 2H), 2.29−2.20 (m, 2H), 2.05−1.93 (m, 6H), 1.42 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 169.0, 79.8, 54.7, 41.1, 35.4, 28.3, 26.3. HRMS (ESI) calcd for C12H23N2O2 [M + H]+: 227.1754, found 227.1748.
Synthesis of (3-Chloro-4-methyl-phenyl)-(3-chloro-propyl)-amine (7).54
To a mixture of 1-bromo-3-chloropropane (10.5 mL, 105.9 mmol) in DMF was added KI (586 mg, 3.5 mmol), and the mixture was stirred at room temperature for 30 min; then, TEA (19.6 mL, 141.2 mmol) and 3-chloro-4-methyl-phenylamine (5.00 g, 35.3 mmol) were added, and the solution was stirred at room temperature for 72 h further. The reaction mixture was filtered, and the filtrate was concentrated in a vacuum. The residue was diluted with EtOAc, washed with water and brine, dried with MgSO4, filtered, and concentrated in a vacuum. Purification by flash chromatography on silica gel (25:1 petroleum ether/ethyl acetate) afforded compound 7 as a yellow oil (5.20 g, 68% yield). 1H NMR (400 MHz, CDCl3) δ 7.01 (d, J = 8.2 Hz, 1H), 6.63 (d, J = 2.4 Hz, 1H), 6.44 (dd, J = 8.2, 2.5 Hz, 1H), 3.65 (t, J = 6.2 Hz, 2H), 3.30 (t, J = 6.6 Hz, 2H), 2.25 (s, 3H), 2.05 (p, J = 6.5 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 147.0, 134.9, 131.4, 124.4 113.1, 111.7, 42.5, 41.1, 31.8, 18.9. HRMS (ESI) calcd for C10H14Cl2N [M + H]+: 218.0498, found 218.0497.
Synthesis of 1-Acetyl-piperidine-4-carboxylic Acid (3-Chloro-4-methyl-phenyl)-(3-chloro-propyl)-amide (8).54
A solution of 1-acetylpiperidine-4-carboxylic acid (6.75 g, 39.4 mmol) in SOCl2 (40 mL) was stirred for 2 h at room temperature. The reaction mixture was diluted with petroleum ether and filtered to afford the acyl chloride as a white solid. Compound 7 (2.87 g, 13.1 mmol) was dissolved in DCM (50 mL), TEA (5.5 mL, 39.4 mmol) and the acyl chloride were added, and then the mixture was stirred at room temperature for 2 h further. The residue was diluted with DCM, washed with water and brine, dried with MgSO4, filtered, and concentrated in a vacuum. Purification by flash chromatography on silica gel (1:1 petroleum ether/ethyl acetate) afforded compound 8 as a white solid (3.74 g, 77% yield, mp 101−103 °C). 1H NMR (400 MHz, CDCl3) δ 7.31 (d, J = 8.0 Hz, 1H), 7.18 (d, J = 2.0 Hz, 1H), 6.97 (dd, J = 8.0, 2.1 Hz, 1H), 4.51 (d, J = 13.3 Hz, 1H), 3.78−3.75 (m, 2H), 3.53 (t, J = 6.6 Hz, 2H), 2.87−2.81 (m, 1H), 2.43 (s, 3H), 2.39−2.28 (m, 2H), 2.05 (s, 3H), 2.02−1.96 (m, 2H), 1.67−1.55 (m, 5H). 13C NMR (126 MHz, CDCl3) δ 174.2, 168.8, 140.9, 136.7, 135.4, 132.1, 128.4, 126.2, 47.7, 45.5, 42.3, 40.7, 39.3, 30.8, 28.8, 28.3, 21.4, 19.8. HRMS (ESI) calcd for C18H25Cl2N2O2 [M + H]+: 371.1288, found 371.1281.
Synthesis of exo-1-Acetyl-piperidine-4-carboxylic Acid [3-(3-Amino-8-aza-bicyclo[3.2.1]oct-8-yl)-propyl]-(3-chloro-4-methylphenyl)-amide (9).
To a mixture of compound 8 (1.70 g, 4.6 mmol), compound 5 (1.04 g, 4.6 mmol), and KI (760 mg, 4.6 mmol) in acetonitrile was added NaHCO3 (1.15 g, 13.7 mmol), and the mixture was stirred at reflux for 12 h. After cooling to room temperature, the mixture was filtered, and the filtrate was concentrated in a vacuum. The residue was diluted with EtOAc, washed with water and brine, dried with MgSO4, filtered, and concentrated in a vacuum. Purification by flash chromatography on silica gel (25:1 DCM/methanol) afforded the product as a white foam (1.72 g, 67% yield). 1H NMR (400 MHz, CDCl3) δ 7.31 (d, J = 8.1 Hz, 1H), 7.20 (d, J = 1.4 Hz, 1H), 7.08 (d, J = 7.7 Hz, 1H), 4.65−4.46 (m, 2H), 3.88−3.60 (m, 4H), 3.49−3.38 (m, 2H), 2.82 (t, J = 13.7 Hz, 1H), 2.57−2.50 (m, 2H), 2.40 (s, 3H), 2.36−2.25 (m, 2H), 2.03 (s, 3H), 2.00−1.95 (m, 2H), 1.84−1.79 (m, 8H), 1.67−1.54 (m, 3H), 1.41 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 174.3, 168.8, 155.3, 140.7, 136.7, 135.3, 132.1, 128.4, 126.6, 79.5, 59.7, 54.7, 48.9, 47.5, 45.5, 41.8, 40.7, 39.4, 36.8, 28.9, 28.4, 28.3, 25.8, 21.4, 19.8. HRMS (ESI) calcd for C30H46ClN4O4 [M + H]+: 561.3202, found 561.3214. Purity: 99.8% (tR = 6.63).
