

Abstract
Autophagy is critical for maintaining cellular function via clearance of excess nutrients and damaged organelles. In pancreatic β-cells, it helps counter the endoplasmic reticulum (ER) stress that impairs insulin secretory capacity during Type 2 diabetes. Chronic exposure of β-cells to saturated fatty acids (FAs) such as palmitate stimulates ER stress and modulates autophagy, but the effects of unsaturated FAs such as oleate, which are also elevated during obesity, are less well understood. We therefore treated MIN6 cells and mouse islets for 8–48 h with either palmitate or oleate, and then monitored autophagic flux, signaling pathways, lysosomal biology, and phospholipid profiles. Compared with palmitate, oleate more effectively stimulated both autophagic flux and clearance of autophagosomes. The flux stimulation occurred independently of ER stress, nutrient-sensing (mTOR) and signaling pathways (protein kinases A, C, and D). Instead the mechanism involved the exchange factor directly activated by cAMP 2 (EPAC2). Oleate reduced cellular cAMP, and its effects on autophagic flux were reproduced or inhibited, respectively, by Epac2 knockdown or activation. Oleate also increased lysosomal acidity and increased phospholipid saturation, consistent with improved autophagosomal fusion with lysosomes. We conclude that a potent stimulation of autophagy might help explain the known benefits of unsaturated FAs in countering the toxicity of saturated FAs in β-cells during obesity and lipid loading.
Keywords: pancreatic islet, lipid, autophagy, cyclic AMP (cAMP), beta cell (B-cell)
Introduction
Macroautophagy (referred to as autophagy hereafter) is a critical degradative mechanism for maintaining cellular function and survival under both normal and stress conditions (1). It is characterized by formation of a double membrane structure, the autophagosome, which engulfs target proteins or organelles (2). Damaged organelles, aggregated protein, and excess nutrients are removed and metabolites recycled. Autophagy is classically activated by starvation, an effect mediated by inhibition of the nutrient sensing protein kinases mammalian target of rapamycin C1 (mTORC1)4 or activation of AMPK (3–5). There is also growing evidence for noncanonical activation pathways for autophagy, which can occur in particular cell types or specialized situations (6).
Pancreatic β-cells play an essential role in maintaining glucose homeostasis by regulating the secretion of insulin, and impaired β-cell function is instrumental in the development of Type 2 diabetes (T2D). Recently, dysregulation of autophagy has emerged as a potential factor that might contribute to this disease setting (7–9). The evidence was based largely on mouse models employing β-cell–specific deletion of the essential autophagy gene, Atg7, which displayed impaired glucose tolerance, especially in the context of high-fat feeding or genetic predispositions to obesity and/or protein aggregation (10–14). Mechanistically, Atg7 deletion inhibited both β-cell mass and insulin secretion, potentially explained by endoplasmic reticulum (ER) stress and mitochondrial defects (10, 11). Several studies have also pointed to an impairment of autophagic clearance in β-cells during T2D, based on alterations in morphology, and accumulation of the autophagosomal marker LC3II (15, 16). However, the latter is generated by the lipidation of the precursor molecule LC3I as part of the proximal signaling cascade, so its accumulation could represent either enhanced flux through the upstream pathway, as well as a reduction in distal clearance. To distinguish between these possibilities we (17), and others (18), have injected mice with chloroquine (CQ) to compromise lysosomal function and hence clearance. Under these conditions high-fat feeding causes an accumulation of LC3II puncta in β-cells. These results suggest that, regardless of what happens in frank diabetes, autophagic flux is activated during obesity or pre-diabetes, and this might potentially help in delaying β-cell failure.
Palmitate and oleate are, respectively, the most prevalent saturated and unsaturated fatty acids (FAs) in the circulation, and both are augmented by high-fat diets. Chronic or excessive exposure to saturated FAs is often considered detrimental to multiple organs and cell types including pancreatic β-cells, whereas unsaturated ones are viewed as less toxic, and potentially beneficial (19). In vitro studies on β-cell autophagy have thus focused predominately on palmitate and have generally (10, 20–25) shown an enhancement of autophagy when conducted in conjunction with lysosomal inhibitors to allow quantification of flux. Mechanistically, this increase is thought to be secondary to the stimulation of ER stress (20, 25) that occurs in response to chronic palmitate treatment of β-cells (26). But there are also indications of an impairment of fusion between autophagosomes and lysosomes potentially explained by changes in lumenal pH or lipid remodelling (22–25). More limited evidence suggests that oleate might enhance LC3II accumulation under steady-state conditions (10, 21, 22). But there have been no detailed studies specifically addressing the effect of oleate on autophagic flux in β-cells. This contrasts with the situation in other cell types, where unsaturated FAs have been shown to promote proximal autophagy via a variety of mechanisms (27–30). Therefore, the aim of the current study was to compare the effects of palmitate and oleate on both proximal and distal autophagy in β-cells, and to address underlying mechanisms.
Results
Oleate and palmitate stimulate autophagic flux in mouse pancreatic β-cells
We sought to establish a threshold for FA-stimulated autophagy in MIN6 cells, as monitored by accumulation of LC3II (Fig. 1A). The steady-state levels of this autophagic marker were unaltered by 48 h pretreatment with either the saturated FA, palmitate, or the unsaturated FA, oleate, at 0.2 or 0.4 mm palmitate alone, or in combination at 0.2 mm. As expected under these conditions, the ER stress marker CHOP was increased by palmitate, with a threshold at 0.4 mm (Fig. S1). Oleate was without effect, even in combination with palmitate (Fig. S1A). As measured in the presence of CQ, however, autophagic flux was selectively enhanced by 0.4 mm oleate (Fig. 1A). In terms of time dependences, neither FA was active at 8 h (Fig. 1B), whereas oleate promoted a transient increase in steady-state LC3II levels at 24 h, and augmented flux by 24 h with a maximal effect at 48 h (Fig. 1, C and D). In many further experiments conducted over several years, with multiple batches and passages of cells, we established that palmitate was more variable in its responses than oleate, such that the unsaturated FA was confirmed as the more effective stimulator of autophagic flux overall (summarized Fig. 1E). Similar results were obtained using isolated mouse islets maintained for 48 h with the different FAs (Fig. 1F).
Figure 1.

Effect of unsaturated and saturated FAs on autophagic flux in MIN6 cells or mouse islets. A–E, levels of the autophagic marker LC3II in MIN6 cells after treatment with (A) oleate or palmitate, alone or combined, at the indicated concentrations for 48 h; or 0.4 mm oleate or 0.4 mm palmitate for (B) 8, (C) 24, or (D and E) 48 h (n = 5–12). F, levels of LC3II in isolated mouse islets with 48 h treatment of 0.4 mm oleate or 0.4 mm palmitate (n = 4). The BSA-treated group was used as the control. Where indicated MIN6 cells or islets were incubated with or without 50 μm CQ for 2 h prior to cell lysis. Data shown are the mean ± S.E. of the densitometric quantification. Statistical analyses were done with one-way ANOVA with Sidak post hoc test. *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001.
Oleate enhances lysosomal size and acidification
Accumulation of LC3II is a function of both its generation via upstream signaling and degradation following fusion of autophagosomes with lysosomes. We first determined whether FAs alter the size of the lysosomal compartment, as assessed using both the dye LysoTracker (Fig. 2A), and staining for the lysosome-associated membrane protein 1 (Lamp-1) (Fig. S2). Indeed lysosomes were markedly expanded but with little distinction between FAs or treatment times, suggesting that changes in the numbers of lysosomes do not account for the differential effects of oleate and palmitate on autophagic flux. To address functional alterations we employed LysoSensor, which exhibits a yellow to blue shift with increases in acidity, which is independent of the size of the uptake compartment. In this manner we determined that both FAs slightly decreased lysosomal pH at 8 h, with more modest effects at 48 h (Fig. 2B). This would suggest improved functionality, because many lysosomal enzymes require low pH for optimal activity.
Figure 2.

FAs enhance lysosomal amount and acidification. MIN6 cells were stained with (A) LysoTracker Red and co-stained with DAPI blue for nuclei (n = 4), or (B) LysoSensor yellow/blue following 8- or 48-h treatments with 0.4 mm oleate (OA) or 0.4 mm palmitate (PA) (n = 4), versus BSA-treated groups as the control (Ctl). Insets show merged images of LysoTracker and DAPI staining. Scale bar = 25 μm. C, quantification of the yellow versus blue intensity ratio is shown as the mean ± S.E. (n = 4). Statistical analyses were by one-way ANOVA with Sidak post hoc test. *, p < 0.05 and **, p < 0.01 versus the time-matched control.
