

Abstract
Chemerin is a leukocyte attractant, adipokine, and antimicrobial protein abundantly produced in the skin epidermis. Despite the fact that most of the bactericidal activity present in human skin exudates is chemerin-dependent, just how chemerin shapes skin defenses remains obscure. Here we demonstrate that p4, a potent antimicrobial human chemerin peptide derivative, displays killing activity against pathogenic methicillin-resistant Staphylococcus aureus strains and suppresses microbial growth in a topical skin infection model. Mechanistically, we show that p4 homodimerization is required for maximal bactericidal activity and that an oxidative environment, such as at the skin surface, facilitates p4 disulfide bridge formation, required for the dimerization. p4 led to rapid damage of the bacterial internal membrane and inhibited the interaction between the membranous cytochrome bc1 complex and its redox partner, cytochrome c. These results suggest that a chemerin p4–based defense strategy combats bacterial challenges at the skin surface.
Keywords: epidermis, antimicrobial peptide (AMP), bacteria, host defense, host–pathogen interaction, chemotaxis, adipokine, Staphylococcus aureus (S. aureus), skin, chemerin, MRSA
Introduction
Chemerin is a natural ligand for the metabotropic receptor CMKLR1 with a growing number of biological functions. Chemerin is best known as a chemotactic factor involved in controlling homing of specific (CMKLR1+) immune cells, such as dendritic cells or macrophages, to sites of inflammation (1–3). Chemerin also plays an important role in other physiological processes, such as the regulation of metabolism, either by influencing the differentiation of fat cells or controlling the production and secretion of insulin by pancreatic cells (4, 5). In addition, chemerin has been implicated in angiogenesis and regulation of vascular function (6, 7).
Chemerin is synthesized as an inactive precursor (prochemerin) that circulates in the bloodstream. Upon proteolytic cleavage of its C terminus, prochemerin is converted into a potent agonist of CMKLR1. The sequence of the chemerin C terminus is crucial for chemotactic activity. Enzymes that activate chemerin include cysteine and serine proteases involved in the blood coagulation cascade and inflammatory processes (8–10).
Chemerin is expressed by many types of epithelial cells (11–13) and, in skin keratinocytes, is up-regulated in response to bacteria and acute-phase cytokines (14). Recombinant chemerin and chemerin-derived peptide 4 (p4), located in a central region of the chemerin protein sequence, are both active against a broad range of microorganisms, including skin-, lung-, and oral cavity–associated bacteria as well as the fungal pathogen Candida albicans (8, 15, 16). Inhibition of bacterial growth by conditioned medium from organ cultures of primary human keratinocytes is largely chemerin-dependent (15), and chemerin deficiency results in higher counts of viable bacteria associated with the epidermis in an experimental model of skin infection (14).
Given the relative abundance of chemerin in the epidermis, chemerin and chemerin-derived peptides may represent important components of the host defense system involved in shaping the skin microbiome and/or may confer protection against skin-invading microbes. Therefore, understanding the modes of action of p4, the most potent antimicrobial chemerin derivative, is of high significance.
Here we demonstrate that p4 is a potent bactericide against pathogenic methicillin-resistant Staphylococcus aureus (MRSA)2 strains. We also show that p4 limits topical microbial growth in vivo and rapidly destroys pathogens via disruption of the microbial cell membrane. Components of the electron transport chain were identified as p4 targets that contributed to the p4 antimicrobial activity. Oxidized conditions boosted the effectiveness of p4 against bacteria by supporting the formation of disulfide-bridged p4 dimers. Thus, we identify a novel redox-mediated pathway that controls host antimicrobial activity at barrier sites.
Results
Chemerin-derived peptide 4 restricts growth of S. aureus in vitro and in experimental topical skin infection
An internal 20-amino acid peptide, Val66-Pro85 (p4), exhibits most of the antimicrobial activity of active chemerin in vitro (15, 16). Among its microbial targets are Gram-positive and Gram-negative bacteria: S. aureus and Escherichia coli, respectively. Because it remains unknown whether p4 is active against antibiotic-resistant bacterial strains, we determined the potency of p4 against MRSA by MDA assay. p4 had bactericidal properties against two tested MRSA strains, ATCC BAA-1707 and clinical isolate E240 (Fig. 1A). Moreover, similar MIC values for the methicillin-sensitive 8325-4 strain and MRSA strains (25 μm versus 12.5 μm for both MRSA strains) indicated that MRSA demonstrates no or low resistance to p4 (Fig. 1A).
Figure 1.
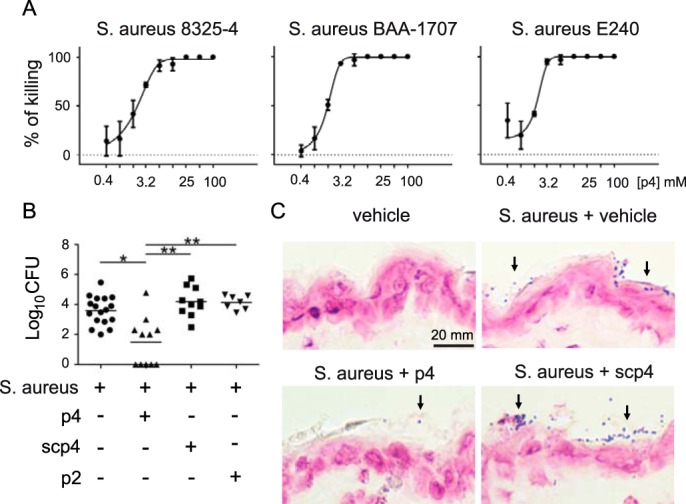
Chemerin-derived p4 peptide is bactericidal in vitro and in vivo. A, the indicated S. aureus strains were incubated with p4 for 24 h. Data show the percentage of killing for the indicated strain. The MIC was defined as the lowest concentration of p4 showing no visible growth (100% of killing). Mean ± S.D. of three independent measurements is shown. B, mice were topically infected with 1 × 107 cfu of S. aureus 8325-4 in the presence of 100 μm peptide p4, scp4, p2, or vehicle. Data points indicate the colony-forming units of bacteria recovered from the skin surface 24 h after application of bacteria, with each data point representing one cavity and a horizontal line indicating the mean value in each group; n = 5 independent experiments. **, p < 0.01; *, p < 0.05 by Kruskal-Wallis test with post hoc Dunn's multiple comparisons test. C, mice were topically treated with vehicle or infected with 1 × 107 cfu S. aureus 8325-4 in the presence of 100 μm peptide p4, scp4, or vehicle for 24 h. Gram-positive S. aureus on the skin surface is indicated by arrows. Data are from one experiment and are representative of three independent experiments.
To determine the role of p4 in restricting bacteria growth in vivo, p4, control peptides, or vehicle were topically administered to mouse skin. The control peptides included a scramble peptide 4 (scp4, Table 1), which, in contrast to p4, was found previously to be nonbactericidal against oral cavity–associated bacteria (16), and peptide p2, which originates from a different region of human chemerin (Glu36-Ala55) and is devoid of antimicrobial activity against E. coli (15). The peptides were allowed to dry on the skin, and mice were then topically infected with S. aureus 8325-4. Bacterial loads that were recovered from the skin surface 24 h later were measured by colony-forming assay (Fig. 1B). Alternatively, skin-associated bacteria were visualized by Gram staining (Fig. 1C). Application of 100 μm p4 significantly reduced the bacterial burden compared with vehicle, 100 μm scp4, or p2 (Fig. 1, B and C). We conclude that p4 is able to kill both antibiotic-resistant and nonresistant S. aureus strains in vitro and restrict the growth of the skin pathogen in situ in the skin environment.
Table 1.
