

Here, we describe the DNA binding and sensor specificity of FarR, a novel TetR family regulator (TFR) in Staphylococcus aureus. Unlike the majority of TFRs that have been characterized, which function to repress a divergently transcribed gene, we find that FarR is needed to promote expression of the divergently transcribed farE gene, encoding a resistance-nodulation-division (RND) family efflux pump that is induced in response to antimicrobial unsaturated fatty acids. Induction of farE was dependent on the function of the fatty acid kinase FakA, which catalyzes the first metabolic step in the incorporation of exogenous unsaturated fatty acids into phospholipid. This represents a novel example of TFR function.
KEYWORDS: Staphylococcus aureus, TetR family regulator, antimicrobial agents, efflux pumps, fatty acids
ABSTRACT
Divergent genes in Staphylococcus aureus USA300 encode the efflux pump FarE and TetR family regulator FarR, which confer resistance to antimicrobial unsaturated fatty acids. To study their regulation, we constructed USA300 ΔfarER, which exhibited a 2-fold reduction in MIC of linoleic acid. farE expressed from its native promoter on pLIfarE conferred increased resistance to USA300 but not USA300 ΔfarER. Complementation of USA300 ΔfarER with pLIfarR also had no effect, whereas resistance was restored with pLIfarER or through ectopic expression of farE. In electrophoretic mobility shift assays, FarR bound to three different oligonucleotide probes that each contained a TAGWTTA motif, occurring as (i) a singular motif overlapping the −10 element of the PfarR promoter, (ii) in palindrome PAL1 immediately in the 3′ direction of PfarR, or (iii) within PAL2 upstream of the predicted PfarE promoter. FarR autorepressed its expression through cooperative binding to PAL1 and the adjacent TAGWTTA motif in PfarR. Consistent with reports that S. aureus does not metabolize fatty acids through acyl coenzyme A (acyl-CoA) intermediates, DNA binding activity of FarR was not affected by linoleoyl-CoA. Conversely, induction of farE required fatty acid kinase FakA, which catalyzes the first metabolic step in the incorporation of unsaturated fatty acids into phospholipid. We conclude that FarR is needed to promote the expression of farE while strongly autorepressing its own expression, and our data are consistent with a model whereby FarR interacts with a FakA-dependent product of exogenous fatty acid metabolism to ensure that efflux only occurs when the metabolic capacity for incorporation of fatty acid into phospholipid is exceeded.
IMPORTANCE Here, we describe the DNA binding and sensor specificity of FarR, a novel TetR family regulator (TFR) in Staphylococcus aureus. Unlike the majority of TFRs that have been characterized, which function to repress a divergently transcribed gene, we find that FarR is needed to promote expression of the divergently transcribed farE gene, encoding a resistance-nodulation-division (RND) family efflux pump that is induced in response to antimicrobial unsaturated fatty acids. Induction of farE was dependent on the function of the fatty acid kinase FakA, which catalyzes the first metabolic step in the incorporation of exogenous unsaturated fatty acids into phospholipid. This represents a novel example of TFR function.
INTRODUCTION
The TetR family of transcriptional regulators (TFR) confers a mechanism of one-component signal transduction employed by bacteria to sense alterations in their environment and convey the appropriate responses (1). To achieve this, sensor and output domains are incorporated into the same polypeptide, as in the prototypic TetR of Escherichia coli, which represses the divergently transcribed gene tetA, encoding a tetracycline efflux pump. Most TFRs characterized to date conform to this type I arrangement, where the TFR and a divergently transcribed gene that is repressed are separated by an intergenic segment of less than 200 bp (1). In the TetR-TetA paradigm, when tetracycline is present, the antibiotic binds to a C-terminal sensor domain of TetR, promoting a conformational change, such that TetR loses affinity for its operator site, leading to derepression of tetA (2). However, there are >200,000 TFR sequences available in protein databases, and among known examples, there is considerable variation, including some that have dual roles as repressor and activator, the ligands that modulate their function, and physiologic processes that are regulated. Thus, while most microbial genomes encode at least one TFR, their functions and mode of regulation are largely undiscovered (1).
In this study, we address the nucleotide binding and sensor specificity of FarR, a novel TFR of Staphylococcus aureus that is needed for expression of a divergently transcribed efflux pump, FarE (3). S. aureus is a Gram-positive bacterium that asymptomatically colonizes the nose and skin of approximately 30% of humans (4), and yet it is also a major cause of human morbidity and mortality. Consequently, it is exposed to a wide range of environmental conditions encountered at different sites of colonization and infection. Prior to our work, the most detailed knowledge of S. aureus TFR function is derived from the plasmid-encoded QacR, a type I TFR that represses a divergently transcribed gene, qacB, encoding an efflux pump specific for antimicrobial quaternary amine compounds (5). However, the function and repertoire of chromosomally encoded TFRs in S. aureus are less well understood, and just one has been defined in terms of its DNA binding specificity, namely, IcaR, which represses divergently transcribed genes that confer synthesis of an intercellular polysaccharide adhesin (6, 7).
In evaluating how S. aureus responds to host-derived unsaturated free fatty acids (uFFA) which are toxic to bacteria (8, 9) and are encountered during colonization and infection (10–12), we isolated fatty acid-resistant (FAR) variants with increased resistance to uFFA, including S. aureus FAR7, which harbored a single nucleotide polymorphism that caused an H121Y substitution in a TFR that we named FarR, as a regulator of resistance to antimicrobial fatty acids (3). Like type I TFRs, farR is divergently transcribed from the farE gene that it regulates, encoding a resistance-nodulation-division (RND) superfamily efflux pump. The expression of farE was selectively induced by unsaturated free fatty acids, and S. aureus FAR7, which expresses an H121Y variant of FarR, exhibited constitutive farE expression, enhanced induced expression, and increased resistance to linoleic acid (3). These observations were consistent with a type I TFR, whereby FarR was expected to repress farE, which would be derepressed in response to antimicrobial fatty acids, or through mutations that impair FarR function. However, additional findings did not support this paradigm. Notably, the type I TFR paradigm stipulates that inactivation of a TFR should cause derepression of the divergently transcribed gene, but this was not the case with farR, such that farE could no longer be induced when farR was disrupted by transposon insertion (3).
The sensor specificity of FarR also remains to be determined. Although the sensor domains of some Gram-positive TFRs bind long-chain acyl coenzyme A (acyl-CoA) metabolites, these examples lead to the derepression of genes required for the degradation of exogenous fatty acids (13, 14). However, this metabolic capacity has not been demonstrated in S. aureus, where the sole fate of exogenous fatty acid is incorporation into phospholipid through a fatty acid kinase FakA-dependent pathway, and a requirement for acyl-CoA in both fatty acid metabolism and phospholipid synthesis has been excluded (8, 15–17). Therefore, the goal of our present study was to define the role of FarR as a novel TFR in terms of its interaction with target promoters and the specificity of its sensor function.
RESULTS
farE cannot confer resistance to antimicrobial fatty acids in the absence of farR.
Although we previously noted that farE could not be induced in the absence of FarR, this was based on a transposon insertion in the 3′ end of farR that would potentially permit the expression of a truncated gene product lacking the C-terminal 37 amino acids (3, 18). Therefore, to precisely define the functions of farE and farR, we constructed USA300 ΔfarER and conducted complementation assays with pLIfarE, pLIfarR, pLIfarR7, or pLIfarER, where the genes are expressed from their native promoters, and pLIfarR7 expresses the variant H121YFarR that promotes increased resistance of S. aureus FAR7 to linoleic acid (3). USA300 exhibited an MIC of 400 µM, compared to 200 µM for USA300 ΔfarER (Table 1; see also Fig. S1 in the supplemental material). When complementation plasmids were tested in USA300 ΔfarER, only pLIfarER was able to restore resistance, permitting robust growth at 500 µM linoleic acid. Although pLIfarE did not complement USA300 ΔfarER, it conferred increased resistance to USA300 (Table 1 and Fig. S1). Since these data supported the contention that farE could not be expressed in the absence of farR, we queried whether ectopic expression of farE would restore resistance, using the Pxyl/tet promoter of pALC2073, which permits a basal level of constitutive expression (19). In USA300 ΔfarER/pALC2073 vehicle, the MIC for linoleic acid was unchanged at 200 µM, compared to >400 µM for USA300 ΔfarER/pALCfarE (Table 1 and Fig. S1). From these data, we conclude that FarE-mediated efflux alone is sufficient to promote resistance to linoleic acid, that farE cannot be expressed in the absence of farR, and that farR has no impact on resistance in the absence of farE.
TABLE 1.
MIC of linoleic acid for USA300 and isogenic variants with complementation plasmids
| Strain | Plasmid | MICa (µM linoleic acid) |
|---|---|---|
| USA300 | pLI50 | 400 |
| pLIfarE | >500 | |
| USA300 ΔfarER | pLI50 | 200 |
| pLIfarE | 200 | |
| pLIfarR | 200 | |
| pLIfarR7 | 200 | |
| pLIfarER | >500 | |
| pALC2073 | 200 | |
| pALCfarE | >400 |
Data for MIC determinations are presented in Fig. S1.