Oleate augments autophagosome–lysosome fusion and phospholipid saturation
We next monitored the capacity for autophagosomes to fuse with lysosomes, using a mRFP-GFP-LC3 construct (31). This appears yellow in autophagosomes, but red upon fusion with the acidic compartment because of the accompanying reduction in pH, which quenches GFP fluorescence. We found that oleate pretreatment significantly increased the ratio of red: yellow puncta at both 8 and 48 h, suggestive of enhanced clearance (Fig. 3, A and B). The more modest effects of palmitate were not statistically significantly different from control. The fusion of membranes is influenced by their fluidity, which in turn depends upon the presence of unsaturated FA side chains in the membrane bilayer (32). We therefore used LC-MS to determine how FA pretreatment of β-cells alters the major lipid components of cellular membranes. Oleate reduced the saturation of both PC and PE by around 30% versus control, with significant but less profound decreases in PI and SM (Fig. 3C). In contrast, palmitate modestly enhanced the saturation of PE (20%), PC (15%), and SM (5%). Neither FA altered the saturation of PS (Fig. 3C), nor the overall mass of any of these lipids (not shown). Although these alterations represent total cellular membrane it is noteworthy that they are much more apparent in those lipids that are enriched in late endosomes/lysosomes (PC, PE, and SM) than in PI and PS, which are depleted in these compartments (32). Taken together these results suggest that oleate, in addition to stimulating upstream pathways, would also promote the clearance of LC3II via a combination of decreases in lysosomal pH and increases in membrane fluidity.
Figure 3.

FAs increase autophagosome-lysosomal fusion. MIN6 cells were transfected with the mRFP-GFP-LC3 construct 24 h prior to treatment with 0.4 mm oleate (OA), 0.4 mm palmitate (PA), or BSA control (Ctl) for a further 48 h. A, representative fluorescence images illustrate the mRFP (red) and mRFP + GFP (yellow) signals; scale bar = 50 μm. Insets are the magnified images, scale bar = 20 μm. Arrows indicate the mRFP signal remaining after autophagosome-lysosomal fusion. B, quantification of red (fused) versus yellow (nonfused) and data shown are the mean ± S.E. of the fluorescence intensity (n = 4). Statistical analyses were with one-way ANOVA with a Sidak post hoc test. **, p < 0.01, and ***, p < 0.001 versus the time-matched control. C, MIN6 cells were incubated with 0.4 mm oleate or 0.4 mm palmitate for 48 h before being subjected to MS analysis of the saturation of lipid species: PC, PE, PI, PS, and SM. Data shown are the mean ± S.E. Statistical analyses were with two-way ANOVA with Tukey's post hoc test. #, p < 0.0001 versus control, *, p < 0.001.
ER stress mediates autophagy in response to palmitate but not oleate
We next sought to determine the mechanisms contributing to the upstream pathway. We first addressed the relevance of ER stress, using the chemical chaperone PBA. As shown previously (33), this protected against induction of the markers CHOP, ATF4, and phospho-PERK in response to palmitate (Fig. S1, B–D), but more importantly it also inhibited LC3II accumulation (Fig. 4A). In keeping with our prior studies with this model (34), oleate did not stimulate ER stress (Fig. S1), nor did PBA block its capacity to augment LC3II (Fig. 4B). The results confirm previous findings that ER stress contributes to the stimulation of autophagic flux due to palmitate (20, 25), but now show that this mechanism does not account for the response due to oleate.
Figure 4.
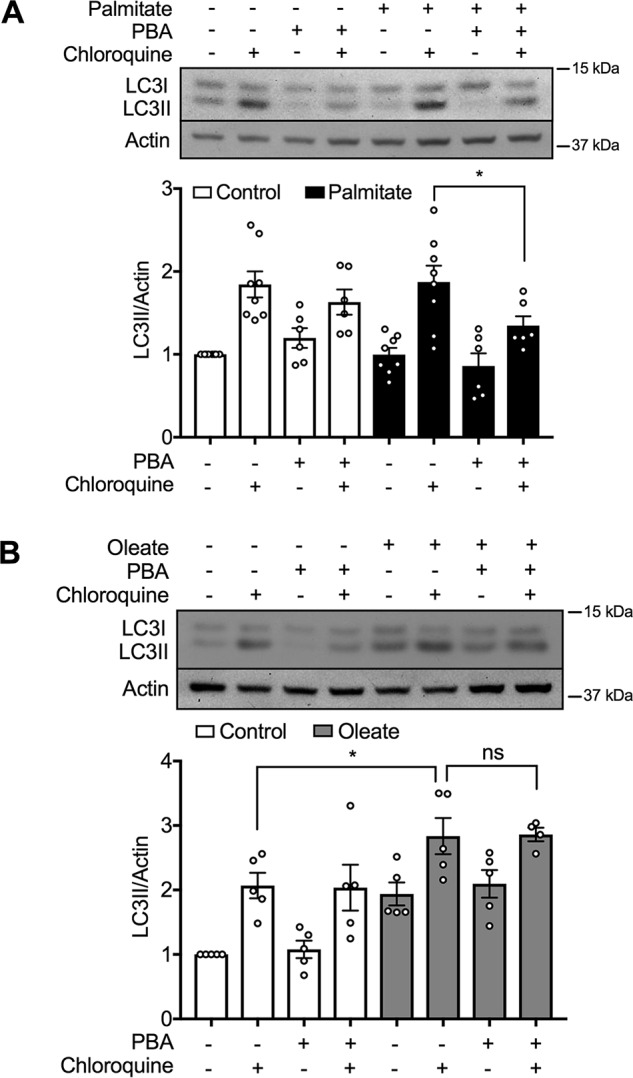
ER stress mediates induction of autophagy by palmitate but not by oleate. A and B, levels of LC3II in MIN6 cells after treatment with (A) 0.4 mm palmitate or (B) 0.4 mm oleate or BSA control, in the presence or absence of 2.5 mm PBA for 48 h, then incubated with or without 50 μm CQ for 2 h prior to cell lysis (n = 6 and 5, respectively). Data shown are the mean ± S.E. of the densitometric quantification. Statistical analyses were with one-way ANOVA with a Sidak post hoc test. *, p < 0.05.
Oleate does not alter the AMPK or mTOR pathways
AMPK and mTOR are key responders to changes in nutritional status and play especially critical roles in multiple facets of β-cell biology (35–38). In particular they are established regulators of autophagy, and, indeed, these pathways have been previously implicated in the induction of autophagy due to oleate in other cell systems (28, 29). In our model, however, we observed no changes in the regulatory phosphorylation sites of AMPK due to FA pretreatment, nor in the phosphorylation status of its substrate, ACC (Fig. S3, A and B). AMPK plays an unusual role in β-cells where, in contrast to most other systems, its inhibition promotes autophagy (39). We were able to confirm this using the AMPK inhibitor, Compound C, which enhanced LC3II accumulation in a manner nonadditive to that of oleate (Fig. 5A). The latter response, however, was unaltered by either AICAR (Fig. 5B) or A769662 (Fig. 5C), respectively, even though these compounds stimulated AMPK as expected (Fig. S3C). This makes it unlikely that changes in AMPK mediate the observed effects of oleate on autophagy. Likewise, neither FA impacted on mTOR activation, as measured by the phosphorylation of either mTOR itself or its downstream substrate, 4EBP (Fig. S4, A and B). We further tested the impact of the mTOR inhibitor, rapamycin, which was without effect in our model, either basally or in the presence of FAs (Fig. S4C). Its action was, however, confirmed directly as reduced phosphorylation of its downstream kinase p70S6K (Fig. S4D). Rapamycin is viewed as a canonical activator of autophagy, although its function in β-cells has almost always been assayed microscopically by visualization of autophagosomes (40, 41) rather than LC3II accumulation as we attempted. Regardless, the fact that we did detect effects of oleate, but not rapamycin using this assay, clearly argues against the involvement of mTOR inhibition in the mechanism of action of the FA.
Figure 5.