Antimicrobial activity of the p4-based peptides
For each peptide, the following properties are specified: the amino acid sequence; the number of amino acid (aa) residues; the total charge at pH 6.0, which models the acidic environment of the water/bacterial membrane interface; and the relative mean hydrophobic moment, determined on the assumption of the spatial structure of the peptide as a twisted β sheet, which corresponds to the periodicity of 160° in the Edmundson projection (15). Peptides with aa substitutes are named based on first and last amino acid residue (the length), aa residue to be substituted, and substituting aa residue (bold). Cysteine with the thiol group blocked by IAA is underlined. Extra aa in the elongated form of p4 are italicized. d-VR15, VR15 comprised only of d-amino acid residues. The AM effect indicates killing activity of the listed peptides at 100 μm relative to p4 (100%), as determined by both MDA and RDA assays. +, >90%; −, <20%; +/−, ∼40%.
| Peptide name | Sequence | No. of aa | Total charge | rHMβ | AM effect (E. coli) |
|---|---|---|---|---|---|
| p4 | VRLEFKLQQTSCRKRDWKKP | 20 | +5 | 0.375 | + |
| scp4 | DPWLKVRKFQTLKQREKRCS | 20 | +5 | 0.033 | − |
| p4-IAA | VRLEFKLQQTSCRKRDWKKP | 20 | +5 | 0.375 | − |
| p4 sister peptides with residue substitutes | |||||
| (VP20)CA | VRLEFKLQQTSARKRDWKKP | 20 | +5 | 0.384 | − |
| (VP20)KA | VRLEFALQQTSCRARDWAAP | 20 | +1 | 0.409 | − |
| (VP20)KR | VRLEFRLQQTSCRRRDWRRP | 20 | +5 | 0.375 | + |
| Longer and shorter p4-based analogs | |||||
| VK23 | VRLEFKLQQTSCRKRDWKKPECK | 23 | +5 | 0.238 | +/− |
| VR15 | VRLEFKLQQTSCRKR- - - - - | 15 | +4 | 0.625 | + |
| RP19 | - RLEFKLQQTSCRKRDWKKP | 19 | +5 | 0.383 | + |
| LP18 | - - LEFKLQQTSCRKRDWKKP | 18 | +4 | 0.348 | − |
| EP17 | - - - EFKLQQTSCRKRDWKKP | 17 | +4 | 0.295 | − |
| FP16 | - - - - FKLQQTSCRKRDWKKP | 16 | +5 | 0.234 | − |
| KP15 | - - - - -KLQQTSCRKRDWKKP | 15 | +5 | 0.139 | − |
| d-VR15 | VRLEFKLQQTSCRKR- - - - - | 15 | +4 | 0.625 | + |
p4 sister peptides reveal a critical role for cysteine and positively charged amino acids for the antimicrobial activity of p4
To define the mechanism by which p4 inhibits bacterial growth, we first tested p4 versus p4 analogs that were designed based on the analysis of variations in cross-species chemerin homology domains. For this analysis, a UniRef50 cluster of amino acid sequences sharing at least 50% sequence identity with the human chemerin sequence (UniProtKB Q99969, RARR2_HUMAN) was identified. The cluster contained 120+ sequences, but eventually the set of chemerin sequences was limited to 44 that had reviewed UniProt Swissprot entries (September 2017). For these 44 amino acid sequences, a multiple sequence alignment was constructed (17). The most strongly conserved amino acid residues in the most strongly conserved region of chemerin are shown in Fig. 2A. The conserved region starts with invariable glycine at position 63 and spans approximately 50 residues to the invariable proline at position 118. In this region, there are 28 invariant (Gly63, Phe65, .., His116, Cys117, Pro118) and eight variable positions at which conservative substitutions are observed ([KR]83, [KR]90, [KR]95, [IV]102, [VI]110, [RQ]113, [MLV]114, and [VI]115). Interestingly, this conserved sequence region comprises the p4 sequence (i.e. residues 66 to 85), where the total number of both invariant and conservatively substituted sites is 14 (Fig. 2A). These sites were targeted in the p4 analogs that included scp4, p4 sister peptides with amino acid substitution(s), or variants either elongated or truncated at the N or C terminus. The peptides were chemically synthesized and analyzed for antimicrobial properties in radial diffusion assays and MDAs against E. coli (Table 1).
Figure 2.

The amino acid and length determinants of p4 antimicrobial activity. A, the sequence logo was determined for the conservative fragment of 44 chemerin ortholog sequences from the UniRef50 database. The logo spans the region between positions 61 and 119 of the human chemerin sequence. The underlying gray line indicates the location of p4. B, SDS-PAGE analysis of the indicated p4-based peptides. Peptides were separated under reducing and nonreducing conditions (left and right panels, respectively, separated by a molecular mass ladder), followed by Coomassie Blue staining. Data are from one experiment and are representative of three independent experiments. C, the sequence of p4 with highlighted amino acid and length determinants of its antimicrobial activity. Critical amino acids are shown in red. The smallest active monomeric form of p4 analog is shown in bold.
Under reducing conditions, p4 migrated as an ∼5-kDa monomer, whereas, under nonreducing conditions, p4 migrated as both an ∼5-kDa monomer and an ∼10-kDa dimer (Fig. 2B). Because the dimeric band disappeared under reducing conditions, these data suggest that dimerization required disulfide cross-linking. The critical role of invariant cysteine at position 77 was demonstrated by the (VP20)CA peptide, in which Cys77 was substituted with alanine. This modification did not influence the peptide net charge and left the relative hydrophobic moment in the β sheet conformation (rHMβ) (15) unchanged (Table 1). However, as expected, substitution of Cys77 with alanine prevented p4 self-association (Fig. 2B) and abrogated the p4 killing activity (Table 1). In addition, when the ability to form disulfide bonds was blocked by treatment of p4 with iodoacetamide (p4-IAA), dimers were not formed, and the antimicrobial activity of p4 was lost (Fig. 2B and Table 1, respectively). Of note, the bactericidal effect did not result solely from a general property of peptides having disulfide bonds because scp4, which also formed C-mediated dimers, was not antimicrobial (Fig. 2B and Table 1). Together, these data suggest that C-mediated dimerization is crucial for maximal effective bacterial killing.
p4 is enriched in lysine residues, which represent 25% of the p4 sequence, suggesting that the cationic nature of p4 and/or the distribution of the charged residues in the p4 sequence contribute to the bactericidal effects of the peptide. Scp4, which has an identical total net charge (+5) but differed substantially in rHMβ compared with p4, did not exhibit antimicrobial activity (Table 1). Although substitution of all lysine with neutral alanine residues reduced the net charge of the p4 peptide to +1 and abrogated its antimicrobial effect, this peptide variant, (VP20)KA, retained its amphipathic character, as evidenced by a high value of rHMβ (Table 1). Replacing lysine residues with basic arginine residues left the physicochemical properties unchanged, and the resulting peptide variant (VP20)KR was still a potent antimicrobial agent (Table 1).
Next we tested whether the length of the peptide was important as well. The chemerin-derived peptide VK23, containing 23 amino acids, partially retained the antibacterial activity (Table 1). In case of truncated forms, the 15-amino acid-long peptide VR15, comprising residues V66-R80 with a +4 net charge and a high rHMβ of 0.625, showed antibacterial activity. On the other hand, the 15-amino acid-long peptide KP15 with +5 net charge and lower rHMβ (0.139) had no activity. Thus, high peptide amphipathicity was important for its antimicrobial potential.
Together, these data suggest that several features enable p4 to act as a potent antimicrobial agent. These include Cys-mediated intermolecular disulfide bonds, a strong positive net charge, and amphipathic features as well as sufficient length. The cationic 14-amino acid-long dimeric peptide is the smallest chemerin derivative equipped with antimicrobial potential (Fig. 2C).
To determine whether the mode of action of p4 relies on its specific interaction with a protein target at the bacterial surface, we assessed the importance of peptide stereochemistry for antimicrobial activity. We compared the antimicrobial potential of the smallest active form of p4 (peptide VR15) with a similar peptide that contained only d-amino acid residues (d-VR15). Both VR15 and d-VR15 were equally potent against E. coli (Table 1). Therefore, it is not likely that p4 binds to a specific site on a protein target but, rather, that the peptide interacts with the lipid bilayer to enter bacteria.