Synthesis of 4–8-(3-(1-Acetyl-N-(3-chloro-4-methylphenyl)-piperidine-4-carboxamido)propyl)-8-azabicyclo[3.2.1]octan-3-yl)-sulfamoyl)benzoic Acid (10).
To a solution of compound 9 (2.62 g, 4.7 mmol) in methanol, HCl (4.8 M, in methanol) was added, and the mixture was stirred at room temperature for 4 h. After the reaction mixture was concentrated in a vacuum, the residue (1.57 g, 3.4 mmol) was dissolved in 1,4-dioxane (40 mL); then, 4-(chlorosulfonyl)benzoic acid (1.50 g, 6.8 mmol) and a solution of Na2CO3 (1.08 g, 10.2 mmol) in H2O (20 mL) were added, and the mixture was stirred at room temperature for 12 h. The residue was evaporated in a vacuum, washed with a mixture of methanol and DCM (9:1 DCM/methanol), and filtered, and the filtrate was concentrated in a vacuum. Purification by flash chromatography on silica gel (10:1 DCM/methanol) afforded compound 10 as a white solid (1.22 g, 40% yield for two steps, mp 182−184 °C). 1H NMR (400 MHz, MeOD) δ 8.01 (d, J = 8.5 Hz, 2H), 7.82 (d, J = 8.5 Hz, 2H), 7.35 (dd, J = 8.5, 5.1 Hz, 2H), 7.11 (dd, J = 8.1, 2.1 Hz, 1H), 4.35−4.26 (m, 1H), 3.86−3.79 (m, 2H), 3.78−3.70 (m, 1H), 3.67−3.60 (m, 2H), 3.56−3.46 (m, 1H), 2.95−2.87 (m, 2H), 2.78 (t, J = 11.4 Hz, 1H), 2.42−2.36 (m, 1H), 2.31 (s, 3H), 2.29−2.24 (m, 1H), 2.16−2.08 (m, 2H), 1.95 (s, 3H), 1.86−1.74 (m, 8H), 1.59 (d, J = 15.2 Hz, 3H), 1.45 (d, J = 16.2 Hz, 1H). 13C NMR (126 MHz, MeOD) δ176.0 (2), 169.7, 143.2, 139.6, 139.0, 136.7, 134.7, 131.9, 129.4, 127.7, 125.9, 125.7, 60.8, 45.9, 44.8, 43.1, 40.0, 38.9, 28.0, 27.5, 23.5, 22.3, 19.3, 18.0. HRMS (ESI) calcd for C32H42ClN4O6S [M + H]+: 645.2508, found 645.2506. Purity: 99.5% (tR = 5.61).
Solid-Phase Synthesis of LJC240−L4.
Rink amide resin (0.1 mmol, 200−400 mesh) was swollen in 10 mL of DMF/DCM (1:1). Fmoc deprotection was achieved using a solution of 20% piperidine in DMF. The Fmoc-protected linker (116 mg, 0.3 mmol) was dissolved in DMF, and DIEA (124 μL, 0.75 mmol) and HBTU (110 mg, 0.29 mmol) were added. The mixture was vibrated for 30 min at room temperature and then added to the reaction vessel, which was vibrated at room temperature for 4 h. Four units of the linker were installed. LJC240−COOH (10, 193 mg, 0.3 mmol) was dissolved in DMF. Then, DIEA (124 μL, 0.75 mmol), HOBt (41 mg, 0.3 mmol), and HBTU (110 mg, 0.29 mmol) were added, and the mixture was vibrated for 30 min and then added to the (linker)4−resin vessel. The reaction vessel was vibrated at room temperature for 12 h. Cleavage was carried out using a cocktail of trifluoroacetic acid/ethanedithiol/H2O (4.75:0.125:0.125, v/v) for 2 h at room temperature. The cleaved-compound solution was concentrated under a gentle N2 stream, and then dissolved in ACN/H2O. The crude product was purified by semipreparative HPLC on an EasySep-1050 with an XBridge Prep C18 reversed-phase column (19 × 150 mm, ACN/H2O/0.1% TFA, tR = 17.7 min, gradient: 10−90% ACN over 30 min, flow rate of 4 mL/min) and finally evaporated in a vacuum to yield the ideal product. 1H NMR (400 MHz, MeOD) δ 8.06−7.99 (m, 4H), 7.51−7.46 (m, 2H), 7.24 (dd, J = 7.9, 2.0 Hz, 1H), 4.45 (d, J = 13.9 Hz, 1H), 4.02 (s, 4H), 3.99 (s, 3H), 3.91−3.75 (m, 3H), 3.75−3.64 (m, 17H), 3.62−3.55 (m, 6H), 3.51−3.39 (m, 5H), 3.06−2.98 (m, 2H), 2.95−2.82 (m, 2H), 2.72 (s, 2H), 2.57−2.49 (m, 1H), 2.44 (s, 3H), 2.43−2.38 (m, 1H), 2.29−2.21 (m, 2H), 2.08 (s, 3H), 2.06 (s, 1H), 2.02−1.87 (m, 7H), 1.77−1.54 (m, 5H), 1.38−1.28 (m, 4H). 13C NMR (126 MHz, MeOD) δ 176.0, 174.0, 171.0, 169.6, 166.9, 143.8, 139.6, 137.8, 136.7, 134.7, 131.8, 127.7, 127.6, 126.2, 125.9, 70.1 (4), 69.7−69.1 (8), 68.9−68.5 (4), 61.2, 45.8, 44.8, 43.1, 40.0, 39.2, 38.9, 37.9, 36.4, 33.5, 28.1, 27.5, 23.4, 22.2, 19.3, 18.0. HRMS (ESI) calcd for C56H87ClN9O17S [M + H]+: 1224.5624, found 1224.5627. Purity: 99.0% (tR = 8.47).