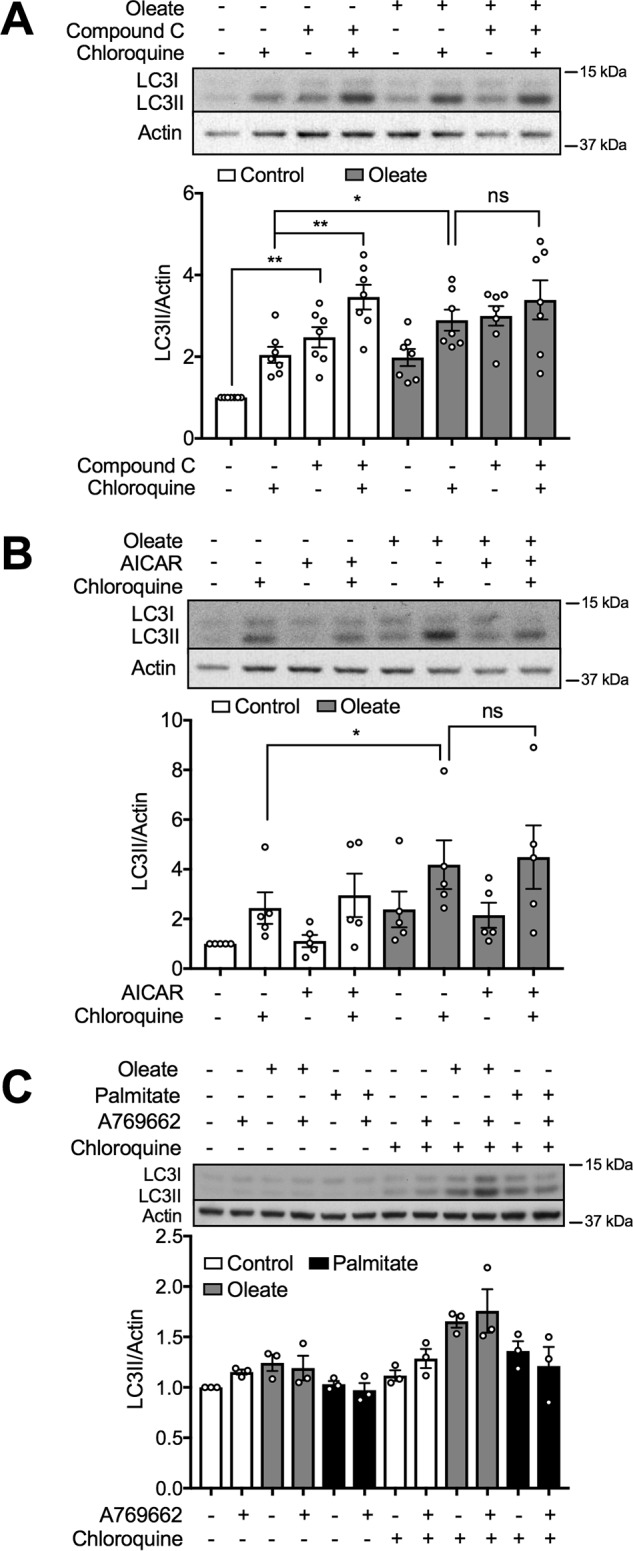
Inhibition of AMPK induces autophagy but oleate-activated autophagy is independent of AMPK. A and B, levels of LC3II in MIN6 cells after 48 h treatment with 0.4 mm oleate in the presence or absence of (A) 5 μm Compound C, or (B) 500 μm AICAR (n = 7 and 5, respectively). C, LC3II levels in MIN6 cells after a 48-h treatment with 10 μm A769662 in the presence of 0.4 mm oleate or 0.4 mm palmitate. The cells were incubated with or without 50 μm CQ for 2 h prior to cell lysis. Controls are (A and C) 0.1% DMSO in BSA or (B) BSA. Data shown are the mean ± S.E. of the densitometric quantification. Statistical analyses were with one-way ANOVA with a Sidak post hoc test. *, p < 0.05 and **, p < 0.01.
Oleate-induced autophagy is independent of GPR40, oxidative stress, PKD, and PKC
We therefore looked to other potential mechanisms, including activation of the cell surface receptor, GPR40 (42, 43), which is abundantly expressed in β-cells and known to be ligated by unsaturated FAs (44). However, a GPR40 antagonist did not impact on oleate-stimulated autophagy (Fig. S5). Other possibilities include oxidative stress, and activation of various protein kinases, particularly PKD, the inhibition of which in β-cells has been previously linked to repression of autophagy (45). However, LC3II generation in response to oleate was unaltered by the antioxidant NAC, nor by inhibitors of PKC (Go-6976) and PKD (CID-755673) (Fig. S6)
Reductions in cAMP contribute to oleate-stimulated autophagy, but via EPAC2 rather than PKA signaling
We investigated cAMP signaling more extensively because of the surprising initial finding that accumulation of this intracellular mediator was reduced by chronic exposure of MIN6 cells to oleate, but not palmitate (Fig. 6A). We therefore assessed the potential role of PKA, the cAMP-stimulated protein kinase. However, the effects of oleate on LC3II were not altered by, respectively, blocking or stimulating PKA, using either the inhibitor H-89 (Fig. 6B), or the long-acting cAMP analogue, 8-Br-cAMP (Fig. 6C). However, the latter is not selective for PKA, but can also activate EPAC, which serves as an exchange factor to stimulate small monomeric G-proteins. We therefore used, 6-Bnz-cAMP-AM, which shows selectivity for PKA activation (46) but this was without any effect on autophagic flux in mouse islets pretreated with oleate (Fig. 6D).
Figure 6.

Oleate reduces cAMP levels and induces autophagy independent of PKA. A, MIN6 cells were treated with 0.4 mm oleate or 0.4 mm palmitate or BSA control, before lysis for assessment of cAMP levels (n = 3). Results are expressed as % of control (18.7 ± 0.3 fmol/mg of protein). B and C, levels of LC3II in MIN6 cells after 48 h treatment with 0.4 mm oleate or BSA control in the presence of (B) 50 μm H-89 (n = 4), or (C) 100 nm 8-Br-cAMP-AM (n = 9). The cells were incubated with or without 50 μm CQ for 2 h prior to cell lysis. D, levels of LC3II in isolated mouse islets with 48 h treatment of 10 μm 6-Bnz-cAMP in the presence or absence of 0.4 mm oleate (n = 4). The islets were incubated with 50 μm CQ for 2 h prior to cell lysis. The BSA-treated group was used as the control. Data shown are the mean ± S.E. of the densitometric quantification. Statistical analyses were with one-way ANOVA with a Sidak post hoc test. *, p < 0.05 as indicated or versus control.
Focusing instead on the EPAC pathway, we found in preliminary studies that the general inhibitor (ESI-09), exhibited off-target effects resulting in disruption of ER stress signaling pathways (not shown). It has also been reported to cause protein denaturation (47). We therefore chose to knockdown Epac2 (also known as Rapgef4), the most abundant and functionally relevant isoform found in β-cells (48). The response to oleate under these conditions was significantly further enhanced (Fig. 7A), although basal accumulation of LC3II was unaltered, possibly because the degree of knockdown was incomplete (Fig. 7B). To further assess this potential mechanism we made use of cell permeable cAMP analogue 8-pCPT-2-O-Me-cAMP-AM (8-CPT-AM), which unlike the 8-Br-cAMP employed above, selectively activates EPAC and not PKA. Using mouse islets incubated in the presence of CQ for measurements of autophagic flux, we observed that activation of EPAC completely blocked the stimulatory effects of oleate pretreatment (Fig. 7C). These findings suggest that inhibition of EPAC2, secondary to reductions in its upstream regulator, cAMP, contributes to the mechanism whereby oleate stimulates autophagic flux in β-cells.
Figure 7.
