

Abstract
Serine-arginine (SR) proteins are essential splicing factors containing a canonical RNA recognition motif (RRM), sometimes followed by a pseudo-RRM, and a C-terminal arginine/serine-rich (RS) domain that undergoes multisite phosphorylation. Phosphorylation regulates the localization and activity of SR proteins, and thus may provide insight into their differential biological roles. The phosphorylation mechanism of the prototypic SRSF1 by serine-arginine protein kinase 1 (SRPK1) has been well-studied, but little is known about the phosphorylation of other SR protein members. In the present study, interaction and kinetic assays unveiled how SRSF1 and the single RRM–containing SRSF3 are phosphorylated by SRPK2, another member of the SRPK family. We showed that a conserved SRPK-specific substrate-docking groove in SRPK2 impacts the binding and phosphorylation of both SR proteins, and the localization of SRSF3. We identified a nonconserved residue within the groove that affects the kinase processivity. We demonstrated that, in contrast to SRSF1, for which SRPK-mediated phosphorylation is confined to the N-terminal region of the RS domain, SRSF3 phosphorylation sites are spread throughout its entire RS domain in vitro. Despite this, SRSF3 appears to be hypophosphorylated in cells at steady state. Our results suggest that the absence of a pseudo-RRM renders the single RRM–containing SRSF3 more susceptible to dephosphorylation by phosphatase. These findings suggest that the single RRM– and two RRM–containing SR proteins represent two subclasses of phosphoproteins in which phosphorylation statuses are maintained by unique mechanisms, and pose new directions to explore the distinct roles of SR proteins in vivo.
Keywords: phosphoryl transfer, protein kinase, protein phosphorylation, serine/threonine protein kinase, RNA splicing, post-translational modification (PTM), localization, speckles, SR protein, SRPK, SRSF, processive phosphorylation, kinase-substrate interaction, post-translational mechanism, regulatory mechanism, nuclear speckle, subcellular localization
Introduction
Serine-arginine (SR)4 proteins are a family of functionally and structurally related splicing factors that are essential for both constitutive and alternative splicing (1). They are so named because of the presence of arginine-serine (RS) dipeptide stretches at their C-terminal regions. SR proteins also contain at least one N-terminal RNA recognition motif (RRM) (e.g. SRSF2 and SRSF3), sometimes followed by a more degenerated pseudo-RRM (e.g. SRSF1 and SRSF6) (2). These essential splicing factors can bind RNA directly, and their arginine/serine-rich (RS) and RRM domains act to promote protein–protein and protein–RNA interactions, facilitating the recruitment and assembly of the spliceosome (3). After the splicing of pre-mRNA, SR proteins continue to participate in various downstream events, including nonsense-mediated RNA decay, mRNA export, and translation (4–6). In addition to their important roles in mRNA processing, SR proteins serve as a link between transcription and splicing (7).
SR proteins localize in the nucleus at the steady state, but some members, such as SRSF1, SRSF3, and SRSF7, can shuttle between the nucleus and cytoplasm (8). Phosphorylation of the RS domain is important for the nuclear import of these proteins via interaction with SR-specific transportin proteins (9, 10). Further phosphorylation facilitates the recruitment of SR proteins from the nuclear speckles to nascent RNA during splicing (11). Although phosphorylation of SR proteins is required for spliceosome assembly, partial dephosphorylation of SR proteins is also critical for splicing catalysis, suggesting that SR proteins modulate splicing by undergoing dynamic cycles of phosphorylation and dephosphorylation (12, 13). The precise biochemical mechanism of how differential phosphorylation of the prototypic SR protein SRSF1 promotes early splicing assembly has been revealed. Cho et al. (14) showed that SR protein kinases (SRPK)-mediated phosphorylation of the N-terminal region of the SRSF1 RS domain enhances its binding to specific splicing enhancers on target RNA. Further phosphorylation of the C-terminal RS domain exposes the RRMs on SRSF1 for the binding of U1snRNP-70K protein, facilitating the formation of the ternary complex required for the recruitment of U1snRNP to the 5′ splice site (14). Besides splicing, hypophosphorylated, but not hyperphosphorylated, SR proteins regulate the export of mature mRNA and translation, which further underscores the significance of the differential phosphorylation of SR proteins in RNA metabolism (15, 16).
The RS domain of SR proteins is predominantly phosphorylated by two families of protein kinases, namely the SRPKs and Cdc2-like kinases (CLKs), although other kinases, such as Akt and DNA topoisomerase, have been implicated in this process (17). SRPKs are constitutively active kinases located in both the cytoplasm and nucleus. They are unusual members of the protein kinase superfamily in that a spacer insert of various lengths bifurcates their kinase domains. Although the spacer is not required for in vitro kinase activity, it plays a critical role in the subcellular localization of SRPKs (18). In the past decade, significant progress has been made in understanding the phosphorylation mechanism of SRSF1 by SRPK1; however, few mechanistic insights into the interaction and phosphorylation of different SR proteins by other SRPKs have been achieved (19). Previously, we investigated the roles of the nonkinase regions and a conserved docking groove of SRPK2, which is highly related to SRPK1, in the recognition and phosphorylation of two different classes of substrate, namely SR protein SRSF1 and SR-like protein acinusS (20). Our findings suggest that the electronegative docking groove in SRPK2, but not its nonkinase regions, is responsible for substrate binding regardless of their identities. Whereas SRPK2 phosphorylates SRSF1, a prototypic SR protein, in a processive manner similar to SRPK1, an electronegative region N-terminal to the phosphorylation site of acinusS restricts SRPK2-mediated phosphorylation to a single specific site despite the presence of multiple RS dipeptides. These results suggest that the phosphorylation mechanisms of different substrates are vastly different despite SRPK2 utilizing the same docking groove for regulation. Moreover, site-specific phosphorylation of RS dipeptides appears to critically regulate the functions of the SR substrates.
All pseudo-RRMs of SR proteins contain a phylogenetically conserved SWQDLKD motif that has been implicated in the interaction with RNA (21). The crystal structure of SRPK1 in complex with SRSF1 reveals that the SWQDLKD motif also directly interacts with the small lobe of the kinase (22). Furthermore, the pseudo-RRM of SRSF1 directly contacts the N-terminal region of the RS domain to regulate the directionality of phosphorylation (23). These findings imply that the subclass of SR proteins containing two RRMs may function and be regulated in a very different manner from their single RRM–containing counterparts. In fact, Cáceres et al. (24) has demonstrated that the RS domain is essential for the proper localization of single RRM–containing SR proteins like SRSF2 and SRSF3, but not the two RRM–containing proteins like SRSF1, to the nuclear speckles, further stressing the importance of studying the RS domain function in the single RRM–containing subclass (24).
SRSF3 contains a single canonical RRM and plays critical roles in many cellular processes. It interacts with RNA cis-elements in a concentration-dependent manner and regulates the alternative splicing of a number of genes, including its own (25–30). In addition to splicing, SRSF3 has also been implicated in pre-mRNA processing, mRNA export, chromatin binding, protein translation, insulin signaling, and a cohort of human diseases (31–36). Although the phosphorylation status of SRSF3 plays an important role in its function in these processes, few efforts have been made to understand its phosphorylation mechanism (25, 31, 34, 37). In this study, we investigated how the docking grooves of SRPK1 and SRPK2 determine the processivity of their phosphorylation mechanisms. We also studied how SRPK2 binds and regulates the phosphorylation of SRSF1 and SRSF3. Whereas SRSF3 could be extensively phosphorylated by SRPK2 in vitro, it remains hypophosphorylated in cells, suggesting that its phosphorylation status at steady state may be maintained by a coordination of SRPKs and cellular phosphatases inside the cells. Furthermore, we showed the docking groove of SRPK2 is important for the subnuclear localization of SRSF3, suggesting that SRPK2-mediated interaction/phosphorylation of SRSF3 might play a regulatory role in its function inside the nucleus.
Results
SRSF1 phosphorylation by SRPK2 occurs in a less processive manner
One hallmark of SRPK1 is its ability to phosphorylate the substrate SRSF1 at multiple sites using a highly efficient and sequential mechanism (22). This reaction happens in a regiospecific manner such that, despite SRSF1 containing ∼20 potential sites for phosphorylation, only the N-terminal RS domain (residues 198–226) that contains 14 phosphorylatable serines is phosphorylated (Fig. 1A) (38). To test whether SRPK2 also phosphorylates SRSF1 at the N-terminal half of the RS domain, we deleted residues 227–248 of SRSF1 and compared its phosphorylation content with that of the full-length substrate. Scintillation counting shows that the deletion did not affect the number of phosphorylation sites (14.4 ± 0.3 sites for SRSF1 and 14.1 ± 0.4 sites for SRSF1ΔRS2; Fig. 1B), suggesting that SRPK2, like SRPK1, preferentially phosphorylates SRSF1 at its N-terminal RS domain. We have previously demonstrated that SRPK2 is able to phosphorylate SRSF1 in a processive manner (20). To examine whether the phosphorylation mechanism of SRPK2 is similar to that of SRPK1, we performed start-trap assays as described previously to compare the reactions carried out by the two kinases (39). In brief, if the reaction is distributive, the excess trap (kinase-dead mutant of the respective SRPK) traps the substrate and inhibits the reaction because the WT kinase dissociates from the substrate after each phosphorylation event. Alternatively, if phosphorylation occurs in a processive manner, the kinase–substrate pair will not dissociate before completion of the phosphorylation event, and the presence of excess trap therefore does not affect the reaction. In the absence of the trap, both kinases phosphorylated SRSF1 at ∼12 sites. However, when traps were added, SRSF1 was phosphorylated at a lower number of sites by SRPK2 (5.2 ± 0.4 sites) when compared with SRPK1 (7.5 ± 0.5 sites) (Fig. 1, C and D).
Figure 1.

