

Abstract
The pathways for biosynthesis of glycogen in bacteria and starch in plants are evolutionarily and biochemically related. They are regulated primarily by ADP–glucose pyrophosphorylase, which evolved to satisfy metabolic requirements of a particular organism. Despite the importance of these two pathways, little is known about the mechanism that controls pyrophosphorylase activity or the location of its allosteric sites. Here, we report pyruvate-bound crystal structures of ADP–glucose pyrophosphorylase from the bacterium Agrobacterium tumefaciens, identifying a previously elusive activator site for the enzyme. We found that the tetrameric enzyme binds two molecules of pyruvate in a planar conformation. Each binding site is located in a crevice between the C-terminal domains of two subunits where they stack via a distinct β-helix region. Pyruvate interacts with the side chain of Lys-43 and with the peptide backbone of Ser-328 and Gly-329 from both subunits. These structural insights led to the design of two variants with altered regulatory properties. In one variant (K43A), the allosteric effect was absent, whereas in the other (G329D), the introduced Asp mimicked the presence of pyruvate. The latter generated an enzyme that was preactivated and insensitive to further activation by pyruvate. Our study furnishes a deeper understanding of how glycogen biosynthesis is regulated in bacteria and the mechanism by which transgenic plants increased their starch production. These insights will facilitate rational approaches to enzyme engineering for starch production in crops of agricultural interest and will promote further study of allosteric signal transmission and molecular evolution in this important enzyme family.
Keywords: glycogen, allosteric regulation, glucose, enzyme structure, pyruvate, allosterism, glucose-1-phosphate adenylyltransferase, glycogen biosynthesis, pyruvate, starch biosynthesis, enzyme evolution, pyrophosphorylase, polyglucan synthesis
Introduction
ADP–glucose pyrophosphorylase (ATP:α-d-glucose-1-phosphate adenylyltransferase; EC 2.7.7.27; ADP–Glc PPase)5 regulates the production of glycogen in bacteria and starch in plants, which are renewable and biodegradable carbon sources. This enzyme catalyzes ATP-dependent conversion of glucose 1-phosphate (Glc1P), producing pyrophosphate and ADP–glucose, which serves as the glucose moiety donor for polyglucan synthesis (1). This allosterically regulated enzyme serves as a major control point for these biosynthetic pathways, and for this reason it has been an attractive target for protein engineering (2, 3). For instance, the Asp mutant of Gly-336 of the Escherichia coli ADP–Glc PPase exists in a preactivated state (4), and when its gene was transformed into a potato (Solanum tuberosum) plant, the resulting tubers had, on average, up to 35% more starch weight compared with control tubers (3).
All known plant and bacterial ADP–Glc PPases are derived from a common ancestor and are predicted to share considerable structural similarities (1). These features are evident in the structures of the enzymes from potato tuber (5), Agrobacterium tumefaciens (6), and E. coli (7, 8). They are tetramers with ∼50-kDa subunits, each having distinct N- and C-terminal domains. The C-terminal domain is characterized as a left-handed parallel β-helix, whereas the catalytic N-terminal domain is composed of mostly parallel but mixed seven-stranded β-sheets surrounded by α-helices (5–7) (reminiscent of the dinucleotide-binding Rossmann fold).
In most species, ADP–Glc PPase is allosterically regulated by intermediates of the main carbon assimilatory pathway of the organism. The A. tumefaciens ADP–Glc PPase is activated by fructose 6-phosphate (Fru6P) as well as pyruvate, and it is inhibited by ADP and AMP (1). Other enzymes of the family have a regulation that is more promiscuous. In some species, ADP–Glc PPases are activated by up to five activators (9, 10). However, it is not clear how many of those activators bind to the same promiscuous site or to different sites. At least in E. coli, kinetic data suggested that there must be two synergistic activator sites for fructose 1,6-bisphosphate (FBP) and pyruvate (11). Pioneer protein chemistry and site-directed mutagenesis experiments suggested that the regulatory site was near the C terminus in plant and cyanobacterial enzymes (12–14) but near the N terminus in nonphotosynthetic bacteria (15, 16). Despite the fact that the exact location of the regulatory site(s) remained elusive for a long time, more recent studies have suggested that the activator-binding site in this family likely resides between the distinct C- and N-terminal domains (5–7) and that the C-terminal domain likely has more influence on the specificity for pyruvate (11, 17). It remains critical to know exactly where the activators bind and how the allosteric signal is transmitted to further understand how glycogen synthesis in bacteria and starch synthesis in plants are controlled.
Here, we report the pyruvate-bound structure of the A. tumefaciens ADP–Glc PPase, identifying a previously unreported activator site for the enzyme. Based on these structural insights, we designed allosteric variants of the A. tumefaciens ADP–Glc PPase to probe the functionality of this site. One mutant mimics the presence of pyruvate, and the other is insensitive. In addition, we present the structure of the WT enzyme bound to ethyl pyruvate, which is a neutral analog of pyruvate but a potent activator of the A. tumefaciens ADP–Glc PPase. These results furnished a better understanding of how glycogen biosynthesis is regulated in bacteria.
Results
Previously, we showed that the P103A mutant of E. coli ADP–Glc PPase fails to respond to its primary activator, FBP (18). As part of our investigation into the allosteric regulation of the A. tumefaciens ADP–Glc PPase, we mutated the homologous proline to an alanine (P96A). In agreement with the effect in the E. coli enzyme, we found that this mutant had virtually no response to the activator Fru6P. However, it retained sensitivity to its other activator, pyruvate (Table 1). For this reason, P96A is an excellent candidate for the study of specific allosteric effects from pyruvate. Here, we obtained pyruvate-bound crystals of P96A and solved the complex structure.
Table 1.
Activation kinetics for the mutant and wildtype A. tumefaciens ADP–Glc PPases
| Enzymea | Activator parametersb |
||||
|---|---|---|---|---|---|
| A0.5 | nH | V0 | Vm | Activationc (Vm/V0) | |
| mm | units/mg | units/mg | -fold | ||
| Pyruvate | |||||
| WT | 0.092 ± 0.007 | 2.2 ± 0.3 | 9 ± 3 | 83 ± 2 | 9.16 |
| K43A | N/Ad | N/Ad | 29 ± 2 | 29 ± 1 | 0.99 |
| P96A | 0.10 ± 0.03 | 1.0 ± 0.4 | 10 ± 2 | 44 ± 2 | 4.25 |
| G329D | 0.180 ± 0.007 | 1.8 ± 0.1 | 72 ± 1 | 76 ± 1 | 1.06 |
| Fru6P | |||||
| WT | 0.17 ± 0.01 | 2.4 ± 0.1 | 9 ± 1 | 90 ± 1 | 10.3 |
| K43A | 2.4 ± 0.2 | 3.5 ± 1.1 | 30 ± 1 | 41 ± 1 | 1.33 |
| P96A | N/Ad | N/Ad | 12 ± 1 | 12 ± 1 | 1.03 |
| G329D | 0.07 ± 0.03 | 1.2 ± 0.5 | 74 ± 1 | 78 ± 1 | 1.05 |
a Assays were performed as described under “Experimental procedures.”
b Parameters are described under “Experimental procedures” for the modified Hill equation. V0 is the activity in absence of activator, Vm is the activity at saturating concentration of activator, A0.5 is the concentration needed to achieve 50% of the activation effect, and nH is the Hill coefficient.
c Calculation performed before rounding the individual parameters.
d Parameter is not applicable because there was no significant activation.