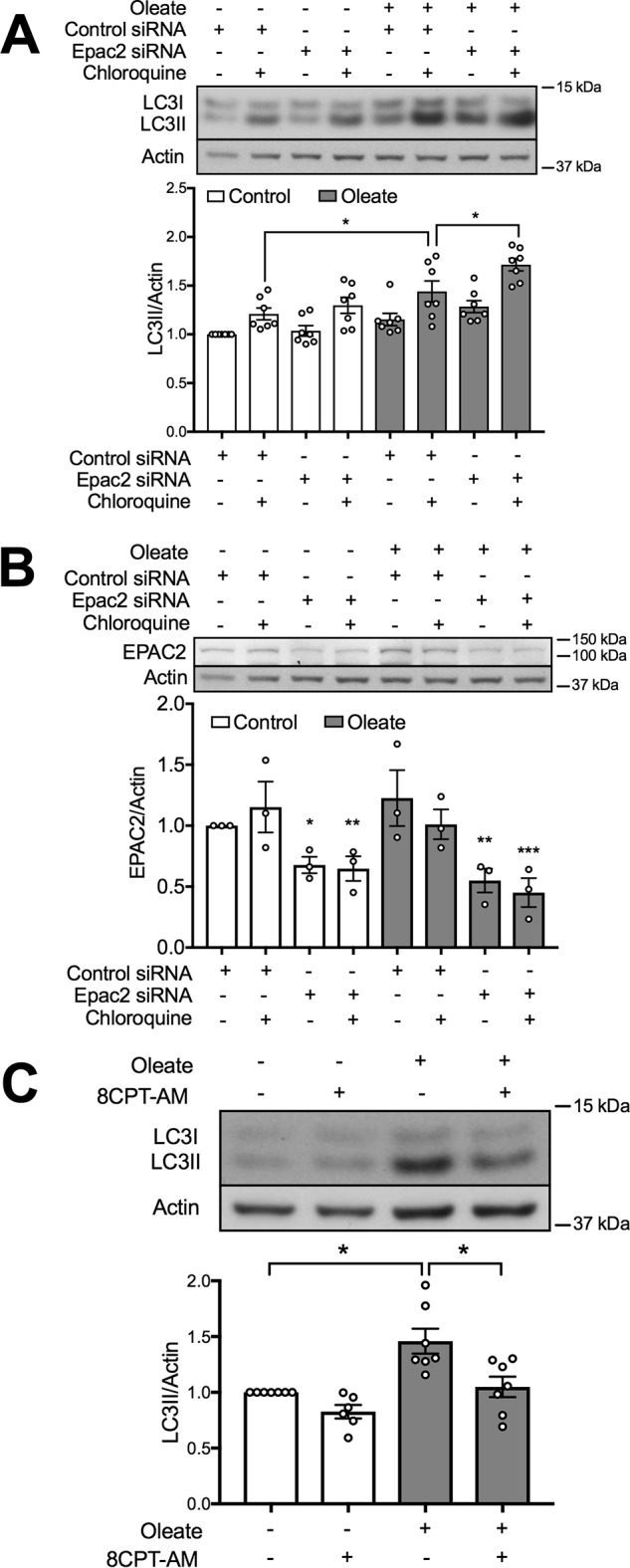
Oleate induces autophagy partially mediated by EPAC2. MIN6 cells were transfected with Epac2 or the control siRNA, and then treated with 0.4 mm oleate. The cells were incubated with or without 50 μm CQ for 2 h prior to cell lysis. Levels of (A) LC3II and (B) EPAC2 were examined in the same gel (same actin blot; n = 7). C, isolated mouse islets were treated with 0.4 mm oleate in the presence or absence of 10 μm 8-CPT-AM or DMSO, then all were incubated with 50 μm CQ for 2 h before cell lysis (n = 4). In all cases, BSA-treated groups served as the control. Data shown are the mean ± S.E. Statistical analyses were with one-way ANOVA with a Sidak post hoc test. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 as indicated or versus control.
Discussion
It is now generally established that obesity and T2D are associated with enhanced accumulation of autophagic markers (7–9, 15). Because most analyses are undertaken under steady-state conditions, however, it has been difficult to establish whether these findings represent a stimulation of autophagy or an inhibition of autophagosomal clearance. More recent studies, incorporating CQ injection in mice, have strongly suggested that autophagic flux is indeed augmented by high-fat feeding (17, 18). Additional findings with knock-out mice would suggest that this most probably represents a beneficial adaptation to obesity, helping β-cells avoid ER stress and the cellular failure that occurs in the transition to overt T2D (10, 11). Therefore understanding the mechanisms whereby FAs stimulate autophagy in β-cells has become an important experimental goal. To date, the overwhelming majority of studies in this area have focused on palmitate, which is surprising from the viewpoint that autophagy is advantageous to β-cells, whereas palmitate is not. Hence we have focused on oleate. Our results show that oleate induced a delayed, proximal autophagic response (24 h), but promoted earlier increases in lysosomal pH and fusion with autophagosomes (8 h). This would constitute a favorable environment prior to signal initiation, such that the resultant, mild increase in steady-state autophagy might contribute to the known protective effects of unsaturated FAs against cellular stress (49, 50). In contrast, canonical stimuli trigger a robust, fast and transient autophagic response, subjected to feedback (51), and often harmful if that feedback is overridden (52).
Although widely studied previously, the effects of palmitate on autophagy in β-cells are somewhat controversial. This is potentially explained by variations in experimental conditions, most notably the final concentration of free FAs, which depends critically on the molar ratio of exogenous FA to BSA, and how these are coupled (53). Our model involves pre-coupling at a ratio of around 3:1 (0.4 mm; 0.9% BSA), which equates to a free concentration in low micromolar, or moderately elevated physiological range (53). In our hands, this constitutes the minimal dose for 48-h treatments of palmitate and oleate to stimulate ER stress or autophagy, respectively (Fig. S1, Fig. 1A). Other variables across the different models include concentrations of glucose and serum, and duration of FA exposure. Species differences also play a role, with rat β-cells being more sensitive to palmitate exposure versus mouse or human β-cells because of their lower expression of desaturase enzymes (54). On the other hand, there is an overwhelming consensus that palmitate promotes β-cell apoptosis and ER stress, in all of these same highly variable models (26), suggesting that there is something inherently different about measuring autophagy as an end point, and unrelated to whether flux or steady-state levels are assayed. In our MIN6 model LC3II accumulation in response to palmitate varied over several years with differing passages and aliquots of cells. One interpretation of this variability is that palmitate exerts multiple and conflicting effects on autophagy, the balance of which determines the overall outcome. One positive input is almost certainly ER stress (20, 25). Thus we found that PBA inhibited autophagic flux in the presence of palmitate, reducing LC3II to levels below those of CQ alone (Fig. 4A). This implies that under conditions in which ER stress is countervailed, the residual effect of palmitate on autophagy is negative. Inhibitory inputs, potentially due to disruption of mitochondrial function (22, 24), might be especially prominent at higher molar ratios of palmitate:BSA and in rat β-cells, whereas in mouse cells and in milder models, activation predominates.
Our most important finding is that, despite its relatively neglected prior status, oleate is a more effective activator of autophagy in β-cells than palmitate in vitro, and thus potentially makes an important contribution in response to high-fat feeding in vivo. Oleate does not activate ER stress (26, 34), making this an unlikely mechanism for triggering autophagy, which we have now formally confirmed using the chemical chaperone PBA. We also ruled out a number of alternative mechanisms based on effects of FAs in other cell systems including PKC activation (27) and reactive oxygen species generation (28). Oleate has previously been shown to activate both GPR40 and PKD in β-cells (44), either of which routes might also stimulate autophagy (42, 43). However, inhibition of either of these pathways did not alter oleate-induced LC3II accumulation. Likewise we found no evidence for modulation of AMPK and mTOR, consistent with investigations of oleate-mediated autophagy on other cell types (28, 29). The fact that FAs did not activate AMPK in MIN6 cells is consistent with our earlier findings using mouse islets (55) but contrasts with other studies (56, 57), again probably explained by higher doses of FA and glucose than we employed. Palmitate treatment of β-cells has been previously reported to inhibit mTORC1 chronically (20), but stimulates in the short-term, in each case at concentrations higher than those we have employed. Notably, oleate was ineffective (58).
Instead our findings suggested a role for a noncanonical activation pathway involving reductions in cAMP and inhibition of EPAC2. This is the predominant isoform expressed in β-cells where its acts in the amplification of insulin secretion (48). Perhaps under these conditions it would be advantageous to suppress autophagic clearance of insulin secretory granules and this has been previously shown to correlate inversely with secretory capacity in β-cells in other experimental settings (59). More broadly, EPAC isoforms have been implicated in both the stimulation (60, 61) and inhibition of autophagy (62), depending on context. Likewise cAMP, acting through PKA, can exert both positive and negative effects (63–65). This probably accounts for our observation that oleate-stimulated autophagic flux was suppressed by the EPAC selective activator, 8-CPT-AM, but by neither 8-Br-cAMP, which targets both EPAC and PKA pathways, nor the selective PKA agonist, 6-Bnz-cAMP-AM. Nevertheless, inhibition of EPAC probably does not account for the whole mechanism because knocking down Epac2 was not sufficient in itself to stimulate autophagy, despite its stimulation overcoming the effects of oleate. Thus we conclude inhibition of EPAC2 is necessary but not sufficient for the activation of autophagy by oleate, suggesting that the FA must also exert additional effects that will require additional studies for elucidation. Likewise, the exact mechanism underlying EPAC2-suppressed autophagy is not clearly known, although EPAC and its downstream effector Rap2b, have been reported to be recruited to phagosomes, where they inhibited autophagy (62).