Although we have not assessed the specific conformation(s) assumed by p4 upon binding the bacterial membrane, the fact that the antibacterial activity of p4 correlates well with relative hydrophobic moments calculated for the β strand conformation (Table 1 and Ref. 15) might indicate that p4 adopts an extended conformation when interacting with bacterial membrane lipids. Unraveling the conformational preferences of both monomeric and dimeric forms of p4 interacting with membrane lipids requires additional studies.
p4 binds to bacteria at either bactericidal or bacteriostatic concentrations, but only high doses of p4 break the inner bacterial cell membrane
E. coli strains exhibit high sensitivity to p4, with MIC = 6.3–12.5 μm (Fig. 3A and Ref. 15). E. coli HB101 exposed to p4 at concentrations above the MIC (12.5–100 μm) was killed rapidly. Over 90% of bacteria were found to be dead within 3 min, and by 30 min, more than 99% of bacteria were dead (Fig. 3B). In contrast to E. coli, p4 did not display any damaging effects against human erythrocytes, as exposure of red blood cells to up to 100 μm p4 for 2 h did not result in hemolysis (Fig. 2C). Likewise, human primary keratinocytes did not significantly change their mitochondrial respiration in response to high doses (12.5–100 μm) of p4 at 2 h, as assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell viability assay (Fig. S1). Similar data were obtained when release of intracellular enzyme lactate dehydrogenase into the conditioned medium was used as a marker of keratinocyte cytotoxicity, although, at the highest dose (100 μm), p4 increased lactate dehydrogenase release ∼2-fold over vehicle control (48% ± 12% versus 21% ± 9%, mean ± S.D.) (Fig. S1).
Figure 3.

p4 exhibits rapid concentration-dependent lytic activity against E. coli. A, E. coli HB101 was incubated with the indicated concentrations of p4 for 2 h. Cell viability was analyzed by MDA assay. n = 3, mean ± S.D. B, E. coli HB101 was incubated with 100 μm p4 or vehicle for the indicated times. Cell viability was analyzed by MDA assay, n = 3; mean ± S.D. C, human erythrocytes were incubated with 1% Triton X-100, the indicated concentration of p4, or vehicle for 2 h. Hemolysis of erythrocytes is shown relative to lysis caused by Triton X-100. n = 3, mean ± S.D. D, E. coli HB101 was incubated with 100 μm p4 or vehicle for the indicated times. Bacterial morphology was assessed by TEM. E, E. coli HB101 was incubated with 100 μm p4 for 5 min. Alterations in bacterial permeability were visualized by fluorescence imaging. Bacteria were treated with FITC-labeled p4 (green), stained with PI (red), and counterstained with Hoechst to visualize DNA (blue). Arrows point to accumulation of p4 at the cell surface. F, β-gal–expressing E. coli JM83 was incubated with the indicated concentrations of p4 for 15 min. The β-gal activity present in supernatants of p4-treated bacteria is shown as a percentage of the vehicle-treated bacteria. n = 3, mean ± S.D. G, E. coli HB101 was treated with p4 for 45 min, followed by TEM. Arrows and asterisks indicate outer membrane perturbations and the discontinuous inner membrane, respectively. H, intracellular localization of p4 is shown by immunogold labeling. E. coli HB101 was treated with biotin-p4 or p4 as a control, fixed, and stained with mouse anti-biotin Abs, followed by anti-mouse Abs conjugated to gold particles. Arrowheads indicate gold particles. The enlarged image (i) demonstrates interaction of p4 with the cell membrane. ***, p < 0.001; **, p < 0.01; *, p < 0.05 by Kruskal-Wallis one-way ANOVA with post hoc Dunn's test. TEM and fluorescence microscopy images are from one experiment and are representative of at least three experiments.
Kinetic studies using TEM (Fig. 3D) or fluorescence microscopy (Fig. 3E) demonstrated that p4-mediated effects on bacteria were rapid, with changes in cell morphology and membrane distortion observed as early as 5 min. p4-triggered alterations progressed over time, and robust ultrastructural lesions accompanied by changes in cytoplasm density and/or condensation of nuclear material were evident in E. coli and S. aureus exposed to p4 but not to vehicle and/or scp4 for 2 h (Fig. 3D and Fig. S2, respectively). Uptake of the membrane-impermeable dye propidium iodide (PI) by E. coli treated with p4 for 5 min suggested that membrane integrity was compromised and that the p4-mediated killing involved rapid disruption of cytoplasmic membrane function (Fig. 3E). To directly demonstrate inner membrane permeabilization, we performed a β-gal leakage assay. Because β-gal is a cytoplasmic enzyme and its substrate ONPG does not cross the inner membrane (18), β-gal activity can be detected in the bacterial conditioned medium only as a result of disintegration of the cytoplasmic membrane. As shown in Fig. 3F, treatment of E. coli JM83 constitutively expressing the lacZ gene with p4 at bactericidal (lethal) concentrations (≥12.5 μm) disrupted the integrity of the inner membrane, as evidenced by β-gal–specific ONPG hydrolysis. TEM analysis confirmed these results in E. coli HB101, revealing cell envelope deformation and a discontinuous inner membrane (Fig. 3G). p4 first appeared to concentrate around the cell membrane, as indicated by accumulation of FITC-labeled p4 (FITC-p4) at the bacterial surface (Fig. 3E). However, TEM revealed that p4 does not localize exclusively at the cell membrane. Peptide tracing using biotinylated p4 demonstrated that p4 was present in the cell walls as well as in the periplasm of the bacteria after 10 min of treatment (Fig. 3H). Together, these data indicate that mechanisms of p4 action likely involve membrane and intracellular off-membrane targets and that p4 at concentrations above its MIC triggers rapid bacterial death by compromising membrane integrity.
In contrast to bactericidal concentrations, membrane permeability was not observed when E. coli was treated with p4 at bacteriostatic concentrations (below its MIC). There was no leakage of β-gal in response to p4 ≤ 6.3 μm (Fig. 3F). Likewise, single-cell analysis using fluorescence microscopy revealed that PI did not penetrate E. coli following treatment with 3 μm FITC-p4 despite staining with FITC-p4 (Fig. 4A). This was in contrast to bacteria treated with 10 μm or 100 μm FITC-p4, where PI was able to enter the cells (Figs. 4A and 3E, respectively). These data suggest that p4 below its MIC inhibits bacterial growth without disrupting cell membrane integrity.
Figure 4.

p4 interacts with bacteria as a monomer or a disulfide-bridged dimer. E. coli HB101 was incubated with lethal (10 μm) or sublethal (3 μm) doses of FITC-p4 for 5 min. A, bacteria were analyzed by fluorescence microscopy following staining with PI (red) to visualize bacterial permeability and Hoechst to visualize DNA (blue). B, interaction of bacteria with the indicated forms of FITC-p4 was analyzed by fluorescence microscopy. C, interaction of bacteria with the indicated forms of FITC-p4 was analyzed by SDS-PAGE, followed by gel imaging. The peptides with or without incubation with bacteria were separated under nonreducing conditions. The fluorescence intensity of FITC-p4 was measured with the ChemiDoc imaging system. The data in each panel are from one experiment and are representative of four independent experiments. D, images of gels from four independent experiments described in C were quantified. Individual data points and the mean ± S.D. are shown as percentage of the indicated forms of FITC-p4 associated with bacteria.