The A. tumefaciens ADP–Glc PPase is a homotetramer, although for purposes of discussion here, it is convenient to refer to the enzyme as a dimer of dimers with subunits A and B forming one dimer and subunits C and D forming the other (Fig. 1). We observed that each dimer of P96A binds one pyruvate with a total stoichiometry of two pyruvates per enzyme tetramer. Pyruvate binds where the C termini of the two subunits from the dimer stack on top of one another, facing the crevice between the C- and N-terminal domains (Fig. 1).
Figure 1.

Complex structure of the A. tumefaciens ADP–Glc PPase (mutant P96A) bound to pyruvate. The ribbon and surface structure represent a homotetramer bound to pyruvate (PDB code 5W5R). The enzyme can also be referred to as a dimer of dimers with subunits A (gray ribbon) and B (yellow ribbon) forming one dimer and subunits C (pink surface) and D (green surface) forming the other. Each dimer binds one pyruvate molecule, shown as purple spheres, and heteroatom oxygen atoms, shown as red spheres, with a stoichiometry of two pyruvate molecules per tetramer.
All three X-ray diffraction data sets, including those of P96A–pyruvate complex, WT–ethyl pyruvate complex, and WT without ligand, were processed in space group P1 (see Table 2). To ensure a good data-to-parameter ratio during refinements, we made the resolution cutoff according to the criteria I/σ(I) > 1.0 and CC½ > 0.5. However, the resolution where I/σ(I) = 2 is also presented in Table 2 for comparison with a more traditional resolution cutoff standard.
Table 2.
Crystallographic data for ADP–Glc PPase P96A–pyruvate, ADP–Glc PPase WT, and ADP–Glc PPase WT–ethyl pyruvate
| P96A–pyruvate | WT (no ligand) | WT–ethyl pyruvate | |
|---|---|---|---|
| Data processing | |||
| Space group | P1 | P1 | P1 |
| Cell dimension | |||
| α, β, γ (°) | 72.0, 78.1, 90.0 | 72.0, 78.1, 89.9 | 107.8, 101.8, 90.0 |
| a, b, c (Å) | 93.4, 140.9, 228.2 | 93.4, 140.6, 228.4 | 93.7, 140.1, 228.8 |
| Resolution (Å) | 1.75 | 1.78 | 1.76 |
| Resolution at I/σ(I) = 2 | 1.85 | 1.88 | 1.82 |
| Rmergea (%) | 5.5 (41.0)b | 3.5 (51.7) | 3.6 (45.4) |
| I/σ(I) | 11.1 (1.27) | 16.2 (1.16) | 16.6 (1.27) |
| Rpimc (%) | 6.6 (54.4) | 4.9 (72.9) | 4.6 (55.6) |
| CC½d | 0.998 (0.672) | 0.998 (0.561) | 0.998 (0.736) |
| Completeness (%) | 95.4 (95.8) | 97.3 (96.1) | 97.1 (91.5) |
| Multiplicity | 2.0 (2.0) | 2.0 (2.0) | 2.1 (1.9) |
| No. reflections | 2,012,923 | 1,967,391 | 2,210,333 |
| No. unique reflections | 1,029,422 | 1,003,131 | 1,035,315 |
| Refinement | |||
| Rworke/Rfreef (%) | 20.08/22.80 | 20.00/22.89 | 16.27/20.65 |
| No. of atoms | |||
| Protein | 64,554 | 64,677 | 64,457 |
| Ligand | 270 | 210 | 778 |
| Water | 7,716 | 11,197 | 11,257 |
| B factors | |||
| Protein | 27.9 | 33.6 | 26.7 |
| Ligand | 23.0 | N/Ag | 26.4 |
| r.m.s.d.h | |||
| Bond lengths (Å) | 0.004 | 0.007 | 0.007 |
| Bond angles (°) | 0.801 | 0.763 | 1.106 |
| Ramachandran plot (%) | |||
| Most favored | 95.2 | 94.3 | 96.0 |
| Allowed | 4.1 | 5.1 | 3.6 |
| Outliers | 0.7 | 0.6 | 0.4 |
aRmerge = Σ|Iobs − Iavg|/ΣIavg.
b The values for the highest-resolution bin are in parentheses.
c Precision-indicating merging R.
d Pearson correlation coefficient of two “half” data sets.
e Rwork = Σ|Fobs − Fcalc /ΣFobs.
f Five percent of the reflection data were selected at random as a test set, and only these data were used to calculate Rfree.
g Not applicable.
h Root mean square deviation.
The P96A–pyruvate complex structure was refined with final Rwork/Rfree values of 20.08/22.8 at a resolution of 1.75 Å. In one asymmetric unit, there are five tetramers of the enzyme (10 dimers) where the pyruvate resolves in nine of 10 dimers. The only unoccupied pyruvate site is likely a crystallographic artifact. We also obtained the structure of the WT enzyme bound to ethyl pyruvate, which in vitro is another effective activator of this enzyme (A0.5 = 0.175 ± 0.008 mm; n = 2.2 ± 0.2; V0 = 9.0 ± 0.9 units/mg; Vm/V0 = 9.3). The WT–ethyl pyruvate complex was refined with Rwork/Rfree values of 16.27/20.65 at a resolution of 1.76 Å. Electron density indicates that ethyl pyruvate is present in all 10 dimers of the asymmetric unit, occupying the same sites as pyruvate. As a structural reference, we obtained a crystal structure of the WT enzyme with no activator bound. This WT structure without ligand was refined with Rwork/Rfree values of 20.00/22.89 at a resolution of 1.78 Å.
Newly observed structural features of the A. tumefaciens ADP–Glc PPase
The overall structure of our A. tumefaciens ADP–Glc PPase in the absence of ligands (Protein Data Bank (PDB) code 5W6J) is very similar to the previously published structure with no ligands (PDB code 3BRK (6)) with some minor but important differences due to higher resolution. As part of the identified heterogeneity among 20 monomers in one asymmetric unit, we were able to resolve, in some subunits, parts of the loop 96–108. This loop has been implicated in regulation in both E. coli and potato tuber homologues (18, 19). We identified that residues 96 and 97 (N-end of this loop) deviate nearly 180° from the direction previously reported. We also observed a clearly defined side chain of Tyr-107 (Fig. S1) where this span of electron density was previously modeled as part of the backbone (6). The proper modeling of Tyr-107 is important because of its proposed involvement in ATP and ADP–Glc binding (20) and allosteric regulation (18) in its homologue in E. coli (Tyr-114). In addition, a loop spanning residues 225–236 is resolved in our WT enzyme structure with no activator bound (Fig. S2). In this loop, Asp-232 is homologous to Asp-239 in E. coli, which is involved in Glc1P binding (21).