SRPK2 phosphorylates SRSF1 in a less processive manner. A, domain organization of SRSF1WT and SRSF1ΔRS2 (aa 1–226) are shown. pRRM, pseudo-RRM. B, phosphorylation of GST-tagged SRSF1WT (1 μm) and SRSF1ΔRS2 (1 μm) by SRPK2 (1.5 μm) in the presence of 100 μm [32P]ATP for 15 min was visualized by autoradiography (top left panel). SDS-PAGE analysis of the SRSF1 proteins used for the kinase assay (bottom left panel). The number of phosphorylation sites was quantitated by scintillation counting. SRSF1 contained 14.4 ± 0.3 phosphorylated sites, whereas SRSF1ΔRS2 contained 14.1 ± 0.4 sites (right panel). Phosphorylation of GST-tagged SRSF1 by SRPK2 (C) and SRPK1 (D) is shown under different trapping conditions. No trap (positive control), trap-start (negative control), and start-trap reactions are indicated in each case. In the start-trap experiment, SRPK1 or SRPK2 (1 μm) was pre-equilibrated with 300 nm SRSF1 and then allowed to react with 100 μm [32P]ATP in the absence (no trap) and the presence (start-trap) of 40 μm SRPK1KD or SRPK2KD (the corresponding kinase-dead mutant), respectively, added at the reaction start time. The kinase-dead mutant was added prior to the reaction start time in the negative control experiment (trap-start). SRSF1 was processively phosphorylated by SRPK2 to 5.2 ± 0.4 sites and SRPK1 to 7.5 ± 0.5 sites. Error bars, S.D. from three independent experiments.
His-601 at SRPK2 docking groove lowers its processivity
To understand the molecular basis of the lower processivity observed in SRPK2, we aligned and compared the amino acid sequences of the docking grooves of SRPK2 and SRPK1 (Fig. 2A). Although the residues within the docking groove are highly conserved between the two kinases, Leu-568 of SRPK1, which is located at the center of the αG helix inside the docking groove, is replaced by His-601 in SRPK2 (Fig. 2B). Histidine contains an imidazole group with a pKa of 6.04 and could be protonated at pH <6 or when it is surrounded by proton donors (i.e. acidic side chains in the docking groove). Thus, we hypothesized that His-601 of SRPK2, which replaces a hydrophobic leucine residue at the corresponding position in SRPK1, may alter the electrostatic potential of the SRPK2 docking groove and result in lower processivity. To test our hypothesis, we mutated His-601 to leucine and performed start-trap assays to compare the phosphorylation activity of the mutant with that of the WT kinase. Our results showed that SRPK2(H601L) mutant increased the number of phosphorylation sites on SRSF1 to 7.8 ± 0.5 in the presence of traps, indicating that His-601 plays an important role governing the kinase processivity (Fig. 2C and Fig. S1).
Figure 2.
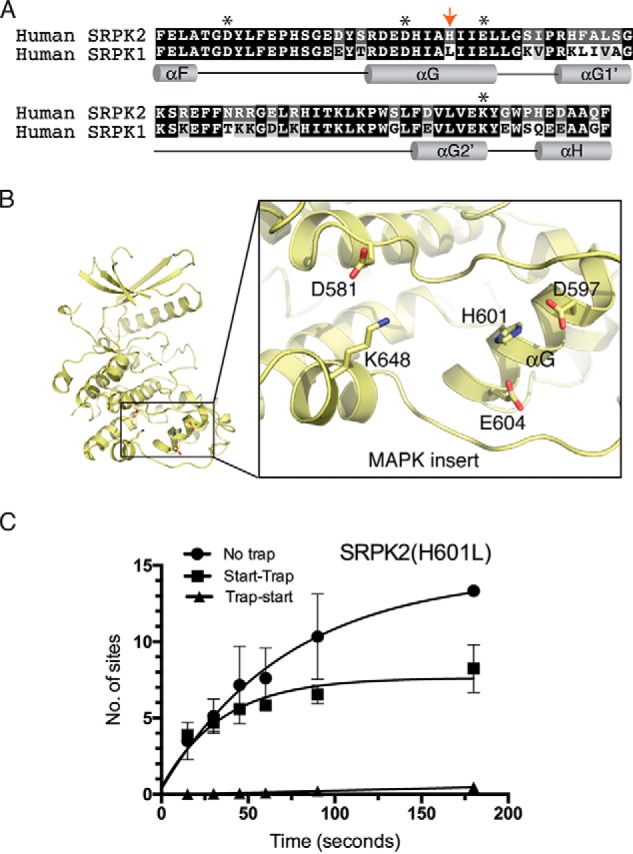
His-601 is responsible for the lower processivity observed in SRPK2. A, alignment and comparison of the amino acid sequences of SRPK2 and SRPK1 docking grooves. Docking groove residues critical for substrate binding and phosphorylation mechanism are denoted by asterisks. His-601 of SRPK2 is denoted by a red arrow. B, His-601 of the helix αG is positioned at the center of the docking groove. C, phosphorylation of SRSF1 by SRPK2(H601L) under no trap, trap-start, and start-trap conditions. The number of phosphorylation sites on SRSF1 has increased to 7.8 ± 0.5 (Fig. 1D). Error bars, S.D. from three independent experiments.
SRSF3 binds to a conserved docking groove of SRPK
The phosphorylation mechanism of SRSF1, a member of the two-RRM subclass, has been studied extensively, but little is known about the phosphorylation of SR proteins containing only one RRM like SRSF3. We therefore studied the mechanism of interaction and phosphorylation of SRSF3 by SRPK2 to see whether the two subclasses are regulated differently. We first determined the Kd of the interaction of SRPK2 with SRSF3 to be 74.5 ± 23.3 nm by microscale thermophoresis (MST) (Fig. 3A). This is comparable with the binding affinity of SRSF1 with SRPK2, suggesting that SRPKs may bind both subclasses of SR proteins with high affinity (20). We next investigated how SRPK2 recognizes SRSF3 by performing pulldown assays with different truncation constructs of SRPK2, where the nonconserved N-terminal and spacer regions were removed individually or in combination. We first confirmed that GSH resin alone or GST protein bound to the resin did not bind any SRPK constructs (Fig. S2). Similar to previous studies on SRSF1 with SRPK1 or SRPK2, the nonkinase regions of SRPK2 were dispensable for the interaction between the kinase and SRSF3 under the condition of the pulldown assay (Fig. 3B). Instead, a conserved docking groove, which has been shown to be important for the interaction of SRSF1 and the SR-like protein acinusS, was critical for SRSF3 binding (20). To further study how the different truncations and mutation might have affected the binding affinity of SRPKs and SRSF3, we determined the Kd of the different constructs with SRSF3 using MST (Fig. 3B and Fig. S3). Our results revealed that whereas the truncation of the N-terminal region did not significantly affect the binding affinity between SRPK2 and SRSF3, the deletion of the spacer region or the combination of both N-terminal and spacer regions reduced the binding affinity to 180 ± 63.1 and 350 ± 72.6 nm, respectively, indicating that both regions, despite not being essential, play a role in the kinase–substrate interaction. Nevertheless, the results confirmed the critical role of the docking groove of SRPK2 in SRSF3 binding. Furthermore, the docking groove was shown to be important for the rapid phosphorylation of SRSF3 in vitro using single-turnover analysis (Fig. 3C). The number of phosphorylation sites on SRSF3 was reduced from 9.5 ± 0.5 to 4.1 ± 0.2 after the docking groove was mutated.
Figure 3.

Docking groove of SRPK2 is important for SRSF3 binding. A, binding affinity of SRSF3WT and SRPK2 analyzed by MicroScale thermophoresis. SRSF3WT was labeled and used at a concentration of 43.5 nm, whereas SRPK2 was titrated to concentrations between 4.5 μm and 0.137 nm. A Kd of 74.5 ± 23.3 nm was determined for this interaction (5-s MST-on time for evaluation). The graph displays data from three independent measurements (error bars represent the S.D.). B, domain organizations of SRPK2 and the different constructs were generated to delineate the region responsible for interaction with GST-SRSF3 (top). GST pulldown assay samples were separated by SDS-PAGE followed by Coomassie Blue staining. Deletions of either the N-terminal extension or spacer, or both, did not affect SRSF3 binding, whereas mutations of the docking groove residues abolished the interaction. SRPK1 was included as a positive control. C, in vitro radioactive kinase assays using SRPK2WT (1.5 μm) or SRPK2DM (1.5 μm) and SRSF3 (500 nm) in the presence of 100 μm [32P]ATP. Samples were resolved by SDS-PAGE and visualized by autoradiography. Mutations of the critical docking groove residues reduce the phosphorylation level of SRSF3 from 9.5 ± 0.5 to 4.1 ± 0.2 sites.
N-terminal RS domain of SRSF3 is required for binding SRPK and its phosphorylation spans the entire RS domain
We next tried to identify the structural determinant(s) of SRSF3 that is important for its interaction with the kinase. Our earlier studies have revealed that the RS domain or arginine-rich region of the substrate binds directly to the docking groove of SRPK. To test whether this is the case for SRSF3 and to identify which region of the RS domain is important for the interaction, we generated different C-terminally truncated mutants of SRSF3 and tested their abilities to bind SRPK2 using pulldown assays (Fig. 4A). Our results show that neither the free GST nor RRM alone interacts with SRPK2. However, the construct that contains the RS domain up to aa 105 could weakly interact with SRPK2, whereas the interaction was nearly fully recovered when residues up to aa 118 were retained (Fig. 4B).
Figure 4.