Our findings are not necessarily in dispute with the recent demonstration that GLP1 augments LC3II accumulation due to glucolipotoxicity (25), because the mechanism of action was not addressed in that study, and so might be independent of cAMP. Other data showing that GLP1 activates both mTORC1 (66) and AMPK (67) would also seem to exclude these protein kinases, suggesting that the mechanism linking GLP1 with autophagy in β-cells is likely to be unusual. It is probably also context dependent, because GLP1 inhibits LC3II accumulation due to tacrolimus dosing (68) in contrast to the situation with glucolipotoxicity (25). It is not surprising that β-cells would display a noncanonical mechanism for mediating the response to oleate, because they display multiple, unusual features in terms of autophagic regulation. Thus, in contrast to many tissues, inhibition of AMPK promotes LC3II accumulation (39), and starvation has even been linked to repression of autophagy (45). Moreover, high-fat feeding stimulates autophagic flux in pancreatic islets, but inhibits it in liver in the very same mice (17). It is also noteworthy that oleate and palmitate differentially regulate cAMP levels in β-cells, another novel finding of our study. The underlying mechanisms will also need to be investigated in future but could relate to the differences in transcriptional programs elicited by the two FAs (69).
In terms of downstream pathways, our results show that oleate and palmitate act very similarly on lysosomal compartment size and in lowering lumenal pH. Palmitate has been previously shown to both enhance (22, 24, 25) and diminish (20) LysoTracker staining in β-cells, accompanied by indications of impaired lysosomal function and fusion with autophagosomes (22–25). Proposed mechanisms included dysregulation of pH gradients (22) or membrane lipid composition (23). All of these studies, however, were conducted in rat β-cells, and employed higher concentrations of FAs and/or glucose than used here. Impaired mitochondrial function and depletion of ATP as observed in at least some of those reports (22, 24) would be expected to compromise lysosomal pH regulation. In contrast, effects of oleate on the size or function of the lysosomal compartment in β-cells have never been addressed, and reported impairments of fusion with autophasomes were based on less direct protocols (22, 24) than the mRFP-GFP-LC3 reporter employed here. In our hands, oleate was better at promoting fusion of lysosomes and autophagosomes than palmitate. This is potentially explained by the observation that although both FAs lower lysosomal pH, which would be expected to be stimulatory, oleate additionally increased the phospholipid desaturation, a critical determinant of membrane fluidity and fusogenicity (32). In contrast, palmitate mildly increased saturation albeit to a lesser extent to that previously reported for rat β-cells, which thus displayed defective autophagic clearance (23).
In conclusion our major findings are that oleate is more effective at promoting autophagic flux than is palmitate; selectively impacts on autophagy via a noncannonical pathway involving the cAMP/EPAC2 pathway; and enhances fusion of lysosomes and autophagosomes consistent with decreases in both lysosomal pH and membrane phospholipid desaturation. Thus effects of unsaturated FAs, which are also present in high-fat diets, need to be taken into consideration when evaluating the autophagy in β-cells in models of obesity and diabetes.
Experimental procedures
Reagents and antibodies
Chemicals and reagents were from Sigma, unless otherwise indicated. Bafilomycin A1 was from Santa Cruz, mRFP-GFP-LC3 construct (31), NAC, PKC inhibitor (Go-6976), JNK Inhibitor II (for both JNK-1 and -2, Calbiochem), PKD inhibitor, Compound C, AICAR, EPAC inhibitor ESI 09, 8-Br-cAMP, and 8-pCPT-2-O-Me-cAMP-AM (Tocris). 6-Bnz-cAMP-AM, control, and Epac2 siRNA are from Dharmacon (Horizon Discovery Group Company, Lafayette, CO). Primary antibodies used in the study were: mouse monoclonal anti-β-actin and rabbit polyclonal anti-CHOP (Santa Cruz), anti-LC3, and anti-Lamp-1. Rabbit polyclonal anti-EPAC2, anti-ATF4, anti-phospho-AMPK, anti-AMPK, anti-phospho-ACC, anti-ACC, anti-phospho-4EBP, anti-4EBP, anti-phospho-mTOR (Ser-2448), anti-mTOR, anti-phospho-PERK, anti-phospho-p70S6K, and anti-p70SK6 (Thr-389) were from Cell Signaling Technology (Danvers, MA).
Animals and islet isolation
All mouse studies were approved by the Garvan/St. Vincent's Hospital Animal Ethics Committee (AEC). C57Bl6 mouse islets were isolated by thermolysin and liberase digestion (Roche Diagnostics, IN) and gradient separation using Ficoll (GE Healthcare, Chalfont St. Giles, UK) as described previously (55).
Cell culture and treatment
The mouse MIN6 insulinoma cell line was routinely passaged and cultured as described previously (70), and was used at passages between 29 and 37 in this study. MIN6 cells were grown at 37 °C and 5% CO2 in Dulbecco's modified Eagle's medium (25 mm glucose), supplemented with 10% fetal calf serum, 10 mm HEPES, 50 units/ml of penicillin, and 50 μg/ml of streptomycin. Isolated islets were cultured in RPMI (11.1 mm glucose) at 37 °C and 5% CO2, supplemented with 10% fetal calf serum, 10 mm HEPES, 2 mm l-glutamine, 50 units/ml of penicillin, and 50 μg/ml of streptomycin, unless other treatments were used as indicated. MIN6 cells or isolated islets were exposed to palmitate or oleate (0.2 or 0.4 mm), which were precoupled to 0.92 g/100 ml of BSA or BSA-only controls in low (5.5 mm) glucose media (69). All treatments were performed with 5.5 mm glucose media. For plasmid transfection, cells were seeded in 12-well plates, and 100 nm mRFP-GFP-LC3 construct was present for 24 h. The media was changed to Dulbecco's modified Eagle's medium (25 mm glucose) and the cells incubated with the treatment indicated for 48 h, changing the media after 24 h. Knockdown experiments with control, or Epac2 siRNA were performed as described previously (71), prior to oleate and CQ treatments.
Western blot analysis
MIN6 cells or islets were lysed and 10 μg of protein was separated by a pre-cast 12% SDS-PAGE gel and transferred to polyvinylidene difluoride membranes. The membranes were incubated with primary and secondary antibodies after blocking with 5% skimmed milk and developed with SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific). Densitometric analysis was performed with the Adobe Photoshop CS4 software.
LysoTracker/LysoSensor staining
MIN6 cells were seeded onto FluoroDishTM (World precision instruments, Sarasota, FL) and lysosomes were visualized with LysoTracker Red DND-99 (ThermoFisher Scientific, L7528) and the lysosomal pH determined by LysoSensor Yellow/Blue DND-160 (ThermoFisher, L7545) as previously described (71). Images were taken by fluorescence microscopy.
Fluorescence microscopy
MIN6 cells were seeded onto the cover slide for treatments. Before subjecting to fluorescent imaging by LysoTracker/LysoSensor signaling, the live cells were washed with PBS three times and then mounted onto the slide (71). For the cells transfected with the mRFP-GFP-LC3 construct or for Lamp-1 staining, they were fixed in 4% paraformaldehyde. For the Lamp-1 staining, fixed cells were incubated with primary antibody followed by the Alexa 555-labeled secondary antibody (Molecular Probes) and then mounted with ProLong® Gold antifade reagent containing DAPI (Molecular Probeds). The fluorescence signals were detected by fluorescence or confocal microscopy, respectively (models DM 5500 or DMI 6000 SP8; Leica Microsystems, Sydney, NSW, Australia).