Expression of farR is autoregulated.
Although our data refute the type I TFR paradigm whereby the TFR functions to repress a divergently transcribed gene, another common trait of TFRs is autorepression (1, 20), which we evaluated using a farR::lux reporter. Although farR::lux was not strongly expressed at any time during growth of USA300 in tryptic soy broth (TSB), it was strongly derepressed in USA300 ΔfarER (Fig. 1A). We also compared farR::lux activity after 3 h of growth in S. aureus USA300, USA300 ΔfarER, and S. aureus FAR7 defined by an H121Y substitution in FarR (3). As expected, farR::lux activity was significantly elevated in FAR7 compared to USA300, and there was another significant increase in USA300 ΔfarER (Fig. 1B). These data indicate that farR is strongly autorepressed by FarR and that H121YFarR is a less effective autorepressor.
FIG 1.

farR is regulated by autorepression. (A) Cultures of USA300 or USA300 ΔfarER harboring pGYfarR::lux were inoculated into triplicate flasks of TSB at OD600 of 0.01, followed by incubation at 37°C with orbital shaking. Growth (OD600), and luminescence (relative light units [RLU]) were determined at hourly intervals. Luminescence data were standardized to 1 OD600 unit (RLU/OD). All data points represent the mean from triplicate cultures. (B) Luminescence values of farR::lux (RLU/OD) in cultures of USA300, USA300 ΔfarER, and FAR7, after growth for 3 h in TSB. Data points represent mean values from triplicate cultures. Statistically significant differences (*, P < 0.05; ***, P < 0.0005) were determined by Tukey’s multiple-comparison test.
Identification of promoters and structural features in the farER intergenic segment farERIS.
The divergent farE and farR genes are separated by a 144-bp intergenic segment (farERIS) (Fig. 2A). A Pustell DNA matrix analysis (21) of this segment identified a 17-nucleotide (nt) palindrome, PAL1, with three mismatches, and a 16-nt PAL2, with 4 mismatches (Fig. 2A). PAL1 is comprised of inverted octanucleotide repeats IR1 (5′-AATATACA-3′) and IR2 (5′-TGTAGATT-3′), separated by a single nucleotide. Comparatively, PAL2 on the minus strand has an identical IR1 and variant IR2a with one nucleotide mismatch (5′-TGTAGtTT-3′), but the 5′-to-3′ orientation of these inverted repeats is juxtaposed, represented by 5′-IR2a-IR1-3′. To further define significant sequence motifs, we identified 15 species of coagulase-negative staphylococci with divergent orthologues of farE and farR, and aligned their intergenic segments to that of S. aureus USA300. This revealed three copies of a conserved TAGWTTA motif, two of which overlap the IR2 and IR2a features of PAL1 and PAL2, while a third precedes the IR1 segment of PAL1 (Fig. 2A and S2).
FIG 2.

Mapping of structural features in farERIS (A) and EMSA to define DNA binding specificity of FarR and H121YFarR (B to E). (A) Nucleotide sequence of farERIS. The TTG and ATG initiation codons of farE and farR on the minus and plus strands, respectively, are in bold type and marked with short arrows. Extended arrows above and below the plus and minus strands define the confirmed PfarR and predicted PfarE promoters, respectively. Nucleotides corresponding to the confirmed PfarR +1 transcription start sites are in larger bold type. Nucleotides comprising the −35 and −10 promoter elements of each promoter are underlined. The spans of nucleotides comprising PAL1, PAL2, and the the IR1, IR2, and IR2a inverted repeats are indicated. Shading identifies a TAGWTTA motif that overlaps the −10 element of PfarR, as well as the IR2 and IR2a features of PAL1 and PAL2. Nucleotide sequences of overlapping oligonucleotide probes OP1, OP2, OP3, OP4, and OP5 used in EMSA are illustrated below farERIS, while gray bars define overlapping probes OP1.1, OP1.2 and OP1.3 that span the OP1 sequence. (B) EMSA with 5 pM of IRDye 800-labeled duplex oligonucleotide probes OP1, OP2, OP3, and OP4, mixed with 0 or 2 µM 6×His-FarR or 6×His-H121Y-FarR. (C) Competition EMSA where 5 pM of IRDye 800-labeled OP1 was mixed with 0, 0.5, or 2 µM 6×His-FarR, without competitor (Nil), or with 50× excess of nonlabeled OP1.1, OP1.2, or OP1.3 duplex probe. The OP1.3 competitor was analyzed on a separate gel with its own Nil control, and the appropriate section of this image was merged with an image of the Nil, OP1.1, and OP1.2 competitors as demarcated by the vertical line that separates OP1.2 and OP1.3. (D) EMSA with 5 pM IRDye 800-labeled OP2 or OP1.3 probe, mixed with 0 or 2 µM 6×His-FarR or 6×His-H121YFarR, as indicated. (E) Competition EMSA where 5 pM of IRDye 800-labeled OP1.3 was mixed with 0, 0.5, or 2 µM 6×His-FarR, without competitor (Nil), or with 50× excess of unlabeled OP1.3 or OP1.3G→A competitor probe.
We then resolved to map these features in relation to promoters for farE and farR. The Berkeley Drosophila Genome Project (BDGP) neural network promoter prediction program (22) identified a putative promoter PfarR spanning nucleotides 4 to 41 of farERIS, and 5′ rapid amplification of cDNA ends (5′-RACE) of RNA extracted from three cultures of S. aureus USA300 identified nucleotides at position 3 or 4 within the IR1 segment of PAL1 as +1 transcription start sites (Fig. 2A and S3). A putative PfarE promoter was also identified, spanning nucleotides 101 to 63 on the minus strand of farERIS. However, several attempts to validate the +1 site by 5′-RACE yielded different nucleotides in the 5′ end of the farE coding sequence (data not shown), suggesting that farE mRNA is highly sensitive to RNase processing.
FarR and H121YFarR exhibit differential binding to oligonucleotide probes spanning farERIS.
To screen the identified structural features for their interactions with FarR, we conducted electrophoretic mobility shift assays (EMSA) with 6×His-FarR and four overlapping IR-dye-labeled duplex oligonucleotide probes spanning farERIS (Fig. 2A and B). Probes OP1, OP2, and OP4 each exhibited a mobility shift in the presence of 6×His-FarR, and each probe has a TAGWTTA motif, either as part of the −10 element of PfarR (OP1) or embedded in PAL1 (OP2) or PAL2 (OP4). Conversely, OP3 lacks a TAGWTTA motif and did not interact with 6×His-FarR. Similar results were obtained with H121YFarR, except that its interaction with OP1 was strongly attenuated (Fig. 2B). These data are supportive of three binding sites in farERIS; two sites associated with PAL1 (OP2) and PAL2 (OP4) are recognized by both native and H121YFarR, while a third site in OP1 is strongly bound only by native FarR.
The −10 and +1 features of PfarR comprise an operator site for FarR.
Since farR::lux was derepressed in S. aureus FAR7, which expresses H121YFarR (Fig. 1B), and H121YFarR exhibited attenuated interaction with OP1 (Fig. 2B), we reasoned that OP1 has an operator site that facilitates autorepression of PfarR. To localize this site, we conducted competition EMSA where IRDye 800-labeled OP1 (IRD800OP1) was mixed with 6×His-FarR and excess nonlabeled overlapping 30mer probes OP1.1, OP1.2, or OP1.3 that span OP1. Of these, only OP1.3 has both the −10 and +1 features of PfarR, and was uniquely able to eliminate binding of FarR to IRD800OP1 (Fig. 2A and C). When EMSA was subsequently conducted with IRD800OP1.3, FarR promoted a strong mobility shift but H121YFarR did not, whereas both proteins interacted equally well with IRDye 800-labeled OP2 (IRD800OP2) (Fig. 2D). To address the significance of the TAGWTTA motif in OP1.3, we synthesized unlabeled duplex probe OP1.3G→A, where TAGTTTA was altered to TAATTTA, which concomitantly alters the −10 element of PfarR from TATAGT, to a consensus TATAAT. When 6×His-FarR and IRD800OP1.3 were mixed with unlabeled OP1.3 or OP1.3G→A in competition EMSA, unlabeled OP1.3 inhibited the mobility shift but OP1.3G→A did not (Fig. 2E), indicating that this nucleotide substitution abolished FarR binding. Therefore, the TAGTTTA motif that overlaps the −10 element of PfarR constitutes an operator site that we designate −10PfarR.
FarR preferentially binds to PAL1.
Although our EMSA indicate that FarR recognizes −10PfarR, this site is proximal to PAL1 where another TAGWTTA motif overlaps the IR2 half-site (Fig. 2A). To delineate the interaction of FarR and H121YFarR with these sites, we performed EMSA with a panel of 339-bp farERIS probes, generated by PCR from pLIfarER or isogenic variants harboring a G→A substitution in −10PfarR (farER1IS), substitutions in the IR1 and IR2 features of PAL1 (farER2IS), or substitutions in both features (farER3IS), as delineated in Fig. 3A. Therefore, each probe is composed of the different 144-bp intergenic segments, flanked by additional nucleotides comprising the 5′ ends of the respective farE and farR genes.