Solid-Phase Synthesis of LJC240−L4−UM15 (11). Solid-Phase Synthesis of Resin-Bound Linker−PT.
Using an Fmoc-based synthesis strategy, the following sequence was assembled using microwave-assisted coupling: (linker)4−ile−asn−asn−ile−AzidoPro−trp−resin. The resin is the rink amide resin (100−200 mesh size), and the synthesis scale was 0.25 mM. An activation scheme (1 mL of 0.5 M DIC/0.5 mL of 0.5 M Oxyma) was used. Deprotection of the Fmoc groups was performed with 5 mL of 20% piperidine and 0.1 M HOBt in DMF.
Coupling LJC240−COOH to the Linker−PT−Resin.
The (linker)4−peptide−resin was swollen by soaking it in 10 mL of DMF/DCM (1:1). LJC240−COOH (10, 1.1 equiv), HBTU (2.2 equiv), HOBt (2.2 equiv), and DIPEA (4.4 equiv) were added to the reaction vessel. The vessel was vibrated for 10 h at room temperature, and the reaction was monitored by the Kaiser test until negative (no blue color) results were observed. The resin-bound complex was then filtered, and the resin was washed thoroughly with DMF, methanol, and DCM.
Click Reaction of the Alkyne (Ethynylferrocene) with the Azide Group on the Pro Residue.
The resin-bound fusion was mixed with 5 mL of ACN, 4 mL of H2O, 1.06 mL of DIPEA, and 0.53 mL of pyridine with 5 equiv of ethynylferrocene, and the reaction vessel was vibrated at room temperature for 12 h. The resin-bound product was washed thoroughly with 5% HCl (2 × 50 mL), DMF (2 × 50 mL), and DCM (2 × 50 mL).
Cleavage of the Fusion from the Resin and Deprotection of the Peptide Side Chains.
The resin-bound product was mixed with 20 mL of a chilled cleavage mixture (95% TFA, 2.5% H20, and 2.5% TIPS) and vibrated for 2.5 h at room temperature. The reaction mixture was filtered, and the leftover resin was washed with 10 mL of the cleavage mixture. The acidic filtrate was concentrated under a gentle N2 stream. Precooled ether (30 mL) was added to the residue, and after rigorous vortexing and centrifugation, the ether layer was decanted. This ether-washing step was repeated two to three times until the ether layer was no longer colored. The residue was then vacuum-dried, redissolved in an ACN/H20/0.1% TFA mixture for RP-HPLC purification (Waters HPLC, absorption at 210 nm) using a Waters C18 prep column. The pure HPLC fractions (≥97% purity as judged by analytical HPLC; 5−95% ACN with 0.1% TFA in 40 min) were lyophilized to give the chimera, 11, as a pale-yellow powder (65% yield calculated on the basis of the cleavage of 0.5 g of resin). The MALDI-TOF-determined mass of the purified chimera was found to be 2212.75 Da, comparable to the calculated mass of 2211.96 Da.
Production of the HIV-1 Envelope-Pseudotyped Virus.
Recombinant envelope-pseudotyped viruses were prepared by cotransfection of an envelope-expression plasmid, either JR-FL (R5-tropic) or HxBc2 (X4-tropic), with an HIV-1-viral-backbone plasmid encoded with a luciferase gene and lacking Env, pNL Luc AM.55 A combination of 4 mg of Env and 8 mg of backbone DNA were transfected into 293T cells using PEI (Fisher Scientific) as a transfection reagent. Cell supernatant containing the recombinant viruses was collected after 48−72 h and purified through a 6−20% iodixanol (Sigma-Aldrich) gradient to remove free viral proteins and exosomes. The gradient was performed at 4 °C for 2 h at 30 000 rpm (SW41 rotor, Beckman ultracentrifuge). The fractions were collected following the spin, validated as previously described, and stored at −80 °C until needed.