Lipid profiling with MS
MIN6 cells were seeded at 1 × 106 cells/well in 6-well plates and then treated with FAs for 48 h. Lipids were extracted from whole cell homogenates and separated via LC before analysis by electrospray ionization-tandem MS as described previously (70, 72). The desaturation index was calculated from the ratio of the totals of saturated and unsaturated FA side chains in each phospholipid.
Intracellular cAMP measurement
MIN6 cells were seeded at 3 × 105 cells/well in 12-well plates and then treated with FAs for 48 h. The cells were washed with cold PBS three times and then lysed with 0.1 m HCl. The intracellular cAMP levels were assessed by the Direct cAMP enzyme-linked immunosorbent assay (ELISA) kit (Enzo Life Sciences, Farmingdale, NY) following the manufacturer's instructions. The cAMP level was normalized to the protein concentration.
Statistical analysis
Data are expressed as mean ± S.E. Multiple comparisons between groups were performed using ANOVA followed by Sidak or Tukey's post hoc test. Unpaired t tests were used when the differences between two groups were analyzed. A p value of less than 0.05 was considered statistically significant.
Author contributions
K. Y. C. and T. J. B. conceptualization; K. Y. C., L. O., N. M., P. J. M., C. B., and T. J. B. data curation; K. Y. C., L. O., N. M., P. J. M., C. B., and T. J. B. formal analysis; K. Y. C. and T. J. B. funding acquisition; K. Y. C. validation; K. Y. C. and T. J. B. investigation; K. Y. C. and T. J. B. visualization; K. Y. C. and T. J. B. methodology; K. Y. C. and T. J. B. writing-original draft; K. Y. C. and T. J. B. project administration; K. Y. C., L. O., N. M., P. J. M., C. B., and T. J. B. writing-review and editing; T. J. B. resources; T. J. B. supervision.
Supplementary Material
This work was supported by Projects Grant 1051658 and Fellowship 1024961 from the National Health and Medical Research Council of Australia (to T. J. B.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6.
- mTOR
- mammalian target of rapamycin
- 4EBP
- eukaryotic initiation factor 4E-binding protein
- 8-CPT-AM
- 8-pCPT-2-O-Me-cAMP-AM
- ACC
- acetyl-CoA carboxylase
- AICAR
- 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside
- ATF4
- activating transcription factor 4
- Atg7
- autophagy-related 7
- CHOP
- C/EBP homologous protein
- CQ
- chloroquine
- EPAC
- exchange protein directly activated by cAMP
- ER
- endoplasmic reticulum
- FA
- fatty acids
- GPR40
- G-protein coupled receptor 40
- Lamp-1
- lysosomal-associated membrane protein 1
- LC3
- microtubule-associated protein 1A/1B light chain 3A
- PBA
- 4-phenylbutyrate
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- PERK
- protein kinase RNA-like endoplasmic reticulum kinase
- PI
- phosphatidylinositol
- PKA
- protein kinase A
- PKC
- protein kinase C
- PKD
- protein kinase D
- PS
- phosphatidylserine
- RFP
- red fluorescent protein
- T2D
- type 2 diabetes
- GFP
- green fluorescent protein
- SM
- sphingomyelin
- ANOVA
- analysis of variance
- DAPI
- 4′,6-diamidino-2-phenylindole
- 8-Br-cAMP
- cyclic 8-bromo-AMP
- 6-Bnz-cAMP
- 6-benzyl-cAMP.
References
- 1. Boya P., Reggiori F., and Codogno P. (2013) Emerging regulation and functions of autophagy. Nat. Cell. Biol. 15, 713–720 10.1038/ncb2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mizushima N., Noda T., Yoshimori T., Tanaka Y., Ishii T., George M. D., Klionsky D. J., Ohsumi M., and Ohsumi Y. (1998) A protein conjugation system essential for autophagy. Nature 395, 395–398 10.1038/26506 [DOI] [PubMed] [Google Scholar]
- 3. Nakatogawa H. (2015) Eating the ER and the nucleus for survival under starvation conditions. Mol. Cell. Oncol. 3, e1073416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Takeshige K., Baba M., Tsuboi S., Noda T., and Ohsumi Y. (1992) Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 119, 301–311 10.1083/jcb.119.2.301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Velázquez A. P., Tatsuta T., Ghillebert R., Drescher I., and Graef M. (2016) Lipid droplet-mediated ER homeostasis regulates autophagy and cell survival during starvation. J. Cell Biol. 212, 621–631 10.1083/jcb.201508102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Codogno P., Mehrpour M., and Proikas-Cezanne T. (2011) Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 13, 7–12 [DOI] [PubMed] [Google Scholar]
- 7. Lee M. S. (2014) Role of islet beta cell autophagy in the pathogenesis of diabetes. Trends Endocrinol. Metab. 25, 620–627 10.1016/j.tem.2014.08.005 [DOI] [PubMed] [Google Scholar]
- 8. Watada H., and Fujitani Y. (2015) Minireview: autophagy in pancreatic beta-cells and its implication in diabetes. Mol. Endocrinol. 29, 338–348 10.1210/me.2014-1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stienstra R., Haim Y., Riahi Y., Netea M., Rudich A., and Leibowitz G. (2014) Autophagy in adipose tissue and the beta cell: implications for obesity and diabetes. Diabetologia 57, 1505–1516 10.1007/s00125-014-3255-3 [DOI] [PubMed] [Google Scholar]
- 10. Ebato C., Uchida T., Arakawa M., Komatsu M., Ueno T., Komiya K., Azuma K., Hirose T., Tanaka K., Kominami E., Kawamori R., Fujitani Y., and Watada H. (2008) Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 8, 325–332 10.1016/j.cmet.2008.08.009 [DOI] [PubMed] [Google Scholar]
- 11. Jung H. S., Chung K. W., Won Kim J., Kim J., Komatsu M., Tanaka K., Nguyen Y. H., Kang T. M., Yoon K. H., Kim J. W., Jeong Y. T., Han M. S., Lee M. K., Kim K. W., Shin J., and Lee M. S. (2008) Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 8, 318–324 10.1016/j.cmet.2008.08.013 [DOI] [PubMed] [Google Scholar]
- 12. Quan W., Hur K. Y., Lim Y., Oh S. H., Lee J. C., Kim K. H., Kim G. H., Kim S. W., Kim H. L., Lee M. K., Kim K. W., Kim J., Komatsu M., and Lee M. S. (2012) Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 55, 392–403 10.1007/s00125-011-2350-y [DOI] [PubMed] [Google Scholar]
- 13. Kim J., Cheon H., Jeong Y. T., Quan W., Kim K. H., Cho J. M., Lim Y. M., Oh S. H., Jin S. M., Kim J. H., Lee M. K., Kim S., Komatsu M., Kang S. W., and Lee M. S. (2014) Amyloidogenic peptide oligomer accumulation in autophagy-deficient beta cells induces diabetes. J. Clin. Invest. 124, 3311–3324 10.1172/JCI69625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shigihara N., Fukunaka A., Hara A., Komiya K., Honda A., Uchida T., Abe H., Toyofuku Y., Tamaki M., Ogihara T., Miyatsuka T., Hiddinga H. J., Sakagashira S., Koike M., Uchiyama Y., et al. (2014) Human IAPP-induced pancreatic beta cell toxicity and its regulation by autophagy. J. Clin. Invest. 124, 3634–3644 10.1172/JCI69866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Masini M., Bugliani M., Lupi R., del Guerra S., Boggi U., Filipponi F., Marselli L., Masiello P., and Marchetti P. (2009) Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 52, 1083–1086 10.1007/s00125-009-1347-2 [DOI] [PubMed] [Google Scholar]
- 16. Abe H., Uchida T., Hara A., Mizukami H., Komiya K., Koike M., Shigihara N., Toyofuku Y., Ogihara T., Uchiyama Y., Yagihashi S., Fujitani Y., and Watada H. (2013) Exendin-4 improves beta-cell function in autophagy-deficient beta-cells. Endocrinology 154, 4512–4524 10.1210/en.2013-1578 [DOI] [PubMed] [Google Scholar]
- 17. Chu K. Y., O'Reilly L., Ramm G., and Biden T. J. (2015) High-fat diet increases autophagic flux in pancreatic beta cells in vivo and ex vivo in mice. Diabetologia 58, 2074–2078 10.1007/s00125-015-3665-x [DOI] [PubMed] [Google Scholar]