The pyruvate-binding region
Each dimer of A. tumefaciens ADP–Glc PPase binds one pyruvate molecule. The unbiased experimental omit map (Fo − Fc), which was obtained before pyruvate was included in refinements, and the final refined map (2Fo − Fc) for the pyruvate atoms are shown in Fig. 2, C and D, respectively. The binding site was found between the C-terminal domains of subunits A/B (or C/D) where they stack on top of one another via the distinct “β-helix” region (Fig. 1B). Consequently, this site comprises the same set of residues from two different subunits (Fig. 2A). The planar pyruvate molecule involves π–π interactions with the peptide bonds of Ser-328 and Gly-329 from both A and B subunits. The planes formed between pyruvate and the peptide bonds are almost parallel with angles that are ∼17° and 11° for subunits A and B, respectively. The distance of pyruvate to each of these planes (represented by the average measurement of each pyruvate atom to the planes) was 3.1 and 3.2 Å for subunits A and B, respectively (Fig. 2A).
Figure 2.

Overall architecture of the pyruvate site. A, the binding site of pyruvate comprises residues from two subunits (PDB code 5W5R). The atoms and the bonds from each subunit are shown as gray and yellow sticks, respectively. Pyruvate is shown as a purple ball-and-stick model. The heteroatoms oxygen and nitrogen are shown in red and blue, respectively. The hydrogen bonding distances (Å) between the enzyme and pyruvate are shown as black dashed lines. B, structure of the WT enzyme with ethyl pyruvate bound (PDB code 5W5J). Ethyl pyruvate is shown as a pink ball-and-stick model. C, Fo − Fc experimental omit map at 3σ superimposed with an atomic model of pyruvate. D, 2Fo − Fc map at 1.5σ superimposed with an atomic model of pyruvate. E, 2Fo − Fc map at 1.5σ superimposed with an atomic model of ethyl pyruvate. Maps were generated as described under “Experimental procedures.”
The location of the Ser-328/Gly-329 peptide bond relative to pyruvate is enforced by interactions with other nearby residues such as Cys-331, Phe-344, and Gly-346 (Fig. S3). Pyruvate also interacts with Lys-43 from both subunits A and B (Fig. 3A). However, the arrangement of Lys-43 in how it binds pyruvate is not symmetrical. Lys-43 from subunit B forms a hydrogen bond with pyruvate via a bidentate interaction with the carboxylate oxygens (Fig. 3B). In contrast, the amino group of Lys-43 from subunit A forms a hydrogen bond with the ketone oxygen of pyruvate (Fig. 3B). The distance from Thr-306 (subunit A) backbone oxygen to Lys-43 (subunit B) side chain nitrogen was 2.9 Å, and this distance was 3.2 Å for that between Lys-43 (subunit A) and Thr-306 (subunit B). Pro-307 and Pro-308 form a hairpin turn that marks the beginning of the C-terminal domain. The backbone of this pair of residues surrounds the regulatory site, and it was within ∼4 Å of the pyruvate ligand.
Figure 3.

Interactions between the functional groups of pyruvate and the enzyme. A, pyruvate binds at the dimer interface formed by two subunits A and B (orange and green ribbons). Ribbon regions marked as 1 (magenta), 2 (magenta), and 3 (cyan) represent the residues that surround the pyruvate-binding site. The surface feature created by these residues from both subunits is shown in violet. Lys-43 and pyruvate are shown as ball-and-stick models. The heteroatoms oxygen and nitrogen are shown in red and blue, respectively. B, interactions among Lys-43, the oxygens of pyruvate, and the backbone. The residues from two subunits are shown as yellow and orange sticks. Pyruvate is shown as a purple ball-and-stick model. Oxygen and nitrogen heteroatoms are shown in red and blue, respectively. Lys-43 residues from both subunits form hydrogen bonds with pyruvate and the backbone. The distances (Å) of these interactions are represented as black dashed lines. C, interactions between the methyl groups and the enzyme. Symmetrical contacts are displayed between the methyl group from pyruvate and the residues from both subunits of a dimer. The surface feature shown was created by the residues surrounding the methyl group.
The methyl group of pyruvate makes symmetrical close contacts with residues from both subunits (Fig. 3C). They were within 3.3 and 3.6 Å of the backbone oxygens of Val-327 of these two subunits and within 3.8 and 3.9 Å of the methyl groups of the nearby Ala-309 residues. In addition, they were 3.8 and 3.9 Å from the backbone nitrogen of Ala-309. As seen in Fig. 3C, all these contact distances are consistent with the van der Waals radii limits, indicating a tightly fit binding pocket for pyruvate (22). In the WT structure with no pyruvate, the structure of the pocket is unaltered with the exception of Lys-43, which forms hydrogen bonds with ordered waters instead of pyruvate. The overall pyruvate-binding pocket is rigid and maintains the same overall architecture to accommodate the ligand.
WT ADP–Glc PPase with ethyl pyruvate bound
Ethyl pyruvate is also an effective activator of the A. tumefaciens ADP–Glc PPase (Fig. 4). Although it has a slightly higher A0.5 compared with that of pyruvate (0.16 and 0.09 mm, respectively), both activators yielded similar specific activities (∼80 units/mg). The only other analog we tested with similar effects to ethyl pyruvate was methyl pyruvate (Fig. 4). Glyoxylate and alanine neither activated (Fig. 4) nor competed with pyruvate (not shown). In the crystal structure of the ethyl pyruvate–bound WT enzyme, the overall binding pattern of ethyl pyruvate is very similar to that of pyruvate (Figs. 1B and 3A). The only significant difference between the structures is the presence of the ethyl moiety that projects toward a relatively open space found within this region. Nonetheless, the ethyl moiety interacts with the protein as seen in Fig. 2B. The final refined map (2Fo − Fc) for the ethyl pyruvate atoms is shown in Fig. 2E.
Figure 4.

Activation of the A. tumefaciens ADP–Glc PPase by pyruvate derivatives. Saturation curves for pyruvate (Pyr), methyl pyruvate (MePyr), ethyl pyruvate (EtPyr), and glyoxylate were obtained as described under “Experimental procedures.”
Lys-43 is critical for pyruvate allosteric effect
The interaction between Lys-43 and pyruvate led to the hypothesis that Lys-43 is an important residue for pyruvate allosteric activation. In an effort to investigate its importance, we generated the K43A mutant. In the absence of an activator and in standard conditions defined under “Experimental procedures,” K43A had an activity of 29 ± 2 units/mg. Upon the addition of pyruvate (up to 15 mm), the specific activity did not significantly increase (29 ± 1 units/mg). However, K43A retained some sensitivity to the other activator, Fru6P. In the presence of saturating concentrations of Fru6P, the activity increased to 41 ± 1 units/mg (Fig. 5).
Figure 5.