Infection-Inhibition Assay.
The LJC240−L4−UM15 (11) bifunctional chimera was evaluated for potency using a standard pseudoviral assay.55,56 Briefly, envelope-pseudotyped viruses were treated for 30 min at 37 °C with the inhibitor and then added to HOS cells expressing both CD4 and an appropriate coreceptor depending on the assay. The infection was carried out for 24 h before a medium change was performed to remove any residual virus or inhibitor. The production of the reporter luciferase was allowed to continue for an additional 24 h before the cells were lysed with passive-lysis buffer (Promega). Cell lysates were transferred to a white well plate and mixed with 1 mM luciferin salt (Anaspec) diluted in 0.1 M potassium phosphate buffer containing 0.1 M magnesium sulfate. The luminescence was measured using a Wallace 1450 Microbeta luminescence reader at a wavelength of 490 nM.
Synergy Analysis.
For the synergy analysis, the cell-infection-inhibition assay was set up as described above with LJC240−L4, UM15, chimera 11, and the combination of LJC240−L4 and UM15 at a 1:30 ratio based on the initially derived IC50 values. The values of percent Bal.01 infection were plotted against the concentration of each inhibitor used in the combination, and the IC50 values were calculated and compared with the IC50 values of the chimera derived from the same experiment. The IC50 values so determined were plotted in an isobologram to assess the synergistic effects of both the noncovalent mixture and the chimera.
HIV Specificity and Cytotoxicity.
HIV specificity was observed by performing an infection-inhibition analysis of the bifunctional chimera against amyotrophic leukemia virus (AMLV) along with pseudotyped HIV-1. In addition, infection-inhibition activities were evaluated with the functionally CD4-independent virus mutant J1HX N197S and compared with those of the nonmutated, CD4-dependent parent virus (J1HX).32 Briefly, the viruses were treated for 30 min at 37 °C with the inhibitor and then added to HOS cells. The infection was carried out for 24 h before a media change was performed to remove any residual virus or inhibitor. The production of the reporter luciferase was allowed to continue for an additional 24 h before the cells were lysed with passive-lysis buffer (Promega), and a luciferase assay was performed to quantify the signal.
The cytotoxicity of the bifunctional chimera, 11, was measured by performing a colorimetric assay designed to measure the relative proliferation rates of cells in culture. Briefly, the cells were treated with the serially diluted bifunctional chimera for 24 h, after which a tetrazolium salt, WST-1, was added to the cells. The viability of the cells was measured by detecting the conversion of the tetrazolium salt, WST-1, into a colored dye (Optical density measurement at 400 nM) by mitochondrial dehydrogenase enzymes functional only in live cells.
Shedding Protocol.
HIV-1 shedding was determined by performing a Western-blot analysis to observe the amount of shed gp120 when the viruses were treated with the bifunctional chimera, 11, and UM15. Pseudovirus BaL.01 was produced and purified as described above. Aliquots (90 μL) of purified BaL.01 pseudovirus were mixed with 10 μL of the compounds (UM15 or LJC240−L4−UM15) at various dilutions or PBS, which served as the negative control. The mixtures were incubated for 18 h at 37 °C and then spun at 15 000 rpm at 4 °C for 2 h. The supernatants (80 μL) of the mixtures were taken and separated by 10% SDS-PAGE gel electrophoresis. Then, Western blotting was used to determine the gp120 contents in the supernatants with a primary anti-gp120 antibody, D7324, and a secondary antibody, anti-sheep HRP. ImageJ was used to determine the Western-blot band densities.
Surface-Plasmon-Resonance-Interaction Analysis.
SPR experiments were performed on a BIACORE 3000 optical biosensor (GE) at 25 °C using standard PBS buffer containing 0.005% tween 20 and 2% DMSO. Three flow cells in a CM5 chip were used for the amine coupling of different ligands using standard 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC)−N-hydroxysuccinamide (NHS) chemistry. Flow cell 1, containing ~2000 RU of immobilized 2B6R antibody, served as a control for flow cells 2 and 3, which contained 2000 RU of CD4 and 17b antibodies, respectively. The protein gp120 (200 nM) in the presence of increasing concentrations of LJC240−L4−UM15 (11) and UM15 was injected over flow cells 1, 2, and 3. After each sample run, bound gp120 was removed with 10 mM HCl (two pulses of 10 s). All experiments were done in sets of three. Data analysis was performed using BIAevaluation V4.1.1 software (GE). To correct for nonspecific binding, response signals from the buffer injection and from the control flow cell were subtracted from all sensorgrams. Inhibition data were determined by calculating the inhibitor concentration required for 50% inhibition of maximal binding (IC50). The inhibition curve was plotted and then fitted using the four-parameter eq 1, as shown below, using Origin Pro 8 graphing software.
| (1) |
where Rhigh is the response at the highest inhibitor concentration, Rlow is the response at the low inhibitor concentration, concn is the concentration of the inhibitor, and A1 and A2 are the fitting parameters.