The oxidized form of p4 with disulfide linkage is the most efficient in restricting growth of E. coli
Because the p4 formulation contained both monomeric and dimeric (stabilized by disulfide linkage) forms of the peptide (Fig. 2B), it was possible that lethal versus bacteriostatic effects of p4 resulted from differential binding of monomeric versus dimeric p4 forms to bacteria under these conditions. Therefore, we next tested whether p4 interacts with bacteria as a monomer and/or dimer. Fluorescence microscopy confirmed that FITC-p4 (a mixture of monomeric and dimeric p4) stained E. coli either under bacteriostatic (3 μm) or bactericidal (10 μm) conditions (Fig. 4A). To increase the oxidation state of the cysteine residues, we treated FITC-p4 with DMSO and then purified the oxidized p4 (oxp4) and the remaining reduced form of p4 (redp4) by HPLC. However, although oxp4 was relatively stable after purification and reconstitution with PBS, redp4 quickly reached the monomer/dimer ratio that resembled the oxp4/redp4 profile in the original p4 formulation (Fig. 4C). Notably, E. coli was found to bind either oxp4 or redp4 (Fig. 4, B–D). Likewise, IAA-treated FITC-p4 that was unable to form disulfide bonds also associated with bacteria (Fig. 4, B–D), although some preference for binding of oxp4 over p4-IAA was noted. This was particularly apparent when different forms of FITC-p4 were tested for association with bacteria by SDS-PAGE (Fig. 4, C and D). Together, these data suggest that p4 can interact with E. coli either as a monomer or a dimer.
Because p4-IAA was found to interact with but not kill E. coli, we hypothesized that p4-IAA cannot initiate an antimicrobial response in the absence of redox activity and/or because it cannot form a disulfide-stabilized dimer. We reasoned that, under the first scenario, both oxp4 and redp4 should be able to restrict bacterial growth because both are able to alter the redox state of cysteine residues. Under the second scenario, oxp4 should be superior to any other form of p4 in inhibiting bacteria growth. Both scenarios were consistent with a critical role of Cys77 for p4 bactericidal activity. To test this hypothesis, we next compared the ability of p4, oxp4, redp4, and p4-IAA or (VP20)CA to restrict the growth of E. coli and S. aureus. Oxp4 exhibited the strongest antimicrobial activity, followed by p4 and redp4 (Fig. 5, A and B). As expected, p4-IAA or (VP20)CA did not significantly limit bacterial growth (Fig. 5, A and B). These data suggest that bacterial killing is primarily mediated by the dimeric, oxidized form of p4.
Figure 5.

p4 bacteriostatic activity depends on oxidation of cysteine residues of p4. A, E. coli HB101 was incubated with 1.5 μm of the indicated forms of p4 for 2 h. B, S. aureus 8325-4 was incubated with the indicated forms of p4 (6 μm) for 2 h. C, FITC-p4 was incubated without bacteria with NAC or H2O2, followed by analysis by SDS-PAGE. D, E. coli HB101 was incubated with 1.5 μm p4 and/or 1.8 mm NAC for 2 h. E, E. coli HB101 was incubated with 1 μm of the indicated forms of p4 and/or 5 μm H2O2 for 2 h. Cell viability, shown as the percentage of a vehicle-treated cells, was analyzed by MDA assay; n = 3, each experiment done in six replicates. Plots show individual data points and mean ± S.D. ***, p < 0.001; **, p < 0.01; ns, nonsignificant by Kruskal-Wallis one-way ANOVA with post hoc Dunn's test.
To assess the contribution of oxidative conditions to the antimicrobial activity of p4, we next evaluated the effect of bacteriostatic doses of p4 on bacteria in the presence of an antioxidant, N-acetyl-l-cysteine (NAC), or an oxidizing agent, hydrogen peroxide (H2O2). Treatment of FITC-p4 with NAC or H2O2 resulted in predictable alterations of the redox status of cysteine residues in p4, as indicated by SDS-PAGE (Fig. 5C). Under similar conditions, the antimicrobial activity of p4 was repressed by NAC (Fig. 5D). In contrast, H2O2 induced a small but significant increase in p4 antimicrobial activity (Fig. 5E). The H2O2-driven increase in p4-mediated bactericidal activity depended on the formation of new intermolecular disulfide bonds within the heterogenous pool of monomeric and dimeric p4 because the fixed oxidation state p4 isoforms oxp4 and p4-IAA were unaffected by H2O2 (Fig. 5E). Overall, these data indicate that oxidation of p4 cysteine residues is a key factor in p4 antimicrobial activity, although the ability to alter the redox state of cysteine residues may still be important for the regulation of p4 antimicrobial function.
The oxidized form of p4 influences the enzymatic activity of cytochrome bc1 by inhibiting interaction between this complex and its redox partner cytochrome c
The bacteriostatic effects of p4 on E. coli suggest that p4 inhibits the growth of bacteria without affecting membrane permeability. Because the cytoplasmic membrane is likely one of the first targets of p4 (Fig. 3, E and H), we speculated that p4 at bacteriostatic concentrations would limit bacterial growth by interfering with cytoplasmic membrane–associated processes such as electron transport chain function. To explore this hypothesis, we next focused on Rhodobacter capsulatus, a Gram-negative bacterium with a well-defined and functionally testable respiratory chain (19). The central component of this chain is the membrane cytochrome bc1 complex. The complex couples electron transfer to proton pumping that drives ATP synthesis. The bc1 complex transfers electrons from the low-potential substrate ubiquinol to a high-potential cytochrome c (20). R. capsulatus possesses an alternative pathway of ubiquinol oxidation that can operate when bacteria grow under oxygenic growth conditions. This alternative pathway is able to bypass the bc1 complex and thus releases bc1 together with its reaction partner, cytochrome c, from their contribution to produce ATP (21). Therefore, genetic deficiency of cytochrome bc1 is nonlethal, which enables the testing of p4 on bc1-dependent electron transport chain function.
R. capsulatus was highly sensitive to p4 (MIC = 5 μm) but much less to the cysteine-deficient (VP20)CA variant (MIC > 80 μm), suggesting that, similar to E. coli, p4 activity against R. capsulatus depends on C-mediated p4 dimerization (Fig. 6A). The antimicrobial effect of p4 was significantly impaired when bacteria were deficient in the bc1 complex (mutant MT-RBC1 knockout, Fig. 6B). Notably, the lack of cytochrome bc1 had an impact on p4-mediated lethality by increasing the MIC from 5 to 20 μm but even more by augmenting bacterial resistance to sublethal p4 levels (Fig. 6B). These data suggested that p4 may limit the growth of R. capsulatus in a way that is associated with cytochrome bc1 activity. Given the electrostatic properties of p4, interactions between cytochrome bc1 and cytochrome c, are a likely target of p4 interference. We directly tested this interference by analyzing the quinol-dependent reduction of cytochrome c by cytochrome bc1 in vitro under steady-state conditions in the presence of p4, oxp4, redp4, or the (VP20)CA variant or p4-IAA as a control. Oxp4 dramatically lowered cytochrome bc1 activity (resulting in 1% of the total bc1 activity, Fig. 6C). In contrast, (VP20)CA or p4-IAA had much smaller effects because approximately 60% of the total bc1 activity remained following treatment. An intermediate or small effect exerted by p4 or redp4, respectively, compared with the strong inhibitory effect of oxp4, indicates that the oxidized form of p4 is required to efficiently block the cytochrome bc1-catalyzed reduction of cytochrome c. The observation that only the dimeric (oxidized) form of p4 exerted such a strong effect implies that it is a specific tertiary arrangement of the electrostatic charges in the dimer that is the prime contributor in impeding electrostatic interactions between proteins. Just the presence of charges in redp4 is not enough.
Figure 6.

p4 affects bacterial growth in a bc1-dependent manner and inhibits cytochrome bc1 activity to reduce cytochrome c in vitro. A, the R. capsulatus WT strain was incubated with the indicated peptides for 24 h. Data indicate percent of killing. Mean ± S.D. of three independent measurements is shown. B, the indicated R. capsulatus strains were incubated with p4 for 24 h. Data indicate percent of killing for the indicated strain. Mean ± S.D. of three independent measurements for each peptide concentration is shown. ***, p < 0.001; **, p < 0.01; *, p < 0.05 by Kruskal-Wallis one-way ANOVA with post hoc Dunn's test. C, enzymatic activities of isolated cytochrome bc1 complexes in the absence and presence of different forms of p4 at concentrations of 400 nm. Conditions were 50 mm Tris (pH 8.0), 1 mm EDTA, 0.01% n-dodecyl-d-maltoside, 20 μm decylubiquinol, and 20 μm cytochrome c. Error bars represent S.D. of the mean of four measurements.