Activation of the WT and K43A mutant of A. tumefaciens ADP Glc PPase. The enzymes were assayed at different concentrations of activators (Fru6P and pyruvate (Pyr)) as described under “Experimental procedures.”
G329D mimics the presence of pyruvate
Based on a model of the mutant G329D of the A. tumefaciens ADP–Glc PPase generated in silico, the carboxylates of the novel Asp residues overlapped with the position of the bound pyruvate ligand in the crystal structure (Fig. 6A). To test the effect of this Asp residue, we produced, expressed, and characterized the G329D mutant. The specific activity of G329D in the absence of pyruvate (∼70 units/mg) was similar to the WT enzyme activated by pyruvate (∼80 units/mg; Table 1). This increased activity agreed with the hypothesis that Asp-329 mimics the presence of pyruvate by inserting carboxylates where the ligand would have its oxygens. Moreover, pyruvate does not seem to have any effect on the preactivated G329D mutant because it does not modify the affinity for substrates (Fig. 6B) or Vmax (70 ± 3 and 66 ± 2 units/mg in the absence and presence of pyruvate, respectively; Fig. S4). This indicates that its binding is not needed for activation and/or that it is blocked by the presence of Asp-329. Interestingly, Fru6P still seems to induce a further increase in apparent ATP affinity to the G329D mutant as it also does to the WT (Fig. 6B).
Figure 6.

Effect of mutation G329D. A, position of carboxylates from Asp-329 in mutant G329D. An overlay of the pyruvate-bound structure (PDB code 5W5R) and a model of G329D is shown. The model was obtained as described under “Experimental procedures.” B, effect of activators on the apparent affinity of substrate ATP for the G329D mutant and WT A. tumefaciens ADP–Glc PPase. Saturation curves for ATP were obtained, and the S0.5 parameters were calculated as described under “Experimental procedures.” Error bars represent S.E. The assays contained 1.5 mm Fru6P or 1.5 mm pyruvate (Pyr) when indicated.
Thermal shift assays of mutants K43A and G329D
To test whether pyruvate binding was retained in mutants K43A and G329D, we assayed the protein stability in the absence and presence of pyruvate by a differential scanning fluorimetry technique as described under “Experimental procedures.” In the WT enzyme, pyruvate increased Tm significantly. The values were 53.9 ± 0.2 and 55.8 ± 0.7 °C in the absence and presence of pyruvate, respectively (Fig. S5). However, that shift was not observed in the mutants K43A and G329D (Fig. S5). Interestingly, the mutation K43A seemed to destabilize the enzyme because the Tm was lower than the WT with two peaks at 36.0 ± 0.3 and 46.7 ± 1.0 °C. In the presence of pyruvate, the curve was nearly identical with peaks at 36.5 ± 0.2 and 46.0 ± 0.4 °C. This indicates that there is no evidence that pyruvate binds and stabilizes the mutant K43A as the WT does. The mutation G329D stabilizes the mutant significantly, but pyruvate does not increase its stability further (Fig. S5). In the absence and presence of pyruvate, the Tm values were 63.4 ± 0.4 and 62.8 ± 0.3 °C, respectively. There is also no evidence that pyruvate binds to G329D as it does to the WT.
Discussion
ADP–Glc PPase plays a vital role in the production of the most abundant reserve polyglucans in the world, starch and glycogen (1, 2). For this reason, it is important to understand how this enzyme is regulated. Despite seeing drastic changes in substrate affinities and specific activities upon the addition of allosteric activators, little is known about how the activators work and where they bind. Here, we present a pyruvate-bound ADP–Glc PPase structure that sheds light on the architecture of the activator-binding site. Previous research into ADP–Glc PPases has suggested that the activator-binding residues lie somewhere between the distinct C-terminal and N-terminal domains (5, 6, 8, 17). In our pyruvate-bound structure, we observed that most of the interactions between pyruvate and the protein involve the C-terminal domain with the exception of the N-terminal Lys-43 (Fig. 2A). This is in agreement with previous results observed with chimeric constructs of the E. coli and A. tumefaciens enzymes. When their N- and C-domains were swapped, the C-domain determined the specificity for pyruvate (17). Lys-43 is present in both enzymes from A. tumefaciens and E. coli (Lys-50). That explains the previous results where swapping the N-domain between those enzymes did not have a significant impact (17).
We found that pyruvate binds in the dimer interface between the C-terminal domains of each subunit (A/B or C/D) (Fig. 1). This binding site is surrounded by like-numbered residues from both subunits (Fig. 2A). The amino groups of both Lys-43 from subunits A and B (or C and D) form hydrogen bonds with the pyruvate oxygens. In agreement, replacement of Lys-43 disrupted the pyruvate allosteric activation (Fig. 5). The planar pyruvate molecule also appears to form π–π interactions as it stacks between the peptide bonds of Ser-328 and Gly-329 of both subunits (Fig. 2A). Moreover, the dipole moments of the separate Ser-328/Gly-329 backbones (from subunits A and B) align well with those present within the pyruvate ligand (Fig. 7). The local geometry of the binding site is likely enforced by other hydrogen bonds in this region, which include backbone interactions of Gly-329 with Gly-346 and the thiol group of Cys-331 (both part of the C-terminal left-handed parallel β-helix). This thiol group is also in hydrogen-bonding range with other residues, including the backbone oxygen of Ser-328 as well as Phe-344 and Val-347 (Fig. S3). This Cys is highly conserved with Ser being another residue present in most of the other ADP–Glc PPases, which underscores the importance of this residue for maintaining the local architecture.
Figure 7.
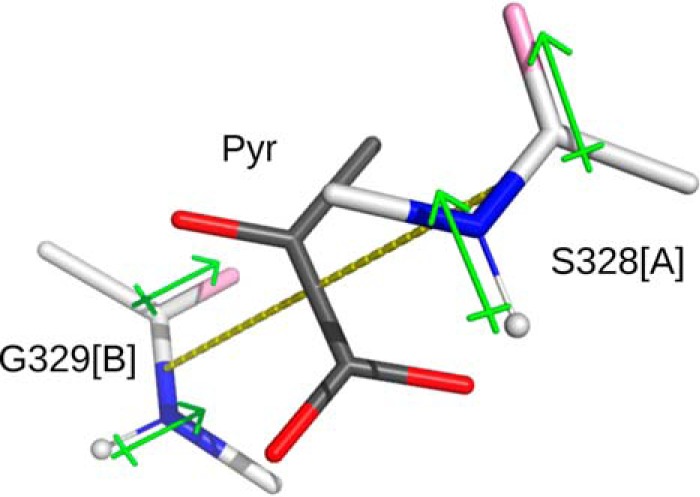
Dipole stacking with pyruvate ligand. Dipoles of residues Ser-328 (subunit A) and Gly-329 (subunit B) surround the ligand pyruvate (Pyr).