Calcium-Mobilization Assay.
CHO cells stably expressing CCR5 and Gα16 were loaded with 2 μmol/L Fluo-4 AM in Hanks’ balanced salt solution (HBSS, containing 5.4 mM KCl, 0.3 mM Na2HPO4, 0.4 mM KH2PO4, 4.2 mM NaHCO3, 1.3 mM CaCl2, 0.5 mM MgCl2, 0.6 mM Mg2SO4, 137 mM NaCl, 5 g/L BSA, 5.6 mM glucose, 250 μM sulfinpyrazone, pH 7.4) at 37 °C for 45 min. After the cells were rinsed with the reaction buffer, HBSS (50 μL) containing known antagonists (the positive control), the compounds of interest, or DMSO (the negative control, final concentration of 1%) were added. After an incubation at room temperature for 10 min, RANTES (25 μL, final concentration 30 nM) was dispensed into the well using a FlexStation II microplate reader (Molecular Devices), and the intracellular calcium change was recorded with an excitation wavelength of 485 nm and an emission wavelength of 525 nm. The half-maximal inhibitory concentrations (IC50) of the compounds were determined with GraphPad Prism software by constructing their dose−response curves.
Cellular-Chemotaxis Assay.
CEM CCR5 cells were resuspended to a density of 2.0 × 106 cells/mL in RPMI-1640 medium containing 0.1% BSA and 10% FBS. The cells were preincubated with maraviroc, LJC240, LJC240−L4, and LJC240−L4−UM15 (11) at the IC50 values indicated in Figure 9 for 15 min at room temperature. The cells (500 μL, 1.0 × 106 cells) were seeded in smooth-walled inserts containing polycarbonate membranes with 8 μm pores in a 24-well tissue-culture plate (BrandTech). Complete medium (1 mL) containing RANTES (10 ng/mL) was added to the lower chamber, and the plate was incubated for 6 h at 37 °C in 5% CO2. The well with complete media only served as a control for RANTES-induced migration. Migrated cells that adhered to the basolateral surface of the insert were dissociated by incubating the inserts in 1 mL of HBSS with 1 mM EDTA for 30 min at 37 °C in 5% CO2. A 1:1 mixture (10 μL) of cells from the basolateral chamber and dissociated cells was then mixed with 10 μL of trypan blue, and the live cells were counted using the Countess automated cell counter (Thermo Scientific). Two-way ANOVA was used to compare the inhibitor-treated groups with the RANTES-only control group, and Student’s t-test was used to compare individual groups. Error bars represent means ± SEM; *p ≤ 0.05, **p ≤ 0.01.
ACKNOWLEDGMENTS
Y.Q.L. received funding from the National Natural Science Foundation of China (81761128022, 81361120410, and 81325020), and I.C. received funding from the National Institutes of Health (R01AI106633 and P01GM56550). We thank Openeye Scientific Software (Santa Fe, NM; http://www.eyesopen.com) for providing a complementary academic license of their software package.
AABBREVIATIONS USED
- ACN
acetonitrile
- Boc
tert-butyloxycarbonyl
- DIC
N,N′-diisopropylcarbodiimide
- DMF
dimethylformamide
- Env
HIV envelope gp160
- Fmoc
9-fluorenylmethoxycarbonyl
- HPLC
high-performance liquid chromatography
- HOBT
hydroxybenzotriazole
- PT
peptide triazole
- SPR
surface plasmon resonance
- tBu
tert-butyl
- TIPS
triisoproylsilane
- Trt
triphenylmethyl
- TFA
trifluoroacetic acid
- WT
wild type
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b00477.