We also noted that p4 and redp4 appear to be redox-active in the presence of high-potential redox-active cofactors, as either p4 or redp4 were able to reduce heme c1 of cytochrome bc1 or heme c of cytochrome c. We observed that 60 μm p4 fully reduced heme c1 on a minute timescale (at a cytochrome bc1 concentration of 6 μm), whereas reduction of heme c occurred approximately 10 times slower (Fig. 7A). Likewise, 6 μm redp4, but to a much lesser extent oxp4 or (VP20)CA peptide, reduced heme c1 on a minute timescale (Fig. 7B). Reduction of the hemes by p4 suggested that p4 alters the redox state of its cysteine residues and forms dimers in the presence of cytochrome bc1. This was found to be the case, as incubation with increasing concentrations of FITC-p4 (6–20 μm) with 6 μm cytochrome bc1 resulted in p4 dimerization (Fig. 7C). It is thus possible that heme c1 of cytochrome bc1, because of its topographic accessibility to externally added ligands penetrating periplasm of the cells, may be one of the redox-active molecules that facilitates the formation of oxp4. In view of these results, it appears that p4 in its reduced form (with a free thiol group) possesses some antioxidant/reductant properties engaging in redox reactions (such as reduction of hemes exemplified here by reductions of heme c1 of cytochrome bc1 or heme c of cytochrome c) associated with its oxidation upon dimer formation.
Figure 7.

p4 and/or redp4 are able to reduce cytochrome c1 or cytochrome c and form dimers in the presence of cytochrome bc1. A, comparison of the ability of p4 to reduce cytochrome c1 of cytochrome bc1 (black) and cytochrome c (white). The concentrations of cytochromes and p4 were 6 μm and 60 μm, respectively. Cytochrome bc1 was fully oxidized by ferricyanide prior addition of p4. Cytochrome c was almost fully oxidized prior addition of p4. Conditions were 50 mm Tris (pH 8.0), 100 mm NaCl, 1 mm EDTA, and 0.01% n-dodecyl-d-maltoside. Measured data points were fitted to the linear function. All linear coefficients of determination (R2) were above 0.95. B, reduction of cytochrome c1 of cytochrome bc1 in the presence of redp4 (black circles), oxp4 (black rectangles), or the (VP20)CA variant (white diamonds). The concentration of cytochrome bc1 and peptides was 6 μm. Cytochrome bc1 was fully oxidized by ferricyanide prior addition of peptides. C, the indicated concentrations of FITC-p4 were incubated with 6 μm cytochrome bc1 for 10 min as described in A. p4 was then analyzed by SDS-PAGE, followed by gel imaging.
Discussion
Chemerin-derived peptide agonists of CMKLR1 helped to reveal the crucial role of the C-terminal moiety of chemerin in directing CMKLR1-mediated cell homing (22). Chemotactic and antimicrobial activities of chemerin are located in different domains of the protein, suggesting that these two activities can be independent of each other (23). Because the antimicrobial region (p4) is localized in the middle of the chemerin sequence, all chemerin isoforms truncated at the C-terminal end can be predicted to be equipped with some level of antimicrobial activity despite their differing potentials to support migration of CMKLR1-positive cells. Indeed, here we demonstrate that either p4 or other p4-based peptides, including the much smaller, highly conservative fragment (VR15), are similarly active as antimicrobial chemerin derivatives. Nevertheless, other truncated or elongated p4 sister peptides did not display antimicrobial potential, or their activity was much weaker compared with p4. Together, these data argue against an unlimited flexibility of chemerin internal regions in restricting bacterial growth. Although chemerin can shield the epidermis against microbial colonization (15), endogenous p4 has not yet been described. However, given its propensity for proteolytic modification (23), the generation of endogenous antimicrobial p4-based peptides in the skin environment is possible. Alternatively, the “antimicrobial p4 domain” can be potentially exposed with or without proteolytic processing of chemerin protein on chemerin-loaded heptahelical receptors. In addition to the signaling receptor CMKLR1, chemerin is known to bind with similar affinity to two atypical receptors, CCRL2 and GPR1 (24, 25), which are expressed on keratinocytes (14). For example, CCRL2, which lacks the ability to convey chemotactic signals upon chemerin binding, can serve to concentrate chemerin on the cell surface and regulate the bioavailability of this chemotactic factor (25). Although the chemerin amino acid sequence or structural determinants required for CCRL2 or GPR1 binding remain largely uncharacterized, CCRL2 and/or GPR1 might be candidates to deliver/enhance chemerin-based antimicrobial protection.
Exogenous p4 was found to exhibit antimicrobial activity against pathogens such as E. coli and S. aureus, including MRSA. Notably, the efficacy of p4 was validated in an in vivo topical skin infection model, which also demonstrated the selectivity of p4 among other peptides in killing S. aureus. In contrast to bacteria, we did not observe any lytic activity of p4 against human erythrocytes. Likewise, administration of p4 in mouse skin did not result in an obvious cytotoxic effect against frontline keratinocytes. Although some cytotoxicity of p4, used at the highest dose, was noted against human keratinocytes in 2D cultures, this activity was negligible compared with the absolute bactericidal activity of p4 against bacteria treated under similar conditions. Given that microbial infection, especially with MRSA strains, poses an emerging health problem, there is a clear need for alternative therapies. We show here that p4 effectively limits MRSA skin infection and thus represents a novel therapeutic approach to combat antibiotic-resistant infections in the clinic.
These studies also provide important mechanistic insights into the antimicrobial activity of chemerin peptide derivatives. First we demonstrate biochemical features important for the antimicrobial activity of p4 that include its cationicity and amphipathicity. The truncated p4 sister peptides also revealed the critical role of N-terminal amino acid residues but not C-terminal residues in p4 for bacterial killing. When five C-terminal residues were removed, the antimicrobial potential of the peptide was not altered (peptide VR15). In contrast, removal of as few as two amino acid residues from the p4 N terminus (peptide LP18) resulted in abrogation of antimicrobial activity. These data suggest that chemerin antimicrobial activity can be narrowed down to an N-terminal fragment of p4, represented by the 15-amino acid-long peptide VR15, whereas the C-terminal domain is dispensable for this function, although it might play other, uncharacterized roles.
Second, our experimental findings indicated that Cys77 in chemerin enabled peptide homodimerization via intermolecular disulfide bridging, which was required for maximal p4 antimicrobial activity. The dependence of p4 activity on a cysteine also suggested a possible redox-regulated mechanism underlying its antimicrobial effects. Because oxidative conditions render bacteria highly susceptible to p4-mediated growth suppression, p4/chemerin is likely most effective in an oxidized environment. For example, high/sufficient oxygen levels at the skin surface, or ROS present at infection sites, can dictate the niche-specific impact on p4- or chemerin-dependent antimicrobial activity. This is supported by our data that show active p4 within the skin environment.
Third, p4 interacted with bacteria as a monomer or dimer but exerted lethality against bacteria primarily in the oxidized (dimer) form. We also showed that p4 rapidly (within minutes of exposure) compromised bacterial viability, which, in cases of lethal doses of p4, led to morphological damage of bacterial cells and breakdown of cell membranes. The rapidity of p4 bactericidal activity suggests that the ability of pathogens to generate resistance to high doses of p4 might be limited. In contrast, the bacteriostatic effect of p4 was not accompanied by permeabilization of cell membranes, indicating that bacterial killing by p4 requires severe membrane distortion.