FIG 3.
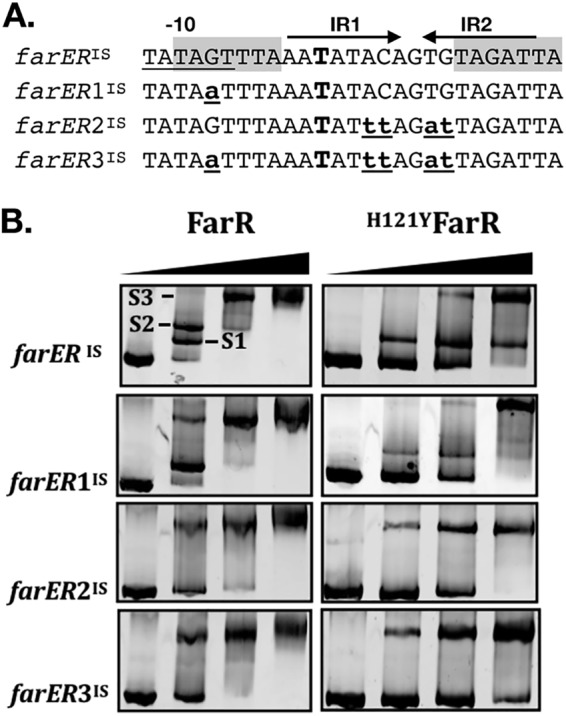
EMSA with 339-bp farERIS segment and derivatives to evaluate impact of nucleotide substitutions in FarR operator sites. (A) Nucleotide sequence showing the variable segments of the 339-bp farERIS, farER1IS, farER2IS, and farER3IS probes. The labeled features above farERIS are as detailed for Fig. 2A. The larger bold type “T” in IR1 indicates the +1 transcription start site of farR. Lowercase underlined nucleotides indicate nucleotide substitutions that differentiate each probe. The different EMSA probes were generated by PCR with primer pair farERIS-F2 and farERIS-R2, using plasmid pLIfarER, pLIfarER1, pLIfarER2, or pLIfarER3 as the template. (B) EMSA was conducted with 5 pM PCR product and 0, 0.5, 1, or 2 µM FarR or H121YFarR, as indicated. The first lane of each panel represents electrophoresis of the farERIS probe without added protein (i.e., 0 µM FarR). In the upper-left panel, the protein-DNA complexes S1, S2, and S3 are labeled. Protein-DNA complexes were directly imaged by ethidium bromide staining.
In EMSA with wild-type farERIS and 0.5 µM FarR, two mobility shifts were detected, with approximately equal amounts of the S1 and slower-migrating S2 products (Fig. 3B). With 1.0 µM FarR, S1 was no longer evident and there was only a trace of S2, accompanied by appearance of a S3 supershift, which was the only product detected at 2.0 µM FarR. In EMSA with farER1IS (G→A substitution in −10PfarR), the S2 complex was eliminated, indicating that this complex is due to binding of FarR to −10PfarR. However, both S1 and S2 were eliminated with farER2IS in which PAL1 is altered, and a similar result occurred with farER3IS, which has substitutions in both −10PfarR and PAL1. From these data, it appears that FarR binds preferentially to PAL1, accounting for the S1 complex, followed by binding to the adjacent −10PfarR site, accounting for S2. This interpretation was supported by EMSA with H121YFarR and wild-type farERIS, where S1 was detected but S2 was not (Fig. 3B), consistent with our observation that H121YFarR bound poorly to the OP1.3 probe that contains −10PfarR (Fig. 2D). Although H121YFarR retained the ability to form the S1 complex with farERIS probe, the S1 complex was again eliminated in EMSA with farER1IS or farER2IS, where only the S3 complex was evident.
Significance of the TAGWTTA motif is context dependent.
EMSA with the larger 339-bp farERIS panel of probes revealed that FarR bound preferentially to PAL1, followed by binding to the adjacent −10PfarR (Fig. 3). Since the S3 supershift only appeared after these sites were occupied and was not affected by substitutions in −10PfarR or PAL1, we surmised that the S3 complex was due to FarR interaction with PAL2, and that this interaction would be of lesser affinity than that with PAL1. We therefore undertook a more detailed assessment of FarR interaction with PAL1 and PAL2, and the significance of the TAGWTTA motif in these features, relative to that of −10PfarR, where binding was eliminated through a single G→A substitution.
When EMSA was conducted with 0.5 µM FarR and probe OP5.1, which is composed of PAL1 extended by four nucleotides at each end, a single mobility shift occurred, with little unbound probe remaining (Fig. 4). With OP5.2 containing a G→A substitution in the TAGWTTA motif of PAL1, complex formation was reduced but not eliminated (Fig. 4B). Therefore, unlike the −10PfarR site, where the TAGWTTA motif is a singular feature and the main determinant of specificity, binding of FarR to the higher-affinity PAL1 is more complex. Consistent with our prediction that FarR interaction with PAL2 would be of a lesser affinity, no comparable complex was observed with probe OP4.1 composed of PAL2 extended by four nucleotides at each end (Fig. 4B). Importantly, although PAL1 and PAL2 are composed of similar or identical inverted repeat half-sites, their positions in PAL2 are juxtaposed compared to PAL1 (Fig. 4A).
FIG 4.
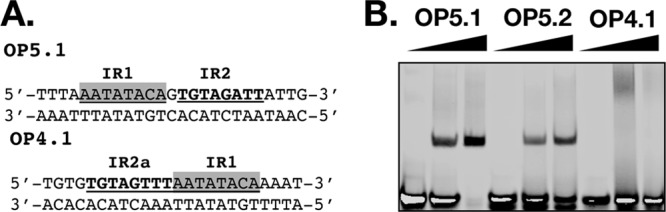
EMSA with minimal PAL1 and PAL2 probes. (A) Composition of PAL1 (OP5.1) and PAL2 (OP4.1) probes. Nucleotides comprising the inverted repeat IR1, IR2, and IR2a components of PAL1 and PAL2 are underlined. The IR1 half-site is shaded, while the IR2 and IR2a half-sites are in bold type. Probe OP5.2 is identical to OP5.1, with the exception of a G→A substitution in the TAGATTA motif that overlaps IR2. The top strand contains a 5′ IRDye 800 addition. (B) EMSA with 0, 0.2, or 0.5 µM FarR mixed with 5 pM OP5.1, OP5.2, or OP4.1, as indicated.
Nucleotide substitutions in −10PfarR and PAL1 alleviate autorepression of farR but do not promote increased resistance to linoleic acid.
Having established that the interaction of FarR with farERIS is altered by nucleotide substitutions in −10PfarR and PAL1, we assessed the impact of these substitutions on expression of FarR protein and resistance to linoleic acid. A Western blot of lysates from E. coli transformed with native pLIfarR or pLIfarR1 harboring a G→A substitution in −10PfarR revealed that pLIfarR1 promoted increased production of FarR protein (Fig. 5A), and reporter gene assays of USA300 transformed with pGYfarR::lux or pGYfarR1::lux confirmed that the G→A substitution in −10PfarR caused derepression of PfarR (Fig. 5B). We also examined production of FarR protein when farR was expressed in the context of pLIfarER and variants harboring nucleotide substitutions in farERIS, as detailed in Fig. 3A. Compared to lysate of E. coli plus wild-type pLIfarER, increased FarR protein was evident in E. coli/pLIfarER1 (Fig. 5A). However, greater FarR production was evident in E. coli/pLIfarER2, consistent with our EMSA data where nucleotide substitutions in PAL1 (farER2IS) abrogated FarR binding to both the PAL1 (S1 shift) and −10PfarR (S2 shift) sites (Fig. 3B). A similar strong derepression of FarR production was noted with pLIfarER3 (Fig. 5A), where both the −10PfarR and PAL1 are altered. Therefore, nucleotide substitutions within PAL1 abrogate FarR binding to both PAL1 and the adjacent −10PfarR, leading to strong derepression of farR expression.
FIG 5.

Influence of nucleotide substitutions in −10PfarR and PAL1 on expression of FarR, and resistance to linoleic acid. (A) Western blot of gel loaded with 25 ng of purified 6×His-FarR or 25 µg of cell lysate protein from E. coli DH5α transformed with pLI50 vehicle, pLIfarR, or pLIfarER and derivatives containing nucleotide substitutions in −10PfarR (pLIfarR1 and pLIfarER1), PAL1 (pLIfarER2), or −10PfarR and PAL1 (pLIfarER3). (B) Luciferase reporter gene assays of S. aureus USA300 transformed with pGYfarR::lux, or pGYfarR1::lux harboring a G→A substitution in −10PfarR. Triplicates of each culture were grown in TSB, with monitoring of OD600 and luciferase activity (relative light units [RLU]) at hourly intervals. Data for RLU determinations are standardized to OD600 units (RLU/OD) to account for differences in cell density. (C) Growth of USA300 ΔfarER complemented with pLI50 vehicle or pLIfarER and derivatives in TSB plus 50 µM linoleic acid. Each data point represents the mean and standard deviation of the results from triplicate cultures.