It is not clear how many enzymes from the ADP–Glc PPase family are activated by pyruvate, which is critical evolutionary information and therefore deserving of further investigation. We recently found that pyruvate plays a relevant role in the regulation of the enzyme from E. coli (11, 23). This was previously overlooked because its synergistic effect was only detected in the presence of the main activator, FBP (11, 23). Possibly other enzymes in this family follow a similar trend, and the structural information provided in this work will serve for better predictions. We identified a conserved motif within the binding region among all enzymes significantly activated by pyruvate (Fig. 8) (1, 11). One exception is Rhodospirillum rubrum, which has an Ala instead of Gly in the homologous position 329. Likely, this extra methyl group does not cause a different spatial arrangement of the backbone in this area.
Figure 8.

Alignment of ADP–Glc PPase protein sequences from different species. All enzymes included are those activated by pyruvate in the literature. Sections 1, 2, and 3 are regions that contact pyruvate. Position 309 makes a direct contact with the methyl from pyruvate, and positions 43 and 329 are the positions on which we performed site-directed mutagenesis. Sequences were collected and aligned as described under “Experimental procedures.”
Pyruvate binds between two subunits of a dimer at a location where like-numbered residues appear to line up. Hence, it would be logical to assume that the residues from each subunit interact symmetrically with the ligand. However, this interaction is not symmetrical because pyruvate itself is not. Lys-43 from subunit A interacts with pyruvate carboxylate oxygens, whereas Lys-43 from subunit B interacts with the pyruvate ketone oxygen. In contrast, a type of pseudosymmetry does exist. If an axis is drawn through the pyruvate molecule from its methyl carbon to its carboxyl carbon and pyruvate is rotated 180° about this axis, the interaction pattern will be reversed. In that case, Lys from subunit B will interact with its carboxylate oxygens, and that of the A subunit will interact with the ketone oxygen. In other words, pyruvate could bind in these two orientations because the electronic densities of the original and flipped forms are similar, and the Lys-43 conformations adjust for the slight asymmetry. Here, we show that an allosteric site may only need to satisfy a certain shape and electronic distribution to bind the ligand. Interestingly, we observed that once pyruvate binds in one direction, the other pyruvate in the other interface binds in the opposite direction. This strongly suggests that a communication between both activator sites exists.
The beginning of the C-terminal domain is marked by a hairpin turn formed by Pro-307 and Pro-308. Considering the location, their distinct structural properties, and that these prolines are highly conserved throughout the ADP–Glc PPase family, they are probably important to maintain the architecture of the pyruvate-binding site. The distances between Ala-309 of subunits A and B and the methyl carbon of pyruvate are 3.8 and 3.8 Å, respectively (Fig. 3C). Both distances indicate van der Waals contacts between methyl groups. These interactions could be important for forming a hydrophobic pocket with a proper shape to accommodate the methyl group of pyruvate. In addition, the side chain of Ala-309 and the propyl group of Val-327 are within 3.8 Å (for either subunit A or B). These, in turn, form close contacts with several other hydrophobic residues that line the inside of the parallel β-helix. Although these interactions do not directly involve the methyl group of pyruvate, they do create a hydrophobic environment to accommodate it. The notion that the methyl group helps to anchor the ligand is supported by kinetic assays in the presence of glyoxylate. Despite having an overall structure similar to pyruvate (only lacking the methyl group), glyoxylate does not induce any noticeable increase in activity (Fig. 4) or substrate apparent affinity for the A. tumefaciens ADP–Glc PPase (data not shown). In addition, glyoxylate (up to 20 mm) does not inhibit the enzyme in either the presence or absence of 0.2 mm pyruvate (not shown). This lack of competition with pyruvate suggests that glyoxylate does not bind to the pyruvate site reported here; otherwise, it should be able to displace it. The differential effect between pyruvate and glyoxylate might be explained by a hydrophobic effect. A comparison between the structures with and without pyruvate shows that the methyl group of pyruvate displaces a water molecule that is surrounded by a hydrophobic environment (Fig. S6). Glyoxylate, which lacks that extra methyl group, may not be able to do it.
Binding of a neutral activator
We obtained a structure of the A. tumefaciens ADP–Glc PPase bound to ethyl pyruvate, which is also a strong activator in vitro (Fig. 4). Pyruvate binds tightly to the enzyme, and it seems that any extra functional group in the molecule may lead to a dramatic steric hindrance. However, ethyl pyruvate can bind because the ethyl group projects toward a relatively open space between the two subunits (A/B or C/D). The ethyl group comes within 4 Å of the ϵ-carbons of Lys-43 of both subunits. However, most of the atoms near the ethyl group were from subunit A, but they were ∼4 Å apart or greater. Otherwise, the interactions between protein and the pyruvate portion of ethyl pyruvate are entirely consistent with those observed in the pyruvate-bound enzyme (Fig. 2, A and B).
In the complex structure with ethyl pyruvate, Lys-43 from both subunits occupies a similar position to the position we observed in the pyruvate-bound complex. In both cases, the amino groups of Lys interact with the oxygens of ethyl pyruvate. An important conclusion from this structure and its comparison with the pyruvate-bound structure is that two hydrogen bonds between the oxygens of pyruvate and residues Gly-329 and Lys-43 seem to be crucial for stabilizing the interaction with the ligand (Fig. 2, A and B). Whether the oxygens are in a carboxylate (negative at neutral pH) or ester (neutral) form does not seem to be important for binding and activation.
Allosteric effect of pyruvate
The involvement of Lys-43 in the allosteric response suggests that neighboring residues could also be important. Residues that interact with Lys-43 may be involved in the transmission of the allosteric signal from pyruvate to the catalytic site. Lys-43 sits just downstream from a “(K/R)RAKPAV” sequence formed by residues 32–38 that has been associated with regulation and catalysis of ADP–Glc PPases (1, 2, 15). The whole loop from residues 20–46 may be important not only for allosteric effects but also for catalysis (Fig. S7). For instance, Arg-33 and Arg-45 are involved in Fru6P activation, whereas Arg-25 is important in catalysis (15). In addition, Lys-35 is a homologue of Lys-43 in the S subunit from potato tuber, which was characterized to be critical for activity (24). Fig. S7 illustrates how the loop from residues 20 to 46 connects the pyruvate site with the catalytic site, depicted by Arg-25, Lys-35, Asp-135, and Asp-269. The latter Asp residues are homologous to Asp-142 and Asp-276 in E. coli, which were shown to be critical for catalysis (21, 25). The previously published structures of ADP–Glc PPases from A. tumefaciens and potato tuber revealed sulfate molecules bound between the C-terminal and N-terminal domains. It had previously been suggested that these sulfates demark the area of the activator site (5, 6). Arg-45 interacts with this sulfate that lies between the N-terminal and C-terminal domains (Fig. S8). When Arg-45 was mutated to alanine, it yielded an enzyme insensitive to Fru6P with only a partial activation by pyruvate (15). In this work, when Lys-43 was mutated to alanine, it yielded an enzyme insensitive to pyruvate with only a partial activation by Fru6P. These two residues are very close in sequence, which highlights the fact that interaction of ligands to this region is responsible for triggering allosteric effects. In the case of the A. tumefaciens enzyme, we can conclude that Lys-43 is responsible for activation by pyruvate and Arg-45 is responsible for activation by Fru6P, but mutation of either of those residues also affects the other.