Finally, p4 at either lethal or sublethal doses targets components of the electron transport chain, such as the bc1 complex in R. capsulatus. p4 strongly inhibited interaction between this complex and its redox partner, cytochrome c. Although bc1 is the most widely occurring electron transfer complex in a variety of respiring and photosynthetic bacteria, the bc1 complex is dispensable for E. coli metabolism (26). However, p4 inhibits growth of E. coli at a similar rate as R. capsulatus, suggesting that the bc1 complex is not the only target of p4. Because the lack of bc1 in R. capsulatus conferred a survival benefit during p4 treatment, it is likely that bc1 contributes to the antimicrobial function of p4; for example, by facilitating formation of p4 dimers. This is supported by our data showing that p4 or redp4 were able to reduce cytochrome c1 of cytochrome bc1, thus becoming oxidized and strongly antimicrobial as a result. We suggest that other high-potential redox-active cofactors of similar topographic accessibility, like heme c of the cytochrome bc1 complex act in a similar way in other bacteria. In view of these observations, we propose that p4 exerts dual effects on bacterial targets. On one hand, dimers of p4 efficiently interfere with electrostatically mediated protein–protein interactions, which can lead to inhibition of physiologic processes, such as electron transfer between cytochrome bc1 and cytochrome c. If such processes were at key and difficult-to-bypass points of physiological paths, this would have a profoundly negative impact on overall cell metabolism. On the other hand, p4 can also engage directly in redox reactions and thus affect the redox status of redox-active compounds. Furthermore, if this reaction favors oxidation of p4 (as demonstrated here by redp4-mediated reduction of hemes), then this would act to increase local working concentrations of p4 dimers, thus amplifying its deleterious effects. All this may again be expected to negatively impact bacterial function, resulting in inhibition of bacterial growth or cell death if the sufficient concentration of p4 dimers is reached to lead to irreversible cell membrane damage. Overall, our findings reveal novel mechanistic insights into the antimicrobial nature of chemerin-derived p4 and opens up new avenues to further exploit chemerin activities in the context of immune defense in the skin.
Experimental procedures
Bacterial strains
The bacterial strains used were E. coli HB101, a conventional laboratory strain; WT S. aureus strain 8325-4 (9); and MRSA strains ATCC BAA-1707 and clinical isolate E240. The MRSA strains were kindly donated by Dr. A. Sabat (University of Groningen, Groningen, The Netherlands). We also used the R. capsulatus pMTS1/MTRbc1 strain with a deletion of the operon coding for cytochrome bc1 and overproducing WT cytochrome bc1 (WT) and the MT-RBC1 knockout strain with a deletion of the operon coding for cytochrome bc1 (19).
Peptides
The chemerin-derived peptides p4 and p2 or p4 sister peptides were chemically synthesized by ChinaPeptide (Shanghai, China) at ≥95% purity. Biotin- or FITC-labeled p4 and peptide d-VR15 comprised only of d-amino acid residues were synthesized by Caslo (Kongens Lyngby, Denmark) at ≥95% or ≥98% purity. Biotin was added directly at the N terminus of p4. For FITC-labeled p4, a C-terminal lysine was added to p4, and FITC was conjugated to the side chain of this C-terminal lysine. Both biotin-labeled and FITC-labeled p4 displayed similar antimicrobial activity as unmodified p4.
Antimicrobial assays
E. coli or S. aureus were grown in brain heart infusion (BHI) broth at 37 °C whereas R. capsulatus was grown protected from light in mineral-peptone-yeast extract at 30 °C. For the microdilution assay (MDA), E. coli in mid-logarithmic phase was harvested and diluted to 4 × 105 cfu/ml with Dulbecco's PBS. Bacteria were incubated with the indicated peptides for 2 h. The number of viable bacteria were enumerated by colony-forming unit counting. For minimal inhibitory concentration (MIC) determination, bacteria were diluted to 4 × 106 cfu/ml with PBS containing 1% (v/v) of medium mixed with a series of 2-fold dilution of p4 or PBS (control) and incubated at 37 °C overnight. The radial diffusion assay was performed as described previously (27) with minor modifications. The underlay agarose gel (15 ml) containing 4 × 106 bacterial cfu (E. coli), 0.03% (w/v) BHI broth, 1% (w/v) low electroendosmosis-type agarose (Lonza), and 0.02% (v/v) Tween 20 (Sigma) diluted in PBS was poured into a Petri dish. After gel solidification, 4-mm-diameter wells were punched, and 8 μl of the peptides was added. After 3 h of incubation, the underlay gel was covered with 15 ml of overlay gel (6% BHI broth and 1% (w/v) agarose). The zone of growth inhibition around each well was measured after 18–24 h of incubation.
Topical skin infection
C57BL6 mice (female, 7–10 weeks old) were housed under pathogen-free conditions in the animal facility at the Faculty of Biochemistry, Biophysics, and Biotechnology of Jagiellonian University. All animal studies were approved by and in compliance with the guidelines of the Second Local Ethical Committee on Animal Testing at the Institute of Pharmacology Polish Academy of Sciences in Krakow. A small dorsal area of the skin was shaved, sterilized with ethanol, and punctured six times at two places using a syringe needle (BD Micro-Fine Plus, 0.3 × 8 mm). Two rubber 8-mm inner diameter rings were subsequently attached using an ethylcyanoacrylate-based adhesive, and peptides or vehicle (sterile water) were topically administered in mouse skin. The peptides were allowed to dry on the skin, and the rings were covered with OpSite (Smith & Nephew). 1 × 107 cfu of S. aureus in a volume of 50 μl (PBS) was thereafter injected through the OpSite into the cavity formed by the rubber rings. The ring injected with sterile PBS was used as a control. After 24 h, bacterial loads were analyzed by enumeration of colony-forming units. The skin within the side of the rings was retrieved, frozen, and fixed in methanol for 1 min, followed by Gram staining (Fluka). Although the data presented are from female animals, similar results were obtained in pilot studies when male mice were used.
Human studies
All human studies were performed in compliance with ethical protocols approved by the Jagiellonian University Institutional Bioethics Committee. The studies abided by the Declaration of Helsinki principles. All participants provided written informed consent to participate in these studies as recommended by the ethical board. Normal human keratinocytes were isolated from the skin of healthy donors as described previously (14).
Cysteine alkylation and oxidation of p4
Formation of disulfide bonds in p4 was blocked by cysteine alkylation. p4 or FITC-p4 (0.3 mm) was incubated in 50 mm Tris-HCl buffer (pH 8) supplemented with 5 mm DTT at 60 °C for 1 h, followed by addition of iodoacetamide (IAA) to a final concentration of 10 mm. Samples were then incubated for 20 min at room temperature and analyzed by SDS-PAGE. Alternatively, an excess of DTT was added to the p4 samples, followed by purification using HPLC and antimicrobial assays. For p4 oxidation, 0.4 mm peptide was incubated in 20% DMSO dissolved in 25 mm ammonium carbonate (pH 8.0) at room temperature for 20 h. After incubation, the samples were diluted, mixed with HCl to a final concentration of 0.17 m, and analyzed or purified by HPLC.
HPLC analysis and peptides identification by MS
p4, IAA-treated p4, and DMSO-treated p4 (either unlabeled or labeled with FITC) were analyzed by HPLC for reduced and oxidized forms of p4 using a Dionex Ultimate 3000 system (Thermo Scientific). The peptides were separated on a Eurosil Bioselect 300-5 C-18 column (5 μm, 4 mm × 250 mm, Knauer) in a two-solvent system (solvent A, 0.1% TFA in water; solvent B, 0.08% TFA in 80% acetonitrile; Merck) in a gradient of 10–50% solvent B over 20 min at a flow rate of 1 ml/min and with spectrophotometric detection at 215 nm. Fractions were collected, evaporated to dryness, resuspended in 30% methanol with 0.1% formic acid, and analyzed using an HCTultra ETDII mass spectrometer (Bruker). Samples were injected directly with a syringe pump (KD Scientific) at a flow rate of 180 μl/h to an electrospray ionization ion source operated in positive ion mode at a capillary voltage of 3.5 kV, nebulizer pressure of 10 p.s.i., drying gas flow of 5 liters/min, and ion source temperature of 300 °C. The ion trap analyzer of the spectrometer was set at both MS and tandem MS mode. The peptide identification was performed using DataAnalysisTM 4.0 software and BiotoolsTM 3.2 software (Bruker).