To evaluate the impact of these substitutions on resistance to linoleic acid, pLIfarER and derivatives were assessed for their ability to promote growth of USA300 ΔfarER in TSB plus 50 µM linoleic acid, which normally imparts a 10- to 12-h lag phase on wild-type USA300. Consistent with the ability of pLIfarER to confer increased resistance, as noted in Table 1, USA300 ΔfarER/pLIfarER grew well in TSB plus 50 µM linoleic acid (Fig. 5C). However, growth of USA300 ΔfarER/pLIfarER1 was impaired, and USA300 ΔfarER harboring either pLI50, pLIfarER2, or pLIfarER3 failed to enter exponential growth over 8 h of incubation. When grown in TSB alone, all cultures grew equally well (Fig. S4), indicating that increased FarR expression did not affect growth in the absence of fatty acid. Therefore, although farR was derepressed as a consequence of nucleotide substitutions in −10PfarR or PAL1, this led to diminished resistance.
Sensor specificity of FarR.
A tenet of TFR function is that the affinity of a TFR for its operator is modulated by small-molecule ligands (1, 23), including TFRs that bind acyl-CoA (13, 24, 25). Since our EMSA data established that PAL1 harbors the initial site of FarR interaction with DNA, we conducted EMSA with 6×HisFarR and probe OP5, composed of PAL1 extended by 6 nucleotides on each end (Fig. 2A), to determine if linoleoyl-CoA or arachidonoyl-CoA could disturb this interaction. However, while linoleic acid and arachidonic acid are both strong inducers of farE (3), their acyl-CoA derivatives did not appear to influence the binding of FarR to OP5 (Fig. 6), consistent with a previous report that S. aureus does not metabolize exogenous uFFA through acyl-CoA intermediates (15). Rather, the first step in metabolism of exogenous uFFA by S. aureus is conversion to an acyl-phosphate through the activity of fatty acid kinase FakA (15, 26). To evaluate the contribution of fakA, we constructed USA300 ΔfakA and compared farE::lux activity in this background relative to that of USA300.
FIG 6.
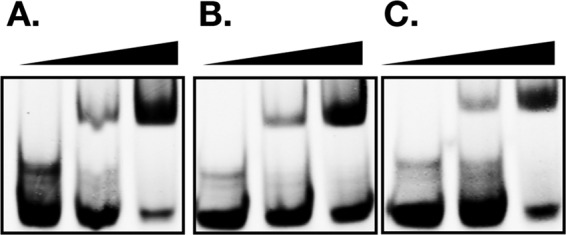
Binding of FarR to OP5 is not diminished by acyl-CoA. EMSA was conducted with 5 pM of IRDye 800-labeled OP5 duplex probe mixed with 0, 0.5, or 2 µM 6×His-FarR only (A) or supplemented with 50 µM linoleoyl-CoA (B) or arachidonoyl-CoA (C).
As previously shown (3), farE::lux activity was negligible during growth of USA300 in TSB but was induced in TSB plus 20 µM linoleic acid (Fig. 7A). Comparatively, USA300 ΔfakA exhibited elevated basal farE::lux activity during growth in TSB, but expression was no longer responsive to linoleic acid and remained well below the induced level of expression in USA300 (Fig. 7A). Since USA300 ΔfakA exhibited elevated noninduced expression of farE, we interrogated wild-type USA300 and USA300 ΔfakA cells to assess what impact this had on bactericidal activity of linoleic acid (Fig. 7B) and growth in the presence of 50 µM linoleic acid (Fig. 7C). For these experiments, USA300 ΔfakA was complemented with pALCfakAON or pALCfakAOFF, where fakA is cloned in the plus or minus orientation, respectively, relative to the Pxyl/tetO promoter in pALC2073. Consequently, USA300 ΔfakA/pALCfakAOFF retains the fakA-null phenotype, while fakA expression is restored in USA300 ΔfakA/pALCfakAON. Prior to being challenged with a 100 µM bactericidal concentration of linoleic acid, these cultures were grown to exponential phase in TSB, conditions under which farE is not expressed in wild-type USA300 but is constitutively expressed in USA300 ΔfakA/pALCfakAOFF. As expected, USA300 exhibited a rapid loss of viability (Fig. 7B), since expression of farE was not induced prior to exposure to 100 µM linoleic acid, whereas significantly greater viability was retained by USA300 ΔfakA/pALCfakAOFF, consistent with the elevated noninduced expression of farE. In contrast, USA300 ΔfakA/pALCfakAON behaved as wild-type USA300 (Fig. 7B), suggesting that restoration of fakA function eliminated the constitutive expression of farE.
FIG 7.

Inactivation of fakA causes constitutive expression of farE and increased resistance to linoleic acid. (A) Assay of farE::lux reporter gene in USA300 and USA300 ΔfakA. Inoculum cultures were grown overnight in TSB and then inoculated at OD600 of 0.01 into TSB only or TSB plus 20 µM LA. Growth (OD600) and luminescence (RLU) were monitored at hourly intervals. Luminescence data were standardized to 1 OD600 unit (RLU/OD). All data points represent the mean of the results from triplicate cultures. (B) Assay of viability in TSB containing 100 µM LA. Cultures of USA300 or USA300 ΔfakA complemented with pALfakAOFF or pALfakAON were grown to mid-exponential phase in TSB and then inoculated into TSB plus 100 µM LA at a cell density of 2 × 106 CFU/ml. Cell viability was immediately determined, and after cultures were placed on an orbital shaker, viability was determined at hourly intervals. Each data point represents the mean and standard deviation of the results from triplicate cultures. Asterisks indicate significant differences in viability between USA300 and USA300/pALfakAOff (**, P < 0.005; ***, P < 0.0005) as determined by multiple t test comparisons of viability at each time point using the Holm-Sidak method. (C) Growth assay of USA300, USA300 ΔfakA, and USA300 ΔfarERfakA in TSB plus 50 µM linoleic acid. Inoculum cultures were grown overnight in TSB and then subcultured into TSB plus 50 µM LA at an initial OD600 of 0.01. Flasks were incubated at 37°C with orbital shaking, and OD600 values were determined at hourly intervals. Each data point represents the mean of the results from triplicate cultures.
To confirm that farE was responsible for enhanced resistance, we constructed a USA300 ΔfarERfakA triple mutant and assessed growth in TSB plus 50 µM linoleic acid. As reported by us previously (3), wild-type USA300 exhibited a 10- to 12-h lag phase (Fig. 7C). However, USA300 ΔfakA initiated growth without a lag phase, while USA300 ΔfarERfakA was unable to grow (Fig. 6C). Therefore, although farE cannot be fully induced in USA300 ΔfakA, it exhibits sufficient elevated expression to promote increased resistance to linoleic acid.
DISCUSSION
The divergent farER arrangement conforms to that of a type I TFR, where in the majority of studied examples, the TFR represses its own expression and the divergently transcribed gene (1), with derepression being achieved in response to a small-molecule ligand that modulates its affinity for operator sites. Our present work describes a variation of this paradigm, with our novel findings including a DNA binding specificity that differs significantly from other S. aureus TFRs (5, 6, 27, 28); farE cannot be expressed in the absence of FarR function, and induction of farE in response to unsaturated fatty acids requires the fatty acid kinase FakA. Since TFRs regulate gene expression in cooperation with a small-molecule ligand and farE is only induced in response to unsaturated fatty acids (3), we expected that FarR function would be modified by a fatty acid metabolite as with TFRs that bind fatty acids or acyl-CoA to repress genes required for β-oxidation of fatty acids (13, 25, 29, 30). However, S. aureus lacks this metabolic capacity, and exogenous uFFA are metabolized by incorporation into phospholipid (15–17). This is initiated through FakA-dependent phosphorylation, and the acyl-phosphate is used by the PlsY acyl-transferase to acylate glycerol-3-phosphate. Since S. aureus does not use acyl-CoA in metabolism of long-chain fatty acids (15, 16), our finding that FakA is required for induction of FarE is consistent with current knowledge on the metabolism of exogenous uFFA in S. aureus. However, we cannot conclude that acyl-phosphate is the relevant physiologic ligand.
In addition to acyl-phosphate, FarR function could also be modulated by an unsaturated acyl-acyl carrier protein (acyl-ACP), since exogenous fatty acids that are phosphorylated by FakA can also be used as a substrate by PlsX and converted to an acyl-ACP (26). Accordingly, although TFRs generally bind a small-molecule ligand, a notable exception is DhaS. DhaS is a TFR that regulates dihydroxyacetone kinase expression, but rather than binding dihydroxyacetone, it interacts with the accessory protein DhaQ that forms a complex with dihydroxyacetone (31). Therefore, a number of physiologic ligands are candidates for the modulation of FarR function, which in addition to acyl-phosphate could include acyl-ACP, acyl-glycerol-3-phosphate, or fatty acid kinase in complex with fatty acid.