UM15−SOSIP (PDB)
LJC240−CCR5 complex (PDB)
Molecular-formula strings (CSV)
REFERENCES
- (1).Wilen CB; Tilton JC; Doms RW Hiv: Cell binding and entry. Cold Spring Harbor Perspect. Med 2012, 2, a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Dorr P; Westby M; Dobbs S; Griffin P; Irvine B; Macartney M; Mori J; Rickett G; Smith-Burchnell C; Napier C; Webster R; Armour D; Price D; Stammen B; Wood A; Perros M Maraviroc (uk-427,857), a potent, orally bioavailable, and selective smallmolecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lieberman-Blum SS; Fung HB; Bandres JC Maraviroc: A CCR5-receptor antagonist for the treatment of hiv-1 infection. Clin. Ther 2008, 30, 1228–1250. [DOI] [PubMed] [Google Scholar]
- (4).Este JA; Telenti A Hiv entry inhibitors. Lancet 2007, 370, 81–88. [DOI] [PubMed] [Google Scholar]
- (5).Yost R; Pasquale TR; Sahloff EG Maraviroc: A coreceptor CCR5 antagonist for management of hiv infection. Am. J. Health-Syst. Pharm 2009, 66, 715–726. [DOI] [PubMed] [Google Scholar]
- (6).Greenberg ML; Cammack N Resistance to enfuvirtide, the first hiv fusion inhibitor. J. Antimicrob. Chemother 2004, 54, 333–340. [DOI] [PubMed] [Google Scholar]
- (7).Eggink D; de Taeye SW; Bontjer I; Klasse PJ; Langedijk JP; Berkhout B; Sanders RW Hiv-1 escape from a peptidic anchor inhibitor through stabilization of the envelope glycoprotein spike. J. Virol. 2016, 90, 10587–10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Nakata H; Steinberg SM; Koh Y; Maeda K; Takaoka Y; Tamamura H; Fujii N; Mitsuya H Potent synergistic anti-human immunodeficiency virus (hiv) effects using combinations of the CCR5 inhibitor aplaviroc with other anti-hiv drugs. Antimicrob. Agents Chemother 2008, 52, 2111–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tremblay CL; Giguel F; Guan Y; Chou TC; Takashima K; Hirsch MS Tak-220, a novel small-molecule CCR5 antagonist, has favorable anti-human immunodeficiency virus interactions with other antiretrovirals in vitro. Antimicrob. Agents Chemother 2005, 49, 3483–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Tremblay CL; Giguel F; Kollmann C; Guan Y; Chou TC; Baroudy BM; Hirsch MS Anti-human immunodeficiency virus interactions of sch-c (sch 351125), a CCR5 antagonist, with other antiretroviral agents in vitro. Antimicrob. Agents Chemother 2002, 46, 1336–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tremblay CL; Kollmann C; Giguel F; Chou TC; Hirsch MS Strong in vitro synergy between the fusion inhibitor t-20 and the CXCR4 blocker amd-3100. JAIDS, J. Acquired Immune Defic. Syndr 2000, 25, 99–102. [DOI] [PubMed] [Google Scholar]
- (12).Doms RW; Peiper SC Unwelcomed guests with master keys: How hiv uses chemokine receptors for cellular entry. Virology 1997, 235, 179–190. [DOI] [PubMed] [Google Scholar]
- (13).Fan X; Zhang HS; Chen L; Long YQ Efficient synthesis and identification of novel propane-1,3-diamino bridged CCR5 antagonists with variation on the basic center carrier. Eur. J. Med. Chem 2010, 45, 2827–2840. [DOI] [PubMed] [Google Scholar]
- (14).Feng DZ; Song YL; Jiang XH; Chen L; Long YQ Forward- and reverse-synthesis of piperazinopiperidine amide analogs: A general access to structurally diverse 4-piperazinopiperidine-based CCR5 antagonists. Org. Biomol. Chem 2007, 5, 2690–2697. [DOI] [PubMed] [Google Scholar]
- (15).Long YQ; Feng DZ; Chen L; Chen RH 1-(3-Amino-Propyl)-Piperidin-4-yl-Amides, Pharmaceutical Compositions, Processes For Their Preparation And Uses. WO2009052708A1, April 30, 2009.
- (16).Zhang HS; Feng DZ; Chen L; Long YQ Discovery of novel (s)-alpha-phenyl-gamma-amino butanamide containing CCR5 antagonists via functionality inversion approach. Bioorg. Med. Chem. Lett 2010, 20, 2219–2223. [DOI] [PubMed] [Google Scholar]
- (17).Cao B; Long YQ; Wei W; Xie X New Imidazopyridine Compound Useful In Pharmaceutical Composition and In Preparation Of Medicine For Treating Dieseases From Hiv Virus Infection, Rheumatoid Arthritis, Inflammation, and Cancer. CN 102675305 B, March 08, 2011.
- (18).Bastian AR; Kantharaju; McFadden K; Duffy C; Rajagopal S; Contarino MR; Papazoglou E; Chaiken I Cell-free hiv-1 virucidal action by modified peptide triazole inhibitors of env gp120. ChemMedChem 2011, 6, 1335–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Rashad AA; Kalyana Sundaram RV; Aneja R; Duffy C; Chaiken I Macrocyclic envelope glycoprotein antagonists that irreversibly inactivate hiv-1 before host cell encounter. J. Med. Chem 2015, 58, 7603–7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Aneja R; Rashad AA; Li H; Kalyana Sundaram RV; Duffy C; Bailey LD; Chaiken I Peptide triazole inactivators of hiv-1 utilize a conserved two-cavity binding site at the junction of the inner and outer domains of env gp120. J. Med. Chem 2015, 58, 3843–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21.) Gopi H; Umashankara M; Pirrone V; LaLonde J; Madani N; Tuzer F; Baxter S; Zentner I; Cocklin S; Jawanda N; Miller SR; Schon A; Klein JC; Freire E; Krebs FC; Smith AB; Sodroski J; Chaiken I Structural determinants for affinity enhancement of a dual antagonist peptide entry inhibitor of human immunodeficiency virus type-1. J. Med. Chem 2008, 51, 2638–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gopi HN; Tirupula KC; Baxter S; Ajith S; Chaiken IM Click chemistry on azidoproline: High-affinity dual antagonist for hiv-1 envelope glycoprotein gp120. ChemMedChem 2006, 1, 54–57. [DOI] [PubMed] [Google Scholar]