Our crystal structure with the activators bound (pyruvate and ethyl pyruvate) do not show a striking conformational difference from the structure without activators bound. There are two possible explanations. 1) Pyruvate exerts its effect by minor changes on the dynamics of the loop 20–46, or 2) the sulfate (from crystallization solution) bound to the structure near Arg-45 keeps the enzyme in a nonactivated form. Both an activator (pyruvate) and inhibitor (sulfate) are present in the structure (Fig. S8). Previously, it was found that the activator Fru6P competes with sulfate (6). Our results show that pyruvate behaves differently. At higher concentrations of sulfate, pyruvate does not activate at the same levels of Vmax (Fig. S9). In other words, pyruvate cannot displace sulfate at higher concentrations, which indicates the existence of a noncompetitive effect. In fact, we see that in the crystal structure both ligands can coexist together. For that reason, it is possible that the structure obtained is not fully activated and represents a T form that has both the activator and the inhibitor bound according to the Monod–Wyman–Changeux model of allosterism (26).
G329D is a preactivated enzyme
In the E. coli ADP–Glc PPase, it was previously found that mutating Gly-336 (homologous to the A. tumefaciens Gly-329) to Asp yielded a preactivated enzyme (4). Moreover, when this gene was transfected into potato plants, it was found that the tubers had, on average, over 30% higher starch content compared with lines that received the E. coli WT ADP–Glc PPase gene (3). At the time of these studies, it was not known why this enzyme had such a unique kinetic profile. When we generated the A. tumefaciens G329D mutant in silico, we observed that the Asp side chains mimic the presence of pyruvate (Fig. 6A). For this reason, the G329D mutant was found to be in a preactivated state (Fig. 6B). In the absence of any allosteric activator, this mutant has a kinetic profile similar to the activated WT enzyme (Table 1 and Fig. 6B). Our recently published work showed that pyruvate is also an activator of the E. coli enzyme (11). A combination of these results and the structure presented here explains why the mutant G336D in the E. coli enzyme is preactivated (4). In addition, it explains why the transgenic plants bearing this gene have an increased production of starch (3). Now that we understand the mechanism by which these genes are highly active, a door is being opened to a more rational approach to enzyme engineering. Increasing starch production in crops of agricultural interest is of great biotechnological importance.
Conclusion
We have established the location of the pyruvate-binding site. It concurs with previous research indicating that the site resides between the C-terminal and N-terminal domains of the enzyme (5, 6, 17). Specifically, our work confirms that the binding domain of the A. tumefaciens allosteric activator pyruvate resides inside this cleft at a point where the two C-terminal domains from subunits A and B stack on top of each other (Fig. 1). The kinetic results of the K43A mutant support crystallographic data in pinpointing the location of the pyruvate-binding site as do the results of the G329D mutant.
Experimental procedures
Chemicals and supplies
All biochemical reagents were purchased from Sigma-Aldrich. Luria broth (LB) medium was purchased from USA Scientific (Ocala, FL). QuikChange Lightning Multi Site-Directed Mutagenesis kits were acquired from Agilent Technologies (Santa Clara, CA). Primers for the production of mutants were acquired from Integrated DNA Technologies (Coralville, IA). The Malachite Green–ammonium molybdate–Tween 20 solution used in the enzyme assays was prepared as described previously (27).
Site-directed mutagenesis and protein expression
The plasmid used to express the A. tumefaciens ADP–Glc PPase gene (pBLH1) was constructed by subcloning the coding region of the enzyme from pETAT (17) using the restriction sites NdeI and SacI to create the N-terminal hexahistidine-tagged protein (Fig. S10). Site-directed mutagenesis was performed on pBLH1 to obtain the K43A, P96A, and G329D mutants (primers used were 5′-CGCGGTTTATTTTGGCGGCGCGGCGCGC-3′, 5′-CTTCGACATTCTGGCGGCTTCGCAGC-3′, and 5′-CGTCGGTCGTCTCGGATGACTGCATCATTTC-3′, respectively). All genetic sequencing was performed by the University of Chicago Comprehensive Cancer Center DNA Sequencing and Genotyping Facility in Chicago, IL. Following protein expression, enzyme purification was performed using a nickel-Sepharose 6 Fast Flow HisTrap FF column (GE Healthcare). Samples were subsequently concentrated to 10 mg/ml in a solution of 50 mm Hepes, pH 8.0, with 10% glycerol with an Amicon Ultra-15 30-kDa centrifugal filter (EMD Millipore Inc.). These served as the concentrated stock enzyme solutions used for protein assays. For crystallographic studies, proteins were subsequently subjected to size-exclusion chromatography using a GE Healthcare HiLoad 16/600 Superdex 200 prep grade column, desalted, and prepared to 10 mg/ml in 50 mm Hepes, pH 7.5. Final enzyme purity was estimated at 95% or greater as determined by apparent band homogeneity (Fig. S11) in SDS-PAGE (28). For protein crystallization, the conditions were 50 mm Hepes with 1.5 m lithium sulfate at pH 7.5 with or without 10 mm pyruvate or ethyl pyruvate.
Enzyme assays
Activity of ADP–Glc PPase was assayed in the direction of ADP–glucose synthesis using the assay developed by Fusari et al. (27) as described previously with minor modifications (18). The unit of enzyme activity is defined as 1.0 μmol of ADP–glucose formed/min. Unless otherwise stated, assay conditions were as follows. In addition to the 10 μl of ADP–Glc PPase enzyme aliquot, the reaction mixtures contained 50 mm Hepes, pH 8.0, 10.0 mm MgCl2, 1.0 mm Glc1P, 1.5 mm ATP, 1.5 units/ml inorganic pyrophosphatase, and 0.2 mg/ml BSA (total volume of 60 μl). Data were plotted as specific enzyme activity (units/mg) versus substrate or effector concentration and fit to a modified Hill equation (29): V = V0 + (Vm − V0)·An/(A0.5n + An). In this equation, V0 is the velocity in the absence of the activator being analyzed, Vm is the velocity at saturating concentrations of the activator, A is the concentration of activator under study, A0.5 is the concentration of activator needed to observe half of the change in velocity (Vm − V0), and n is the Hill coefficient. For substrate saturation curves, the Hill equation used was V = Vm·Sn/(S0.5n + Sn) where S is the concentration of substrate and S0.5 is the concentration of S needed to reach half of the Vmax. Fitting was performed with the Levenberg–Marquardt nonlinear least-squares algorithm provided by the computer program OriginTM 8.0. The errors, rounded to one significant figure, were obtained with that algorithm. All saturation curves were performed at least twice with kinetic parameters reproducible within ±10%.