SDS-PAGE
Samples were resolved by a gradient 6/16% SDS-PAGE gel based on a method described by Schägger and von Jagow (28). Bands were detected by Coomassie Brilliant Blue staining or FITC fluorescence detection (Bio-Rad Chemidoc MP Imager). For Coomassie Blue–stained gels and FITC detection, 5 μg or 140 ng of peptide was loaded per lane, respectively.
Fluorescence microscopy
E. coli HB101 bacteria (1 × 108 cfu) were incubated with FITC-p4 and the membrane-impermeable dye PI (Thermo Fisher) in PBS for 5 min. Cells were washed three times with PBS to remove the peptide, attached to slides by cytospin centrifugation, fixed in 3.7% paraformaldehyde (Sigma-Aldrich), and counterstained with Hoechst 33258 dye (Life Technologies). Images were captured with a fluorescence microscope (Eclipse, Nikon) and analyzed using NIS-Elements (Nikon) software.
TEM
E. coli or S. aureus (5 × 108) were treated with p4, scp4, or vehicle (PBS) for up to 2 h at 37 °C. E. coli cell pellets were fixed in 2% glutaraldehyde in 0.1 m sodium cacodylate buffer (pH 7.4) overnight at 4 °C, whereas S. aureus pellets were washed three times in PBS for 5 min and fixed overnight in 2.5% glutaraldehyde in PBS at 4 °C. E. coli was then post-fixed in 1% osmium tetroxide in 0.1 m cacodylic buffer for 1 h at room temperature and stained en bloc with 2% uranyl acetate aqueous solution for 1 h at room temperature. S. aureus was post-fixed with 1% osmium tetroxide for 2 h at 4 °C. Samples were embedded in epoxy resin (PolyBed 812, Polysciences, Inc.) after dehydration in a graded ethanol series (50–100%) and in propylene oxide. Ultrathin sections (65 nm) were cut using an ultramicrotome (Leica EM UC7) and post-stained with uranyl acetate and lead citrate. Specimens were observed using a transmission electron microscope (JEOL JEM2100) operating at an accelerating voltage of 80 kV.
Immunogold labeling
Ultrathin sections of E. coli on nickel grids were incubated with 4% sodium metaperiodate for 10 min, followed by 1% aqueous periodic acid for 10 min and 1% fish skin gelatin in PBS for 2 h. Sections were incubated with primary mouse anti-biotin A Ab (clone 3D6.6) in 1% fish skin gelatin overnight at 4 °C, followed by secondary antibodies (12 nm colloidal gold-donkey anti-mouse Abs, both from Jackson ImmunoResearch Laboratories) for 2 h at room temperature. Sections were fixed in 1% glutaraldehyde for 5 min, stained with uranyl acetate, and examined using TEM.
Cell lysis
Hemolytic activity was measured by incubating human erythrocytes (2.8% v/v) with p4 in PBS for 2 h at 37 °C. Samples were centrifuged, and absorbance of the supernatants was measured at 540 nm. For the β-gal leakage assay, the β-gal reporter E. coli JM83 strain was incubated with p4 in PBS for 15 min. β-Gal activity in the supernatants was evaluated using O-nitrophenyl-β-d-galactopyranoside (ONPG, Thermo Fisher) as the substrate.
Enzymatic activity measurements of cytochrome bc1
Th activity of R. capsulatus cytochrome bc1 isolated complexes was determined spectroscopically by measuring the quinol-dependent reduction of cytochrome c under steady-state conditions at 550 nm as described previously (29). Assays were conducted in 50 mm Tris (pH 8.0), containing 1 mm EDTA and 0.01% n-dodecyl-d-maltoside. The final concentrations of substrates were 20 μm decylubiquinol (a reduced form of 2,3-dimethoxy-5-methyl-6-decyl-1,4-benzoquinone from Sigma-Aldrich) and 20 μm cytochrome c (bovine heart cytochrome c, Sigma-Aldrich). The final concentration of cytochrome bc1 was 7 nm and that of the peptides 0.4 to 4 μm. The enzymatic reaction was started by injection of cytochrome bc1 preincubated with peptides for 30–60 s to the reaction mixture containing the substrates. The background of nonenzymatic reduction of cytochrome c was monitored for 20 s after injection of DBH2 and prior injection of enzyme. Turnover rates were calculated from the initial linear parts of the time-dependent reduction of cytochrome c curves, and the background rate was subtracted.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software) and Statistica 13.1 (Statsoft, Dell Software). For multiple comparison statistics, one-way ANOVA with post hoc Tukey's test was used. To compare the control with each of the other samples, one-way ANOVA with post hoc Dunnett's test was used. Levene, Brown-Forsythe, and Shapiro's tests were used to verify whether ANOVA assumptions were met. If one or more assumptions of a parametric test was violated, then Kruskal-Wallis one-way ANOVA with post hoc Dunn's test was used instead.
Author contributions
U. G., B. B., A. Z., P. B., A. B., K. M., O. B., and A. M. formal analysis; U. G., B. B., A. Z., P. B., A. B., O. B., A. M., and P. K. investigation; U. G., B. B., A. Z., P. B., A. B., K. M., O. B., A. M., P. K., A. K., E. P., A. O., and B. A. Z. methodology; K. M. software; A. K., E. P., A. O., B. A. Z., and J. C. conceptualization; A. K., E. P., A. O., and J. C. supervision; B. A. Z. writing-review and editing; J. C. resources; J. C. funding acquisition; J. C. validation; J. C. visualization; J. C. writing-original draft.
Supplementary Material
Acknowledgments
The Faculty of Biochemistry, Biophysics, and Biotechnology of the Jagiellonian University is a partner of the Leading National Research Center (KNOW) supported by the Polish Ministry of Science and Higher Education.
This work was supported by Polish National Science Center Grant UMO 2014/12/W/NZ6/00454 (to J. C., E. P., and B. A. Z.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1 and S2 and supporting text.
- MRSA
- methicillin-resistant Staphylococcus aureus
- MDA
- microdilution assay
- MIC
- minimal inhibitory concentration
- IAA
- iodoacetamide
- PI
- propidium iodide
- ONPG
- O-nitrophenyl-β-d-galactopyranoside
- NAC
- N-acetyl-l-cysteine
- Ab
- antibody
- ANOVA
- analysis of variance
- TEM
- transmission electron microscopy.