Although our data demonstrate that FakA is needed to induce farE in response to linoleic acid, paradoxically, USA300 ΔfakA exhibited an elevated basal level of farE expression sufficient to promote increased resistance to linoleic acid. This may be due to the pleiotropic phenotype ascribed to USA300 ΔfakA, including an elevated pool of nonesterified fatty acids, leading to reduced transcription of the SaeRS two-component regulator and defective production of Hla toxin (32–34). Broader metabolic changes were also noted, including altered carbon and amino acid metabolism (35). An elevated basal expression of farE in response to accumulation of cellular metabolites would be consistent with observations that other RND family efflux pumps also exhibit increased expression in response to an accumulation of cellular metabolites (36–39).
Based on knowledge of TFR function, interaction of FarR with its cognate ligand should modulate its affinity for relevant operator sites, and we identified a TAGWTTA motif as a canonical sequence in all EMSA probes that were bound by FarR. We propose that three occurrences of this motif, each in a distinct context, function to promote respective repression and activation functions. PAL1 contains the primary operator site for autorepression, where the TAGWTTA motif overlaps the IR2 feature (Fig. 2A). Binding of FarR to PAL1 promotes cooperative binding to the adjacent TAGWTTA motif, comprising the −10PfarR operator site, and nucleotide substitutions in PAL1 abrogate FarR binding to both PAL1 and −10PfarR, leading to strong derepression of farR expression (Fig. 3 and 5). These findings are compliant with current knowledge of type I TFR function, where the TFR serves to autorepress its expression.
Cooperative binding of FarR to this primary repressor site that regulates farR expression resembles that described for AlkX repression of AlkW, a gene required for alkane metabolism in Gram-positive Dietzia spp. (30). In this example, AlkX bound to a long 48-bp inverted repeat, causing two distinct mobility shifts, where binding of an AlkX dimer to a preferred operator half-site facilitated recruitment of a second AlkX dimer to an adjacent site. A different mode of cooperative binding was described for QacR of S. aureus, a TFR that represses expression of the QacA efflux pump. In this example, QacR binds to a 28-bp operator as a dimer of dimers, and only one mobility shift was evident irrespective of the amount of QacR used in EMSA (27, 40). For AlkW, it was proposed that this cooperative mode allows for functional repression at lower protein concentrations, so as to better facilitate bacterial adaptation in harsh environments (30).
While our data provide a mechanistic basis for autorepression of PfarR by FarR, we have yet to define the means by which FarR promotes farE expression. Since farE cannot be expressed in the absence of farR and cannot be induced in the absence of fakA, our model predicts that FarR should promote expression of farE in response to a FakA-dependent metabolite of exogenous unsaturated fatty acid, by binding to PAL2, which is upstream of the predicted PfarE promoter (Fig. 2A). Since our EMSA was conducted in the absence of exogenous ligand, our initial finding that FarR bound equally well to larger probes containing PAL1 or PAL2 (Fig. 2B) appeared to be inconsistent with such an expectation. However, additional EMSA revealed that FarR retained the ability to bind a minimal PAL1 probe (OP5.1) but not PAL2 (OP4.1) (Fig. 4B), consistent with an expectation that a small-molecule ligand would be required to facilitate specific binding of FarR to the minimal PAL2 site. Such an example was noted with FabR, a TFR that regulates fabB required for synthesis and metabolism of unsaturated fatty acids in E. coli (41). Notably, binding of FabR to a minimal canonical motif required oleoyl-CoA ligand, but binding also occurred independently of ligand when this minimal motif was extended by additional nucleotides.
Our data have revealed that FarR binds to three different locations within farERIS, each harboring a TAGWTTA motif presented in a different context, either as a singular feature in −10PfarR or overlapping the IR2 or IR2a half-sites of PAL1 and PAL2. Further adding to the complexity, although PAL1 and PAL2 are composed of identical (IR1) or similar (IR2/IR2a) inverted repeat half-sites, the order of these repeats is juxtaposed in PAL2 relative to PAL1. Although there are few mechanistic examples to compare with other TFRs that activate gene expression, two instances reflect our present findings, whereby the TFR recognizes a canonical motif that occurs in different contexts. SczA is a streptococcal TFR which in the absence of zinc represses a divergently transcribed efflux pump, SczD, by binding a TGTTCA motif that is part of an inverted repeat; however, in the presence of zinc, it activates SczD by binding to an upstream TGTTCA motif embedded in an imperfect palindrome (42). Similarly, LuxR/HapR in Vibrio spp. may either repress or activate gene expression through recognition of a TATTGATA motif that can have multiple occurrences in target promoters (43, 44). This motif resembles the IR2/IR2a half-site TGTAGWTT in PAL1 and PAL2 and the −10PfarR site TATAGTTT. Therefore, the ability to recognize a common motif in a context-dependent manner may be a general trait of TFRs that function as both repressor and activator.
Based on our present data, combined with consideration of these examples and knowledge of S. aureus fatty acid metabolism and its environmental niche, we propose that expression of farE is tightly regulated so as to permit expression only when the availability of unsaturated fatty acid exceeds the metabolic capacity for incorporation into phospholipid. Although uFFA are toxic to S. aureus if allowed to accumulate in the membrane as free fatty acids, their FakA-dependent esterification into phospholipid represents a means of detoxification (8), which concurrently allows S. aureus to optimize its resources by utilizing host-derived fatty acids for phospholipid synthesis. However, sustained efflux of uFFA would be wasteful of resources, such that farE may only be expressed when there is an appropriate balance of FarR and its cognate ligand. This may account for our finding that strong derepression of farR through nucleotide substitutions in PAL1 is detrimental to resistance, presumably because the physiologic ligand is limiting, and the majority of FarR would be in a ligand-free form. Our future work will focus on resolving these mechanistic considerations, including a detailed analysis of how FarR interacts with the PAL2 site in the presence or absence of ligand.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacteria and plasmids that were used or constructed in this study are listed in Table S1 in the supplemental material. S. aureus cultures were maintained as frozen stocks (−80°C) in 20% glycerol and streaked on TSB agar when required. TSB was supplemented, when needed, with 10 µg/ml erythromycin or chloramphenicol for propagation of strains bearing resistance markers. E. coli strains were grown on LB agar or LB broth supplemented with 100 µg/ml ampicillin when needed. Unless otherwise stated, all cultures were grown at 37°C, and liquid cultures were incubated on an orbital shaking platform at 180 rpm. When required for growth analyses, inoculum cultures of S. aureus were prepared by transferring cells from a single colony into 13-ml polypropylene tubes containing 3 ml of TSB supplemented with antibiotic, as required, followed by overnight incubation. After determination of optical density at 600 nm (OD600), aliquots were subcultured into 25 ml of TSB in 125-ml flasks to achieve an initial OD600 of 0.01. To supplement media with fatty acids, a 5 mM stock concentration was first prepared in TSB containing 0.1% dimethyl sulfoxide (DMSO) and then diluted into TSB plus 0.1% DMSO to achieve the desired concentration of fatty acids.
Strain and plasmid construction.
Genetic manipulation of S. aureus was conducted following established guidelines (45) and as described in our previous work (46–48). All recombinant plasmids were constructed as shuttle vectors in E. coli DH5α. The integrity of plasmids isolated from E. coli was confirmed by nucleotide sequencing of the cloned DNA fragments, prior to electroporation into S. aureus USA300 or isogenic derivatives, using S. aureus RN4220 as an intermediate host. Primers used for PCR amplification of gene segments required for plasmid construction are listed in Table S2, based on the reference genome of USA300 FPR3757 (49).
USA300 ΔfarER containing an in-frame markerless deletion of farE (SAUSA300_2489) and farR (SAUSA300_2490) was generated using pKOR1 (50). Briefly, ∼1-kbp segments that flank farER were amplified by PCR with primers farE-UP-attB1 and farE-UP-SacII for the upstream segment and farR-DW-SacII and farR-DW-attB2 for the downstream segment. The PCR products were digested with SacII, ligated, and incorporated into pKOR1 by treatment with BP Clonase II (Invitrogen), generating pKORfarER. USA300 pKORfarER was then subjected to a two-step temperature shift and antisense counterselection, as previously described (50), generating USA300 ΔfarER. A similar strategy was used to construct USA300 ΔfakA, containing an in-frame deletion of the fatty acid kinase gene fakA, which has also been named dak2 or vfrB (16, 32, 51).