RS domain of SRSF3 is important for interaction with SRPK2. A, domain organizations of SRSF3WT and SRSF3ΔRS (aa 1–85) are shown. 18 potentially phosphorylatable serines that are present in the context of either RS or SR dipeptides are denoted in gray boldface type. B, GST pulldown assay samples were separated by SDS-PAGE and stained by Coomassie Blue. Deletion of the RS domain of GST-tagged SRSF3 completely abolished its interaction with SRPK2 (left). GST-tagged SRSF3(1–105) failed to bind SRPK2 with similar efficiency as other C-terminally truncated constructs, indicating that residues C-terminal to aa 105 are critical for the interaction. Asterisks denote minor impurity bands. C, RS/SR dipeptides C-terminal to aa 105 were phosphorylated by SRPK2. WT or mutant SRSF3 (500 nm) was phosphorylated by SRPK2 (1.5 μm) using 100 μm [32P]ATP until the end point was reached (15 min). Radiolabeled protein bands corresponding to the phosphorylated SRSF3 constructs were excised and quantified by scintillation counting. Error bars, S.D.
The RS domain of SRSF3 contains 18 serines that are present in the context of either RS or SR dipeptides, which may potentially be phosphorylated by SRPK2 (Fig. 4A). We next tested which regions of this RS domain are phosphorylated by SRPK2 by performing in vitro kinase assays using the different SRSF3 truncation mutants under single-turnover conditions with excess kinase (1.5 μm kinase versus 500 nm substrate). In particular, the construct SRSF3 (1–137), which contains an extra phosphorylatable site at Ser-130 when compared with SRSF3(1–128), was specifically designed to test whether the kinase distinguishes between SR and RS dipeptides during phosphorylation. The results indicate that SRSF3 was phosphorylated at ∼11–12 sites, which were located C-terminal to residue 105 and spanned the rest of the RS domain (Fig. 4C). The observation that SRSF3(1–137) was phosphorylated at one additional site when compared with SRSF3(1–128) suggests that SRPK2 phosphorylates serine in the context of either SR or RS dipeptide.
SRSF3 is processively phosphorylated by SRPK2
To investigate whether SRSR3, like SRSF1, can be phosphorylated by SRPK2 processively, we performed a start-trap kinase assay using 1 μm SRPK2, 300 nm substrate, and a 40-fold excess SRPK2KD as trap. Our results show that SRPK2 phosphorylated SRSF3 at 10.2 ± 0.6 sites at 5 min in the absence of trap in contrast to 4.6 ± 0.5 sites in the presence of it, suggesting that only a few RS dipeptides in SRSF3 are modified processively (Fig. 5A). We further tested whether the sites being processively phosphorylated were located at the N-terminal or C-terminal region of the RS domain by performing the start-trap kinase assay using SRSF3(1–146) and SRSF3(1–128) constructs (Fig. 5, B and C). We observed that although SRSF3(1–146) was phosphorylated at 3.1 ± 0.3 sites in the presence of the traps, SRSF3(1–128) could not be phosphorylated at multiple sites in the start-trap assay. This suggests that either the processively phosphorylated sites are located C-terminal to residues 128, or this region plays a regulatory role in the processive phosphorylation mechanism.
Figure 5.

SRSF3 is processively phosphorylated by SRPK2. In the start-trap experiment, SRPK2WT (1 μm) was pre-equilibrated with 300 nm GST-tagged SRSF3WT (A), SRSF3(1–146) (B), or SRSF3(1–128) (C) and then allowed to react with 100 μm [32P]ATP in the absence (no trap) and the presence (start-trap) of 40 μm SRPK2KD added at the start of the reaction. SRPK2KD was added prior to the reaction start time in the control experiment (trap-start). The number of phosphorylation sites was determined by scintillation counting. SRSF3WT was phosphorylated at 10.2 ± 0.6 sites in the absence of trap and processively phosphorylated at 4.6 ± 0.5 sites by SRPK2. In contrast, SRSF3(1–146) was phosphorylated at 5.6 ± 0.2 and 3.1 ± 0.3 sites in the presence and absence of trap respectively, whereas SRSF3(1–128) was phosphorylated at 3.1 ± 0.1 sites with no trap and 1.1 ± 0.4 sites in the presence of trap. Asterisks denote minor impurity bands. D, docking groove of SRPK2 is essential for processive phosphorylation of SRSF3. SRSF3 was phosphorylated at 4.3 ± 0.2 sites by SRPK2DM, but the reaction was abolished in the presence of SRPK2KD. Error bars, S.D. from three independent experiments.
We next repeated the start-trap assay using SRPK2DM to test the role of its docking groove in the processive phosphorylation of SRSF3. As speculated, mutation of the critical docking groove residues in SRPK2 completely abolished its processivity on SRSF3 (Fig. 5D).
SRPK2 phosphorylates SRSF3 in a C-to-N direction
Previously, Ma et al. (40) used an ATP-dependent phosphorylation mapping strategy to demonstrate that SRSF1 phosphorylation by SRPK1 occurs in a C-to-N direction. We applied a similar strategy to investigate the mode of phosphorylation of SRSF1 by SRPK2. In this method, several lysine residues on SRSF1 were mutated to arginines to remove the cut sites of the endoproteinase Lys-C, followed by the reintroduction of a single lysine at position 214 of the RS domain (mutant R214K). Liquid scintillation counting was used to confirm that the mutant protein phosphorylated by SRPK2 in the presence of radiolabeled ATP was phosphorylated to the same extent as its WT counterpart (data not shown), suggesting that the mutation did not alter the phosphorylation mechanisms. A limited (0.5 μm) or excess (100 μm) amount of [32P]ATP was then added to reactions containing the mutant substrate and SRPK2 and allowed to react under single-turnover conditions until the reactions reach an end point (SRPK2 > substrate; 15 min). Scintillation counting confirmed that SRSF1 could be phosphorylated at nearly two sites at low ATP concentration and about 14 sites at high ATP concentration, as expected (Fig. S4A). Lys-C was then added to cleave the phosphorylated products to generate N- and C-terminal fragments with different amounts of radiolabeled phosphorylation sites, enabling the detection of their ratio at different ATP concentrations. We demonstrated that at a low ATP concentration of 0.5 μm, the stronger band corresponds to the C-terminal fragment of the RS domain (N/C ratio = 0.2) (Fig. 6A and Fig. S4C). However, the presence of the phosphorylated N-terminal half of RS domain was surprising, as there should be no more than two phosphorylation sites on each substrate. We speculate that this could be due to the lower processivity observed in SRPK2, which might have allowed it to phosphorylate the substrate in a less orderly manner; alternatively, the initiation site of the processive phosphorylation mechanism of SRPK2 might be closer to the cleavage site than we anticipated, allowing the N-terminal halves of some substrates to be phosphorylated before ATP was depleted. Nonetheless, at 100 μm ATP, the relative ratio of 32P incorporation into the N- and C-terminal fragments increased to 0.7 (Fig. 6A and Fig. S4C), a value close to the anticipated ratio of 1 based on the position of R214K within the RS1 domain. This suggests that the phosphorylation of SRSF1 by SRPK2 prefers C-terminal initiation to N-terminal initiation when ATP is limited, which is consistent with the observation for SRPK1.
Figure 6.

Both SRSF1 and SRSF3 are phosphorylated at the C-terminal region first. A, left, cleavage construct of SRSF1. Positions of lysine are denoted by arrowheads. The C-terminal His-tagged SRSF1(R214K) construct contains five Lys-to-Arg mutations in the RRM2 and a single Arg-to-Lys mutation at position 214 of the RS domain. Cleavage of the construct by Lys-C produces two major N-terminal and C-terminal fragments of 19 and 5 kDa, respectively. Asterisk, minor impurity band. Right, Lys-C cleavage of SRPK2-phosphorylated SRSF1(R214K) at 0.5 μm (low) and 100 μm (high) ATP concentrations. The autoradiogram was scanned, and the ratio of N-terminal band to C-terminal fragments (N/C) was determined by a PhosphorImager. B, left, cleavage construct of SRSF3. The C-terminal His-tagged SRSF3(R131K) construct contains three Lys-to-Arg mutations and a single Arg-to-Lys mutation at position 131 of the RS domain. Lys-C cleavage produces two major N-terminal and C-terminal fragments of 13 and 4 kDa, respectively. Right, Lys-C cleavage of SRPK2-phosphorylated SRSF3(R131K) at low and high ATP concentrations.
Because the phosphorylation of SRSF3 spans the entire RS domain and nearly half of the phosphorylation sites are located C-terminal to residue 128, we introduced a lysine-to-arginine mutation at residue 131 to bifurcate the RS domain during Lys-C treatment. Similar to the aforementioned strategy for SRSF1, the SRSF3 mutant was first phosphorylated by SRPK2 in single-turnover reactions at low and high ATP concentrations, which generated substrates that were phosphorylated at about 2 and 12 sites, respectively, and then followed by a Lys-C cleavage step (Fig. S4, B, D, and E). Like SRSF1, two bands corresponding to the N-terminal and C-terminal fragments of the mutant were observed after Lys-C cleavage, with the C-terminal fragment showing a stronger band. The N/C ratio increases from 0.3 at low ATP concentration to 0.7 at high ATP concentration, which is again approaching our estimated ratio of 1 owing to the location of R131K at the RS domain (both N- and C-terminal halves contain similar numbers of sites according to our results) (Fig. 6B). This indicates that SRPK2 also prefers to phosphorylate SRSF3 in a C-to-N direction. Taken together, our data provide evidence that despite SRSF3 lacking the pseudo-RRM, it is phosphorylated in the same direction as SRSF1 by SRPK2, and the processively phosphorylated sites are likely located at the C terminus.