- (23).Kamanna K; Aneja R; Duffy C; Kubinski P; Rodrigo Moreira D; Bailey LD; McFadden K; Schon A; Holmes A; Tuzer F; Contarino M; Freire E; Chaiken IM Non-natural peptide triazole antagonists of hiv-1 envelope gp120. ChemMedChem 2013, 8, 322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Umashankara M; McFadden K; Zentner I; Schon A; Rajagopal S; Tuzer F; Kuriakose SA; Contarino M; Lalonde J; Freire E; Chaiken I The active core in a triazole peptide dual-site antagonist of hiv-1 gp120. ChemMedChem 2010, 5, 1871–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Julien JP; Cupo A; Sok D; Stanfield RL; Lyumkis D; Deller MC; Klasse PJ; Burton DR; Sanders RW; Moore JP; Ward AB; Wilson IA Crystal structure of a soluble cleaved hiv-1 envelope trimer. Science 2013, 342, 1477–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Tan Q; Zhu Y; Li J; Chen Z; Han GW; Kufareva I; Li T; Ma L; Fenalti G; Li J; Zhang W; Xie X; Yang H; Jiang H; Cherezov V; Liu H; Stevens RC; Zhao Q; Wu B Structure of the CCR5 chemokine receptor-hiv entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Huang CC; Lam SN; Acharya P; Tang M; Xiang SH; Hussan SS; Stanfield RL; Robinson J; Sodroski J; Wilson IA; Wyatt R; Bewley CA; Kwong PD Structures of the CCR5 n terminus and of a tyrosine-sulfated antibody with hiv-1 gp120 and CD4. Science 2007, 317, 1930–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Moraca F; Acharya K; Melillo B; Smith AB III; Chaiken I; Abrams CF Computational evaluation of hiv-1 gp120 conformations of soluble trimeric gp140 structures as targets for de novo docking of first- and second-generation small-molecule cd4 mimics. J. Chem. Inf. Model 2016, 56, 2069–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tallarida RJ An overview of drug combination analysis with isobolograms. J. Pharmacol. Exp. Ther 2006, 319, 1–7. [DOI] [PubMed] [Google Scholar]
- (30).Emileh A; Tuzer F; Yeh H; Umashankara M; Moreira DR; Lalonde JM; Bewley CA; Abrams CF; Chaiken IM A model of peptide triazole entry inhibitor binding to hiv-1 gp120 and the mechanism of bridging sheet disruption. Biochemistry 2013, 52, 2245–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tuzer F; Madani N; Kamanna K; Zentner I; LaLonde J; Holmes A; Upton E; Rajagopal S; McFadden K; Contarino M; Sodroski J; Chaiken I Hiv-1 env gp120 structural determinants for peptide triazole dual receptor site antagonism. Proteins: Struct., Funct., Genet 2013, 81, 271–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kolchinsky P; Kiprilov E; Bartley P; Rubinstein R; Sodroski J Loss of a single n-linked glycan allows CD4-independent human immunodeficiency virus type 1 infection by altering the position of the gp120 v1/v2 variable loops. J. Virol 2001, 75, 3435–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Pancera M; Zhou T; Druz A; Georgiev IS; Soto C; Gorman J; Huang J; Acharya P; Chuang GY; Ofek G; Stewart-Jones GB; Stuckey J; Bailer RT; Joyce MG; Louder MK; Tumba N; Yang Y; Zhang B; Cohen MS; Haynes BF; Mascola JR; Morris L; Munro JB; Blanchard SC; Mothes W; Connors M; Kwong PD Structure and immune recognition of trimeric prefusion hiv-1 env. Nature 2014, 514, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Borsi V; Calderone V; Fragai M; Luchinat C; Sarti N Entropic contribution to the linking coefficient in fragment based drug design: A case study. J. Med. Chem 2010, 53, 4285–4289. [DOI] [PubMed] [Google Scholar]
- (35).Ji C; Kopetzki E; Jekle A; Stubenrauch KG; Liu X; Zhang J; Rao E; Schlothauer T; Fischer S; Cammack N; Heilek G; Ries S; Sankuratri S CD4-anchoring hiv-1 fusion inhibitor with enhanced potency and in vivo stability. J. Biol. Chem 2009, 284, 5175–5185. [DOI] [PubMed] [Google Scholar]