- 18. Sheng Q., Xiao X., Prasadan K., Chen C., Ming Y., Fusco J., Gangopadhyay N. N., Ricks D., and Gittes G. K. (2017) Autophagy protects pancreatic beta cell mass and function in the setting of a high-fat and high-glucose diet. Sci. Rep. 7, 16348 10.1038/s41598-017-16485-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cnop M., Igoillo-Esteve M., Cunha D. A., Ladrière L., and Eizirik D. L. (2008) An update on lipotoxic endoplasmic reticulum stress in pancreatic beta-cells. Biochem. Soc. Trans. 36, 909–915 10.1042/BST0360909 [DOI] [PubMed] [Google Scholar]
- 20. Choi S. E., Lee S. M., Lee Y. J., Li L. J., Lee S. J., Lee J. H., Kim Y., Jun H. S., Lee K. W., and Kang Y. (2009) Protective role of autophagy in palmitate-induced INS-1 beta-cell death. Endocrinology 150, 126–134 10.1210/en.2008-0483 [DOI] [PubMed] [Google Scholar]
- 21. Komiya K., Uchida T., Ueno T., Koike M., Abe H., Hirose T., Kawamori R., Uchiyama Y., Kominami E., Fujitani Y., and Watada H. (2010) Free fatty acids stimulate autophagy in pancreatic beta-cells via JNK pathway. Biochem. Biophys. Res. Commun. 401, 561–567 10.1016/j.bbrc.2010.09.101 [DOI] [PubMed] [Google Scholar]
- 22. Las G., Serada S. B., Wikstrom J. D., Twig G., and Shirihai O. S. (2011) Fatty acids suppress autophagic turnover in beta-cells. J. Biol. Chem. 286, 42534–42544 10.1074/jbc.M111.242412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Janikiewicz J., Hanzelka K., Dziewulska A., Kozinski K., Dobrzyn P., Bernas T., and Dobrzyn A. (2015) Inhibition of SCD1 impairs palmitate-derived autophagy at the step of autophagosome-lysosome fusion in pancreatic beta-cells. J. Lipid Res. 56, 1901–1911 10.1194/jlr.M059980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mir S. U., George N. M., Zahoor L., Harms R., Guinn Z., and Sarvetnick N. E. (2015) Inhibition of autophagic turnover in beta-cells by fatty acids and glucose leads to apoptotic cell death. J. Biol. Chem. 290, 6071–6085 10.1074/jbc.M114.605345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zummo F. P., Cullen K. S., Honkanen-Scott M., Shaw J. A. M., Lovat P. E., and Arden C. (2017) Glucagon-like peptide 1 protects pancreatic beta-cells from death by increasing autophagic flux and restoring lysosomal function. Diabetes 66, 1272–1285 10.2337/db16-1009 [DOI] [PubMed] [Google Scholar]
- 26. Biden T. J., Boslem E., Chu K. Y., and Sue N. (2014) Lipotoxic endoplasmic reticulum stress, beta cell failure, and type 2 diabetes mellitus. Trends Endocrinol. Metab. 25, 389–398 10.1016/j.tem.2014.02.003 [DOI] [PubMed] [Google Scholar]
- 27. Tan S. H., Shui G., Zhou J., Li J. J., Bay B. H., Wenk M. R., and Shen H. M. (2012) Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin). J. Biol. Chem. 287, 14364–14376 10.1074/jbc.M111.294157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mei S., Ni H. M., Manley S., Bockus A., Kassel K. M., Luyendyk J. P., Copple B. L., and Ding W. X. (2011) Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J. Pharmacol. Exp. Ther. 339, 487–498 10.1124/jpet.111.184341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Niso-Santano M., Malik S. A., Pietrocola F., Bravo-San Pedro J. M., Marino G., Cianfanelli V., Ben-Younès A., Troncoso R., Markaki M., Sica V., Izzo V., Chaba K., Bauvy C., Dupont N., Kepp O., et al. (2015) Unsaturated fatty acids induce non-canonical autophagy. EMBO J. 34, 1025–1041 10.15252/embj.201489363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng C. I., Lee Y. H., Chen P. H., Lin Y. C., Chou M. H., and Kao Y. H. (2017) Free fatty acids induce autophagy and LOX-1 upregulation in cultured aortic vascular smooth muscle cells. J. Cell. Biochem. 118, 1249–1261 10.1002/jcb.25784 [DOI] [PubMed] [Google Scholar]
- 31. Kimura S., Noda T., and Yoshimori T. (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 10.4161/auto.4451 [DOI] [PubMed] [Google Scholar]
- 32. van Meer G., Voelker D. R., and Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 10.1038/nrm2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Akerfeldt M. C., Howes J., Chan J. Y., Stevens V. A., Boubenna N., McGuire H. M., King C., Biden T. J., and Laybutt D. R. (2008) Cytokine-induced beta-cell death is independent of endoplasmic reticulum stress signaling. Diabetes 57, 3034–3044 10.2337/db07-1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Laybutt D. R., Preston A. M., Akerfeldt M. C., Kench J. G., Busch A. K., Biankin A. V., and Biden T. J. (2007) Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 50, 752–763 10.1007/s00125-006-0590-z [DOI] [PubMed] [Google Scholar]
- 35. Rutter G. A., and Leclerc I. (2009) The AMP-regulated kinase family: enigmatic targets for diabetes therapy. Mol. Cell. Endocrinol. 297, 41–49 10.1016/j.mce.2008.05.020 [DOI] [PubMed] [Google Scholar]
- 36. Fu A., Eberhard C. E., and Screaton R. A. (2013) Role of AMPK in pancreatic beta cell function. Mol. Cell. Endocrinol. 366, 127–134 10.1016/j.mce.2012.06.020 [DOI] [PubMed] [Google Scholar]
- 37. Xie J., and Herbert T. P. (2012) The role of mammalian target of rapamycin (mTOR) in the regulation of pancreatic beta-cell mass: implications in the development of type-2 diabetes. Cell Mol. Life Sci. 69, 1289–1304 10.1007/s00018-011-0874-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yuan T., Lupse B., Maedler K., and Ardestani A. (2018) mTORC2 signaling: a path for pancreatic beta cell's growth and function. J. Mol. Biol. 430, 904–918 10.1016/j.jmb.2018.02.013 [DOI] [PubMed] [Google Scholar]
- 39. Han D., Yang B., Olson L. K., Greenstein A., Baek S. H., Claycombe K. J., Goudreau J. L., Yu S. W., and Kim E. K. (2010) Activation of autophagy through modulation of 5′-AMP-activated protein kinase protects pancreatic beta-cells from high glucose. Biochem. J. 425, 541–551 10.1042/BJ20090429 [DOI] [PubMed] [Google Scholar]
- 40. Bartolome A., Guillen C., and Benito M. (2012) Autophagy plays a protective role in endoplasmic reticulum stress-mediated pancreatic beta cell death. Autophagy 8, 1757–1768 10.4161/auto.21994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Riahi Y., Wikstrom J. D., Bachar-Wikstrom E., Polin N., Zucker H., Lee M. S., Quan W., Haataja L., Liu M., Arvan P., Cerasi E., and Leibowitz G. (2016) Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia 59, 1480–1491 10.1007/s00125-016-3868-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wauson E. M., Dbouk H. A., Ghosh A. B., and Cobb M. H. (2014) G protein-coupled receptors and the regulation of autophagy. Trends Endocrinol. Metab. 25, 274–282 10.1016/j.tem.2014.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eisenberg-Lerner A., and Kimchi A. (2012) PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ. 19, 788–797 10.1038/cdd.2011.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ferdaoussi M., Bergeron V., Zarrouki B., Kolic J., Cantley J., Fielitz J., Olson E. N., Prentki M., Biden T., MacDonald P. E., and Poitout V. (2012) G protein-coupled receptor (GPR)40-dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1. Diabetologia 55, 2682–2692 10.1007/s00125-012-2650-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goginashvili A., Zhang Z., Erbs E., Spiegelhalter C., Kessler P., Mihlan M., Pasquier A., Krupina K., Schieber N., Cinque L., Morvan J., Sumara I., Schwab Y., Settembre C., and Ricci R. (2015) Insulin granules. Insulin secretory granules control autophagy in pancreatic beta cells. Science 347, 878–882 10.1126/science.aaa2628 [DOI] [PubMed] [Google Scholar]