SRSF3 is hypophosphorylated at steady state
To investigate the phosphorylation state of cellular SRSF3, we generated new SRSF3 mutant constructs in which all serines C-terminal to residue 85 (SRSF3(85SA)), 118 (SRSF3(118SA)), or 128 (SRSF3(128SA)) were mutated to alanines. These mutant proteins and SRSF3WT, as well as SRSF1 as a control, were transiently overexpressed in HEK293 cells and isolated for mobility comparison by using SDS-PAGE, with or without treatment with SRPK2 or calf intestinal alkaline phosphatase (CIP). In accordance with previous findings, SRSF1 is hypophosphorylated in cells at steady state, as the mobility of the untreated sample is between the dephosphorylated sample and the SRPK2-phosphorylated counterpart (Fig. 7) (11). We observed that the mobility of SRSF3(85SA) and SRSF3(118SA) was not affected by the treatment with SRPK2 or CIP, which reaffirms that the phosphorylation sites of SRSF3 are located C-terminal to residue 118 (Fig. 7). In contrast, SRPK2-treated SRSF3WT migrated significantly more slowly than its untreated counterpart, and the mobility of both SRSF3(128SA) and SRSF3WT increased upon CIP treatment. These results suggest that cellular SRSF3, like SRSF1, is hypophosphorylated in its steady state and that the region between residues 118 and 128 is phosphorylated in vivo.
Figure 7.

SRSF3 is hypophosphorylated at steady state. HEK293 cells were transiently transfected with plasmids encoding Myc-SRSF1WT, Myc-SRSF3WT, Myc-SRSF3(85SA), Myc-SRSF3(118SA), or Myc-SRSF3(128SA), where serines located C-terminal to aa 85, 118, or 128 were all mutated to alanines, respectively. Lysates were collected 24 h post-transfection and subjected to SRPK2-mediated phosphorylation or CIP-mediated dephosphorylation or left untreated. Samples were separated by a 15% Tris-Tricine SDS-polyacrylamide gel followed by Western blotting with α-Myc mAb. The mobility of either Myc-SRSF3(85SA) or Myc-SRSF3(118SA) was similar with or without treatment of SRPK2 or CIP. The mobility of untreated Myc-SRSF1WT, Myc-SRSF3(128SA), or Myc-SRSF3WT is lower than that of the SRPK2-treated sample but higher than that of the CIP-treated sample. The asterisk indicates the presence of a slower-migrating band in the untreated sample of Myc-SRSF3(128SA).
Phosphorylated RS domain of SRSF3 is dephosphorylatable after SRPK2 release
We showed that the entire stretch of the SRSF3 RS domain could be phosphorylated by SRPK2 in vitro; however, SRSF3 is hypophosphorylated in its steady state. Thus, SRPK-mediated phosphorylation is unlikely to be the sole event that determines the observed phosphoryl content of SRSF3 in vivo. Previous studies by Serrano et al. (23) have demonstrated that the pseudo-RRM in SRSF1 directly contacts the RS domain to regulate its phosphorylation mechanism and phosphoryl content by shielding it from phosphatases. In addition, the canonical RRM of SRSF1 contains a putative phosphatase-binding motif that regulates the interaction and dephosphorylation activity of PP1γ (41). Because SRSF3 lacks both interaction due to the absence of a pseudo-RRM and the conserved RVXF PP1-binding sequence, we speculated that the hypophosphorylation state of SRSF3 is the result of increased sensitivity to dephosphorylation by phosphatases. To test this, we monitored how the phosphoryl contents of SRSF1 and SRSF3 are affected in the presence of the protein phosphatase 1 catalytic subunit α (PP1α), which coexists with both SRSF1 and SRSF3 in the human spliceosomal B complex (42). We first determined whether PP1α is capable of dephosphorylating phosphorylated RS dipeptide by monitoring the phosphorylation of acinusS in the presence of PP1α. AcinusS was chosen because it is an SR-like protein that contains only one SRPK2-mediated phosphorylation site (20). Our result showed that PP1α could dephosphorylate acinusS despite the absence of its physiological regulators (Fig. S5). Next we phosphorylated the SR proteins (1 μm) by SRPK2 (100 nm) in the absence and presence of excess PP1α (300 nm) to test whether both SRSF1 and SRSF3 serve as substrates of the phosphatase. Our results show that the addition of PP1α reduced the phosphoryl content of both substrates by about 20% when compared with the control reactions after the phosphorylation levels approached equilibrium, indicating the SR proteins are substrates of PP1α (Fig. 8A and Fig. S6). Because it has been shown that SRPK–SR protein complexes dissociate slowly compared with the fast forward rate of multisite phosphorylation, SRPK2 might remain bound to the substrates after phosphorylation and interfere with the dephosphorylation reaction. To ensure dissociation of SRPK2 after phosphorylation, we performed the PP1α treatment in the presence of excess SRSF1(BD) (400 nm), which would compete with the docking interaction with SRPK2 and dislodge all of the complexes (Fig. 8, B and C). In this experiment, SRSF1 and SRSF3 were first phosphorylated by SRPK2. Next, unreacted ATP was removed using desalting columns, and the kinase activity was inhibited by the SRPK inhibitor SRPIN340 (100 μm), which is potent against the phosphorylation of SRSF1 and SRSF3 by SRPK2 (Fig. S7) (43). PP1α was then added to the samples with or without the presence of SRSF1(BD).
Figure 8.

Presence of pseudo-RRM affects dephosphorylation. A, 1 μm SRSF1 or SRSF3 was phosphorylated by SRPK2 (100 nm) in the absence or presence of PP1α (300 nm). The bar graph shows the phosphorylation levels of SRSF1 and SRSF3 in the presence of both SRPK2 and PP1α relative to that of SRPK2 only. Error bars, S.D. from three independent experiments. Data were analyzed by unpaired t test. **, p < 0.005; *, p < 0.05. SRSF1 (B) or SRSF3 (C) was first phosphorylated by SRPK2 for 5 min, and then the reaction was quenched by removing the ATP using a desalting column. The phosphorylated SR proteins were next treated with PP1α in the absence or presence of SRSF1(BD). The domain organization of SRSF1(BD) is shown in B. Only SRSF3 was dephosphorylated by PP1 under the experimental conditions. D, GST-SRSF1 was first phosphorylated by SRPK2 in the presence of [32P]ATP for 5 min, followed by Lys-C treatment to generate a hypophosphorylated RS domain (p-SRSF1(RS)). A pulldown assay using His-tagged pseudo-RRM of SRSF1(His-SRSF1(pRRM)) as bait was performed. No interaction was detected between radiolabeled p-SRSF1(RS) and His-SRSF1(pRRM).
Intriguingly, our results show that, in contrast to Fig. 8A, both SRSF1 and SRSF3 did not undergo dephosphorylation when PP1α was added after the phosphorylation by SRPK2 had completed (Fig. 8, B and C). Only SRSF3 became dephosphorylatable by PP1α in the presence of SRSF1(BD) (Fig. 8C). Together, the data suggest that the RS domain may remain accessible by the phosphatase during reaction turnover but become protected, either via interaction with the kinase or by a stabilized folded structure, after the completion of the reaction (44, 45). Upon dissociation of SRPK2, SRSF3, but not SRSF1, becomes dephosphorylatable.
Previous studies have shown that neither phosphorylated RS1 nor the unphosphorylated RS2 domain of SRSF1 directly interacts with its pseudo-RRM independently. However, there are reports that the RS domain can adopt a new and stabilized folded structure after SRPK1-mediated phosphorylation (44, 45). Therefore, we speculated that the phosphorylated sites on SRSF1 were protected by an alternative binding mechanism between its pseudo-RRM and a potentially different structured phospho-RS domain. To test this, SRSF1, which contains a lysine (Lys-193) between its RRMs and the RS domain, was phosphorylated by SRPK2 to ∼14 sites using [32P]ATP (determined by scintillation counting; data not shown) and treated with Lys-C to generate a hypophosphorylated RS domain (p-SRSF1(RS)). Pulldown assays were then performed to test whether His-tagged pseudo-RRM of SRSF1 (His-SRSF1(pRRM)) would interact with the hypophosphorylated RS domain (Fig. 8D). In contrast to our speculation, His-SRSF1(pRRM) failed to bind to the hypophosphorylated RS domain, confirming that the phosphorylated sites of SRSF1 were not protected via stable interaction with its pseudo-RRM.
SRPK2 docking groove is important for the regulation of SRSF3 subnuclear localization
Because the docking groove of SRPK2 plays a critical role in the regulation of interaction and phosphorylation of SRSF3, the docking groove mutant (SRPK2DM) provides a unique tool to investigate the direct role of SRPK2 on SRSF3 function in vivo. It is well-accepted that the subcellular localization of SR proteins is regulated by SRPKs-mediated phosphorylation; we therefore attempted to study the localization of SRSF3 in the presence of SRPK2WT or SRPK2DM. We first transiently transfected HEK293 cells with Myc-tagged SRSF3 along with an empty FLAG-tagged vector to determine the localization of SRSF3 by indirect immunofluorescence. In contrast to previous studies, where overexpressed SR proteins always localized to the nuclear speckles, we observed that SRSF3, although predominantly localized inside the nucleus, was found to concentrate at the nuclear speckles in less than 30% of HEK293 cells. We next examined how SRPK2 affects SRSF3 subcellular localization by repeating the indirect immunofluorescence study in a SRPK2-knockout HEK293 cell line (HEK(K2X)) that was generated by our group using CRISPR-Cas9.5 Interestingly, we observed a moderate increase in the number of cells that contained speckles, suggesting that SRPK2 may play a role in breaking up SRSF3 from the nuclear speckles (Fig. 9). To test our hypothesis, we co-transfected the HEK(K2X) cells with Myc-SRSF3 and either FLAG-tagged SRPK2WT or SRPK2DM. In accordance with our speculation, SRPK2-knockout cells reconstituted with the WT kinase showed a significant decrease in the number of speckle-containing cells when compared with the control HEK(K2X) cells co-transfected with Myc-SRSF3 and an empty FLAG-tagged vector. On the contrary, knockout cells reconstituted with SRPK2DM failed to dissociate SRSF3 from the nuclear speckles. Our studies thus implicated the role of the SRPK2 docking groove in the regulation of SRSF3 cellular activity.
Figure 9.