Plasmids pLI50 and pALC2073 were used for gene complementation (52, 53). Previously described pLIfarR and pLIfarE were used to generate pLIfarER. This was achieved by digesting pLIfarE with KpnI and SacII to excise a 2.5-kb fragment containing all but the 5′ end of farE. This fragment was ligated to pLIfarR that was digested with the same enzymes to generate pLIfarER. To construct pLIfarR1, which harbors a G→A substitution in the −10 promoter element of PfarR, pLIfarR was used as the template in a PCR with Phusion polymerase and complementary plus and minus strand mutagenic primers farR1-P and farR1-M, following protocols described in the QuikChange site-directed mutagenesis manual (Stratagene). pLIfarR1 was further modified by digestion with KpnI and SacII, followed by ligation to a 2.5-kb farE fragment from pLIfarE to generate pLIfarER1. Similar techniques were employed to construct pLIfarR2 (mutagenic primers farR2-P and farR2-M) and pLIfarER2 with nucleotide substitutions in the PAL1 feature. pLIfarR3 with nucleotide substitutions in both the PfarR and PAL1 features was constructed using pLIfarR1 as the template with mutagenic primers farR3-P and farR3-M.
To construct the pALCfakA, where fakA is expressed from the Pxyl/tetO promoter of pALC2073, the promoterless fakA gene was amplified by PCR with primers fakA-pALC-F and fakA-pALC-R, digested with KpnI, and ligated into KpnI-digested pALC2073. Similarly, pALCfarE was constructed by amplifying the promoterless farE gene with primers farE-pALC-F and farE-pALC-R, digestion of the product with SacI, and ligation into SacI-digested pALC2073. The pGYlux reporter gene vector (54) was used as described previously (3) to construct a pGYfarR::lux promoter fusion. Briefly, a 248-bp segment spanning the 5′ ends of the divergent farE and farR genes was amplified by PCR of S. aureus genomic DNA with primers far::lux-F and farR::lux-R, followed by digestion with BamHI and SalI, and ligation into pGYlux to create pGYfarR::lux. The same primers were used in PCR with pLIfarR1 as the template, and the amplified PCR product was cloned into pGYlux to create pGYfarR1::lux, in which the −10 motif of PfarR harbors a G→A substitution. For the expression of recombinant 6×His-FarR and 6×His-H121YFarR, the FarR open reading frame was amplified by PCR from genomic DNA of S. aureus USA300 and S. aureus FAR7, respectively, using primers 6HfarR-F and 6HfarR-R. The PCR amplicons were digested with SacI and HindIII and ligated into SacI-HindIII-digested pQE30, creating pQE-FarR and pQE-H121YFarR.
Assays of bactericidal activity and reporter gene function.
In preparation for bactericidal activity of antimicrobial fatty acids, overnight cultures were subcultured into 25 ml of TSB to prepare noninduced cells or in TSB containing 20 μM subinhibitory fatty acid to assay for inducible resistance. For complementation analyses, antibiotic was included in the overnight and initial subcultures but was omitted when cells were inoculated for assay of bactericidal activity. For the bactericidal assay, inoculum cultures were grown to mid-exponential phase (OD600, 0.5) and then subcultured into triplicate or quadruplicate flasks of fresh TSB (OD600, 0.01; approximately 2 × 106 CFU/ml) containing 100 μM linoleic acid. These cultures were then incubated with shaking at 37°C, and aliquots were withdrawn at hourly intervals for preparation of serial dilutions in sterile TSB. Aliquots of 10 μl were then spotted in quadruplicate onto TSB agar, and colonies were counted after 24 h of incubation. The mean from each quadruplicate technical replicate was entered as a single data point for each flask, from which the mean and standard deviation from the biologic replicate flasks were determined.
For assay of reporter gene activity, S. aureus cultures harboring pGYlux and derivatives were subcultured into triplicate or quadruplicate flasks of TSB or TSB supplemented with fatty acids to achieve an initial OD600 of 0.01. The cultures were incubated at 37°C with orbital shaking, and samples were withdrawn at hourly intervals for OD600 determinations. Concurrently, four 200-μl aliquots of each sample were added to 96-well white opaque flat-bottom plates (Greiner Bio-one), and each well was supplemented with 20 μl of 0.1% (vol/vol) decanal in 40% ethanol. Luminescence measurements were immediately taken on a Synergy H4 hybrid reader (BioTek, Winooski, VT), with 1 s of integration and a gain of 200. Data values were recorded as relative light units (RLU), corrected for background by subtraction of values recorded from cultures harboring empty pGYlux. Data points were standardized for differences in growth by dividing RLU values by the recorded OD600 values of the cultures when samples were withdrawn.
Determination of MIC.
Prior to determination of MIC to linoleic acid, inoculum cultures were grown to mid-exponential phase in 125-ml flasks containing 25 ml of TSB supplemented with antibiotics as appropriate for plasmid maintenance. Cultures containing pALC2073 vector and derivatives were additionally supplemented with 20 ng/ml anhydrotetracycline to stimulate expression of the Pxyl/tet promoter. These cultures were then inoculated at OD600 of 0.01 into triplicate 20- by 150-mm glass culture tubes containing 3 ml of TSB supplemented with 0.1% DMSO and indicated concentrations of linoleic acid. Cultures were incubated at 37°C with vigorous shaking, and OD600 values were determined after 24h.
RNA isolation and 5′ rapid amplification of cDNA ends.
RNA was isolated from S. aureus cells (USA300 or FAR7) grown to mid-exponential phase (OD600, 0.5) in TSB supplemented with 20 μM linoleic acid. Cells equivalent to 3.0 OD600 units were harvested from triplicate cultures, and RNA extraction was performed using the E.Z.N.A. total RNA kit, with the addition of 0.25 µg/ml lysostaphin to the lysis solution. RNA integrity was visualized by agarose gel electrophoresis, and after quantification on a NanoDrop ND1000 UV-Vis spectrophotometer, cDNA synthesis was initiated using Superscript II reverse transcriptase (Invitrogen) and gene-specific primer farR-GSP1. The original mRNA template was subsequently removed by treatment with a mixture of RNase H (0.5 units) and RNase T1 (50 units) at 37°C for 30 min. The cDNA was then purified using the BioArray cDNA purification kit (Enzo Life Sciences), as per manufacturer’s instructions, to eliminate unincorporated farR-GSP1 and dinucleoside triphosphates (dNTPs). A d(C) tail was added to the 3′ end of the purified cDNA using 20 units of terminal transferase (Roche) and 0.5 mM CTP. This was followed by two consecutive cycles of PCR using respective abridged anchor primer AAP and abridged universal amplification primer AUAP as forward primers and respective nested reverse primers farR-GSP2 and farR-GSP3. Following the second cycle of PCR, the amplification products were digested with SalI, using restriction sites incorporated into AUAP and farR-GSP3, and then ligated into pUC18 for transformation into E. coli DH5α. After plating on LB-ampicillin (LB-Amp) supplemented with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) and isopropyl thio-β-d-galactopyranoside (IPTG), white colonies were selected for sequencing using M13-F and M13-R primers that flank the multiple-cloning site of pUC18. The +1 transcription start site was identified as the first nucleotide following the poly(C) or poly(G) tail, depending on the orientation of the cloned insert.
Expression and purification of recombinant protein.
Recombinant 6×His-FarR and 6×His-H121YFarR were expressed and purified from E. coli M15/pREP. Bacterial cultures were grown at 37°C in LB (Sigma-Aldrich) supplemented with 100 μg/ml ampicillin and 50 μg/ml kanamycin, to an OD600 of 0.8, before the addition of 0.1 mM IPTG and incubation at room temperature with shaking for an additional 18 h. Cells were harvested by centrifugation and resuspended in binding buffer (20 mM sodium phosphate, 0.5 M NaCl, 40 mM imidazole [pH 7.4]), lysed in a cell disruptor (Constant System Ltd.) at 25,000 lb/in2, and then centrifuged for 50 min at 50,000 × g in a Beckman Coulter Optima L-900K ultracentrifuge, after which the soluble fraction was filtered through a 0.45-μm Acrodisc syringe filter (Pall Laboratory). The lysate was applied onto a 1-ml His-Trap nickel affinity column (GE Healthcare) equilibrated with binding buffer. After washing extensively with binding buffer, bound His-tagged protein was eluted over a linear imidazole gradient up to 0.5 M imidazole in 20 mM sodium phosphate buffer (pH 7.4). Column fractions were assessed by SDS-polyacrylamide gel electrophoresis to check for purity, and fractions containing FarR protein were pooled and dialyzed in 20 mM sodium phosphate and 0.5 M NaCl (pH 7.4) at 4°C. Protein concentration was determined by Bradford assay using Bio-Rad protein assay reagent.
Electrophoretic mobility shift assays.