References
- 1. Vermi W., Riboldi E., Wittamer V., Gentili F., Luini W., Marrelli S., Vecchi A., Franssen J. D., Communi D., Massardi L., Sironi M., Mantovani A., Parmentier M., Facchetti F., and Sozzani S. (2005) Role of ChemR23 in directing the migration of myeloid and plasmacytoid dendritic cells to lymphoid organs and inflamed skin. J. Exp. Med. 201, 509–515 10.1084/jem.20041310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wittamer V., Franssen J. D., Vulcano M., Mirjolet J. F., Le Poul E., Migeotte I., Brézillon S., Tyldesley R., Blanpain C., Detheux M., Mantovani A., Sozzani S., Vassart G., Parmentier M., and Communi D. (2003) Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 198, 977–985 10.1084/jem.20030382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zabel B. A., Silverio A. M., and Butcher E. C. (2005) Chemokine-like receptor 1 expression and chemerin-directed chemotaxis distinguish plasmacytoid from myeloid dendritic cells in human blood. J. Immunol. 174, 244–251 10.4049/jimmunol.174.1.244 [DOI] [PubMed] [Google Scholar]
- 4. Goralski K. B., McCarthy T. C., Hanniman E. A., Zabel B. A., Butcher E. C., Parlee S. D., Muruganandan S., and Sinal C. J. (2007) Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J. Biol. Chem. 282, 28175–28188 10.1074/jbc.M700793200 [DOI] [PubMed] [Google Scholar]
- 5. Takahashi M., Okimura Y., Iguchi G., Nishizawa H., Yamamoto M., Suda K., Kitazawa R., Fujimoto W., Takahashi K., Zolotaryov F. N., Hong K. S., Kiyonari H., Abe T., Kaji H., Kitazawa S., et al. (2011) Chemerin regulates β-cell function in mice. Sci. Rep. 1, 123 10.1038/srep00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bozaoglu K., Curran J. E., Stocker C. J., Zaibi M. S., Segal D., Konstantopoulos N., Morrison S., Carless M., Dyer T. D., Cole S. A., Goring H. H., Moses E. K., Walder K., Cawthorne M. A., Blangero J., and Jowett J. B. (2010) Chemerin, a novel adipokine in the regulation of angiogenesis. J. Clin. Endocrinol. Metab. 95, 2476–2485 10.1210/jc.2010-0042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Watts S. W., Dorrance A. M., Penfold M. E., Rourke J. L., Sinal C. J., Seitz B., Sullivan T. J., Charvat T. T., Thompson J. M., Burnett R., and Fink G. D. (2013) Chemerin connects fat to arterial contraction. Arterioscler. Thromb. Vasc. Biol. 33, 1320–1328 10.1161/ATVBAHA.113.301476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kulig P., Kantyka T., Zabel B. A., Banas M., Chyra A., Stefanska A., Tu H., Allen S. J., Handel T. M., Kozik A., Potempa J., Butcher E. C., and Cichy J. (2011) Regulation of chemerin chemoattractant and antibacterial activity by human cysteine cathepsins. J. Immunol. 187, 1403–1410 10.4049/jimmunol.1002352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kulig P., Zabel B. A., Dubin G., Allen S. J., Ohyama T., Potempa J., Handel T. M., Butcher E. C., and Cichy J. (2007) Staphylococcus aureus-derived staphopain B, a potent cysteine protease activator of plasma chemerin. J. Immunol. 178, 3713–3720 10.4049/jimmunol.178.6.3713 [DOI] [PubMed] [Google Scholar]
- 10. Zabel B. A., Allen S. J., Kulig P., Allen J. A., Cichy J., Handel T. M., and Butcher E. C. (2005) Chemerin activation by serine proteases of the coagulation, fibrinolytic, and inflammatory cascades. J. Biol. Chem. 280, 34661–34666 10.1074/jbc.M504868200 [DOI] [PubMed] [Google Scholar]
- 11. Albanesi C., Scarponi C., Pallotta S., Daniele R., Bosisio D., Madonna S., Fortugno P., Gonzalvo-Feo S., Franssen J. D., Parmentier M., De Pità O., Girolomoni G., and Sozzani S. (2009) Chemerin expression marks early psoriatic skin lesions and correlates with plasmacytoid dendritic cell recruitment. J. Exp. Med. 206, 249–258 10.1084/jem.20080129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luangsay S., Wittamer V., Bondue B., De Henau O., Rouger L., Brait M., Franssen J. D., de Nadai P., Huaux F., and Parmentier M. (2009) Mouse ChemR23 is expressed in dendritic cell subsets and macrophages, and mediates an anti-inflammatory activity of chemerin in a lung disease model. J. Immunol. 183, 6489–6499 10.4049/jimmunol.0901037 [DOI] [PubMed] [Google Scholar]
- 13. Nagpal S., Patel S., Jacobe H., DiSepio D., Ghosn C., Malhotra M., Teng M., Duvic M., and Chandraratna R. A. (1997) Tazarotene-induced gene 2 (TIG2), a novel retinoid-responsive gene in skin. J. Invest. Dermatol. 109, 91–95 10.1111/1523-1747.ep12276660 [DOI] [PubMed] [Google Scholar]
- 14. Banas M., Zegar A., Kwitniewski M., Zabieglo K., Marczynska J., Kapinska-Mrowiecka M., LaJevic M., Zabel B. A., and Cichy J. (2015) The expression and regulation of chemerin in the epidermis. PLoS ONE 10, e0117830 10.1371/journal.pone.0117830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Banas M., Zabieglo K., Kasetty G., Kapinska-Mrowiecka M., Borowczyk J., Drukala J., Murzyn K., Zabel B. A., Butcher E. C., Schroeder J. M., Schmidtchen A., and Cichy J. (2013) Chemerin is an antimicrobial agent in human epidermis. PLoS ONE 8, e58709 10.1371/journal.pone.0058709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Godlewska U., Brzoza P., Sroka A., Majewski P., Jentsch H., Eckert M., Eick S., Potempa J., Zabel B. A., and Cichy J. (2017) Antimicrobial and attractant roles for chemerin in the oral cavity during inflammatory gum disease. Front. Immunol. 8, 353 10.3389/fimmu.2017.00353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- 18. Oliva B., Gordon G., McNicholas P., Ellestad G., and Chopra I. (1992) Evidence that tetracycline analogs whose primary target is not the bacterial ribosome cause lysis of Escherichia coli. Antimicrob. Agents Chemother. 36, 913–919 10.1128/AAC.36.5.913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Atta-Asafo-Adjei E., and Daldal F. (1991) Size of the amino acid side chain at position 158 of cytochrome b is critical for an active cytochrome bc1 complex and for photosynthetic growth of Rhodobacter capsulatus. Proc. Natl. Acad. Sci. U.S.A. 88, 492–496 10.1073/pnas.88.2.492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sarewicz M., and Osyczka A. (2015) Electronic connection between the quinone and cytochrome c redox pools and its role in regulation of mitochondrial electron transport and redox signaling. Physiol. Rev. 95, 219–243 10.1152/physrev.00006.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trumpower B. L. (1990) Cytochrome bc1 complexes of microorganisms. Microbiol. Rev. 54, 101–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wittamer V., Grégoire F., Robberecht P., Vassart G., Communi D., and Parmentier M. (2004) The C-terminal nonapeptide of mature chemerin activates the chemerin receptor with low nanomolar potency. J. Biol. Chem. 279, 9956–9962 10.1074/jbc.M313016200 [DOI] [PubMed] [Google Scholar]
- 23. Zabel B. A., Kwitniewski M., Banas M., Zabieglo K., Murzyn K., and Cichy J. (2014) Chemerin regulation and role in host defense. Am. J. Clin. Exp. Immunol 3, 1–19 [PMC free article] [PubMed] [Google Scholar]
- 24. Barnea G., Strapps W., Herrada G., Berman Y., Ong J., Kloss B., Axel R., and Lee K. J. (2008) The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. U.S.A. 105, 64–69 10.1073/pnas.0710487105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zabel B. A., Nakae S., Zúñiga L., Kim J. Y., Ohyama T., Alt C., Pan J., Suto H., Soler D., Allen S. J., Handel T. M., Song C. H., Galli S. J., and Butcher E. C. (2008) Mast cell-expressed orphan receptor CCRL2 binds chemerin and is required for optimal induction of IgE-mediated passive cutaneous anaphylaxis. J. Exp. Med. 205, 2207–2220 10.1084/jem.20080300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kracke F., Vassilev I., and Krömer J. O. (2015) Microbial electron transport and energy conservation: the foundation for optimizing bioelectrochemical systems. Front. Microbiol. 6, 575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lehrer R. I., Rosenman M., Harwig S. S., Jackson R., and Eisenhauer P. (1991) Ultrasensitive assays for endogenous antimicrobial polypeptides. J. Immunol. Methods 137, 167–173 10.1016/0022-1759(91)90021-7 [DOI] [PubMed] [Google Scholar]
- 28. Schägger H., and von Jagow G. (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166, 368–379 10.1016/0003-2697(87)90587-2 [DOI] [PubMed] [Google Scholar]
- 29. Sarewicz M., Borek A., Cieluch E., Swierczek M., and Osyczka A. (2010) Discrimination between two possible reaction sequences that create potential risk of generation of deleterious radicals by cytochrome bc1: implications for the mechanism of superoxide production. Biochim. Biophys. Acta 1797, 1820–1827 10.1016/j.bbabio.2010.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.