Localization study of SRSF3 in HEK293 and HEK(K2X) cells. Each cell line was transfected with pCMV-Myc-SRSF3. pCMV-Tag-2c-FLAG empty vector was transfected to HEK and HEK(K2X) as control. pCMV-Tag-2c-FLAG_SRPK2WT/DM was transfected to HEK(K2X) to generate HEK(K2X)-K2WT and HEK(K2X)-K2DM, respectively. Anti-FLAG antibody and Alexa 594 secondary antibody were used to visualize FLAG-SRPK2WT or FLAG-SRPK2DM. Anti-Myc antibody and Alexa 488 secondary antibody were used to visualize Myc-SRSF3. The percentage of cells containing nuclear speckles was evaluated in three independent experiments for each tested plasmid construct. A total of at least 150 cells were counted for each tested construct. HEK(K2X) has a higher percentage of speckle-containing cells than HEK293. HEK(K2X)-K2WT shows significantly fewer speckle-containing cells, when compared with HEK(K2X)-K2DM, which has a percentage similar to that of HEK(K2X). Results were analyzed with Student's t test and presented as mean ± S.D. (error bars) of three independent experiments. *, p < 0.05; **, p < 0.01.
Discussion
Members of the SRPK family contain a unique docking groove that is important for the regulation of the binding and phosphorylation of different classes of substrate, including the classical SR protein SRSF1 and SR-like proteins such as Npl3p and acinusS (11, 20, 46). In this study, we demonstrated that SRPK2 utilizes the same docking groove for the interaction and phosphorylation of both subclasses of SR proteins, namely SR proteins with a single canonical RRM and SR proteins with both a canonical RRM and a pseudo-RRM. Similar to SRPK1, SRPK2 is capable of processively phosphorylating SRSF1 at multiple sites in a C-to-N-terminal direction. However, the number of sites being phosphorylated by SRPK2 before substrate dissociation is lower than that phosphorylated by SRPK1. Mutational studies revealed that the presence of a chargeable histidine residue at the center of the docking groove of SRPK2 provides a plausible explanation for the observed difference. As shown in our previous and current studies, docking groove residues that are critical for the interaction with SR and SR-like substrates are mostly negatively charged, producing a negative electrostatic surface potential within the substrate binding cleft (Fig. S8). The replacement of a nonpolar leucine residue in SRPK1 with a protonatable histidine in SRPK2 might cause electrostatic repulsion with the arginine-rich RS domain of SR substrates, resulting in a weakened sliding docking interaction and early substrate release. Such a difference in processivity may be more prominent in substrates that contain more negatively charged residues at their docking regions. During a splicing event, this lowered processivity of SRPK2 may result in phosphorylated SR proteins that are functionally distinguishable from those produced by SRPK1 and therefore differentiate their functions in vivo.
SR proteins have wide-ranging roles in the regulation of RNA metabolism. Although different SR proteins can interchangeably restore constitutive splicing in in vitro splicing assays, conditional knockout experiments have shown that individual SR proteins play distinct biological roles. For instance, hepatocyte-specific knockout of SRSF3 in mice demonstrates that the SR protein is critical for hepatocyte differentiation and glucose and lipid metabolism (47). By contrast, SRSF1 plays an important role during functional remodeling in developing heart tissue by regulating the splicing of a restricted set of genes including Ca2+/calmodulin-dependent kinase IIδ (CaMKIIδ) (48). These findings demonstrate that the studies of the function and regulation of each individual SR protein are important for deciphering their unique biological roles in splicing and disease development. We hereby elucidated the phosphorylation mechanism of SRSF3 by SRPK2. Concurring with our previous findings, the current study demonstrated that the conserved docking groove of SRPK2 is also important for the interaction and phosphorylation of SRSF3. However, unlike previous observations for SRSF1, Tra2β, and acinusS, in which phosphorylation occurs at a specific region or site, the phosphorylation sites in SRSF3 are distributed over the entire RS domain. In addition, whereas SRSF3 is also phosphorylated by SRPK2 in a C-to-N manner, the processivity of the reaction is lower than that of SRSF1 and is limited to the C-terminal half of the RS domain. This observation is consistent with our hypothesis that the presence of acidic residue(s) at the docking interface may lower processivity because SRSF3 contains 6 acid residues that locate intermittently along its RS domain, in contrast to the absence of acidic side chains in the SRSF1 RS domain.
Besides the kinase docking groove, the mechanism and content of phosphorylation of SRPK substrate is also regulated by structural elements in the substrate itself. For instance, an acidic region immediately preceding the docking motif of the SR-like protein acinusS prevents the substrate from sliding at the docking groove of SRPK2 and restricts the phosphorylation at a single specific site (20). On the other hand, Serrano et al. (23) demonstrated that the pseudo-RRM of SRSF1 is important for the directionality and content of phosphorylation. The interaction between an acidic cluster within the pseudo-RRM and the RS domain, in particular, prevents dephosphorylation of the RS domain by phosphatases, suggesting a novel role of SR protein pseudo-RRM in vivo. The same research team later found that the canonical RRM of SRSF1, which contains a putative PP1-binding motif, also plays a regulatory role during its RS domain dephosphorylation by interacting with and allosterically inhibiting PP1γ (41). SRSF3 does not contain either structural element to protect its RS domain from dephosphorylation. Our finding that, after SRPK2 dissociation, SRSF3, but not SRSF1, underwent PP1α-mediated dephosphorylation supports this notion. Nonetheless, we cannot rule out the possibility that the canonical RRM of SRSF3 may also play a role in dephosphorylation regulation, as we only observed a 30% decrease in phosphoryl content after PP1α treatment. It is important to note that the presence of pseudo-RRM of SRSF1(BD) did not prevent phospho-SRSF3 from dephosphorylation, indicating that the pseudo-RRM–mediated protection could be RS domain–specific.
Because SRSF3 was found to be hypophosphorylated in cells at steady state, our findings together suggest that the phosphoryl content of the SRSF3 RS domain after an SRPK-mediated event might be maintained by dynamic phosphorylation and dephosphorylation in vivo due to the lack of protection from the pseudo-RRM and PP1-binding motif. This contrasts with the phosphorylation of SRSF1 by SRPKs, in which the phosphoryl content appears to be regulated by a regiospecific phosphorylation mechanism and protection from both canonical and pseudo-RRMs. The aforementioned cluster of acidic residues that is important for SRSF1 phosphoryl-content protection is present in all pseudo-RRMs but not the canonical RRMs of SR proteins (23). We therefore propose that the regulatory mechanisms of phosphorylation observed in SRSF3 and SRSF1 may be applicable to all SR proteins that belong to the same subclass (i.e. the phosphoryl content of SR proteins containing two RRMs is protected from dephosphorylation, at least transiently, after an SRPK-mediated phosphorylation event, whereas members of the single-RRM subclass of SR proteins, due the lack of pseudo-RRM, may undergo an additional level of regulation where cellular phosphatases could reversibly regulate their subcellular/subnuclear localization and cellular activities) (Fig. 10). Although we showed that the pseudo-RRM of SRSF1 did not interact stably with the hypophosphorylated RS domain, we cannot rule out that transient interaction between the two domains may shield the phosphorylated serines from the phosphatase active site. The pseudo-RRM, alternatively, may exhibit molecular chaperone-like function to facilitate the stable folding of the phosphorylated RS domain as predicted and prevent dephosphorylation (44, 45). Intriguingly, the dissociation of the RS domain from the pseudo-RRM promotes PP1 binding, suggesting that the pseudo-RRM may potentially facilitate or stabilize the PP1–RRM interaction. Further mechanistic studies on the pseudo-RRMs of other SR proteins may help to elucidate their importance in the regulation of SR proteins and subsequently splicing.
Figure 10.

Model of phosphorylation of two subclasses of SR protein. A, after phosphorylation of SR protein that contains two RRMs, the presence of the pseudo-RRM prevents the phosphoryl content, at least transiently, from dephosphorylation by protein phosphatases. RS dipeptides colored in gray indicate that the C-terminal region, in the case of SRSF1, is regulated by a regiospecific mechanism and remains unphosphorylated. B, phosphorylation sites of single RRM–containing SR protein are accessible by phosphatases, allowing dynamic regulation of the phosphoryl content by both phosphatases and SRPK. A dashed arrow indicates that the dephosphorylated substrate may be subjected to rephosphorylation by SRPK.
The subcellular localization of SR proteins has been shown to be governed by a number of kinases, including SRPKs and CLKs. Whereas the nuclear kinase CLK1 phosphorylates SP dipeptides of the RS2 domain of SRSF1 to relocalize it from speckles to the nucleoplasm, it remains unclear how members of the SRPK family, which strongly prefer phosphorylation of RS dipeptides, accomplish similar task when overexpressed in cells (49). New light has been shed on this by two recent studies that show that SRPK1, after being localized to the nucleus, facilitates the dissociation of SRSF1 from the nuclear speckles by the formation of an SRPK1–CLK1 complex. The presence of SRPK1 facilitates the release of SRSF1 from CLK1 and enhances further phosphorylation of SP dipeptides, resulting in the mobilization of SRSF1 from nuclear speckles to nucleoplasm (50, 51). In this study, we observed that SRPK2 also plays a role in the mobilization of SRSF3 from the speckles to nucleoplasm. Such activity depended on the presence of an intact substrate docking groove in the kinase, suggesting that the direct interaction or phosphorylation of SRSF3 by SRPK2 is important for its subnuclear localization. Whereas the nuclear localization of SRPK2 is stimulated by epidermal growth factor in a similar manner as SRPK1, there is no evidence that SRPK2 will interact with CLK inside the nucleus. Furthermore, whereas SP dipeptides are present in SRSF3, they are not concentrated at a specific region of the RS domain as in the case of SRSF1. Importantly, our findings also indicate that SRSF3 is not phosphorylated in a region-specific manner, suggesting that it might not follow the same SRPK–CLK phosphorylation pathway of SRSF1. Future studies focusing on the mechanism of phosphorylation of single RRM–containing SR proteins by CLK will help to resolve the unanswered questions.