Sequence alignment of ADP–Glc PPases and structural alignment of NDP–Glc PPases
The sequences of known plant and bacterial ADP–Glc PPases were from the NCBI website. Sequences from A. tumefaciens, Nitrosomonas europaea ATCC 19718, Rhodobacter sphaeroides, Rhodomicrobium vannielii ATCC 17100, E. coli K12, Rhodobacter capsulatus, and Rhodospirillum rubrum were from accession numbers AAD03473.1, CAD85941.1, AAD53958.1, ADP70402.1, NP_417888.1, KQB17540, and AAC71050.2, respectively. The software used for the alignments was Unipro UGENE, and the MUSCLE algorithm was used for the fitting (30).
Data collection and processing
Monochromatic data sets were collected at the 19-ID beamline-Structural Biology Center (SBC), Advanced Photon Source (APS) at Argonne National Laboratory (ANL). Diffraction data were collected at a wavelength of 0.98 Å at 100 K using a Quantum 315r charge-coupled device (CCD) detector from Area Detector Systems Corp. (ADSC). All collected data sets were indexed, integrated, and scaled using HKL3000 (31) or the CCP4 program suite (32). The best data sets were processed in space group P1 at resolutions of 1.78, 1.75, and 1.76Å for the WT, P96A–pyruvate, and WT–ethyl pyruvate structures, respectively. Data collection statistics are in Table 2.
Structure determination, model building, and refinement
To identify the structural heterogeneity in each subunit, data processing, structure determination, model building, and refinement were all carried out in P1 space group as previously mentioned. All structures were solved by molecular replacement using PHASER in the Phenix software suit (33) The initial search model was a single subunit of a previously published structure of ADP–Glc PPase from A. tumefaciens (PDB code 3BRK). The solutions of molecular replacement were then refined in Phenix through rigid-body, isotropic, and anisotropic refinements. A twin law (h, −k, −h−l) was generated using Xtriage in Phenix and added during refinement. After each round of refinement, all models were rebuilt in Coot (34) and refined using Phenix (33), and the final models were analyzed and figures were made using the programs UCSF Chimera (35) and PyMOL (36). Final refinement statistics are presented in Table 2. The experimental omit map (Fo − Fc) was generated after molecular replacement and rigid-body and restrained refinements. No water, ligand, or twin law was added during these refinements. Hence, there is no bias toward the existence of the ligand. A model of the tetramer with the G329D mutation was built with the program Modeller 9.11 (37). As a template, we used the atomic coordinates of the P96A mutant of the ADP–Glc PPase structure with the pyruvate bound.
Thermal shift assay
Thermal shift assays were performed as described (38) using the Step One Real-Time PCR SystemTM (Thermo Fisher Scientific) and Step OneTM software. The final volume for the assay was 20 μl and contained 20 mm Hepes, pH 7.5, 4× SYPRO Orange Dye (Sigma-Aldrich), 0.02 mm purified protein, 2 mm ATP, and 2 mm MgCl2. The assays were run in triplicates, and where indicated pyruvate (1 mm) was added to the mixture. A control (blank) with no protein was also performed for all the samples. A continuous temperature scanning from 25.0 to 99.0 °C was performed with a ramp increment of 0.37 °C min−1.
Author contributions
B. L. H., R. M., A. A. I., D. L., and M. A. B. conceptualization; B. L. H., R. M., H. P. P., M. D. A. D., and R. W. data curation; B. L. H., D. L., and M. A. B. formal analysis; B. L. H., D. L., and M. A. B. validation; B. L. H., D. L., and M. A. B. visualization; B. L. H., R. M., H. P. P., M. D. A. D., R. W., A. A. I., D. L., and M. A. B. methodology; B. L. H., R. M., D. L., and M. A. B. writing-original draft; R. M., A. A. I., D. L., and M. A. B. writing-review and editing; A. A. I. and M. A. B. investigation; M. A. B. supervision; M. A. B. funding acquisition; M. A. B. project administration.
Supplementary Material
Acknowledgments
For some of this work, we thank Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by the University of Chicago Argonne, LLC, for the United States Department of Energy, Office of Biological and Environmental Research under Contract DE-AC02-06CH11357. We thank John C. Hawkins (Writing Center, Loyola University Chicago) for assistance in proofreading the manuscript.
This work was supported by National Science Foundation Grant MCB 1616851 (to M. A. B.), Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) Grants PICTs-2014-3362 and -2015-0634 (to M. D. A. D.) and PICTs-2014-3256 and -2015-1767 (to A. A. I.), UNL CAID'11, and PIO-CONICET-YPF. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S11.
The atomic coordinates and structure factors (codes 5W5R, 5W5T, and 5W6J) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ADP–Glc PPase
- ADP–glucose pyrophosphorylase
- Glc1P
- glucose 1-phosphate
- Fru6P
- fructose 6-phosphate
- FBP
- fructose 1,6-bisphosphate.
References
- 1. Ballicora M. A., Iglesias A. A., and Preiss J. (2003) ADP-glucose pyrophosphorylase, a regulatory enzyme for bacterial glycogen synthesis. Microbiol. Mol. Biol. Rev. 67, 213–225 10.1128/MMBR.67.2.213-225.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ballicora M. A., Iglesias A. A., and Preiss J. (2004) ADP-glucose pyrophosphorylase: a regulatory enzyme for plant starch synthesis. Photosynth. Res. 79, 1–24 10.1023/B:PRES.0000011916.67519.58 [DOI] [PubMed] [Google Scholar]
- 3. Stark D. M., Timmerman K. P., Barry G. F., Preiss J., and Kishore G. M. (1992) Regulation of the amount of starch in plant tissues by ADP glucose pyrophosphorylase. Science 258, 287–292 10.1126/science.258.5080.287 [DOI] [PubMed] [Google Scholar]
- 4. Meyer C. R., Bork J. A., Nadler S., Yirsa J., and Preiss J. (1998) Site-directed mutagenesis of a regulatory site of Escherichia coli ADP-glucose pyrophosphorylase: the role of residue 336 in allosteric behavior. Arch. Biochem. Biophys. 353, 152–159 10.1006/abbi.1998.0648 [DOI] [PubMed] [Google Scholar]
- 5. Jin X., Ballicora M. A., Preiss J., and Geiger J. H. (2005) Crystal structure of potato tuber ADP-glucose pyrophosphorylase. EMBO J. 24, 694–704 10.1038/sj.emboj.7600551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cupp-Vickery J. R., Igarashi R. Y., Perez M., Poland M., and Meyer C. R. (2008) Structural analysis of ADP-glucose pyrophosphorylase from the bacterium Agrobacterium tumefaciens. Biochemistry 47, 4439–4451 10.1021/bi701933q [DOI] [PubMed] [Google Scholar]
- 7. Cifuente J. O., Comino N., Madariaga-Marcos J., López-Fernández S., García-Alija M., Agirre J., Albesa-Jové D., and Guerin M. E. (2016) Structural basis of glycogen biosynthesis regulation in bacteria. Structure 24, 1613–1622 10.1016/j.str.2016.06.023 [DOI] [PubMed] [Google Scholar]