- 46. Christensen A. E., Selheim F., de Rooij J., Dremier S., Schwede F., Dao K. K., Martinez A., Maenhaut C., Bos J. L., Genieser H. G., and Døskeland S. O. (2003) cAMP analog mapping of Epac1 and cAMP kinase: discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J. Biol. Chem. 278, 35394–35402 10.1074/jbc.M302179200 [DOI] [PubMed] [Google Scholar]
- 47. Rehmann H. (2013) Epac-inhibitors: facts and artefacts. Sci. Rep. 3, 3032 10.1038/srep03032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Seino S., Takahashi H., Takahashi T., and Shibasaki T. (2012) Treating diabetes today: a matter of selectivity of sulphonylureas. Diabetes Obes. Metab. 14, 9–13 10.1111/j.1463-1326.2011.01507.x [DOI] [PubMed] [Google Scholar]
- 49. Cnop M., Hannaert J. C., Hoorens A., Eizirik D. L., and Pipeleers D. G. (2001) Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes 50, 1771–1777 10.2337/diabetes.50.8.1771 [DOI] [PubMed] [Google Scholar]
- 50. Busch A. K., Gurisik E., Cordery D. V., Sudlow M., Denyer G. S., Laybutt D. R., Hughes W. E., and Biden T. J. (2005) Increased fatty acid desaturation and enhanced expression of stearoyl coenzyme A desaturase protects pancreatic beta-cells from lipoapoptosis. Diabetes 54, 2917–2924 10.2337/diabetes.54.10.2917 [DOI] [PubMed] [Google Scholar]
- 51. Kroemer G., Mariño G., and Levine B. (2010) Autophagy and the integrated stress response. Mol. Cell 40, 280–293 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kroemer G., and Levine B. (2008) Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 9, 1004–1010 10.1038/nrm2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Prentki M., Vischer S., Glennon M. C., Regazzi R., Deeney J. T., and Corkey B. E. (1992) Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J. Biol. Chem. 267, 5802–5810 [PubMed] [Google Scholar]
- 54. Lai E., Bikopoulos G., Wheeler M. B., Rozakis-Adcock M., and Volchuk A. (2008) Differential activation of ER stress and apoptosis in response to chronically elevated free fatty acids in pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 294, E540–E550 10.1152/ajpendo.00478.2007 [DOI] [PubMed] [Google Scholar]
- 55. Cantley J., Burchfield J. G., Pearson G. L., Schmitz-Peiffer C., Leitges M., and Biden T. J. (2009) Deletion of PKCepsilon selectively enhances the amplifying pathways of glucose-stimulated insulin secretion via increased lipolysis in mouse beta-cells. Diabetes 58, 1826–1834 10.2337/db09-0132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang X., Zhou L., Li G., Luo T., Gu Y., Qian L., Fu X., Li F., Li J., and Luo M. (2007) Palmitate activates AMP-activated protein kinase and regulates insulin secretion from beta cells. Biochem. Biophys. Res. Commun. 352, 463–468 10.1016/j.bbrc.2006.11.032 [DOI] [PubMed] [Google Scholar]
- 57. Shaked M., Ketzinel-Gilad M., Cerasi E., Kaiser N., and Leibowitz G. (2011) AMP-activated protein kinase (AMPK) mediates nutrient regulation of thioredoxin-interacting protein (TXNIP) in pancreatic beta-cells. PLoS ONE 6, e28804 10.1371/journal.pone.0028804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hatanaka M., Maier B., Sims E. K., Templin A. T., Kulkarni R. N., Evans-Molina C., and Mirmira R. G. (2014) Palmitate induces mRNA translation and increases ER protein load in islet beta-cells via activation of the mammalian target of rapamycin pathway. Diabetes 63, 3404–3415 10.2337/db14-0105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Marsh B. J., Soden C., Alarcón C., Wicksteed B. L., Yaekura K., Costin A. J., Morgan G. P., and Rhodes C. J. (2007) Regulated autophagy controls hormone content in secretory-deficient pancreatic endocrine beta-cells. Mol. Endocrinol. 21, 2255–2269 10.1210/me.2007-0077 [DOI] [PubMed] [Google Scholar]
- 60. Laurent A. C., Bisserier M., Lucas A., Tortosa F., Roumieux M., De Régibus A., Swiader A., Sainte-Marie Y., Heymes C., Vindis C., and Lezoualc'h F. (2015) Exchange protein directly activated by cAMP 1 promotes autophagy during cardiomyocyte hypertrophy. Cardiovasc. Res. 105, 55–64 10.1093/cvr/cvu242 [DOI] [PubMed] [Google Scholar]
- 61. Ugland H., Naderi S., Brech A., Collas P., and Blomhoff H. K. (2011) cAMP induces autophagy via a novel pathway involving ERK, cyclin E and Beclin 1. Autophagy 7, 1199–1211 10.4161/auto.7.10.16649 [DOI] [PubMed] [Google Scholar]
- 62. Mestre M. B., and Colombo M. I. (2012) cAMP and EPAC are key players in the regulation of the signal transduction pathway involved in the α-hemolysin autophagic response. PLoS Pathog. 8, e1002664 10.1371/journal.ppat.1002664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen M. L., Yi L., Jin X., Liang X. Y., Zhou Y., Zhang T., Xie Q., Zhou X., Chang H., Fu Y. J., Zhu J. D., Zhang Q. Y., and Mi M. T. (2013) Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy 9, 2033–2045 10.4161/auto.26336 [DOI] [PubMed] [Google Scholar]
- 64. Shahnazari S., Namolovan A., Mogridge J., Kim P. K., and Brumell J. H. (2011) Bacterial toxins can inhibit host cell autophagy through cAMP generation. Autophagy 7, 957–965 10.4161/auto.7.9.16435 [DOI] [PubMed] [Google Scholar]
- 65. Holen I., Gordon P. B., Strømhaug P. E., and Seglen P. O. (1996) Role of cAMP in the regulation of hepatocytic autophagy. Eur. J. Biochem. 236, 163–170 10.1111/j.1432-1033.1996.00163.x [DOI] [PubMed] [Google Scholar]
- 66. Carlessi R., Chen Y., Rowlands J., Cruzat V. F., Keane K. N., Egan L., Mamotte C., Stokes R., Gunton J. E., Bittencourt P. I. H., and Newsholme P. (2017) GLP-1 receptor signalling promotes beta-cell glucose metabolism via mTOR-dependent HIF-1α activation. Sci. Rep. 7, 2661 10.1038/s41598-017-02838-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kondo M., Tanabe K., Amo-Shiinoki K., Hatanaka M., Morii T., Takahashi H., Seino S., Yamada Y., and Tanizawa Y. (2018) Activation of GLP-1 receptor signalling alleviates cellular stresses and improves beta cell function in a mouse model of Wolfram syndrome. Diabetologia 61, 2189–2201 10.1007/s00125-018-4679-y [DOI] [PubMed] [Google Scholar]
- 68. Lim S. W., Jin L., Jin J., and Yang C. W. (2016) Effect of exendin-4 on autophagy clearance in beta cell of rats with tacrolimus-induced diabetes mellitus. Sci. Rep. 6, 29921 10.1038/srep29921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Busch A. K., Cordery D., Denyer G. S., and Biden T. J. (2002) Expression profiling of palmitate- and oleate-regulated genes provides novel insights into the effects of chronic lipid exposure on pancreatic beta-cell function. Diabetes 51, 977–987 10.2337/diabetes.51.4.977 [DOI] [PubMed] [Google Scholar]
- 70. Boslem E., Weir J. M., MacIntosh G., Sue N., Cantley J., Meikle P. J., and Biden T. J. (2013) Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic beta-cells. J. Biol. Chem. 288, 26569–26582 10.1074/jbc.M113.489310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pearson G. L., Mellett N., Chu K. Y., Cantley J., Davenport A., Bourbon P., Cosner C. C., Helquist P., Meikle P. J., and Biden T. J. (2014) Lysosomal acid lipase and lipophagy are constitutive negative regulators of glucose-stimulated insulin secretion from pancreatic beta cells. Diabetologia 57, 129–139 10.1007/s00125-013-3083-x [DOI] [PubMed] [Google Scholar]
- 72. Boslem E., MacIntosh G., Preston A. M., Bartley C., Busch A. K., Fuller M., Laybutt D. R., Meikle P. J., and Biden T. J. (2011) A lipidomic screen of palmitate-treated MIN6 beta-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem. J. 435, 267–276 10.1042/BJ20101867 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.