We have recently entered a new era of understanding of eukaryotic pre-mRNA splicing due to the solving of multiple high-resolution cryo-EM structures of the spliceosome. These structural models of spliceosomes are captured at different steps of the splicing reaction and are obtained from different species, including Saccharomyces cerevisiae, Schizosaccharomyces pombe, and, most recently, humans, providing invaluable structural and mechanistic insights into the assembly and catalytic steps of the spliceosome. However, due to the highly dynamic nature of the spliceosome, these structures do not contain any members of the SR protein family, despite their essential roles in spliceosome assembly. Although the Ser/Arg-related nuclear matrix protein SRm300 is present in both cryo-EM structures of the C* complex, its RS domain cannot be located due to its flexibility (52, 53). Therefore, how SR proteins interact with the pre-mRNA during splicing and how they are involved in the structural organization of the spliceosome remain ambiguous. Further analysis of the phosphorylation/dephosphorylation mechanism of the RS domain using biochemical and biophysical methods, and structural studies using X-ray crystallography or NMR, may remain the major means of studying the regulation and function of individual SR proteins during splicing.
Experimental procedures
Protein expression and purification
Proteins of all SRPK2 and SRSF1 constructs were expressed and purified as described previously (11, 20). WT and different truncation constructs of SRSF3 were cloned into BamHI and XhoI cut sites of a pET28a-GST vector, which was constructed in our laboratory by inserting the gene of GSH S-transferase between the NdeI and BamHI cut sites of pET28a. Different SRSF3 expression constructs were transformed into BL21 (DE3) pLysS strain Escherichia coli. Large-scale cultures were grown at 37 °C in Terrific Broth with 100 μg/ml kanamycin and 50 μg/ml chloramphenicol, respectively. Protein expression was induced with 0.4 mm isopropyl 1-thio-β-d-galactopyranoside at 30 °C for 5 h. Pelleted cells were lysed by sonication in 50 mm Tris-HCl, pH 8.0, 400 mm NaCl, 10 mm imidazole, 15% glycerol, 2 mm phenylmethylsulfonyl fluoride, 3 mm benzamidine hydrochloride, and protease inhibitor mixture (Sigma-Aldrich). The soluble fraction was loaded onto a self-packed Ni-NTA affinity column (Qiagen); washed with 100 ml of lysis buffer and 100 ml of washing buffer (50 mm Tris-HCl, pH 8.0, 400 mm NaCl, 60 mm imidazole, 15% glycerol), respectively; and then eluted by 20 ml of the buffer containing 200 mm imidazole. The eluate then was loaded onto a self-packed GST affinity column (GenScript) and washed with 30 ml of 50 mm Tris-HCl, pH 8.0, 400 mm NaCl, 15% glycerol, and 1 mm DTT. Finally, the protein was eluted by 15 ml of the buffer containing 60 mm GSH. The eluate was dialyzed overnight against buffer containing 50 mm glycine, pH 9.5, 400 mm NaCl, 50 mm arginine, 50 mm glutamate, 1 mm DTT, and 15% glycerol. The dialysate was concentrated, frozen in liquid nitrogen, and stored at −80 °C.
In vitro kinase assays of SRSF3 constructs
Purified kinase (1.5 μm) and different SRSF3 constructs (0.5 μm) were incubated in kinase reaction buffer (50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 10 mm MgCl2, 1 mg/ml BSA, and 10% glycerol) at room temperature for 15 min. The phosphorylation reaction was initiated by adding 100 μm unlabeled ATP and 0.5 μCi of [32P]ATP at 25 °C. Reactions were quenched by the addition of Laemmli buffer and boiling at 98 °C for 1 min. The reactions were resolved on a 12.5% polyacrylamide gel, which was subsequently dried on Whatman filter paper (GE Healthcare) and then exposed to autoradiography film (Fujifilm). To quantify the phosphorylation sites, bands of phosphorylated substrate were excised from the dried gel, dissolved in OptiPhase HiSafe 2 solution (PerkinElmer Life Sciences), and counted using a Beckman LS 6500 scintillation counter.
Processivity assays
No trap assay
300 nm of substrate was incubated with 1 μm of SRPK2WT in kinase reaction buffer (50 mm Tris, pH 7.5, 200 mm NaCl, 10 mm MgCl2, 1 mg/ml BSA, and 10% glycerol) at 25 °C. Reactions were initiated by the addition of 100 μm [32P]ATP and then quenched by Laemmli buffer together with subsequent boiling at corresponding time points.
Start-trap assays
300 nm substrate was incubated with 1 μm SRPK2WT in kinase reaction buffer at 25 °C for 10 min. Phosphorylation was then initiated by the simultaneous addition of 100 μm [32P]ATP and 40 μm SRPK2KD (trap). Reactions were finally quenched by Laemmli buffer and subsequent boiling at different time points.
Trap-start assays
300 nm substrate was preincubated with 40 μm SRPK2KD in kinase reaction buffer at 25 °C for 10 min. Phosphorylation was then initiated by the simultaneous addition of 100 μm [32P]ATP and 1 μm SRPK2WT. Reactions were finally quenched by Laemmli buffer and subsequent boiling at different time points. All reaction mixtures were analyzed by SDS-PAGE, followed by drying the gel and exposing it to autoradiography film. Phosphorylation content was determined by excising the corresponding bands and subjecting them to liquid scintillation counting.
ATP-dependent footprinting assays
250 nm substrate protein was incubated with 500 nm SRPK2WT, and the phosphorylation was performed as described above for 15 min, except that the reactions were initiated by adding varying amounts of [32P]ATP (0.5 and 100 μm, respectively). Sample proteolysis was then carried out through the addition of 100 ng of Lys-C (Promega) in digestion buffer (50 mm Tris-HCl, pH 8.5, 2 mm EDTA) and allowed to incubate for 4 h at 37 °C. Reactions were loaded onto a 18% SDS gel, dried, and exposed to an imaging plate (Fujifilm). Results were scanned by the multipurpose image scanner FLA-9000 (Fujifilm), and the intensities of corresponding bands were quantified by Multi Gauge version 3.2 software.
MST analysis
MST measurements were performed using Monolith NT.115 with the Pico RED detector. Before labeling the target protein SRSF3WT, buffer was exchanged to the MST buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 0.05% Tween 20) with ZebaTM Spin desalting columns (Thermo Fisher Scientific). SRSF3WT was then labeled using the MonolithTM protein labeling kit RED-NHS (NanoTemper Technologies) according to the manufacturer's instructions in the supplied labeling buffer. Labeled SRSF3WT was diluted to the indicated concentration and was mixed to 16 serially diluted titrations of the unlabeled ligand (one of the SRPK constructs) in a 1:1 volume ratio. The complexes were then loaded into NT.115 standard treated capillaries (NanoTemper Technologies) for binding measurements. Data were analyzed using MO.Affinity Analysis software (NanoTemper Technologies) at the standard MST-on time of 5 s.
Pulldown assays of SRSF3 with SRPK2
GST-His–tagged SRSF3WT and its variants (600 nm) were incubated with untagged SRPK2WT (900 nm), 1 mm ATP, and 2 mm MgCl2 in 300 μl of binding buffer (GST pulldown assays: 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 2 mm DTT, 1% Triton X-100; Ni-NTA pulldown assays: 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 50 mm imidazole, 1% Triton X-100) for 1 h at 4 °C. 30 μl of GSH-agarose (GenScript) or Ni-NTA resins (Qiagen) was then added to the mixture and allowed to incubate for 1 h at 4 °C. Afterward, the resins were washed three times with 350 μl of binding buffer. Laemmli buffer was added to the resins and boiled for 5 min at 98 °C to elute the bound protein. Samples were resolved by SDS-PAGE (15% gel) and visualized by Coomassie Blue staining.
Phosphorylation state of SRSF3
HEK293 cells were proliferated at log phase and seeded in 6-well plates for 24 h before transfection. Cells were then transfected with 2 μg of plasmid of Myc-tagged SRSF3 constructs per well for 24 h and were lysed on ice with radioimmune precipitation buffer (Cell Signaling Technology). Cell lysates were then evenly divided into three groups: an untreated group, a SRPK2-treated group, and a CIP-treated group. The SRPK2-treated group was incubated with 5 mm SRPK2WT in the presence of 1 mm ATP and 2 mm MgCl2 for 15 min at 25 °C, the CIP-treated group was incubated with 2 μl of alkaline phosphatase, calf intestinal (New England Biolabs) for 2 h at 37 °C. The Myc-tagged SRSF3 constructs were detected by Western blotting using α-Myc antibodies.
SR protein dephosphorylation
1 μm SRSF1 or SRSF3 was incubated with 100 nm SRPK2 in kinase reaction buffer (100 mm Mops, pH 7.4, 50 mm Hepes, 100 mm NaCl, 2 mm DTT, 0.01% Tween 20, 1 mm MnCl2, 10 mm MgCl2, and 5 mg/ml BSA) at 25 °C, with or without 300 nm PP1α phosphatase (Abcam). Afterwards, 25 μm unlabeled ATP and 0.5 μCi of [32P]ATP was added and allowed to react for 5 min. Reactions were quenched by the addition of SDS-PAGE loading buffer and boiling at 98 °C for 1 min and were resolved on a 12.5% polyacrylamide gel, which was subsequently dried on Whatman filter paper (GE Healthcare). Results were scanned by multipurpose image scanner FLA-9000 (Fujifilm), and the intensity of corresponding bands was quantified by Multi Gauge version 3.2 software.