For EMSA, IRDye 800-labeled duplex and single-stranded nucleotide probes or PCR primers, as detailed in Table S1, were purchased from IDT. Complementary single-stranded oligonucleotides were mixed at 100 μM concentration in 10 mM Tris (pH 8.0) and 0.1 mM EDTA, heated at 95°C for 5 min, and then cooled at room temperature for 45 min to allow duplex formation. Each EMSA reaction was conducted in a 25-μl volume consisting of EMSA buffer (20% glycerol, 30 mM Tris-HCl [pH 8.0], 1 mM MnCl2, 120 mM KCl, 1 mM MgCl2, 16 mM dithiothreitol) supplemented with 5 pM of IRDye 800-labeled probe, up to 2 μM recombinant 6×His-FarR or 6×His-H121YFarR, 240 μg/ml bovine serum albumin (BSA), and 15.2 μg/ml poly[d(I-C)] in EMSA buffer. After incubation at room temperature for 60 min, individual samples were applied to a 6% polyacrylamide gel prepared in Tris-borate-EDTA buffer and electrophoresed in the same buffer system for 45 min at 120 V. The gels were then imaged using an Odyssey imager (Li-Cor Biosciences). In competition EMSA, unlabeled competitor probes harboring specific nucleotide substitutions were added in 50-fold excess relative to labeled probes.
Production of polyclonal antibodies and Western blotting.
The production of FarR-specific polyclonal antibodies in New Zealand White rabbits was contracted to ProSci Incorporated (Poway, CA). Each of two animals was initially immunized with 200 µg of 6×His-FarR emulsified in complete Freund’s adjuvant, followed by 100 μg of protein administered in incomplete Freund’s adjuvant, at 2-week intervals for 6 weeks. The animals were then bled for collection of antiserum 2 weeks after the final immunization.
For preparation of cell lysates, E. coli DH5α harboring pLIfarR, pLIfarER, and derivatives were grown to mid-exponential phase in LB, and cells were harvested by centrifugation at 5,000 × g for 20 min, washed in ice cold 50 mM Tris-HCl, 5 mM EDTA, and 150 mM NaCl (pH 8.0), and then resuspended at 1/10 the original culture volume, in the same buffer supplemented with 1% (vol/vol) Triton X-100, 0.5% (vol/vol) SDS, and Pierce complete protease inhibitor cocktail. The cell suspensions were incubated for 2 h at room temperature on a rocking platform, followed by centrifugation at 5,000 × g for 20 min. The clarified cell lysate was then assayed for determination of total protein. For Western blots, samples containing 25 μg of total cell lysate protein were subjected to SDS-PAGE using a 12% polyacrylamide resolving gel. After transfer of proteins to FluoroTrans polyvinylidene difluoride (PVDF) membrane (Pall Life Sciences), the membranes were blocked by incubation in phosphate-buffered saline (PBS) supplemented with 5% nonfat skim milk powder and 10% horse serum (Sigma). Wash buffer and antibody dilution buffer consisted of PBS plus 0.05% Tween 20, and PBS-Tween supplemented with 2% skim milk powder. Primary anti-FarR antiserum was used at a dilution of 1:5,000, followed by secondary IRDye 800-conjugated goat anti-rabbit IgG (Jackson Immunoresearch Laboratories, Inc.). Membranes were imaged using an Odyssey imager (Li-Cor Biosciences).
Data analyses.
Data points for growth, viability, reporter gene assays, and murine infection models were plotted and analyzed using GraphPad Prism version 7. Significant differences at specific time points were determined using GraphPad statistics functions.
Supplementary Material
ACKNOWLEDGMENTS
We declare no potential conflicts of interest.
This work was supported by grants to M.J.M. and D.E.H. from the Natural Sciences and Engineering Research Council of Canada. J.E.T.S. and R.C.K. were recipients of Ontario Graduate Scholarship awards.
M.J.M. and D.E.H. contributed to the design of the study; H.A., R.C.K., K.A.F., and J.E.T.S. were responsible for data acquisition; H.A., D.E.H., and M.J.M. contributed to the analysis and interpretation of data; H.A. and M.J.M. shared in writing of the manuscript; and D.E.H. provided critical evaluation.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00602-18.
REFERENCES
- 1.Cuthbertson L, Nodwell JR. 2013. The TetR family of regulators. Microbiol Mol Biol Rev 77:440–475. doi: 10.1128/MMBR.00018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kisker C, Hinrichs W, Tovar K, Hillen W, Saenger W. 1995. The complex formed between Tet repressor and tetracycline-Mg2+ reveals mechanism of antibiotic resistance. J Mol Biol 247:260–280. doi: 10.1006/jmbi.1994.0138. [DOI] [PubMed] [Google Scholar]
- 3.Alnaseri H, Arsic B, Schneider JET, Kaiser JC, Scinocca ZC, Heinrichs DE, McGavin MJ. 2015. Inducible expression of a resistance-nodulation-division-type efflux pump in Staphylococcus aureus provides resistance to linoleic and arachidonic acids. J Bacteriol 197:1893–1905. doi: 10.1128/JB.02607-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL. 2005. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis 5:751–762. doi: 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 5.Grkovic S, Brown MH, Schumacher MA, Brennan RG, Skurray RA. 2001. The staphylococcal QacR multidrug regulator binds a correctly spaced operator as a pair of dimers. J Bacteriol 183:7102–7109. doi: 10.1128/JB.183.24.7102-7109.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeng WY, Ko TP, Liu CI, Guo RT, Liu CL, Shr HL, Wang AHJ. 2008. Crystal structure of IcaR, a repressor of the TetR family implicated in biofilm formation in staphylococcus epidermidis. Nucleic Acids Res 36:1567–1577. doi: 10.1093/nar/gkm1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jefferson KK, Pier DB, Goldmann DA, Pier GB. 2004. The teicoplanin-associated locus regulator (TcaR) and the intercellular adhesin locus regulator (IcaR) are transcriptional inhibitors of the ica locus in Staphylococcus aureus. J Bacteriol 186:2449–2456. doi: 10.1128/JB.186.8.2449-2456.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parsons JB, Yao J, Frank MW, Jackson P, Rock CO. 2012. Membrane disruption by antimicrobial fatty acids releases low-molecular-weight proteins from Staphylococcus aureus. J Bacteriol 194:5294–5304. doi: 10.1128/JB.00743-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bibel DJ, Miller SJ, Brown BE, Pandey BB, Elias PM, Shinefield HR, Aly R. 1989. Antimicrobial activity of stratum corneum lipids from normal and essential fatty acid-deficient mice. J Invest Dermatol 92:632–638. doi: 10.1111/1523-1747.ep12712202. [DOI] [PubMed] [Google Scholar]
- 10.Do TQ, Moshkani S, Castillo P, Anunta S, Pogosyan A, Cheung A, Marbois B, Faull KF, Ernst W, Chiang SM, Fujii G, Clarke CF, Foster K, Porter E. 2008. Lipids including cholesteryl linoleate and cholesteryl arachidonate contribute to the inherent antibacterial activity of human nasal fluid. J Immunol 181:4177–4187. doi: 10.4049/jimmunol.181.6.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drake DR, Brogden KA, Dawson DV, Wertz PW. 2008. Thematic review series: skin lipids. Antimicrobial lipids at the skin surface. J Lipid Res 49:4–11. doi: 10.1194/jlr.R700016-JLR200. [DOI] [PubMed] [Google Scholar]
- 12.Shryock TR, Dye ES, Kapral FA. 1992. The accumulation of bactericidal lipids in staphylococcal abscesses. J Med Microbiol 36:332–336. doi: 10.1099/00222615-36-5-332. [DOI] [PubMed] [Google Scholar]
- 13.Fujihashi M, Nakatani T, Hirooka K, Matsuoka H, Fujita Y, Miki K. 2014. Structural characterization of a ligand-bound form of Bacillus subtilis FadR involved in the regulation of fatty acid degradation. Proteins 82:1301–1310. doi: 10.1002/prot.24496. [DOI] [PubMed] [Google Scholar]
- 14.Yeo HK, Park YW, Lee JY. 2017. Structural basis of operator sites recognition and effector binding in the TetR family transcription regulator FadR. Nucleic Acids Res 45:4244–4254. doi: 10.1093/nar/gkx009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parsons JB, Frank MW, Jackson P, Subramanian C, Rock CO. 2014. Incorporation of extracellular fatty acids by a fatty acid kinase-dependent pathway in Staphylococcus aureus. Mol Microbiol 92:234–245. doi: 10.1111/mmi.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parsons JB, Broussard TC, Bose JL, Rosch JW, Jackson P, Subramanian C, Rock CO. 2014. Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc Natl Acad Sci U S A 111:10532–10537. doi: 10.1073/pnas.1408797111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenway DL, Dyke KG. 1979. Mechanism of the inhibitory action of linoleic acid on the growth of Staphylococcus aureus. J Gen Microbiol 115:233–245. doi: 10.1099/00221287-115-1-233. [DOI] [PubMed] [Google Scholar]