- 8. Comino N., Cifuente J. O., Marina A., Orrantia A., Eguskiza A., and Guerin M. E. (2017) Mechanistic insights into the allosteric regulation of bacterial ADP-glucose pyrophosphorylases. J. Biol. Chem. 292, 6255–6268 10.1074/jbc.M116.773408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cereijo A. E., Asencion Diez M. D., Dávila Costa J. S., Alvarez H. M., and Iglesias A. A. (2016) On the kinetic and allosteric regulatory properties of the ADP-glucose pyrophosphorylase from Rhodococcus jostii: an approach to evaluate glycogen metabolism in oleaginous bacteria. Front. Microbiol. 7, 830 10.3389/fmicb.2016.00830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Asención Diez M. D., Peirú S., Demonte A. M., Gramajo H., and Iglesias A. A. (2012) Characterization of recombinant UDP- and ADP-glucose pyrophosphorylases and glycogen synthase to elucidate glucose-1-phosphate partitioning into oligo- and polysaccharides in Streptomyces coelicolor. J. Bacteriol. 194, 1485–1493 10.1128/JB.06377-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Asención Diez M. D., Aleanzi M. C., Iglesias A. A., and Ballicora M. A. (2014) A novel dual allosteric activation mechanism of Escherichia coli ADP-glucose pyrophosphorylase: the role of pyruvate. PLoS One 9, e103888 10.1371/journal.pone.0103888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ballicora M. A., Fu Y., Nesbitt N. M., and Preiss J. (1998) ADP-glucose pyrophosphorylase from potato tubers. Site-directed mutagenesis studies of the regulatory sites. Plant Physiol. 118, 265–274 10.1104/pp.118.1.265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sheng J., Charng Y. Y., and Preiss J. (1996) Site-directed mutagenesis of lysine382, the activator-binding site, of ADP-glucose pyrophosphorylase from Anabaena PCC 7120. Biochemistry 35, 3115–3121 10.1021/bi952359j [DOI] [PubMed] [Google Scholar]
- 14. Ball K., and Preiss J. (1994) Allosteric sites of the large subunit of the spinach leaf ADPglucose pyrophosphorylase. J. Biol. Chem. 269, 24706–24711 [PubMed] [Google Scholar]
- 15. Gómez-Casati D. F., Igarashi R. Y., Berger C. N., Brandt M. E., Iglesias A. A., and Meyer C. R. (2001) Identification of functionally important amino-terminal arginines of Agrobacterium tumefaciens ADP-glucose pyrophosphorylase by alanine scanning mutagenesis. Biochemistry 40, 10169–10178 10.1021/bi002615e [DOI] [PubMed] [Google Scholar]
- 16. Gardiol A., and Preiss J. (1990) Escherichia coli E-39 ADPglucose synthetase has different activation kinetics from the wild-type allosteric enzyme. Arch. Biochem. Biophys. 280, 175–180 10.1016/0003-9861(90)90533-5 [DOI] [PubMed] [Google Scholar]
- 17. Ballicora M. A., Sesma J. I., Iglesias A. A., and Preiss J. (2002) Characterization of chimeric ADPglucose pyrophosphorylases of Escherichia coli and Agrobacterium tumefaciens. Importance of the C-terminus on the selectivity for allosteric regulators. Biochemistry 41, 9431–9437 10.1021/bi025793b [DOI] [PubMed] [Google Scholar]
- 18. Hill B. L., Wong J., May B. M., Huerta F. B., Manley T. E., Sullivan P. R., Olsen K. W., and Ballicora M. A. (2015) Conserved residues of the Pro103-Arg115 loop are involved in triggering the allosteric response of the Escherichia coli ADP-glucose pyrophosphorylase. Protein Sci. 24, 714–728 10.1002/pro.2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Figueroa C. M., Kuhn M. L., Falaschetti C. A., Solamen L., Olsen K. W., Ballicora M. A., and Iglesias A. A. (2013) Unraveling the activation mechanism of the potato tuber ADP-glucose pyrophosphorylase. PLoS One 8, e66824 10.1371/journal.pone.0066824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee Y. M., and Preiss J. (1986) Covalent modification of substrate-binding sites of Escherichia coli ADP-glucose synthetase. Isolation and structural characterization of 8-azido-ADP-glucose-incorporated peptides. J. Biol. Chem. 261, 1058–1064 [PubMed] [Google Scholar]
- 21. Bejar C. M., Jin X., Ballicora M. A., and Preiss J. (2006) Molecular architecture of the glucose 1-phosphate site in ADP-glucose pyrophosphorylases. J. Biol. Chem. 281, 40473–40484 10.1074/jbc.M607088200 [DOI] [PubMed] [Google Scholar]
- 22. Batsanov S. (2001) van der Waals radii of elements. Inorg. Mater. 37, 871–885 10.1023/A:1011625728803 [DOI] [Google Scholar]
- 23. Ebrecht A. C., Solamen L., Hill B. L., Iglesias A. A., Olsen K. W., and Ballicora M. A. (2017) Allosteric control of substrate specificity of the Escherichia coli ADP-glucose pyrophosphorylase. Front. Chem. 5, 41 10.3389/fchem.2017.00041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ballicora M. A., Dubay J. R., Devillers C. H., and Preiss J. (2005) Resurrecting the ancestral enzymatic role of a modulatory subunit. J. Biol. Chem. 280, 10189–10195 10.1074/jbc.M413540200 [DOI] [PubMed] [Google Scholar]
- 25. Frueauf J. B., Ballicora M. A., and Preiss J. (2001) Aspartate residue 142 is important for catalysis by ADP-glucose pyrophosphorylase from Escherichia coli. J. Biol. Chem. 276, 46319–46325 10.1074/jbc.M107408200 [DOI] [PubMed] [Google Scholar]
- 26. Monod J., Wyman J., and Changeux J. P. (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118 10.1016/S0022-2836(65)80285-6 [DOI] [PubMed] [Google Scholar]
- 27. Fusari C., Demonte A. M., Figueroa C. M., Aleanzi M., and Iglesias A. A. (2006) A colorimetric method for the assay of ADP-glucose pyrophosphorylase. Anal. Biochem. 352, 145–147 10.1016/j.ab.2006.01.024 [DOI] [PubMed] [Google Scholar]
- 28. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 29. Figueroa C. M., Esper M. C., Bertolo A., Demonte A. M., Aleanzi M., Iglesias A. A., and Ballicora M. A. (2011) Understanding the allosteric trigger for the fructose-1,6-bisphosphate regulation of the ADP-glucose pyrophosphorylase from Escherichia coli. Biochimie 93, 1816–1823 10.1016/j.biochi.2011.06.029 [DOI] [PubMed] [Google Scholar]
- 30. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
- 32. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., and Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 10.1107/S0907444902016657 [DOI] [PubMed] [Google Scholar]
- 34. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 35. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 36. DeLano W. L. (2015) The PyMOL Molecular Graphics System, Version 1.8, Schrödinger, LLC, New York [Google Scholar]
- 37. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- 38. Niesen F. H., Berglund H., and Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 10.1038/nprot.2007.321 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.