Phosphatase treatment of phosphorylated SR proteins with SRSF1(BD)
1 μm SRSF1 or SRSF3 was incubated with 100 nm SRPK2 in kinase reaction buffer at 25 °C for 5 min, in the presence of 25 μm unlabeled ATP and 0.5 μCi of [32P]ATP. Reactions were stopped by removing ATP using ZebaTM spin desalting columns (Thermo Fisher Scientific) and the addition of the SRPK inhibitor SRPIN340. 300 nm PP1α phosphatase was then added to dephosphorylate substrates at 37 °C for 5, 15, or 30 min with or without 400 nm SRSF1(BD) (residues 105–226). Reactions were quenched by the addition of Laemmli buffer and boiling at 98 °C for 1 min and were resolved on a 12.5% polyacrylamide gel, which was subsequently dried on Whatman filter paper. Results were scanned by multipurpose image scanner FLA-9000, and the intensity of corresponding bands was quantified by Multi Gauge version 3.2 software.
Radioactive pulldown assay
GST-tagged full-length SRSF1 (10 μm) was phosphorylated by SRPK2 (0.5 μm) with 500 μm [γ-32P]ATP and kinase buffer (80 mm NaCl, 50 mm Tris, pH 7.5, 10 mm MgCl2, and 2% glycerol). After a 5-min incubation at room temperature, 15 mm EDTA was added to the reaction to stop further phosphorylation. The samples were added into a desalting column along with nickel beads to remove EDTA and His-tagged SRPK2. Lys-C enzyme (Promega) was added to the supernatant to digest p-SRSF1, leaving only the phosphorylated RS domain of SRSF1 intact. Benzamidine (2 mm) and phenylmethylsulfonyl fluoride (10 mm) were added to inhibit Lys-C activity after an overnight incubation at 37 °C. His-tagged SRSF1(pRRM) (10 μm) and nickel beads were incubated with the digested fraction in a binding buffer (0.1% Nonidet P-40, 50 mm Tris (pH 7.5), 75 mm NaCl, and 5 mm imidazole). After 1 h at room temperature, the resin was washed four times with 200 μl of binding buffer, and SDS dye was added to the resin and boiled for 5 min. The samples were run on a 15% Tricine gel. Dried gels were then exposed with X-ray film (FUJIFILM).
Cell culture and transfection
Dulbecco's modified Eagle's medium with 10% FBS and 1% penicillin and streptomycin were used to grow HEK293 and HEK(K2X) cells. SRPK2WT/DM and SRSF3 were transfected using Lipofectamine LTX (Life Technologies, Inc., product no. 15338030) following the manufacturer's protocol. After a 48-h transfection, cells were fixed for image analysis.
Immunofluorescence staining and immunofluorescence imaging analysis
Transfected HEK293 and HEK(K2X) cells were cultured on glass coverslips. Cells were fixed with 3.7% formaldehyde for 10 min, washed three times with PBS, and permeabilized with PBS-T (PBS with 1% BSA and 0.3% Triton X-100) for 3 min. Cells were then incubated in anti-FLAG rabbit antibody (Sigma, product no. F7425-2MG) and anti-Myc(9B11) mouse antibody (Cell Signaling, product no. 2278S) in PBS with 1% BSA overnight at 4 °C. The next day, cells were washed three times with PBS-BSA and then incubated in Alexa 594 rabbit secondary antibody (Invitrogen, product no. 21207) and Alexa 488 mouse secondary antibody (Invitrogen, product no. A21202) for 3 h at room temperature. The cells were then washed and mounted onto microscope slides. The samples were examined with a Nikon NI-U upright fluorescence microscope.
Statistical analysis
All data are presented as the mean ± S.D. of three independent experiments. Statistical analysis was performed using the unpaired Student's t test, and the calculated p values are indicated in the figure legends. A p value of <0.05 was considered statistically significant.
Author contributions
Y. L., W. H. S., T. C. K. L, and J. C. K. N. designed the research; Y. L., W. H. S., K. W. Y. Y., H. L., C. Z., S. W. C. W., Q. L., C. O. K. L., and G. H. C. C. performed the research; J. C. K. N. analyzed the data and wrote the paper.
Supplementary Material
Acknowledgments
We thank Professor Joseph Adams (University of California, San Diego, La Jolla, CA) for the SRSF1(K214R) plasmid. We also thank Professor Gourisankar Ghosh (University of California, San Diego, La Jolla, CA) for helpful discussions.
This work was supported by Hong Kong SAR Research Grant Council General Research Fund Grants 473310 and 14122615 and The Chinese University of Hong Kong direct grant scheme Grant 3132811 (to J. C. K. N.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S8.
K. W. Y. Yung, R. P. H. Yip, P. W. T. Lee, C. P. Chan, and J. C. K. Ngo, unpublished data.
- SR
- serine-arginine or serine/arginine-rich
- RS
- arginine-serine or arginine/serine-rich
- SP
- serine-proline
- RRM
- RNA recognition motif
- SRPK
- serine-arginine protein kinase
- CLK
- Cdc2-like kinase
- MST
- microscale thermophoresis
- aa
- amino acid(s)
- CIP
- calf intestinal alkaline phosphatase
- Ni-NTA
- nickel-nitrilotriacetic acid
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
References
- 1. Long J. C., and Caceres J. F. (2009) The SR protein family of splicing factors: master regulators of gene expression. Biochem. J. 417, 15–27 10.1042/BJ20081501 [DOI] [PubMed] [Google Scholar]
- 2. Manley J. L., and Krainer A. R. (2010) A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes Dev. 24, 1073–1074 10.1101/gad.1934910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xiao S. H., and Manley J. L. (1997) Phosphorylation of the ASF/SF2 RS domain affects both protein-protein and protein-RNA interactions and is necessary for splicing. Genes Dev. 11, 334–344 10.1101/gad.11.3.334 [DOI] [PubMed] [Google Scholar]
- 4. Zhang Z., and Krainer A. R. (2004) Involvement of SR proteins in mRNA surveillance. Mol. Cell 16, 597–607 10.1016/j.molcel.2004.10.031 [DOI] [PubMed] [Google Scholar]
- 5. Sanford J. R., Gray N. K., Beckmann K., and Cáceres J. F. (2004) A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 18, 755–768 10.1101/gad.286404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang Y., Gattoni R., Stévenin J., and Steitz J. A. (2003) SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol. Cell 11, 837–843 10.1016/S1097-2765(03)00089-3 [DOI] [PubMed] [Google Scholar]
- 7. Das R., Yu J., Zhang Z., Gygi M. P., Krainer A. R., Gygi S. P., and Reed R. (2007) SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol. Cell 26, 867–881 10.1016/j.molcel.2007.05.036 [DOI] [PubMed] [Google Scholar]
- 8. Cáceres J. F., Screaton G. R., and Krainer A. R. (1998) A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev. 12, 55–66 10.1101/gad.12.1.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lai M. C., Lin R. I., and Tarn W. Y. (2001) Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc. Natl. Acad. Sci. U.S.A. 98, 10154–10159 10.1073/pnas.181354098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lai M. C., Lin R. I., Huang S. Y., Tsai C. W., and Tarn W. Y. (2000) A human importin-β family protein, transportin-SR2, interacts with the phosphorylated RS domain of SR proteins. J. Biol. Chem. 275, 7950–7957 10.1074/jbc.275.11.7950 [DOI] [PubMed] [Google Scholar]
- 11. Ngo J. C., Chakrabarti S., Ding J. H., Velazquez-Dones A., Nolen B., Aubol B. E., Adams J. A., Fu X. D., and Ghosh G. (2005) Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Mol. Cell 20, 77–89 10.1016/j.molcel.2005.08.025 [DOI] [PubMed] [Google Scholar]
- 12. Prasad J., Colwill K., Pawson T., and Manley J. L. (1999) The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol. Cell. Biol. 19, 6991–7000 10.1128/MCB.19.10.6991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cao W., Jamison S. F., and Garcia-Blanco M. A. (1997) Both phosphorylation and dephosphorylation of ASF/SF2 are required for pre-mRNA splicing in vitro. RNA 3, 1456–1467 [PMC free article] [PubMed] [Google Scholar]
- 14. Cho S., Hoang A., Sinha R., Zhong X. Y., Fu X. D., Krainer A. R., and Ghosh G. (2011) Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1–70K snRNP protein determines early spliceosome assembly. Proc. Natl. Acad. Sci. U.S.A. 108, 8233–8238 10.1073/pnas.1017700108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lai M. C., and Tarn W. Y. (2004) Hypophosphorylated ASF/SF2 binds TAP and is present in messenger ribonucleoproteins. J. Biol. Chem. 279, 31745–31749 10.1074/jbc.C400173200 [DOI] [PubMed] [Google Scholar]
- 16. Sanford J. R., Ellis J. D., Cazalla D., and Cáceres J. F. (2005) Reversible phosphorylation differentially affects nuclear and cytoplasmic functions of splicing factor 2/alternative splicing factor. Proc. Natl. Acad. Sci. U.S.A. 102, 15042–15047 10.1073/pnas.0507827102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou Z., and Fu X. D. (2013) Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 122, 191–207 10.1007/s00412-013-0407-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ding J. H., Zhong X. Y., Hagopian J. C., Cruz M. M., Ghosh G., Feramisco J., Adams J. A., and Fu X. D. (2006) Regulated cellular partitioning of SR protein-specific kinases in mammalian cells. Mol. Biol. Cell 17, 876–885 10.1091/mbc.e05-10-0963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghosh G., and Adams J. A. (2011) Phosphorylation mechanism and structure of serine-arginine protein kinases. FEBS J. 278, 587–597 10.1111/j.1742-4658.2010.07992.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liang N., Zeng C., Tao K. P., Sou W. H., Hsia H. P., Qu D., Lau S. N., and Ngo J. C. (2014) Primary structural features of SR-like protein acinusS govern the phosphorylation mechanism by SRPK2. Biochem. J. 459, 181–191 10.1042/BJ20131091 [DOI] [PubMed] [Google Scholar]