- 18.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corrigan RM, Foster TJ. 2009. An improved tetracycline-inducible expression vector for Staphylococcus aureus. Plasmid 61:126–129. doi: 10.1016/j.plasmid.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Ramos JL, Martinez-Bueno M, Molina-Henares AJ, Teran W, Watanabe K, Zhang X, Gallegos MT, Brennan R, Tobes R. 2005. The TetR family of transcriptional repressors. Microbiol Mol Biol Rev 69:326–356. doi: 10.1128/MMBR.69.2.326-356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pustell J, Kafatos FC. 1982. A high speed, high capacity homology matrix: zooming through SV40 and polyoma. Nucleic Acids Res 10:4765–4782. doi: 10.1093/nar/10.15.4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reese MG. 2001. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput Chem 26:51–56. doi: 10.1016/S0097-8485(01)00099-7. [DOI] [PubMed] [Google Scholar]
- 23.Yu Z, Reichheld SE, Savchenko A, Parkinson J, Davidson AR. 2010. A comprehensive analysis of structural and sequence conservation in the TetR family transcriptional regulators. J Mol Biol 400:847–864. doi: 10.1016/j.jmb.2010.05.062. [DOI] [PubMed] [Google Scholar]
- 24.Derouiche A, Bidnenko V, Grenha R, Pigonneau N, Ventroux M, Franz-Wachtel M, Nessler S, Noirot-Gros MF, Mijakovic I. 2013. Interaction of bacterial fatty-acid-displaced regulators with DNA is interrupted by tyrosine phosphorylation in the helix-turn-helix domain. Nucleic Acids Res 41:9371–9381. doi: 10.1093/nar/gkt709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agari Y, Agari K, Sakamoto K, Kuramitsu S, Shinkai A. 2011. TetR-family transcriptional repressor Thermus thermophilus FadR controls fatty acid degradation. Microbiology 157:1589–1601. doi: 10.1099/mic.0.048017-0. [DOI] [PubMed] [Google Scholar]
- 26.Parsons JB, Frank MW, Subramanian C, Saenkham P, Rock CO. 2011. Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc Natl Acad Sci U S A 108:15378–15383. doi: 10.1073/pnas.1109208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schumacher MA, Miller MC, Grkovic S, Brown MH, Skurray RA, Brennan RG. 2002. Structural basis for cooperative DNA binding by two dimers of the multidrug-binding protein QacR. EMBO J 21:1210–1218. doi: 10.1093/emboj/21.5.1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cue DR, Lei MG, Lee C. 2012. Genetic regulation of the intercellular adhesion locus in staphylococci. Front Cell Infect Microbiol 2:38. doi: 10.3389/fcimb.2012.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agari Y, Sakamoto K, Kuramitsu S, Shinkai A. 2012. Transcriptional repression mediated by a TetR family protein, PfmR, from Thermus thermophilus HB8. J Bacteriol 194:4630–4641. doi: 10.1128/JB.00668-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang J-L, Nie Y, Wang M, Xiong G, Wang Y-P, Maser E, Wu X-L. 2016. Regulation of alkane degradation pathway by a TetR family repressor via an autoregulation positive feedback mechanism in a Gram-positive Dietzia bacterium. Mol Microbiol 99:338–359. doi: 10.1111/mmi.13232. [DOI] [PubMed] [Google Scholar]
- 31.Christen S, Srinivas A, Bähler P, Zeller A, Pridmore D, Bieniossek C, Baumann U, Erni B. 2006. Regulation of the Dha operon of Lactococcus lactis: a deviation from the rule followed by the TetR family of transcription regulators. J Biol Chem 281:23129–23137. doi: 10.1074/jbc.M603486200. [DOI] [PubMed] [Google Scholar]
- 32.Bose JL, Daly SM, Hall PR, Bayles KW. 2014. Identification of the Staphylococcus aureus vfrAB operon, a novel virulence factor regulatory locus. Infect Immun 82:1813–1822. doi: 10.1128/IAI.01655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krute CN, Rice KC, Bose JL. 2017. VfrB is a key activator of the Staphylococcus aureus SaeRS two-component system. J Bacteriol 199:e00828-16. doi: 10.1128/JB.00828-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ericson ME, Subramanian C, Frank MW, Rock CO. 2017. Role of fatty acid kinase in cellular lipid homeostasis and SaeRS-dependent virulence factor expression in Staphylococcus aureus. mBio 8:e00988-17. doi: 10.1128/mBio.00988-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeMars ZR, Bose JL. 2018. Redirection of metabolism in response to fatty acid kinase in Staphylococcus aureus. J Bacteriol 200:e00345-18. doi: 10.1128/JB.00345-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor DL, Ante VM, Bina XR, Howard MF, Bina JE. 2015. Substrate-dependent activation of the Vibrio cholerae vexAB RND efflux system requires vexR. PLoS One 10:e0117890. doi: 10.1371/journal.pone.0117890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bina XR, Howard MF, Taylor-Mulneix DL, Ante VM, Kunkle DE, Bina JE. 2018. The Vibrio cholera RND efflux systems impact virulence factor production and adaptive responses via periplasmic sensor proteins. PLoS Pathog 14:e1006804. doi: 10.1371/journal.ppat.1006804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruiz C, Levy SB. 2014. Regulation of acrAB expression by cellular metabolites in Escherichia coli. J Antimicrob Chemother 69:390–399. doi: 10.1093/jac/dkt352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buckner MMC, Blair JMA, La Ragione RM, Newcombe J, Dwyer DJ, Ivens A, Piddock LJV. 2016. Beyond antimicrobial resistance: evidence for a distinct role of the AcrD efflux pump in Salmonella biology. mBio 7:e01916-16. doi: 10.1128/mBio.01916-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grkovic S, Brown MH, Roberts NJ, Paulsen IT, Skurray RA. 1998. QacR is a repressor protein that regulates expression of the Staphylococcus aureus multidrug efflux pump QacA. J Biol Chem 273:18665–18673. doi: 10.1074/jbc.273.29.18665. [DOI] [PubMed] [Google Scholar]
- 41.Feng Y, Cronan JE. 2011. Complex binding of the FabR repressor of bacterial unsaturated fatty acid biosynthesis to its cognate promoters. Mol Microbiol 80:195–218. doi: 10.1111/j.1365-2958.2011.07564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kloosterman TG, Van Der Kooi-Pol MM, Bijlsma JJ, Kuipers OP. 2007. The novel transcriptional regulator SczA mediates protection against Zn2+ stress by activation of the Zn2+-resistance gene czcD in Streptococcus pneumoniae. Mol Microbiol 65:1049–1063. doi: 10.1111/j.1365-2958.2007.05849.x. [DOI] [PubMed] [Google Scholar]
- 43.van Kessel JC, Ulrich LE, Zhulin IB, Bassler L. 2013. Analysis of activator and repressor functions reveals the requirements for transcriptional control by LuxR, the master regulator of quorum sensing in Vibrio harveyi. mBio 4:e00378-13. doi: 10.1128/mBio.00378-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ball AS, Chaparian RR, van Kessel JC. 2017. Quorum sensing gene regulation by LuxR/HapR master regulators in Vibrios. J Bacteriol 199:e00105-17. doi: 10.1128/JB.00105-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Novick RP. 1991. Genetic systems in staphylococci. Methods Enzymol 204:587–636. doi: 10.1016/0076-6879(91)04029-N. [DOI] [PubMed] [Google Scholar]
- 46.Cadieux B, Vijayakumaran V, Bernards MA, McGavin MJ, Heinrichs DE. 2014. Role of lipase from community-associated methicillin-resistant Staphylococcus aureus strain USA300 in hydrolyzing triglycerides into growth-inhibitory free fatty acids. J Bacteriol 196:4044–4056. doi: 10.1128/JB.02044-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nickerson N, Ip J, Passos DT, McGavin MJ. 2010. Comparison of Staphopain A (ScpA) and B (SspB) precursor activation mechanisms reveals unique secretion kinetics of proSspB (staphopain B), and a different interaction with its cognate staphostatin, SspC. Mol Microbiol 75:161–177. doi: 10.1111/j.1365-2958.2009.06974.x. [DOI] [PubMed] [Google Scholar]
- 48.Arsic B, Zhu Y, Heinrichs DE, McGavin MJ. 2012. Induction of the Staphylococcal proteolytic cascade by antimicrobial fatty acids in community acquired methicillin resistant Staphylococcus aureus. PLoS One 7:e45952. doi: 10.1371/journal.pone.0045952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367:731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 50.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 51.Li M, Rigby K, Lai Y, Nair V, Peschel A, Schittek B, Otto M. 2009. Staphylococcus aureus mutant screen reveals interaction of the human antimicrobial peptide dermcidin with membrane phospholipids. Antimicrob Agents Chemother 53:4200–4210. doi: 10.1128/AAC.00428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee CY, Iandolo JJ. 1986. Lysogenic conversion of staphylococcal lipase is caused by insertion of the bacteriophage L54a genome into the lipase structural gene. J Bacteriol 166:385–391. doi: 10.1128/jb.166.2.385-391.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bateman BT, Donegan NP, Jarry TM, Palma M, Cheung AL. 2001. Evaluation of a tetracycline-inducible promoter in Staphylococcus aureus in vitro and in vivo and its application in demonstrating the role of sigB in microcolony formation. Infect Immun 69:7851–7857. doi: 10.1128/IAI.69.12.7851-7857.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mesak LR, Yim G, Davies J. 2009. Improved lux reporters for use in Staphylococcus aureus. Plasmid 61:182–187. doi: 10.1016/j.plasmid.2009.01.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.