- (36).Kagiampakis I; Gharibi A; Mankowski MK; Snyder BA; Ptak RG; Alatas K; LiWang PJ Potent strategy to inhibit hiv-1 by binding both gp120 and gp41. Antimicrob. Agents Chemother 2011, 55, 264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kopetzki E; Jekle A; Ji C; Rao E; Zhang J; Fischer S; Cammack N; Sankuratri S; Heilek G Closing two doors of viral entry: Intramolecular combination of a coreceptor- and fusion inhibitor of hiv-1. Virol. J 2008, 5, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Parajuli B; Acharya K; Yu R; Ngo B; Rashad AA; Abrams CF; Chaiken IM Lytic inactivation of human immunodeficiency virus by dual engagement of gp120 and gp41 domains in the virus env protein trimer. Biochemistry 2016, 55, 6100–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Quinlan BD; Joshi VR; Gardner MR; Ebrahimi KH; Farzan M A double-mimetic peptide efficiently neutralizes hiv-1 by bridging the CD4- and coreceptor-binding sites of gp120. J. Virol 2014, 88, 3353–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zhao B; Mankowski MK; Snyder BA; Ptak RG; Liwang PJ Highly potent chimeric inhibitors targeting two steps of hiv cell entry. J. Biol. Chem. 2011, 286, 28370–28381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Martinez-Munoz L; Barroso R; Dyrhaug SY; Navarro G; Lucas P; Soriano SF; Vega B; Costas C; Munoz-Fernandez MA; Santiago C; Frade JMR; Franco R; Mellado M CCR5/CD4/CXCR4 oligomerization prevents hiv-1 gp120iiib binding to the cell surface. Proc. Natl. Acad. Sci. U. S. A 2014, 111, E1960–E1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Jiang X; Feyertag F; Meehan CJ; McCormack GP; Travers SA; Craig C; Westby M; Lewis M; Robertson DL Characterizing the diverse mutational pathways associated with r5-tropic maraviroc resistance: Hiv-1 that uses the drug-bound CCR5 coreceptor. J. Virol 2015, 89, 11457–11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Rashad AA; Acharya K; Haftl A; Aneja R; Dick A; Holmes AP; Chaiken I Chemical optimization of macrocyclic hiv-1 inactivators for improving potency and increasing the structural diversity at the triazole ring. Org. Biomol. Chem 2017, 15, 7770–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).De Clercq E The bicyclam amd3100 story. Nat. Rev. Drug Discovery 2003, 2, 581–587. [DOI] [PubMed] [Google Scholar]
- (45).Long YQ; Cao B; Xie X; Wei W Imidazopyridines as CXCR4 Antagonists and Their Preparation, Pharmaceutical Compositions and Use In The Treatment Of Diseases. CN 102675305, September 19, 2012. [Google Scholar]
- (46).Openeye Scientific Software Home Page. http://www.eyesopen.com.
- (47).Madhavi Sastry G; Adzhigirey M; Day T; Annabhimoju R; Sherman W Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- (48).Acharya K; Rashad AA; Moraca F; Klasse PJ; Moore JP; Abrams C; Chaiken I Recognition of hiv-inactivating peptide triazoles by the recombinant soluble env trimer, bg505 sosip.664. Proteins: Struct., Funct., Genet 2017, 85, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Murphy R; Repasky M; Zhou ZY; Abel R; Krilov G; Tubert-Brohman I; Sherman W; Farid R; Friesner RA Evaluation of docking and scoring accuracy using a new version of schrodinger’s glide xp and induced fit docking (ifd) methodologies. Presented at the 241st ACS National Meeting & Exposition, Anaheim, CA, March 27−31, 2011; Paper 93. [Google Scholar]
- (50).Zhao GL; Lin S; Korotvička A; Deiana L; Kullberg M; Córdova A Asymmetric synthesis of maraviroc (uk-427,857). Adv. Synth. Catal 2010, 352, 2291–2298. [Google Scholar]
- (51).Haycock-Lewandowski SJ; Wilder A; Åhman J Development of a bulk enabling route to maraviroc (uk-427,857), a ccr-5 receptor antagonist. Org. Process Res. Dev 2008, 12, 1094–1103. [Google Scholar]
- (52).Asano S; Gavrilyuk J; Burton DR; Barbas CF 3rd Preparation and activities of macromolecule conjugates of the CCR5 antagonist maraviroc. ACS Med. Chem. Lett 2014, 5, 133–137.24563723 [Google Scholar]
- (53).Kiely JS; Hutt MP; Culbertson TP; Bucsh RA; Worth DF; Lesheski LE; Gogliotti RD; Sesnie JC; Solomon M; Mich TF Quinolone antibacterials: Preparation and activity of bridged bicyclic analogues of the c7-piperazine. J. Med. Chem. 1991, 34, 656–663. [DOI] [PubMed] [Google Scholar]
- (54).Hu S; Wang Z; Hou T; Ma X; Li J; Liu T; Xie X; Hu Y Design, synthesis, and biological evaluation of novel 2-methylpiperazine derivatives as potent CCR5 antagonists. Bioorg. Med. Chem. 2015, 23, 1157–1168. [DOI] [PubMed] [Google Scholar]
- (55).Bastian AR; Contarino M; Bailey LD; Aneja R; Moreira DR; Freedman K; McFadden K; Duffy C; Emileh A; Leslie G; Jacobson JM; Hoxie JA; Chaiken I Interactions of peptide triazole thiols with env gp120 induce irreversible breakdown and inactivation of hiv-1 virions. Retrovirology 2013, 10, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).McFadden K; Fletcher P; Rossi F; Kantharaju; Umashankara M; Pirrone V; Rajagopal S; Gopi H; Krebs FC; Martin-Garcia J; Shattock RJ; Chaiken I Antiviral breadth and combination potential of peptide triazole hiv-1 entry inhibitors. Antimicrob. Agents Chemother. 2012, 56, 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]