- 21. Cléry A., Sinha R., Anczuków O., Corrionero A., Moursy A., Daubner G. M., Valcárcel J., Krainer A. R., and Allain F. H. (2013) Isolated pseudo-RNA-recognition motifs of SR proteins can regulate splicing using a noncanonical mode of RNA recognition. Proc. Natl. Acad. Sci. U.S.A. 110, E2802–E2811 10.1073/pnas.1303445110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ngo J. C., Giang K., Chakrabarti S., Ma C. T., Huynh N., Hagopian J. C., Dorrestein P. C., Fu X. D., Adams J. A., and Ghosh G. (2008) A sliding docking interaction is essential for sequential and processive phosphorylation of an SR protein by SRPK1. Mol. Cell 29, 563–576 10.1016/j.molcel.2007.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Serrano P., Aubol B. E., Keshwani M. M., Forli S., Ma C. T., Dutta S. K., Geralt M., Wüthrich K., and Adams J. A. (2016) Directional phosphorylation and nuclear transport of the splicing factor SRSF1 is regulated by an RNA recognition motif. J. Mol. Biol. 428, 2430–2445 10.1016/j.jmb.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cáceres J. F., Misteli T., Screaton G. R., Spector D. L., and Krainer A. R. (1997) Role of the modular domains of SR proteins in subnuclear localization and alternative splicing specificity. J. Cell Biol. 138, 225–238 10.1083/jcb.138.2.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walsh C. M., Suchanek A. L., Cyphert T. J., Kohan A. B., Szeszel-Fedorowicz W., and Salati L. M. (2013) Serine arginine splicing factor 3 is involved in enhanced splicing of glucose-6-phosphate dehydrogenase RNA in response to nutrients and hormones in liver. J. Biol. Chem. 288, 2816–2828 10.1074/jbc.M112.410803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang Y., Horikawa I., Ajiro M., Robles A. I., Fujita K., Mondal A. M., Stauffer J. K., Zheng Z. M., and Harris C. C. (2013) Downregulation of splicing factor SRSF3 induces p53β, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 32, 2792–2798 10.1038/onc.2012.288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sen S., Talukdar I., and Webster N. J. G. (2009) SRp20 and CUG-BP1 modulate insulin receptor exon 11 alternative splicing. Mol. Cell. Biol. 29, 871–880 10.1128/MCB.01709-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jumaa H., and Nielsen P. J. (1997) The splicing factor SRp20 modifies splicing of its own mRNA and ASF/SF2 antagonizes this regulation. EMBO J. 16, 5077–5085 10.1093/emboj/16.16.5077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yu Q., Guo J., and Zhou J. (2004) A minimal length between tau exon 10 and 11 is required for correct splicing of exon 10. J. Neurochem. 90, 164–172 10.1111/j.1471-4159.2004.02477.x [DOI] [PubMed] [Google Scholar]
- 30. Ajiro M., Jia R., Yang Y., Zhu J., and Zheng Z. M. (2016) A genome landscape of SRSF3-regulated splicing events and gene expression in human osteosarcoma U2OS cells. Nucleic Acids Res. 44, 1854–1870 10.1093/nar/gkv1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Loomis R. J., Naoe Y., Parker J. B., Savic V., Bozovsky M. R., Macfarlan T., Manley J. L., and Chakravarti D. (2009) Chromatin binding of SRp20 and ASF/SF2 and dissociation from mitotic chromosomes is modulated by histone H3 serine 10 phosphorylation. Mol. Cell 33, 450–461 10.1016/j.molcel.2009.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lou H., Neugebauer K. M., Gagel R. F., and Berget S. M. (1998) Regulation of alternative polyadenylation by u1 snRNPs and SRp20. Mol. Cell. Biol. 18, 4977–4985 10.1128/MCB.18.9.4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang Y., and Steitz J. A. (2001) Splicing factors SRp20 and 9G8 promote the nucleocytoplasmic export of mRNA. Mol. Cell 7, 899–905 10.1016/S1097-2765(01)00233-7 [DOI] [PubMed] [Google Scholar]
- 34. Saeki K., Yasugi E., Okuma E., Breit S. N., Nakamura M., Toda T., Kaburagi Y., and Yuo A. (2005) Proteomic analysis on insulin signaling in human hematopoietic cells: identification of CLIC1 and SRp20 as novel downstream effectors of insulin. Am. J. Physiol. 289, E419–E428 [DOI] [PubMed] [Google Scholar]
- 35. Corbo C., Orrù S., and Salvatore F. (2013) SRp20: an overview of its role in human diseases. Biochem. Biophys. Res. Commun. 436, 1–5 10.1016/j.bbrc.2013.05.027 [DOI] [PubMed] [Google Scholar]
- 36. Bedard K. M., Daijogo S., and Semler B. L. (2007) A nucleo-cytoplasmic SR protein functions in viral IRES-mediated translation initiation. EMBO J. 26, 459–467 10.1038/sj.emboj.7601494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang Y., and Steitz J. A. (2005) SRprises along a messenger's journey. Mol. Cell 17, 613–615 10.1016/j.molcel.2005.02.020 [DOI] [PubMed] [Google Scholar]
- 38. Ma C. T., Hagopian J. C., Ghosh G., Fu X. D., and Adams J. A. (2009) Regiospecific phosphorylation control of the SR protein ASF/SF2 by SRPK1. J. Mol. Biol. 390, 618–634 10.1016/j.jmb.2009.05.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aubol B. E., Chakrabarti S., Ngo J., Shaffer J., Nolen B., Fu X. D., Ghosh G., and Adams J. A. (2003) Processive phosphorylation of alternative splicing factor/splicing factor 2. Proc. Natl. Acad. Sci. U.S.A. 100, 12601–12606 10.1073/pnas.1635129100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma C. T., Velazquez-Dones A., Hagopian J. C., Ghosh G., Fu X. D., and Adams J. A. (2008) Ordered multi-site phosphorylation of the splicing factor ASF/SF2 by SRPK1. J. Mol. Biol. 376, 55–68 10.1016/j.jmb.2007.08.029 [DOI] [PubMed] [Google Scholar]
- 41. Aubol B. E., Hailey K. L., Fattet L., Jennings P. A., and Adams J. A. (2017) Redirecting SR protein nuclear trafficking through an allosteric platform. J. Mol. Biol. 429, 2178–2191 10.1016/j.jmb.2017.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cvitkovic I., and Jurica M. S. (2013) Spliceosome Database: a tool for tracking components of the spliceosome. Nucleic Acids Res. 41, D132–D141 10.1093/nar/gks999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fukuhara T., Hosoya T., Shimizu S., Sumi K., Oshiro T., Yoshinaka Y., Suzuki M., Yamamoto N., Herzenberg L. A., Herzenberg L. A., and Hagiwara M. (2006) Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc. Natl. Acad. Sci. U.S.A. 103, 11329–11333 10.1073/pnas.0604616103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hamelberg D., Shen T., and McCammon J. A. (2007) A proposed signaling motif for nuclear import in mRNA processing via the formation of arginine claw. Proc. Natl. Acad. Sci. U.S.A. 104, 14947–14951 10.1073/pnas.0703151104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xiang S., Gapsys V., Kim H. Y., Bessonov S., Hsiao H. H., Möhlmann S., Klaukien V., Ficner R., Becker S., Urlaub H., Lührmann R., de Groot B., and Zweckstetter M. (2013) Phosphorylation drives a dynamic switch in serine/arginine-rich proteins. Structure 21, 2162–2174 10.1016/j.str.2013.09.014 [DOI] [PubMed] [Google Scholar]
- 46. Lukasiewicz R., Velazquez-Dones A., Huynh N., Hagopian J., Fu X. D., Adams J., and Ghosh G. (2007) Structurally unique yeast and mammalian serine-arginine protein kinases catalyze evolutionarily conserved phosphorylation reactions. J. Biol. Chem. 282, 23036–23043 10.1074/jbc.M611305200 [DOI] [PubMed] [Google Scholar]
- 47. Sen S., Jumaa H., and Webster N. J. (2013) Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat. Commun. 4, 1336 10.1038/ncomms2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu X., Yang D., Ding J. H., Wang W., Chu P. H., Dalton N. D., Wang H. Y., Bermingham J. R. Jr., Ye Z., Liu F., Rosenfeld M. G., Manley J. L., Ross J. Jr., Chen J., Xiao R. P., et al. (2005) ASF/SF2-regulated CaMKIIδ alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 120, 59–72 10.1016/j.cell.2004.11.036 [DOI] [PubMed] [Google Scholar]
- 49. Koizumi J., Okamoto Y., Onogi H., Mayeda A., Krainer A. R., and Hagiwara M. (1999) The subcellular localization of SF2/ASF is regulated by direct interaction with SR protein kinases (SRPKs). J. Biol. Chem. 274, 11125–11131 10.1074/jbc.274.16.11125 [DOI] [PubMed] [Google Scholar]
- 50. Aubol B. E., Keshwani M. M., Fattet L., and Adams J. A. (2018) Mobilization of a splicing factor through a nuclear kinase-kinase complex. Biochem. J. 475, 677–690 10.1042/BCJ20170672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aubol B. E., Wu G., Keshwani M. M., Movassat M., Fattet L., Hertel K. J., Fu X. D., and Adams J. A. (2016) Release of SR proteins from CLK1 by SRPK1: a symbiotic kinase system for phosphorylation control of pre-mRNA splicing. Mol. Cell 63, 218–228 10.1016/j.molcel.2016.05.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bertram K., Agafonov D. E., Dybkov O., Haselbach D., Leelaram M. N., Will C. L., Urlaub H., Kastner B., Lührmann R., and Stark H. (2017) Cryo-EM structure of a pre-catalytic human spliceosome primed for activation. Cell 170, 701–713.e11 10.1016/j.cell.2017.07.011 [DOI] [PubMed] [Google Scholar]
- 53. Bertram K., Agafonov D. E., Liu W. T., Dybkov O., Will C. L., Hartmuth K., Urlaub H., Kastner B., Stark H., and Lührmann R. (2017) Cryo-EM structure of a human spliceosome activated for step 2 of splicing. Nature 542, 318–323 10.1038/nature21079 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.